

Abstract
Tumors thrive in an immunosuppressive microenvironment that impedes antitumor innate and adaptive immune responses. Thus, approaches that can overcome immunosuppression and engage antitumor immunity are needed. This study defines the adjuvant and cancer immunotherapy potential of the recombinant polio:rhinovirus chimera, PVSRIPO. PVSRIPO is currently in clinical trials against recurrent WHO grade IV malignant glioma, a notoriously treatment-refractory cancer. Cytopathogenic infection of neoplastic cells releases the proteome and exposes pathogen- and damage-associated molecular patterns. At the same time, sublethal infection of antigen-presenting cells, such as dendritic cells and macrophages, yields potent, sustained type I interferon-dominant activation in an immunosuppressed microenvironment and promotes the development of tumor antigen-specific T cell responses in vitro and antitumor immunity in vivo. PVSRIPO’s immune adjuvancy stimulates canonical innate anti-pathogen inflammatory responses within the tumor microenvironment that culminate in dendritic cell and T cell infiltration. Our findings provide mechanistic evidence that PVSRIPO functions as a potent intratumor immune adjuvant that generates tumor antigen-specific CTL responses.
INTRODUCTION
The tumor microenvironment favors tumor immune escape by suppressing production, activation and/or function of antitumor T cells. Therapeutic interception of inhibitory immune receptors (immune checkpoints) that block T cell activation has yielded unprecedented clinical benefit. However, a limitation of immune checkpoint inhibitors, specifically those that target PD1, is their reliance on pre-existing antitumor immune responses and the presence of CD8 T cells in the tumor (1). Moreover, resistance to checkpoint blockade can occur through multiple immunosuppressive pathways; particularly when blocking only one of many repressive nodes (2). Thus, strategies to reverse tumor immunosuppression, stimulate antigen presentation, and induce antitumor T cell responses are needed. Tumor antigen-specific approaches, such as dendritic cell (DC) vaccines, aim to elicit antigen-specific antitumor immunity (3). However, such approaches are ultimately limited by profound tumor immunosuppression (4, 5). A reversal of tumor immunosuppression, the activation of antigen presentation, and the generation of antitumor immunity might be achieved by provoking canonical innate antiviral responses within tumors.
Indeed, viral targeting of tumors may engage immune responses in a pattern and intensity that mirrors natural infection, including innate immune activation capable of instigating the transition to adaptive immunity. Antiviral effector responses (especially CD8 T cells), geared to stop viral spread by killing infected cells, engage the very mechanisms capable of killing cancer cells. However, the utility of viruses for this purpose depends on their ability to effectively stimulate adaptive immunity. This is important, because viruses commonly use elaborate strategies to restrict their inflammatory footprint and prevent antigen presentation as a means to avoid immune rejection and establish long-term host relationships (6). We hypothesized that intratumoral administration of an oncolytic poliovirus (PV) is a rational approach to accomplish these goals due to: 1) targeting of CD155 on malignant cells and antigen-presenting cells (APCs); 2) neoplasia-specific cytopathogenicity (7); 3) extreme genetic austerity with a lack of immune evasion/suppression capacity.
To test our hypothesis, we defined mechanisms of cancer immunotherapy with PVSRIPO, a non-pathogenic PV engineered for neuronal incompetence by recombination with human rhinovirus type 2 (8). PVSRIPO has potent cytopathogenicity in cancer cells, owing to widespread expression of the PV receptor CD155 in solid neoplasms (9). It also targets CD155 present on monocytic-lineage cells (10). Incidentally, CD155 is a ligand for CD226, TIGIT, and CD96 with roles in immune response modulation (11). We show chronic, sublethal PVSRIPO infection of primary human DCs and macrophages, which produces persistent, type I interferon (IFN)-dominant activation that is resistant to cancer cell-mediated immunosuppression. PVSRIPO cytotoxicity in malignant cells combined with pro-inflammatory DC activation results in tumor-associated antigen presentation and induction of antitumor cytotoxic T lymphocyte (CTL) responses in human in vitro assays. Single intratumoral injection of PVSRIPO yields systemic antitumor CTL responses in a syngeneic, immunocompetent rodent tumor model. This response is preceded by rapid recruitment of neutrophils to tumors, followed by infiltration of DCs and T cells, indicative of an immune-engaged tumor microenvironment.
RESULTS
PVSRIPO targets human cancer cells
PVSRIPO is the live-attenuated type 1 (Sabin) PV vaccine containing an internal ribosomal entry site (IRES) from its relative, human rhinovirus type 2 (HRV2) (8). The HRV2 IRES is inherently neuron-incompetent by virtue of cell type-specific ribonucleoprotein complexes intercepting ribosome recruitment (12, 13). Thus, PVSRIPO does not cause encephalo- or poliomyelitis in primates (14).
Constitutively active Raf-ERK1/2-MNK1/2 signals favor selective PVSRIPO translation and cytotoxicity in cancer cells (15-17). To explore PVSRIPO oncolysis in cancers beyond gliomas (7), we examined the course of PVSRIPO translation and cytopathogenicity in melanoma, prostate, and breast cancer cell lines (Fig. 1). Viral translation (exemplified by 2C and its 2BC/P2 precursors) yields cytopathogenic viral proteases, whose actions, such as early cleavage of the translation initiation scaffold eIF4G, suppress host protein synthesis (18) (Fig. 1A). This coincided with other signs of host cell damage, including PARP cleavage. PVSRIPO-induced eIF2α(S51) phosphorylation is likely due to the double-stranded (ds) RNA-activated protein kinase (Fig. 1A). At ≥48 hours post-infection (hpi), host protein synthesis inhibition and cell lysis diminished all proteins (see loss of tubulin loading control at late hpi; Fig. 1A). This cytopathogenicity pattern mirrors PVSRIPO’s effects in glioma cells (7) (fig. S1A). Flow cytometry analysis of MHC class I and CD155 surface expression pre- and post-PVSRIPO confirmed CD155 expression in all cell lines (Fig. 1B). MHC class I was present on all cell lines pre/post infection (Fig. 1B). It is unclear if a decline in surface CD155 after infection of most cell lines (Fig. 1B) reflects global loss of protein from samples or a specific host response.
Fig. 1. PVSRIPO causes cytopathogenicity in cancer cell lines.

Melanoma (DM6, DM440), prostate (LNCaP, DU145), and breast cancer (MDA-MB231, SUM149) cell lines were infected with PVSRIPO at multiplicities of infection (MOIs) of 0.1 or 10. (A) Lysates were collected at the denoted hpi and tested by immunoblot for markers of direct viral cytotoxicity (eIF4G cleavage), host cell demise (PARP cleavage), viral translation (viral proteins P2/2BC/2C), and the innate antiviral response [p-eIF2α(S51)]. Global reduction of host (tubulin) and viral proteins at later time points reflects gross sample loss upon lytic destruction of cells. (B) Cells were infected (MOI=0.1) and harvested (48 hpi) for analysis of MHC class I and CD155 expression by flow cytometry (light gray, uninfected cells stained with isotype control; dark gray, uninfected cells stained for MHC or CD155; red, infected cells stained for MHC or CD155). All experiments were repeated twice, and representative results are shown.
PVSRIPO oncolysis releases the cancer cell proteome and dsRNA
To unravel inflammatory perturbations due to PVSRIPO oncolysis, we examined the release of damage/pathogen-associated molecular patterns (D/PAMPs) and tumor antigens in two cell lines: DM6 melanoma and MDA-MB231 triple-negative breast cancer (Fig. 2). Analyses of cell pellets or supernatants by silver stain revealed that PVSRIPO infection releases the proteome (Fig. 2A), resulting in the appearance of tumor-associated antigens MART1 (DM6) or CEA (MDA-MB231) in the supernatant (Fig. 2B). DAMPs, such as heat shock protein (HSP) 60/70/90, were detected in supernatants ≥24 hpi with PVSRIPO (Fig. 2B). HSPs have been shown to mediate anti-cancer immunity against HSP-bound peptides (19). Another DAMP, HMGB1 (20), was released only by infected DM6 cells, possibly due to variable expression (21). Loss of loading controls from cell pellets into the supernatant for cytoplasmic (tubulin), nuclear (PARP), mitochondrial (HSP60), and plasma membrane (Na+/K+ ATPase) subcellular fractions indicates sweeping destruction affecting all cellular compartments (Fig. 2B). We also examined the release of dsRNA from infected cells. dsRNA is a PV replicative intermediate (22), and an agonist for pattern recognition receptors toll-like receptor (TLR) 3 and melanoma-derived antigen (MDA) 5 (23). Dot blots of mock vs. virus-infected cancer cell lysates probed with an anti-dsRNA antibody revealed dsRNA presence in the supernatant of infected cells, indicating that PVSRIPO replication intermediates are released after cell lysis (Fig. 2C). Performing these assays in DU54 glioblastoma cells revealed similar results (fig. S1B, C). Thus, PVSRIPO-mediated killing of malignant cells exposes host tumor antigens, DAMPs, and PAMPs.
Fig. 2. PVSRIPO oncolysis releases tumor antigens, dsRNA, and DAMPs.
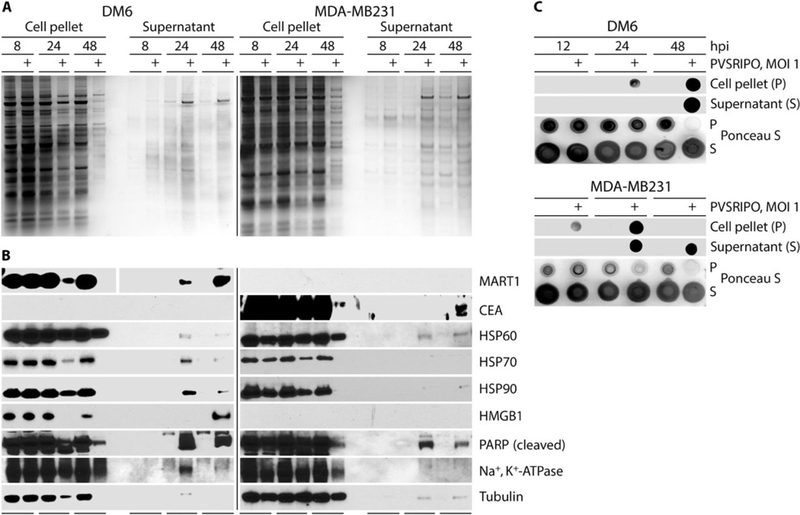
DM6 and MDA-MB231 cells were infected with PVSRIPO. (A) Silver stain was performed on oncolysates/corresponding supernatants. (B) Immunoblots from oncolysates/corresponding supernatants for tumor antigens (MART1/CEA), DAMPs (HSPs, HMGB1), HSP60 (mitochondrial), Na+/K+ ATPase (membrane-associated), PARP (nucleus), and tubulin (cytoplasm). (C) Immunoblot for dsRNA was performed from oncolysates/ corresponding supernatants of DM6 and MDA-MB231 cells (48 hpi); Ponceau S stain serves as loading control. Loss of total protein late during PVSRIPO infection corresponds with cell lysis. All experiments were repeated twice, and a representative series is shown.
PVSRIPO activates dendritic cells
The presence of PAMPs and DAMPs in PVSRIPO oncolysate may recruit and/or activate APCs and innate cell populations, which could promote adaptive antitumor immunity. Because APCs, particularly DCs, are essential enablers of tumor-specific responses (24, 25), we tested the effects of PVSRIPO oncolysis on human DCs. This is especially relevant, because APCs express CD155 and are susceptible to productive and lethal infection with PV (26). DM6 supernatant was collected 48 h after mock or PVSRIPO infection (hereafter referred to as ‘oncolysate’) and co-incubated with human monocyte-derived DCs (24 h). Untreated immature (iDCs) and cytokine cocktail-treated mature DCs (mDCs) were used as negative and positive controls (27), respectively, to compare DC activation phenotypes. In addition, we tested corresponding mock controls and oncolysates in which PVSRIPO was removed using a 100 kDa cut-off filter before co-incubation with DCs (Fig. 3A, B).
Fig. 3. PVSRIPO-induced oncolysate mediates DC activation.
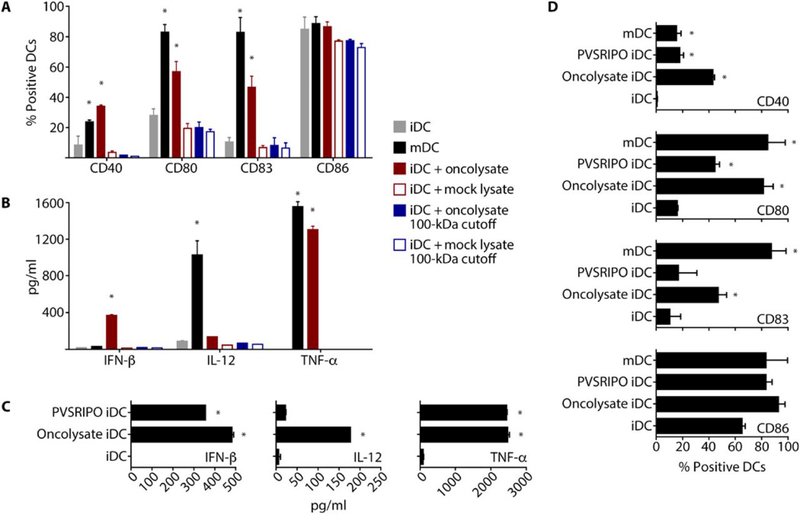
Human DCs (immature DCs, iDCs; mature DCs, mDCs) were generated, and iDCs were treated as indicated. iDCs and mDCs were used as controls, as indicated in the figure. (A and B) Supernatant from untreated or PVSRIPO-infected (MOI=0.1; 48 hpi) DM6 melanoma cells was either unfiltered or filtered through a 100 kD cut-off filter. iDCs were treated with the resulting supernatants (24 h). Flow cytometry (A) and ELISA (B) were used to assess DC activation/maturation phenotype, viability, and pro-inflammatory cytokine production. (C and D) Human DCs were treated with DM6 oncolysate produced as in (A) or PVSRIPO at a titer equal to the amount detected in DM6 oncolysate. Supernatants were assessed for cytokine production by ELISA (C) and cell phenotype by flow cytometry (D). (A and D) For representative flow cytometry data see fig. S2. Experiments were repeated three times with cells from 2 donors; error bars denote SEM, and asterisks denote significant (p<0.05) ANOVA protected Tukey’s HSD test compared to mock controls.
Exposure to the oncolysate (24 h) increased the percentage of DCs expressing costimulation/activation markers CD40, CD80 (B7-1), and CD83 (Fig. 3A), and increased the production of cytokines IFN-β and TNF-α (Fig. 3B). IL-12 induction was modest. Virus removal through the 100 kDa cut-off filter mitigated these effects (Fig. 3A, B). The costimulatory molecule CD86 (B7-2) is high on both iDCs and mDCs, as previously shown (27), and, as expected, none of the treatments affected CD86 expression. A representative flow cytometry analysis illustrating DC phenotype is depicted in fig. S2. Our findings suggest that PVSRIPO oncolysate activates DCs, and that the presence of (infectious) virus contributes to this effect. Stimulation of iDCs with PVSRIPO oncolysate or purified virus (adjusted to the same concentrations of plaque-forming units; pfu) revealed similar levels of IFN-β and TNF-α secretion (Fig. 3C). As shown in Fig. 3B, we observed modest IL-12 production in DC-stimulated PVSRIPO oncolysate, however, IL-12 production was negligible with PVSRIPO alone at 24 hpi (Fig. 3C). Analysis of costimulatory/activation markers at 24 hpi demonstrated that CD40/CD80/CD83 induction was less pronounced with PVSRIPO infection alone compared to exposure to PVSRIPO oncolysate (Fig. 3D), indicating that non-viral oncolysate components contribute to DC activation. Altogether, our data suggest that DC activation caused by PVSRIPO oncolysis occurs predominately via direct infection, which induces a potent type I IFN response. This is of relevance to cancer immunotherapy, because type I IFN responses are essential for DC function and the transition to adaptive immune responses (28-31).
PVSRIPO infection of DCs induces sustained type I IFN-dominant responses, costimulatory molecule expression, and cytokine production
To decipher the effects of PVSRIPO on DCs over time, we examined markers of cytopathogenicity and activation status after iDC infection at MOIs of 1 or 10 (Fig. 4). In contrast to DM6 and MDA-MB231 cancer cells, PVSRIPO did not cause lysis of DCs as measured by LDH release (Fig. 4A; magnified view in fig. S3A), and viral propagation was only marginally productive in DCs (Fig. 4B; magnified view in fig. S3B). Viral translation was active in DCs, evident as detectable viral protein 2C at 8 hpi (Fig. 4C), but declined over time. Despite active viral translation (implying viral protease release) there was no eIF4G cleavage, even at an MOI of 10 (Fig. 4C; compare to Fig. 1A). Thus, PVSRIPO infects DCs, but DCs are resistant to viral cytopathogenic damage/host protein synthesis inhibition. An earlier study using the same DC differentiation technique and infection procedure demonstrated efficient viral growth, host protein synthesis shut-down, and DC killing with wild-type PV type 1 (Mahoney) and the type 1 (Sabin) vaccine (26). Thus, our data imply that, similar to neuron-lineage cells (32), the foreign HRV2 IRES in PVSRIPO mediates loss of cytopathogenicity and intrinsic host resistance in DCs.
Fig. 4. PVSRIPO infection of DCs is sublethal, marginally productive, and induces sustained pro-inflammatory cytokine production.
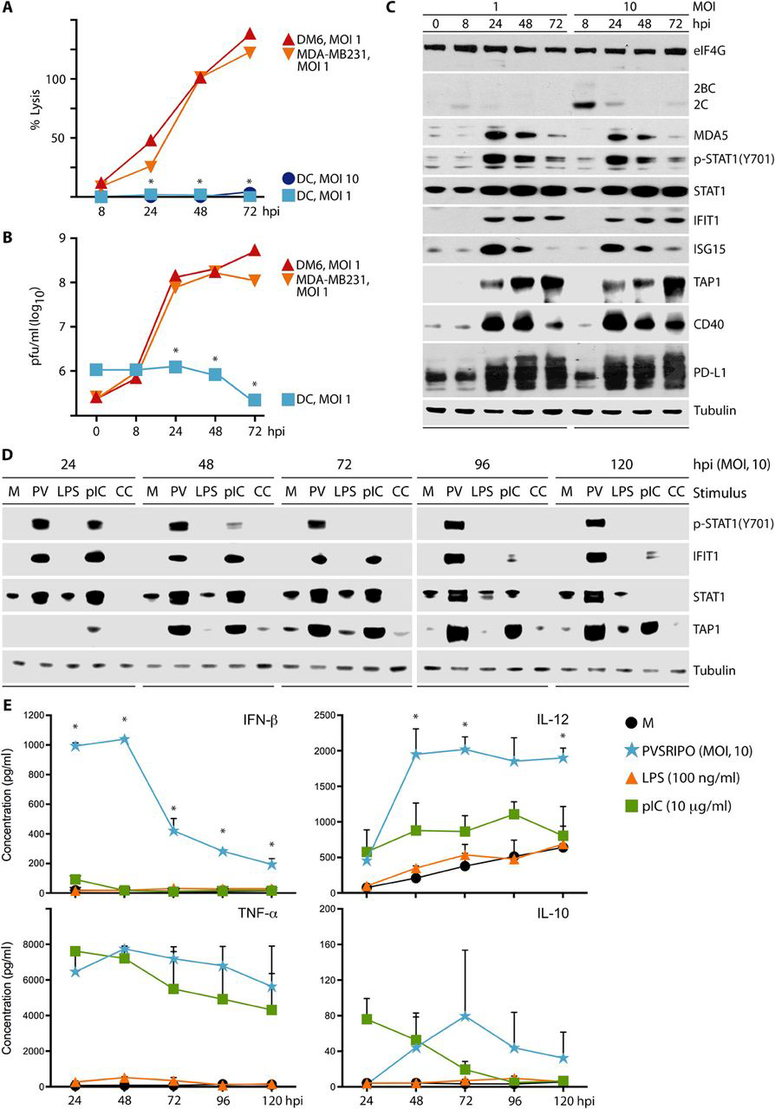
Analysis of percent lysis (as measured by LDH release) (A) and viral progeny (B) after PVSRIPO infection of DCs compared to PVSRIPO infection of DM6 and MDA-MB231 cells. (A and B) Asterisks denote statistical significance comparing pooled cancer cell line data to DC values by ANOVA protected HSD test; error bars depict SEM. (C) Dose-dependent PVSRIPO infection (viral protein 2C), type I IFN responses, and lack of cytotoxicity (PARP and eIF4G) in primary human DCs shown by immunoblot. (D and E) iDCs were untreated (mock; M) or treated with PVSRIPO (PV; MOI=10), LPS (100 ng/ml), poly(I:C) (10 μg/ml), or maturation cytokine cocktail (CC) as shown. Cells and supernatant were harvested at the designated intervals. (D) Cell lysates were analyzed by immunoblot for the IFN response and DC activation proteins. (E) ELISA was used to measure IFN-β, TNF-α, IL-12, and IL-10 in DC supernatants after treatment. Data represent cumulative cytokine release at the designated time point. The mean of 2 experiments is shown for each time point. Asterisks denote PVSRIPO mediated cytokine production that is significant compared to all other groups using ANOVA protected Tukey’s HSD test. Repeat assays and magnified view of these data are presented in fig. S3.
Accordingly, rather than viral cytotoxicity, the most prominent effect of PVSRIPO infection in DCs was a robust, durable type I IFN response, indicated by STAT1(Y701) phosphorylation with STAT1 and IFN-stimulated gene (ISG) induction (IFIT1, ISG15; Fig. 4C). We observed substantial up-regulation of TAP1 (Fig. 4C), a protein required for MHC class I-restricted antigen processing in cells, likely induced by IFN-β (33). These changes, indicative of an enhanced DC activation status, were accompanied by induction of CD40 (Fig. 4C). Earlier work showed that IFN-β triggers PD-L1 (or B7-H1) expression on APCs (34). Consequently, PVSRIPO infection induced PD-L1 in DCs, in a pattern coinciding with ISG expression/IFN signaling (Fig. 4C).
We next compared the adjuvant effect of PVSRIPO on DCs with standard methods of maturing and activating human iDCs via TLR stimulation pathways with lipopolysaccharide (LPS), poly(I:C), or maturation cytokine cocktail [CC; used in clinical applications of DC-based vaccines (27)]. We treated DCs with PVSRIPO/TLR stimuli over a period of 5 days (Fig. 4D, E). Compared to TLR stimuli, PVSRIPO infection was distinguished by potent, sustained STAT1(Y701)-phosphorylation and ISG induction throughout the 5-day interval (Fig. 4D; repeat data shown in fig. S3C). This coincided with IFN-β release at levels exceeding all other stimuli (Fig. 4E). Poly(I:C) exhibited some IFN-β production, albeit at reduced levels and duration; neither LPS nor CC caused IFN-β production (Fig. 4D, E), but induced STAT3(Y705) phosphorylation and TNF-α production (fig. S3D, E). PVSRIPO infection of DCs also led to higher levels and sustained production of IL-12 relative to poly(I:C) or the other stimuli (Fig. 4E). Of note, IL-12 production spiked after 24 h, which explains modest IL-12 release shown in Fig. 3B and C, where analyses were conducted at 24 h only. CC-stimulated mDCs were not assayed for cytokine production in Fig. 4E, because the cocktail contains TNF-α; we tested them for IFN-β and IL-12 production, and confirmed lack of type I IFN activity (Fig. 4D)
PVSRIPO infects and activates immunosuppressed dendritic cells and macrophages
APC activation is hampered in the immunosuppressive context of the tumor microenvironment. We tested the effects of PVSRIPO and poly(I:C) on iDCs cultured for 24 h in plain AIM-V medium or in cancer cell-conditioned medium (AIM-V medium harvested from DM6 melanoma cells after 48 h in culture; DM6CM). DM6CM contained a range of cytokines implicated in APC suppression and other immune cell functions (table S1). iDCs incubated in DM6CM showed reduced basal levels of CD40, CD80, and CD86 compared to iDCs incubated in plain AIM-V (Fig. 5A; similar to mock-infected oncolysate in Fig. 3A). Despite this, PVSRIPO infection enhanced expression of CD40, CD80, and CD86 in DM6CM-exposed iDCs, equivalent to the effects on iDCs cultured in plain AIM-V medium (Fig. 5A). In contrast, co-stimulatory molecule expression upon poly(I:C) treatment was markedly reduced in DM6CM-cultured iDCs compared to AIM-V cultured iDCs (Fig. 5A; representative flow cytometry analysis is shown in fig. S4).
Fig. 5. PVSRIPO-mediated APC activation occurs in immunosuppressive conditions.
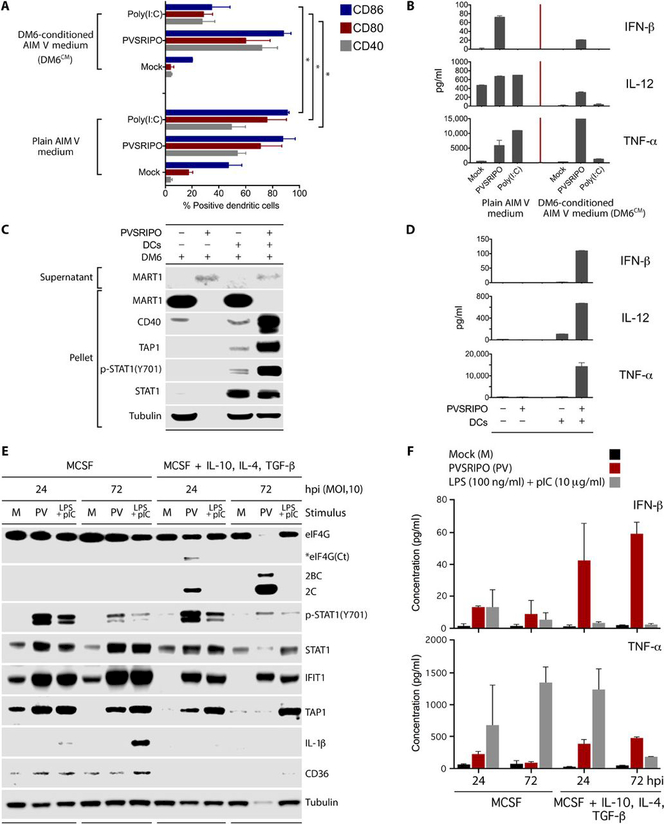
. (A and B) DCs were cultured in the presence of AIM-V or DM6CM (24 h) and untreated (mock) or treated with PVSRIPO (MOI=10) or poly(I:C) (10 μg/ml) (48 h). (A) Cells were analyzed for activation markers by flow cytometry. Data bars represent the mean of two independent experiments, and error bars denote SEM. Asterisks depict significance as determined by ANOVA protected Tukey’s HSD test. For representative flow cytometry data, see fig. S4. (B) Supernatant from (A) was tested for pro-inflammatory cytokine production by ELISA. (C and D) DM6 cells were cultured alone or with DCs and mock-/PVSRIPO infected (MOI=10). Supernatant was tested for lytic release of MART1 by immunoblot; cell pellets were tested for markers of DM6 cells (MART1) and DC activation [CD40, TAP1, p-STAT1(Y701), STAT1] (C). Supernatants from (C) were assessed for pro-inflammatory cytokine production (D). (E, F) Negatively selected human monocytes were differentiated with MCSF (25 ng/ml) or MCSF + IL-10, TGF-β, and IL-4 (all at 20 ng/ml) for 7 days. The cells were infected with PVSRIPO (MOI=10) or treated with combined poly(I:C) (10 μg/ml) and LPS (100 ng/ml) as shown. Cell lysates were retained for immunoblot (E), and supernatants were used for ELISA (F). (B, C, E) Experiments were repeated three times, and representative data are shown. (D, E) Data bars represent the mean of two independent experiments, and error bars indicate SEM.
As in prior analyses (Fig. 4E), only PVSRIPO infection of DCs, but not poly(I:C) stimulation, caused potent, sustained IFN-β release (Fig. 5B). DCs cultured in AIM-V medium released IL-12 and TNF-α after PVSRIPO infection or poly(I:C) addition (Fig. 5B). In contrast, DCs cultured in DM6CM did not produce appreciable amounts of IFN-β, IL-12, or TNF-α after poly(I:C) treatment, but retained IFN-β, IL-12, and TNF-α release after PVSRIPO infection (Fig. 5B). Although PVSRIPO-induced IFN-β and IL-12 production was tempered in DM6CM-cultured DCs, compared to AIM-V-cultured DCs, TNF-α production was actually enhanced (Fig. 5B). These data suggest that although DM6CM-suppressed DCs are resistant to poly(I:C) stimulation, they retain sensitivity to PVSRIPO-mediated activation.
To extend these findings, we investigated the effect of PVSRIPO on DM6 cells with and without DC co-culture. Regardless of the presence of DCs, infection of DM6 cells with PVSRIPO resulted in complete cell lysis (48 hpi) as indicated by the shift of MART1 (a melanoma-specific antigen) from the cell pellet to the supernatant fraction of infected cultures (Fig. 5C; also shown in Fig. 2B). Retention of the tubulin immunoblot signal after PVSRIPO infection only in DC:DM6 co-cultures (48 hpi) was due to the inherent resistance of DCs to PVSRIPO killing. Consistent with type I IFN-dominant DC activation upon PVSRIPO infection, we observed potent up-regulation of CD40, TAP1, and p-STAT1(Y701) in DC:DM6 co-cultures (48 hpi) (Fig. 5C). Similar to infection of DCs alone (Fig. 4E), PVSRIPO infection of DC:DM6 co-cultures resulted in the release of IFN-β, IL-12, and TNF-α (Fig. 5D). DC:DM6 co-culture reduced viral titers in culture supernatants ~70-fold (fig. S5A). These data suggest that: 1) DCs remain unharmed in the presence of active PVSRIPO lysis of co-cultured cancer cells; 2) PVSRIPO activates DCs, even in the presence of cancer cell-mediated immunosuppression; 3) DC co-culture does not prevent PVSRIPO from killing tumor cells; and 4) the principal source for pro-inflammatory cytokines in DC:DM6 co-cultures are PVSRIPO-infected DCs.
Because tumor-associated macrophages (TAMs) are implicated in tumor immunosuppression (35), we investigated the effects of PVSRIPO infection on macrophages in vitro. Purified human monocytes were differentiated to macrophages with MCSF alone, or with MCSF plus TGF-β, IL-10, and IL-4 (Fig. 5E). The latter induced an immunosuppressive phenotype in vitro (36, 37), associated with reduced basal expression of IFIT1, TAP1, and CD36, a scavenger receptor associated with macrophage activity (38). As in DCs, PVSRIPO infection of macrophages yielded robust type-I IFN responses and TNF-α production that was paradoxically enhanced by TGF-β/IL-10/IL-4 treatment (Fig. 5E). Increased IFN-β release coincided with enhanced viral translation and overt cytopathogenicity (eIF4G cleavage, tubulin loss) upon prolonged infection (Fig. 5E, F). Viral translation or cytopathogenicity were not detected in PVSRIPO-infected macrophages derived with MCSF only (Fig. 5E). Treatment of macrophages with combined poly(I:C) and LPS induced IL1-β, IFN-β, and TNF-α responses in MCSF-derived macrophages (Fig. 5E, F). However, TGF-β/IL-10/IL-4 immunosuppression of macrophages reduced IL1-β, IFN-β, and TNF-α production upon treatment with combined poly(I:C)/LPS at 72 hrs (Fig. 5E, F). Analysis of human monocytes without differentiation revealed similar type-I IFN responses to PVSRIPO infection as in MCSF-derived macrophages (fig. S5B). These findings suggest that PVSRIPO infection of macrophages (and monocytes) yields type I IFN-dominant activation that is paradoxically enhanced by an immunosuppressive macrophage phenotype.
PVSRIPO oncolysate-treated DCs induce antigen-specific cytotoxic T cells in vitro
Our experiments thus far show that PVSRIPO effectively damages tumor cells, releases tumor antigens, and simultaneously induces DC and macrophage activation, including in the context of cancer immunosuppression. Therefore, to test the downstream effects of PVSRIPO tumor cell killing with simultaneous infection and DAMP/PAMP-activation of DCs, we examined whether human DCs exposed to PVSRIPO oncolysate pick up released tumor antigen, present it, and prime autologous T cells using an in vitro DC-T cell stimulation assay (Fig. 6A).
Fig. 6. PVSRIPO oncolysate-pulsed DCs generate tumor antigen-specific CTL immunity in vitro.
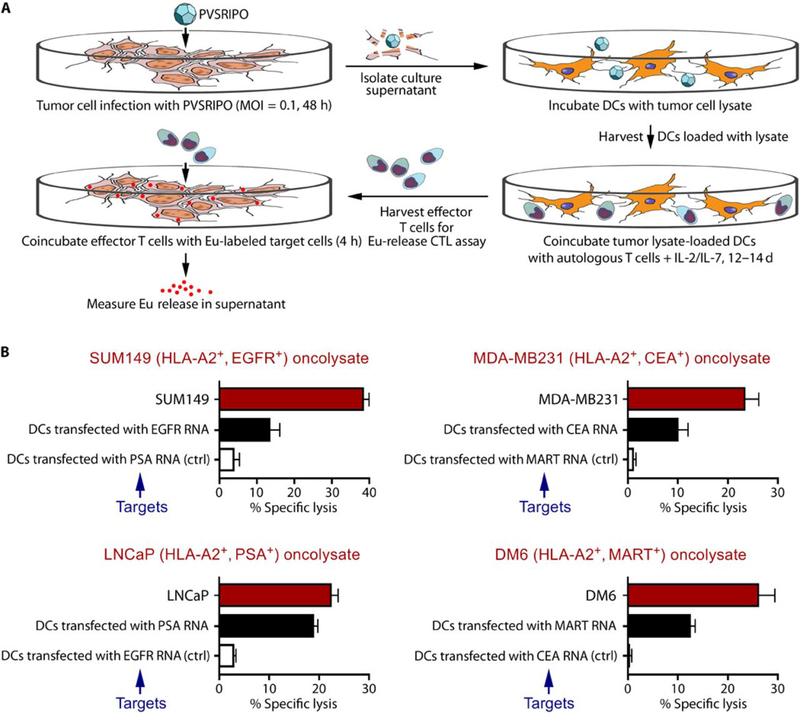
Primary human DCs co-incubated with SUM149, MDA-MB231, LNCaP, or DM6 oncolysate stimulate tumor antigen-specific T cell responses in vitro. (A) Schema of the assay. (B) T cells were co-cultured with oncolysate-pulsed autologous DCs, and the stimulated effector T cells were then harvested and tested in a CTL assay against the corresponding tumor cells (red bars), autologous DCs transfected with RNA that encodes for a relevant tumor antigen (black bars; positive control), or autologous DCs transfected with RNA that encodes for an irrelevant tumor antigen (white bars; negative control). Each bar represents mean % specific lysis and standard deviation (SD) of triplicate samples. Statistical significance comparing autologous DCs expressing either the relevant or irrelevant tumor antigen for each panel in (B) was calculated using paired two-tailed Student’s t test. A probability of less than 0.05 (p<0.05) is considered statistically significant. Panel SUM149 DC targets, p=0.04; Panel LNCaP DC targets, p=0.0008; panel MDA-MB231 DC targets, p=0.01; Panel DM6 DC targets, p=0.01.
Experiments were conducted using HLA-A2+ peripheral blood mononuclear cells (PBMCs) obtained from healthy donors (n=2) and human HLA-A2+ tumor cell lines: SUM149 inflammatory breast cancer, MDA-MB231 triple-negative breast cancer, DM6 melanoma, and LNCaP prostate cancer (Fig. 6B). These cell lines express class I MHC, required for CD8+ T cell recognition of antigens (Fig. 1B). DCs were first treated with supernatant from mock- or PVSRIPO-infected tumor cells (48 hpi), harvested, and then co-cultured with autologous T cells (non-adherent fraction of PBMCs). After 12-14 days of DC:T cell co-culture in vitro, the cells were harvested and assayed for tumor antigen-specific cytotoxic reactivity using a standard 4 h europium (Eu)-release CTL assay (Fig. 6A). To assess CTL reactivity, T cells (effectors) were cultured with the following Eu-labeled target cells: (i) a tumor cell line yielding the PVSRIPO oncolysate used for DC loading; (ii) DCs transfected with mRNA encoding a tumor antigen known to be expressed by the tumor cell line (positive control); or (iii) DCs transfected with mRNA encoding an irrelevant tumor antigen not expressed by the tumor cell line (negative control) (Fig. 6B). The use of autologous antigen-expressing DCs as positive and negative control targets allowed determination of specific CTL reactivity to known tumor-associated antigens as we have done in previous studies (39, 40), by eliminating MHC mismatch between T cells and target cells. CTL-mediated killing of the target cells was assessed by measuring Eu - release in the supernatant.
DCs treated with PVSRIPO-oncolysate produced CTL responses that effectively lysed the original cancer lines as well as the positive control (DCs expressing a relevant tumor antigen), but not the negative control (DCs expressing an irrelevant tumor antigen; Fig. 6B). Antigen presentation by PVSRIPO oncolysate-treated DCs did not require the additional maturation step with the cytokine cocktail (CC), which is routinely used (27, 39, 41, 42) to stimulate effector T cells in such in vitro assays. We hypothesized that DC maturation/activation in this instance was due to infection of DCs with PVSRIPO present in the oncolysate. Because the oncolysate represents the entire repertoire of tumor-associated antigens, oncolysate-stimulated T cells likely target multiple tumor antigens, which may explain the greater extent of tumor cell lysis versus target DCs expressing select tumor-associated antigens. Of note, SUM149 breast cancer oncolysate-stimulated T cells did not lyse LNCaP prostate cancer cells and vice-versa (fig. S6) Moreover, DCs loaded with supernatant from control lysate (mock infected cell supernatant) did not stimulate antigen-specific T cells as indicated by minimal lysis of target cells (fig. S6).
Together, our findings define a concerted range of activities for PVSRIPO in mediating cancer cytopathogenicity, DAMP/PAMP release, and antigen release for uptake by APCs (such as DCs), while simultaneously infecting and activating APCs to bolster co-stimulatory molecule expression, antigen processing and presentation, and cytokine production. Ultimately these processes influence the generation of antitumor T cell populations that are capable of lysing cancer cells directly.
PVSRIPO induces antitumor T cell immunity in an immunocompetent murine melanoma model
The PVs are exclusively human pathogens; only old-world primates develop poliomyelitis after experimental infection (chimpanzees are more susceptible than other species). Enteroviridae (PV is their flagship) have long been associated with Hominids, and the human innate antiviral response is shaped by thousands of years of evolving virus:host relations during co-speciation (43). Moreover, aggressive rodent syngeneic/xenotransplantation tumor models are notoriously deficient in recapitulating human tumor:stroma relations (44). Given these shortcomings, we sought to confirm our findings in principle, addressing whether PVSRIPO therapy induces antitumor T cell immunity in an immunocompetent murine tumor model in vivo. We chose B16-F10.9 melanoma expressing chicken ovalbumin (OVA) as a model antigen (B16-F10.9-OVA); these cells were transduced with (human) CD155 to generate B16-F10.9-OVA-CD155 (the mouse CD155 homolog does not function as a PV receptor), and we used (human) CD155-transgenic (tg) C57Bl/6 mice (45) as tumor hosts. PVSRIPO replication is deficient in murine cells due to inherent host restrictions of the HRV2 IRES, but serial passage of PVSRIPO in CD155-transduced mouse cells selects for mouse-competent variants (46). Using an approach similar to Jahan et al. (46), we adapted PVSRIPO by serial passage in explant mouse astrocytoma cells, yielding mRIPO. mRIPO exhibited patterns of viral translation, cytopathogenicity, and eIF2α(S51) phosphorylation in the B16-F10.9-OVA-CD155 model resembling PVSRIPO in human melanoma cells (compare Fig. 1A, 7A). Also, mRIPO infection of bone marrow-derived DCs from CD155-tg mice induced sustained IFN-β production at levels exceeding stimulation with LPS, resembling PVSRIPO infection of human DCs (fig. S7).
Fig. 7. mRIPO therapy restricts tumor growth and produces antigen-specific antitumor immunity.
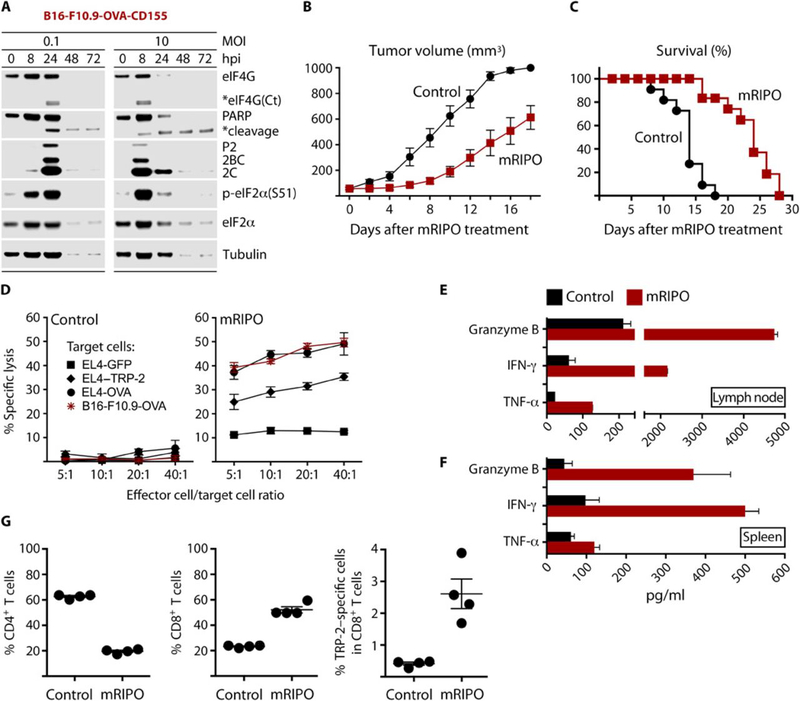
(A) Cytopathogenicity profile of mRIPO in B16-F10.9-OVA-CD155 cells. (B and C) Subcutaneous B16-F10.9-OVA-CD155 tumors were injected with either control (DMEM) or mRIPO (20 μl) when they reached a volume of ~50–100 mm3. Tumor volume was measured (n=11 per group) on the days indicated (B); mice were euthanized when tumors reached 1000 mm3 (C). Data are from two pooled assays. Tumor growth curves were compared using multiple t-tests with Holm-Sidak multiple comparison post-test; p≤0.005 starting on day 6. Comparison of survival curves between the 2 groups was performed using the log-rank test (Mantel–Cox test), p<0.0001. Median survival day: control=14; mRIPO=24. (D) Tumor-draining inguinal lymph nodes were harvested from mice (n=4) 7 days after treatment with DMEM or mRIPO and restimulated with antigen-expressing cells for 5 days. Restimulated cells were tested for lytic activity against B16-F10.9-OVA cells or EL4 cells electroporated with RNA encoding GFP (control), TRP-2 (melanoma antigen), or OVA. (E) Supernatant from the CTL assay (D) was tested for markers of T cell activation and lytic activity by ELISA. (F) Tumor-bearing mice were treated with DMEM or mRIPO, and spleens were harvested 14 days after treatment (n=4). Splenocytes from individual mice were co-cultured with B16-F10.9-OVA-CD155 cells (5:1 ratio; 48 h); supernatant was tested as in (E). (G) Tumor-draining inguinal lymph nodes were harvested after treatment with DMEM or mRIPO and individually restimulated in vitro. After 5 days, restimulated cells were analyzed for CD4 and CD8 T cells by flow cytometry. TRP-2-specific response was assessed using a H-2Kb TRP-2 tetramer (right panel). TRP-2 specific responses were compared using student t test, with p<0.05 considered significant. Fig. S8D shows representative flow cytometry analyses of T cells and TRP-2-specific T cells (out of 4 tested per group).
B16-F10.9-OVA-CD155 cells were implanted in the flank of CD155-tg C57Bl/6 mice and treated with a single intratumoral injection of control (DMEM) or mRIPO (107 pfu). mRIPO therapy delayed tumor growth as measured by tumor volume (Fig. 7B) and increased overall survival (Fig. 7C). Loss of intact PARP and eIF2α(S51) phosphorylation after mRIPO treatment was observed in tumor homogenates early after treatment (fig. S8A), mirroring viral cytotoxicity observed in vitro (Fig. 7A, Fig. 1A, fig. S1A). To determine whether such therapy produced antitumor T cells, we obtained draining inguinal lymph nodes from mice 7 days after treatment with control or mRIPO. Lymph node cells were restimulated in vitro using tumor antigen-expressing cells. After 5 days, effector cells were harvested and tested for cytotoxicity against: EL4 thymoma cells expressing (i) GFP (control); (ii) OVA; (iii) TRP-2 (B16 melanoma antigen); or B16-F10.9-OVA-CD155 cells (Fig. 7D). Effector T cells from control-treated mice exhibited minimal cytotoxicity against the target cells (Fig. 7D). Effector T cells isolated from mRIPO-treated mice killed target cells expressing OVA or TRP-2 or both antigens (B16-F10.9-OVA-CD155 cells) and not EL4 cells expressing GFP, indicating antigen-specific killing (Fig. 7D). Analysis of cytokines in supernatant harvested after 5-day T cell restimulation revealed granzyme B, IFN-γ, and TNF-α induction only in the mRIPO-treated group (Fig. 7E), which corroborates cytotoxic T cell activity observed in Fig. 7D. To test induction of systemic antitumor responses, we harvested spleens 14 days after intratumoral treatment with control (DMEM) or mRIPO. Splenocytes were co-cultured with B16-F10.9-OVA-CD155 cells, and only splenocytes from mRIPO-treated mice released T cell effector cytokines (Fig. 7F). The generation of systemic antitumor T cells was preceded by enhanced IFN-stimulated gene expression in tumor lysates and a reduction in viral titers over time (fig. S8B, C).
To validate the presence of melanoma-specific T cells using another approach, we used a TRP-2-specific tetramer to detect TRP-2-specific CD8 T cells. Tumor-draining inguinal lymph nodes were harvested from control and mRIPO-treated mice and stimulated for 5 days in vitro with tumor antigen-expressing cells. After 5 days, the effector cells were harvested and individual lymph nodes were analyzed using flow cytometry for the presence of CD4, CD8, and TRP-2-specific CD8 T cells. Representative flow cytometry data are shown in fig. S8D. Analysis of lymph node cells after restimulation revealed that mRIPO-treated mice had a higher ratio of CD8 T cells to CD4 T cells, indicating CD8 T cell expansion relative to the control group (Fig. 7G, 2 left panels; T cell numbers are shown in fig. S8E). TRP-2-specific CD8+ T cells were detected only in mRIPO-treated mice (Fig. 7G, right panel, and fig. S8E), corroborating data in Fig. 7D that suggest the generation of TRP-2 specific CTLs in mRIPO-treated mice. Altogether, our data in human and murine systems indicate that PVSRIPO’s cytopathogenic targeting of malignant cells combined with pro-inflammatory activation of APCs induces productive antitumor T cell responses.
PVSRIPO therapy induces acute neutrophil influx followed by DC and T cell infiltration
Intratumoral administration of mRIPO results in delayed tumor growth, improved survival, and systemic tumor-specific CTLs (Fig. 7). To address events that may contribute to the generation of antitumor CTL responses after mRIPO (vs. control) treatment, we analyzed the immune cell content of tumors longitudinally for 6 days. Analysis of mRIPO-treated tumors revealed considerable infiltration of CD45.2+ immune cells as compared to control-treated tumors (Fig. 8A). This was accompanied by early intratumoral release of IFN-β, TNF-α, IL-12, and IFN-γ in response to mRIPO treatment (Fig. 8B).
Fig. 8. mRIPO elicits neutrophil influx followed by DC and T cell infiltration into tumors.
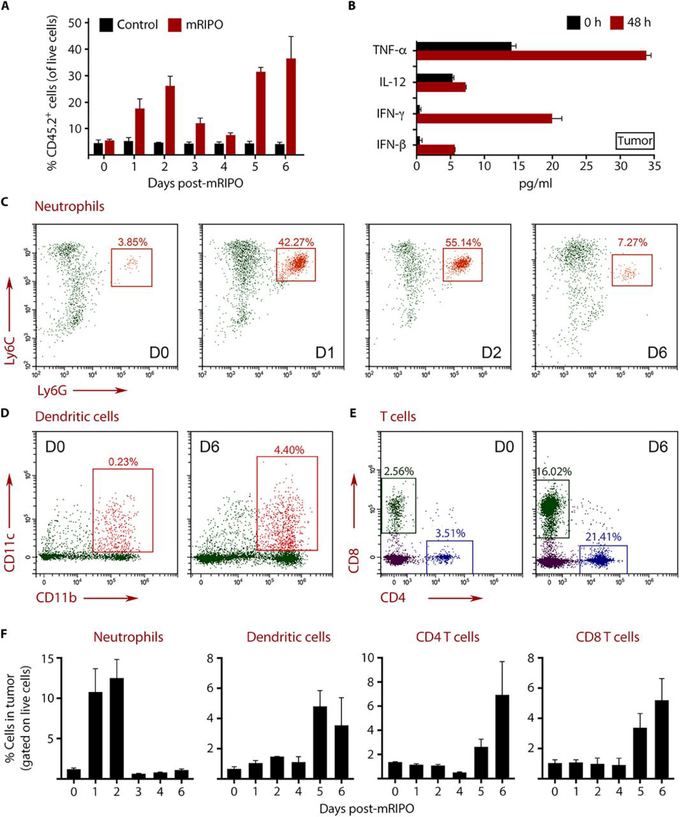
B16-F10.9-OVA-CD155 tumors were implanted subcutaneously, and when the tumor volume reached ~100 mm3, they were injected with DMEM (control) or mRIPO. Tumors were harvested after injection as indicated, digested to single cell suspensions, and analyzed by flow cytometry. (A) Analysis of percentage of CD45.2+ immune cells in the tumor after DMEM (control) or mRIPO treatment. Each bar represents 3 mice analyzed individually. (B) Cytokine concentrations in tumor homogenates. (C to E) Analysis of tumor-infiltrating neutrophils (C), DCs (D), and T cells (E) at the indicated days after mRIPO injection. (F) Longitudinal analysis of neutrophil, DC, CD4+ T cell, and CD8+ T cell infiltration is depicted as a percentage of total live cells in the tumor. Each bar represents 3 mice analyzed individually. Error bars represent SEM. The flow cytometry gating strategy is shown in fig. S9 and S10.
Analyses of tumors before mRIPO treatment (day 0) revealed that myeloid cells (CD45+ CD11b+) were the predominant immune cells in the tumor (fig. S9). Although monocyte/macrophage populations were present on the day of treatment, their numbers remained relatively unchanged throughout the course of treatment (fig. S9). We previously reported striking neutrophil infiltration of tumors after PVSRIPO injection in xenograft models of prostate and breast cancer (47). Confirming these findings, mRIPO-treated tumors demonstrated a marked influx of neutrophils (CD45.2+CD11b+CD11c-Ly6C+Ly6G+) (Fig. 8C). Neutrophils are a central component of acute inflammatory responses that facilitate the transition from innate to adaptive tumor immunity (48, 49). Neutrophil numbers peaked on days 1 and 2 and declined rapidly thereafter, indicative of a classic acute inflammatory response (Fig. 8C). Neutrophil influx was followed by DC (CD45.2+CD11b+CD11c+classII+F4/80-) and T cell (CD45.2+CD4+ or CD8+) infiltration into mRIPO-treated tumors (Fig. 8D-F) on days 5 and 6.
Immune cell infiltration into the tumor occurred in the presence of infectious virus in the tumor (fig. S8C) as in our in vitro oncolysate studies (Fig. 3-6). The gating strategy used for analysis of tumor-infiltrating immune cells is shown in fig. S9 and S10. Fig. 8F depicts the timeline of neutrophil, DC, CD4 T cell, and CD8 T cell influx over 6 days (data represent the percentage of total live cells in tumor). These data suggest that mRIPO tumor infection sets in motion an inflammatory cascade causing the recruitment of DCs and T cells into the tumor (Fig. 8), which ultimately results in the generation of a productive antitumor T cell response (Fig. 7). This confirms key observations in human systems (Fig. 3-6), indicating that combined PVSRIPO oncolysis and APC infection induces type I IFN dominant responses, antigen presentation activity, and antitumor CTL responses (fig. S11).
DISCUSSION
Disruption of the immunological masking of tumors and expansion of the antitumor immune repertoire are eminent objectives for cancer immunotherapy. The inspiration for using PVSRIPO to this end had its origin in specific cytopathogenicity for cells derived from virtually any solid cancer, due to widespread CD155 expression in solid malignancy (7). PVSRIPO was granted Breakthrough Therapy Designation by FDA/CBER in May 2016, due to promising early clinical results against glioblastoma. Here we define PVSRIPO’s immunotherapeutic potential, in terms of its ability to engage tumors immunologically and to induce tumor antigen-specific antitumor immunity. Our studies suggest that, in addition to lytic damage to malignant cells, this potential rests on non-cytotoxic infection of APCs/DCs.
PVSRIPO infection of primary human DCs did not produce cytopathogenicity or cell killing, but induced type I IFN responses that exceeded standard TLR-stimulants in potency and duration. This was accompanied by up-regulation of markers of DC antigen-presenting and costimulatory functions. We believe that this pattern of APC stimulation may be specific to PVSRIPO and translation restrictions imparted by the foreign HRV2 IRES, because earlier reports indicated productive, cytotoxic propagation and killing of DCs by wild type PV (26). Such activities of wild type PV are prototypical for human pathogenic virus:host relations, where DC targeting is very common and may be a mechanism to prevent the generation of adaptive antiviral immunity by impairing DC function (50). Thus, pro-inflammatory conditioning of APCs/DCs may be a consequence of PVSRIPO’s recombinant IRES.
Sustained IFN responses induced in DCs by PVSRIPO may be key to engaging the adaptive arm (especially CTLs) of the immune system (28-31). Indeed, type I IFNs are critical antitumor immunity-stimulating cytokines because they are essential for DC-mediated rejection of tumors in mice (57). Moreover, type I IFN production by DCs after pathogen encounter is critical for regulating immune responses (52-54). PD-L1 induction after PVSRIPO infection of DCs, an expected consequence of the IFN response, points towards potential nodes of synergy with anti-PD-L1/PD-1 checkpoint inhibition.
The finding that intratumoral PVSRIPO administration induces immune cell infiltration and antitumor CTLs is critical for its clinical application and the selection of rational immune-modulating combinations. PVSRIPO adjuvant effects (infectious PVSRIPO oncolysate) promote DC activation and DC-mediated stimulation of tumor-specific CTLs that recognize target tumor antigens such as CEA, MART1, EGFR, and PSA in assays with human PBMCs.
In an immunocompetent murine model of melanoma, single intratumoral injection of mouse-adapted PVSRIPO delayed tumor growth. This effect was associated with a rapid influx of neutrophils followed by DC and T cell invasion. As such, neutrophils can impose either tumor suppressive or supportive functions (55); however, upon pathogen challenge, neutrophils recruit other immune cell populations, including DCs and T cells (48, 49, 56). We speculate that neutrophil influx follows the release of inflammatory cytokines and chemokines by monocytes and macrophages, which were the primary myeloid cells in the tumor at the time of treatment (48, 57). Presumably, PVSRIPO infects myeloid cells in the tumor, because infectious virus was detected in tumors as late as day 7 (fig. S8C), after DC infiltration into tumors had occurred.
A limitation of this study is the use of murine models and in vitro systems because they lack the complexity and virus:host relationships that dictate the extent and quality of antitumor immunity generated by PVSRIPO therapy in patients. Genetically homogeneous mice and tumors do not recapitulate the range of responses to PVSRIPO infection that may determine antitumor efficacy in patients with heterogeneous genetics and tumor microenvironment composition. Additionally, virtually all humans are immunized against polio as children and consequently, patients have pre-existing immunity to polio, which is challenging to recapitulate in mice and outside the scope of current studies. Development of murine models to study this will be needed for future investigations. This study outlines the adjuvant potential of PVSRIPO that drives the generation of antitumor immune responses. PVSRIPO-induced antitumor CTL responses occurred regionally (tumor-draining lymph nodes) and systemically (spleen), were tumor antigen-specific, and were associated with T cell infiltration in PVSRIPO-treated tumors. Such immune engagement and CTL initiation is desirable for current immunotherapies and would conceivably complement other modalities including immune checkpoint blockade or adoptive tumor-infiltrating lymphocyte (TIL)/CAR T lymphocyte therapy, by enhancing and maintaining TIL/CAR T cell activation.
MATERIALS AND METHODS
Study Design:
This study sought to define the cancer immunotherapy potential of PVSRIPO, a recombinant poliovirus, using in vitro and in vivo experiments. For in vitro studies, experimental findings were confirmed using multiple cell lines and cells from healthy volunteers (n=3). Cells were obtained from human subjects after written informed consent using protocols approved by the Duke University Institutional Review Board. For in vivo tumor volume measurements, treatment groups were randomized by cage and tumor volume at the time of treatment, to ensure equal mean and range of tumor volumes within each group. Measurements were obtained without knowledge of treatment information; however, measurements were recorded by the same person who administered treatment. To test the antitumor immune response after PVSRIPO therapy in vivo, mice were randomized by tumor size, with equal mean tumor volume between the control and PVSRIPO groups. Information on experimental repeats is presented in figure legends.
Cell lines and virus
DM6 and DM440 cell lines were a gift of Douglas Tyler (University of Texas, Medical Branch, Galveston, TX). SUM149 cells were a gift of Neil Spector (Duke University, Durham, NC). All other cell lines were obtained from ATCC. All lines, except for SUM149, were grown in 10% FBS in DMEM (Invitrogen); SUM149 cells were grown in 10% FBS in Ham’s DMEM-F12 medium (Lonza). PVSRIPO and mRIPO were grown in HeLa cells as previously described (75) and purified using a 0.45 μM syringe filter followed by concentration and filtration through a 100 kDa filter (Millipore); titers were measured by plaque assay (58). PBMCs and monocytes were obtained and processed as described in Supplemental Materials and Methods.
Immunoblot, ELISA, LDH release assay, and flow cytometry
Immunoblots were performed as described previously (15) using antibodies listed in Supplemental Materials and Methods. Dot blots were performed by adding an equal amount (2 μl) of cell pellet lysate or supernatant directly onto a nitrocellulose membrane, air dried, and processed identically to immunoblots. ELISA was performed using the manufacturer’s recommendations for TNF-α, IL-12, Granzyme B (all R&D Biosystems); IFN-γ (Invitrogen); and IFN-β (PBL Biosciences). LDH release assays were performed per the manufacturer’s instructions (Thermo-Fisher). DM6 conditioned medium (DM6CM) was analyzed by cytokine array immunoblot (RayBiotech Inc.), following the manufacturer’s instructions. To analyze DC phenotype, DCs were blocked with anti-human CD16/32 Fc block (eBioscience) and stained with the following antibodies: CD83, CD86, CD197 (CCR7), CD80, CD64, HLA-DR (Beckman Coulter), and MHC-I (BD Biosciences); antibodies against CD11c, CD14, CD11b (Beckman Coulter) were used to confirm differentiation and purity. To examine tumor immune cell infiltration, tumors were harvested at the indicated time points. Tumors were minced and incubated in RPMI-1640 containing 1.67 Wunsch units/ml Liberase (Sigma) and 100 units/ml DNAse (Sigma). Cells were filtered, blocked with anti-mouse CD16/CD32 Fc block, and stained with the following panels of antibodies and appropriate isotype controls: panel 1: live/dead cell stain, Ly6G, Ly6C, CD45.2, CD11c, CD11b, F4/80, MHC-II; (Biolegend) Panel 2: live/dead cell stain, NK1.1, CD11b, CD45.2, CD8, CD4, B220 (Biolegend). Data were acquired on a Cytoflex B5-R3-V5 (Beckman Coulter) flow cytometer and analyzed using FlowJo (Tree Star, Inc.) software.
Virus infection assays and oncolysate preparation
Five hundred thousand cells were plated in 35 mm dishes, and infected at the designated MOI by adding virus to medium; DC and cancer cell infections were done in AIM-V medium or growth medium, respectively. For cell protein and dsRNA release experiments, cells were washed 5 times with serum-free DMEM before infection in serum-free DMEM. Oncolysates for DC and T cell assays were prepared by adding virus to a 75% confluent 100 mm dish in AIM-V medium (48 h, MOI = 0.1), followed by centrifugation to remove cell debris. Mock controls were generated similarly, only without virus addition. For virus removal from oncolysates using 100 kDa cutoff centrifuge filters, filters were centrifuged after addition of the oncolysate, and flow-through was used in experiments after confirming the absence of virus by plaque assay.
Generation and RNA electroporation of DCs
Cloning of tumor antigen cDNA and production of tumor antigen-encoding RNA were performed using standard techniques (see Supplemental Materials and Methods). Cytokines were obtained from R&D Systems unless otherwise noted. iDCs were generated from PBMCs as previously described (27) using AIM-V medium + 800 U/ml human GM-CSF (Berlex Laboratories) and 500 U/ml human IL-4. iDCs were harvested on day 6. GM-CSF and IL-4 were maintained during all DC treatments. To generate PVSRIPO oncolysate-loaded DCs, 1 ml of lysate (described above) was added to 1×106 iDCs (24 h). To generate mDCs, a cytokine cocktail of TNF-α (10 ng/ml), IL-1β (10 ng/ml), IL-6 (1000 U/ml), and PGE2 (1 μg/ml) (27) was added to iDCs (18–20 h). For RNA transfection, DCs were electroporated as described previously (59). RNA-electroporated DCs were cultured overnight and used as targets in Eu-release CTL assays the following day.
In vitro stimulation of T cells with DCs treated with PVSRIPO oncolysate
PBMCs were thawed and resuspended in PBS and treated with DNase I (Sigma) at 200 U/mL for 20 minutes at 37°C. DNase I-treated PBMCs were incubated for 1 h at 37°C, 5% CO2 in a humidified incubator. Non-adherent cells were harvested and stimulated with DCs loaded with PVSRIPO-induced oncolysate at a responder cell to stimulator DC ratio of 10:1 in the presence of 25 ng/ml IL-7. All stimulations were done in RPMI 1640 with 10% FCS, 2 mM L-glutamine, 20 mM HEPES, 1 mM sodium pyruvate, 0.1 mM MEM non-essential amino acids, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 5×10-5 M β-mercaptoethanol (CTL stimulation medium). The responder cell concentration was 2×106 cells/ml. IL-2 was added at 100 U/ml on day 3 and at 50 U/ml every 4-5 days. T cells were maintained at 1-2×106 cells/ml in CTL stimulation medium. In some assays, T cells were restimulated with PVSRIPO oncolysate loaded DCs at a responder to stimulator ratio of 10:1 after 7 days. T cells were harvested on day 12-14, counted, and used as effector cells in a CTL assay.
In vitro CTL assay
Tumor cell lines and RNA-electroporated DCs were used as targets. Cells were harvested, washed to remove all traces of medium and Eu-labeled (Supplemental Materials and Methods). Ten-thousand Eu-labeled targets (T) and serial dilutions of effector cells (E) at varying E:T ratios were incubated in 200 μl of CTL stimulation medium without antibiotics in 96-well V-bottom plates. The plates were centrifuged and incubated (4 h). Supernatant (50 μl was harvested and added to enhancement solution (150 μl Wallac, Perkin-Elmer) in 96-well flat-bottom plates. Eu release was measured by time-resolved fluorescence using the VICTOR3 Multilabel Counter (Perkin-Elmer). Specific cytotoxic activity was determined using the formula: % specific release = [(experimental release - spontaneous release)/(total release - spontaneous release)] × 100. Spontaneous release in target cells was <30% of total release by detergent. Spontaneous release in target cells was determined by incubating the target cells in medium without T cells. All assays were done in triplicate.
Generation of murine cancer cell lines and mouse-adapted PVSRIPO (mRIPO)
Human rhinovirus IRESes are functionally impaired in mouse cells, and mice do not express a functional PV receptor. Thus, we generated mouse-adapted PVSRIPO (mRIPO) and human CD155-expressing murine cancer cell lines. A CD155 pcDNA3 expression plasmid was kindly provided by Eckard Wimmer (Stony Brook University, NY), and pHIV-luciferase was a gift of Bryan Welm (Addgene plasmid #21375). CD155 was subcloned into pHIV-luciferase, and lentivirus was produced (see Supplemental Materials and Methods). KB158R, a genetically engineered murine astrocytoma line [derived from a mutant Nf1 and Trp53 mouse model (60)]; a gift of John Sampson (Duke University, NC)] was lentivirally transduced with CD155 followed by selection (10 μg/ml blasticidin). A stable CD155-expressing clone was identified by immunoblot. The same approach was used to produce B16-F10.9-OVA-CD155. To generate mRIPO, KB158R-CD155 expressing cells were infected with PVSRIPO and washed three times with serum-free DMEM to remove unbound virus. After 48 h, supernatant was collected and used to infect HeLa cells to amplify any propagated virus. Amplified virus was added to fresh KB158R-CD155 cells, and this process was repeated 20 times. The resulting virus was prepared and purified as described above, and sequenced. This revealed seven genetic adaptations in the 5’UTR of mRIPO, five of which were identical to those observed after serial passage adaptation of a PV:HRV2 IRES construct to mouse competence (46).
B16-F10.9-OVA-CD155 immunotherapy model
CD155-tg mice were derived from a homozygous colony provided by Julie Pfeiffer (UT Southwestern, TX) and were bred in-house for all studies. For tumor initiation, 5×105 B16-F10.9-OVA-CD155 cells were implanted into the right flank of 6–8 week CD155-tg C57Bl/6 mice. Mice were treated when tumor volumes reached ~50–100 mm3. DMEM (control) or mRIPO (1-5×107 pfu) were injected in a volume of 20 μl into tumors via a Hamilton syringe. Tumor volume was determined by caliper measurements using the equation (L×W×W)/2; survival endpoint was tumor volume >1000 mm3. All mouse experiments were performed under approved Duke University IACUC protocols and represent combined data from two independent experiments.
In vivo induction of CTLs, CTL assay
Seven days after treatment of tumor-bearing CD155-tg C57Bl/6 mice with DMEM or mRIPO, tumor-draining inguinal lymph nodes were harvested (n = 4/group), pooled, processed into a single cell suspension, and plated in a 6-well plate (3×106 cells/well). Cells were cultured in CTL stimulation medium (see above) with 20 U/ml IL-2 for 5 days. Lymph node responder cells were stimulated with 3×105 activated B cells expressing TRP-2 (1.5×105) and OVA antigen (1.5×105). B cell activation is described in Supplemental Materials and Methods. EL4 thymoma cells electroporated with TRP-2, OVA, or GFP-encoding mRNA or B16-F10.9-OVA-CD155 cells were used as targets. For tetramer analysis, tumor-draining inguinal lymph nodes (n = 4/group) were processed and cultured individually in a 24-well plate (5×105 cells/well) in CTL stimulation medium. Activated B cells were co-incubated with 2.5 μg/ml TRP-2 peptide (aa 180-188, SVYDFFVWL H-2Kb epitope; 2 h) and used as stimulators. TRP-2 peptide-pulsed B cells were harvested, washed, and 5×104 B cells were cultured with 5×105 lymph node responder cells for 5 days. For studies with splenocytes, spleens were harvested 14 days after DMEM or mRIPO treatment; red blood cells were lysed using ammonium chloride Tris buffer. Splenocytes (107) were cultured with 2×106 B16-F10.9-OVA-CD155 tumor cells in RPMI-10% FCS in a 6-well tissue culture plate (48 h). Supernatant was harvested and analyzed by ELISA.
Statistical analysis
Data are summarized as means ± SEM. Statistical significance in immune assays comparing two groups was calculated using paired two-tailed Student’s t test. Statistical analysis was conducted using GraphPad Prism version 6. A probability of <0.05 (p<0.05) was considered statistically significant. Specific tests were performed as described in figure legends.
Supplementary Material
One-Sentence Summary.
PVSRIPO infection of tumors elicits antitumor immunity by damaging neoplastic cells and activating type I IFN responses in antigen-presenting cells.
Acknowledgments:
The authors thank David Snyder, Katelyn Rudemiller, and Victoria Frazier for assistance with the animal studies. We are also grateful to Dr. Dani Bolognesi for reading the manuscript and insightful comments. We thank Dr. Julie Pfeiffer (UT Southwestern, TX) for human CD155 transgenic mice, Dr. Douglas Tyler (University of Texas, Medical Branch, TX) for DM6 and DM440 cell lines, Dr. Neil Spector (Duke University, NC) for SUM149 cell line, Dr. Eckard Wimmer (Stony Brook University, NY) for pcDNA3-CD155 cDNA, Dr. Bryan Welm (University of Utah, UT) for pHIV lentivirus plasmid (Addgene plasmid #21375), and Dr. John Sampson (Duke University, NC) for the KB158R murine glioma cell line.
Funding: This work was supported by PHS grant CA197264 (DDB); PHS grants CA124756 and CA190991, grants from the Lefkofsky Family Foundation, from Hope & Gavin Wolfe, and the BLAST Glioblastoma Foundation (MG); Department of Defense (DoD) breast cancer research program (BCRP) award W81XWH-16-1-0354 and discretionary support from the Department of Surgery (SKN).
Footnotes
Author contributions: SKN, MCB, MG contributed to the conception and design of the study. SKN, MCB, EKH, MG administered the project and carried out statistical analyses. SKN, MCB, EKH, DB, MG developed the methodology and empirical approaches. The investigations were carried out by SKN, MCB, EKH, DB, ED, MM. SKN, MG, VC and DDB contributed reagents to conduct this study. The original manuscript draft was composed by SKN, MCB, MG and all authors participated in editing and revising the text and figures. The project was supervised by SKN, MG.
Competing Interests: MCB, VC, DDB, MG and SKN own intellectual property related to this research, which has been licensed to Istari Oncology, Inc. MG and DDB are cofounders and equity holders in Istari Oncology, Inc. SKN, MCB, DDB, MG are inventors on patent application PCT/US2017/039953 held/submitted by Duke University that covers the composition and methods for activating antigen presenting cells with chimeric poliovirus.
Data and materials availability: Materials are available to noncommercial researchers through a material transfer agreement with Duke University Medical School upon filing a request with Drs. Smita Nair or Matthias Gromeier.
REFERENCES AND NOTES
- 1.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF, Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 5, 200ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou W, Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5, 263-274 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Palucka K, Banchereau J, Cancer immunotherapy via dendritic cells. Nat Rev Cancer 12, 265-277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilboa E, DC-based cancer vaccines. J Clin Invest 117, 1195-1203 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH, Therapeutic cancer vaccines. J Clin Invest 125, 3401-3412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ploegh HL, Viral strategies of immune evasion. Science 280, 248-253 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E, Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci U S A 97, 6803-6808 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gromeier M, Alexander L, Wimmer E, Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A 93, 2370–2375 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takai Y, Miyoshi J, Ikeda W, Ogita H, Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol 9, 603-615 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Freistadt MS, Fleit HB, Wimmer E, Poliovirus receptor on human blood cells: a possible extraneural site of poliovirus replication. Virology 195, 798-803 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Martinet L, Smyth MJ, Balancing natural killer cell activation through paired receptors. Nat Rev Immunol 15, 243-254 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Merrill MK, Gromeier M, The double-stranded RNA binding protein 76:NF45 heterodimer inhibits translation initiation at the rhinovirus type 2 internal ribosome entry site. J Virol 80, 6936-6942 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neplioueva V, Dobrikova EY, Mukherjee N, Keene JD, Gromeier M, Tissue type-specific expression of the dsRNA-binding protein 76 and genome-wide elucidation of its target mRNAs. PLoS One 5, e11710 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobrikova EY, Goetz C, Walters RW, Lawson SK, Peggins JO, Muszynski K, Ruppel S, Poole K, Giardina SL, Vela EM, Estep JE, Gromeier M, Attenuation of neurovirulence, biodistribution, and shedding of a poliovirus:rhinovirus chimera after intrathalamic inoculation in Macaca fascicularis. J Virol 86, 2750-2759 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown MC, Bryant JD, Dobrikova EY, Shveygert M, Bradrick SS, Chandramohan V, Bigner DD, Gromeier M, Induction of viral, 7-methyl-guanosine cap-independent translation and oncolysis by mitogen-activated protein kinase-interacting kinase-mediated effects on the serine/arginine-rich protein kinase. J Virol 88, 13135-13148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown MC, Dobrikov MI, Gromeier M, Mitogen-activated protein kinase-interacting kinase regulates mTOR/AKT signaling and controls the serine/arginine-rich protein kinase-responsive type 1 internal ribosome entry site-mediated translation and viral oncolysis. J Virol 88, 13149-13160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown MC, Gromeier M, MNK Controls mTORC1:Substrate Association through Regulation of TELO2 Binding with mTORC1. Cell Rep 18, 1444-1457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW, Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem 257, 14806-14810 (1982). [PubMed] [Google Scholar]
- 19.Srivastava P, Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol 20, 395-425 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G, Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17, 97-111 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Lotze MT, Tracey KJ, High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5, 331-342 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Richards OC, Martin SC, Jense HG, Ehrenfeld E, Structure of poliovirus replicative intermediate RNA. Electron microscope analysis of RNA cross-linked in vivo with psoralen derivative. J Mol Biol 173, 325-340 (1984). [DOI] [PubMed] [Google Scholar]
- 23.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S, Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101-105 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Mellman I, Coukos G, Dranoff G, Cancer immunotherapy comes of age. Nature 480, 480-489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinman RM, Linking innate to adaptive immunity through dendritic cells. Novartis Found Symp 279, 101-109; discussion 109,-113, 216-109 (2006). [PubMed] [Google Scholar]
- 26.Wahid R, Cannon MJ, Chow M, Dendritic cells and macrophages are productively infected by poliovirus. J Virol 79, 401-409 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nair S, Archer GE, Tedder TF, Isolation and generation of human dendritic cells. Curr Protoc Immunol Chapter 7, Unit7 32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G, Type I interferons in anticancer immunity. Nat Rev Immunol 15, 405-414 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu YX, Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 25, 37-48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu YX, Weichselbaum RR, STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843-852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Husemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Tureci O, Sahin U, Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396-401 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Campbell SA, Lin J, Dobrikova EY, Gromeier M, Genetic determinants of cell type-specific poliovirus propagation in HEK 293 cells. J Virol 79, 6281-6290 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiffer R, Baron J, Dagtekin G, Jahnen-Dechent W, Zwadlo-Klarwasser G, Differential regulation of the expression of transporters associated with antigen processing, TAP1 and TAP2, by cytokines and lipopolysaccharide in primary human macrophages. Inflamm Res 51, 403-408 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M, Wiendl H, Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol 155, 172-182 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol, (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mia S, Warnecke A, Zhang XM, Malmstrom V, Harris RA, An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand J Immunol 79, 305-314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oswald IP, Gazzinelli RT, Sher A, James SL, IL-10 synergizes with IL-4 and transforming growth factor-beta to inhibit macrophage cytotoxic activity. J Immunol 148, 3578-3582 (1992). [PubMed] [Google Scholar]
- 38.Silverstein RL, Febbraio M, CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2, re3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nair SK, De Leon G, Boczkowski D, Schmittling R, Xie W, Staats J, Liu R, Johnson LA, Weinhold K, Archer GE, Sampson JH, Mitchell DA, Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin Cancer Res, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pruitt SK, Boczkowski D, de Rosa N, Haley NR, Morse MA, Tyler DS, Dannull J, Nair S, Enhancement of anti-tumor immunity through local modulation of CTLA-4 and GITR by dendritic cells. Eur J Immunol 41, 3553-3563 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dannull J, Haley NR, Archer G, Nair S, Boczkowski D, Harper M, De Rosa N, Pickett N, Mosca PJ, Burchette J, Selim MA, Mitchell DA, Sampson J, Tyler DS, Pruitt SK, Melanoma immunotherapy using mature DCs expressing the constitutive proteasome. J Clin Invest 123, 3135-3145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH, Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol 27, 3135-3142 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Van Blerkom LM, Role of viruses in human evolution. Am J Phys Anthropol Suppl 37, 14-46 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreiber K, Rowley DA, Riethmuller G, Schreiber H, Cancer immunotherapy and preclinical studies: why we are not wasting our time with animal experiments. Hematol Oncol Clin North Am 20, 567-584 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Koike S, Taya C, Kurata T, Abe S, Ise I, Yonekawa H, Nomoto A, Transgenic mice susceptible to poliovirus. Proc Natl Acad Sci U S A 88, 951-955 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jahan N, Wimmer E, Mueller S, A host-specific, temperature-sensitive translation defect determines the attenuation phenotype of a human rhinovirus/poliovirus chimera, PV1(RIPO). J Virol 85, 7225-7235 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holl EK, Brown MC, Boczkowski D, McNamara MA, George DJ, Bigner DD, Gromeier M, Nair SK, Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolaczkowska E, Kubes P, Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13, 159-175 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Leliefeld PH, Koenderman L, Pillay J, How Neutrophils Shape Adaptive Immune Responses. Front Immunol 6, 471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen JL, Morris CR, Solheim JC, Virus evasion of MHC class I molecule presentation. J Immunol 171, 4473-4478 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD, Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208, 1989-2003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C, Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424, 324-328 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Cella M, Salio M, Sakakibara Y, Langen H, Julkunen I, Lanzavecchia A, Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J Exp Med 189, 821-829 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reis e Sousa C, Dendritic cells as sensors of infection. Immunity 14, 495-498 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Uribe-Querol E, Rosales C, Neutrophils in Cancer: Two Sides of the Same Coin. J Immunol Res 2015, 983698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mantovani A, Cassatella MA, Costantini C, Jaillon S, Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11, 519-531 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Kim ND, Luster AD, The role of tissue resident cells in neutrophil recruitment. Trends Immunol 36, 547-555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dulbecco R, Production of Plaques in Monolayer Tissue Cultures by Single Particles of an Animal Virus. Proc Natl Acad Sci U S A 38, 747-752 (1952). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee J, Boczkowski D, Nair S, Programming human dendritic cells with mRNA. Methods Mol Biol 969, 111-125 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T, Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 26, 109-113 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Abdel-Wahab Z, Kalady MF, Emani S, Onaitis MW, Abdel-Wahab OI, Cisco R, Wheless L, Cheng TY, Tyler DS, Pruitt SK, Induction of anti-melanoma CTL response using DC transfected with mutated mRNA encoding full-length Melan-A/MART-1 antigen with an A27L amino acid substitution. Cell Immunol 224, 86-97 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Cisco RM, Abdel-Wahab Z, Dannull J, Nair S, Tyler DS, Gilboa E, Vieweg J, Daaka Y, Pruitt SK, Induction of human dendritic cell maturation using transfection with RNA encoding a dominant positive toll-like receptor 4. J Immunol 172, 7162-7168 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Lee J, Boczkowski D, Nair S, Engineering B cells with mRNA. Methods Mol Biol 969, 101-110 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Lee J, Dollins CM, Boczkowski D, Sullenger BA, Nair S, Activated B cells modified by electroporation of multiple mRNAs encoding immune stimulatory molecules are comparable to mature dendritic cells in inducing in vitro antigen-specific T-cell responses. Immunology 125, 229-240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nair S, Boczkowski D, Moeller B, Dewhirst M, Vieweg J, Gilboa E, Synergy between tumor immunotherapy and antiangiogenic therapy. Blood 102, 964-971 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Nair S, McLaughlin C, Weizer A, Su Z, Boczkowski D, Dannull J, Vieweg J, Gilboa E, Injection of immature dendritic cells into adjuvant-treated skin obviates the need for ex vivo maturation. J Immunol 171, 6275-6282 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Nair SK, Heiser A, Boczkowski D, Majumdar A, Naoe M, Lebkowski JS, Vieweg J, Gilboa E, Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 6, 1011-1017 (2000). [DOI] [PubMed] [Google Scholar]
- 68.Blomberg K, Granberg C, Hemmila I, Lovgren T, Europium-labelled target cells in an assay of natural killer cell activity. I. A novel non-radioactive method based on time-resolved fluorescence. J Immunol Methods 86, 225-229 (1986). [DOI] [PubMed] [Google Scholar]
- 69.Rose S, Misharin A, Perlman H, A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry A 81, 343-350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghasemlou N, Chiu IM, Julien JP, Woolf CJ, CD11b+Ly6G- myeloid cells mediate mechanical inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A 112, E6808-6817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, Gabrilovich DI, Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol 160, 1224-1232 (1998). [PubMed] [Google Scholar]
- 72.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP, Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 92, 4150-4166 (1998). [PubMed] [Google Scholar]
- 73.Li G, Kim YJ, Broxmeyer HE, Macrophage colony-stimulating factor drives cord blood monocyte differentiation into IL-10(high)IL-12absent dendritic cells with tolerogenic potential. J Immunol 174, 4706-4717 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Hu J, Jo M, Eastman BM, Gilder AS, Bui JD, Gonias SL, uPAR induces expression of transforming growth factor beta and interleukin-4 in cancer cells to promote tumor-permissive conditioning of macrophages. Am J Pathol 184, 3384-3393 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Michielsen AJ, Hogan AE, Marry J, Tosetto M, Cox F, Hyland JM, Sheahan KD, O'Donoghue DP, Mulcahy HE, Ryan EJ, O'Sullivan JN, Tumour tissue microenvironment can inhibit dendritic cell maturation in colorectal cancer. PLoS One 6, e27944 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rose-John S, IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci 8, 1237-1247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, Kamimura D, Ueda N, Iwakura Y, Ishihara K, Murakami M, Hirano T, IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 173, 3844-3854 (2004). [DOI] [PubMed] [Google Scholar]
- 78.The UniProt C, UniProt: the universal protein knowledgebase. Nucleic Acids Res 45, D158-D169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.