

Abstract
VIM2 is a carbapenem-hydrolyzing metallo β–lactamase (MBL) found in clinical isolates of ESKAPE pathogens. For drug development, currently, there is a lack of lead compounds with optimal therapeutic potential. Here we report the discovery of 1-hydroxypyridine-2(1H)-thiones-6-carboxylic acid (3) as a potent VIM2 inhibitor with a Ki of 13 nM and low cytotoxicity CC50 of 97.4 μM. We further showed this inhibitor can restore the antibiotic activity of amoxicillin against VIM-2 producing E. coli in whole cells assays. Its potential mode of binding was examined by molecular modeling and its stability in mouse and human plasma studies was assessed. Overall, 3 exhibits a remarkable 0.99 ligand efficiency against VIM2, a 6,900 fold difference in cytotoxicity from its parent compound and a therapeutic index (TI) of 880, making it a promising lead candidate for combination antibacterial therapy development.
Introduction
Combination antibacterial therapy with β–lactamase inhibitors, remains a viable strategy for overcoming β–lactam drug resistance. With the recent approval of Avibactam, currently four β–lactamase inhibitors are available for combination antibacterial therapy with β–lactam antibiotics.[1] With the continuing emergence of β–lactam drug resistance worldwide, the discovery of other β–lactamase inhibitors remains an urgent area of research.
Expression of β–lactamase from innate or acquired bla gene is the leading mechanism of β–lactam drug resistance. β–lactamase enzymatically cleaves the β–lactam ring, rendering the β–lactam antibiotics inactive. Based on their mechanistic action of hydrolysis, β– lactamases are commonly referred to as serine (class A, C and D) or metallo β–lactamase (MBLs) (class B).[2] IMP, VIM and NDM belong to the subclass B1 of the metallo β–lactamase subfamily, consisting of, in most enzymes, dinuclear zinc metal cofactors necessary for enzyme catalysis. There is currently a lack of lead compounds with optimal therapeutic potential against MBLs available for development. To identify novel classes of metallo β–lactamase inhibitors (MBLi), we utilized VIM2, a carbapenemase commonly found in clinical isolates of ESKAPE pathogens, as the biochemical screening platform for MBLi discovery.
L-Captopril is an angiotensin converting enzyme inhibitor approved by the FDA for the treatment of hypertension and congestive heart failure. For over a decade, L-captopril and its stereoisomer have been shown to exhibit broad spectrum inhibitory activity against various MBLs.[3, 4] Its inhibitory potency stems from its thiol group which is viewed as a pharmacological liability for non-specific zinc binding against other metalloenzymes and is prone to inactivation by metabolic oxidation[5]. As such, captopril has never been further pursued clinically as a β-lactamase inhibitor for combination antibacterial therapy. To date, its use has been limited to laboratory for improving our understanding of MBL inhibition in antibacterial discovery. Recent structural characterization of Captopril stereoisomer against IMP-1, Bc1I, and VIM-2 has shown a common mode of binding involving bridge chelation between deprotonated thiolate ion with the two zinc ions (Figure S1).[4] To achieve unique nanomolar inhibition against VIM2 as compared to other MBLs, a salt bridge interaction between its carboxylate to the ionized Arg205 and hydrogen bonding to N210 sidechain is essential. Identifying a novel scaffold that replaces the thiol as the zinc binding group (ZBG) would provide a significant advance in the design of potent MBLi.
1-Hydroxypyridine-2-thione (1,2-HPT), also referred to as pyrithione, is a heterocyclic thiohydroxamic acid [6] that forms a five-membered complex via its oxygen and sulfur atoms with zinc. Zinc pyrithione (ZPT), 2a, can be isolated from Chinese herbal roots Polyalthia nemoralis [7] and has been shown to possess a broad range of antimicrobial activities[8–14]. Most recently, we have reported compounds with the 1,2-HPT moiety as zinc specific chelating inhibitors of VanX for the re-sensitization of vancomycin against vancomycin resistant Enterococcus faecium (VREF)[15] and as selective inhibitors of HDAC8 for their potential treatment of leukemia.[10] These earlier successes against zinc enzymes, in particular in selectivity, have prompted us to further explore the application 1,2-HPT as potential MBLi for overcoming β–lactam drug resistance in ESKAPE pathogens. For comparison purposes, we included L-captopril and other representative compounds consisting of the hydroxamic and cyclic hydroxamic acid moieties as alternative ZBGs (Figure 1).
Figure 1.
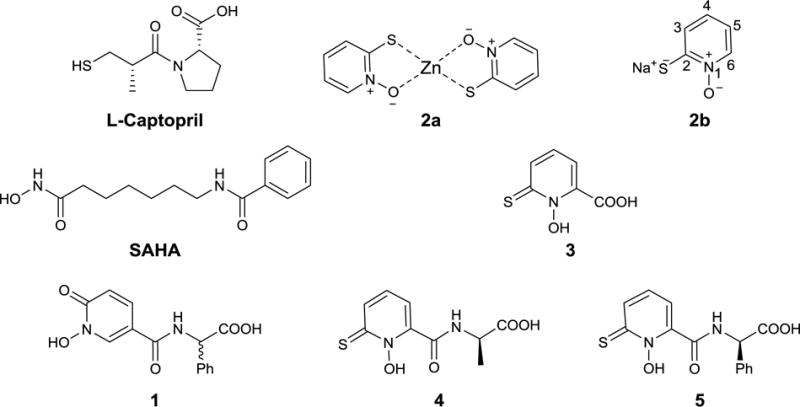
Structures of L-Captopril, SAHA, 1-hydroxypyridine-2(1H)-ones (1) and 1-hydroxypyridine-2(1H)-thiones analogues (2-5).
Results and Discussion
The synthesis of compound 1 is shown in Scheme 1, starting with 6-chloropyridine 3-carboxylic acid 6. N-oxidation with H2O2 in trifluoro acetic acid anhydride (TFAA) did not lead to a complete reaction, and substantial amounts of starting material were remained after 2-3 additional equivalents of hydrogen peroxide were introduced. Further, separation of N-oxide from starting acid was cumbersome and led to unacceptable yields. Therefore, we started the synthesis with methyl ester of 7. The N-oxidation reaction was carried out using urea-hydrogen peroxide addition complex in trifluoro acetic acid anhydride.[16] The 6-chloro N-oxide 8 was converted into 6-oxo compound 9 using TFAA keeping the carboxylic acid ester intact. The product was then benzylated with benzyl bromide in the presence of potassium carbonate. The methyl ester of N-benzyl compound 10 was hydrolyzed with NaOH in methanol. Initially we tried coupling with (R) methyl 2-amino-2-phenylacetate hydrochloride 12 under a variety of peptide-coupling conditions. Racemization occurred in almost all cases and in addition resulted in poor yields after chromatography. Racemization occurred with all reactions, but good yields could be accomplished with a coupling reagent combination- EDC, HOBt and DIPEA in DMF. Attempts to debenzylate with Pd/C/H2 were unsuccessful; almost all conditions lead to the benzyloxy group being cleaved, resulting in the 6-hydroxy pyridine derivative. However, first hydrolysis of the ester to the carboxylic acid with LiOH followed by hydrogenation, gave the desired compound 1 in acceptable yields with 80% enantiomer excess (chiral HPLC).
Scheme 1.
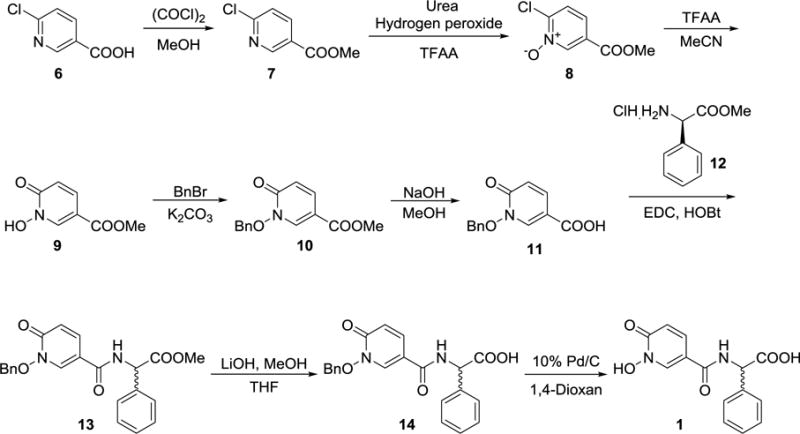
Synthesis of compound 1
Figure 2 shows steady-state kinetics for the hydrolysis of nitrocefin by VIM-2. The determined Km and kcat was 23.0 μM and 212 s−1, respectively, which is comparable values previously published.[17] As shown in Table 1, both clavulanate and tazobactam, two of the FDA-approved β–lactamase inhibitors, were included as controls for the preliminary single dose inhibition assay screening, with neither compounds exhibiting more that 25% inhibition against VIM2. Both compound 3 and L-captopril exhibited a remarkable 98% inhibition of VIM2. Poor inhibition was observed for SAHA and compound 1, suggesting that both hydroxamic acid and cyclic hydroxamic acid are poor starting pharmacophores for MBLi design. Interestingly, 1,2-HPT from a zinc pyrithione salt showed relatively weak inhibition activity of 15% as compared to the sodium pyrithione salt, which exbibited a comparable inhibition activity of 93% to compound 3. This difference is likely a result of available unchelated 1,2-HPT for VIM2 inhibition. Moderate inhibitory activities were also observed for 4 and 5 between the methyl and phenyl substitution, indicating inhibitory affinity can be enhanced by the addition of aromatic ring at the non-zinc binding group.
Figure 2.
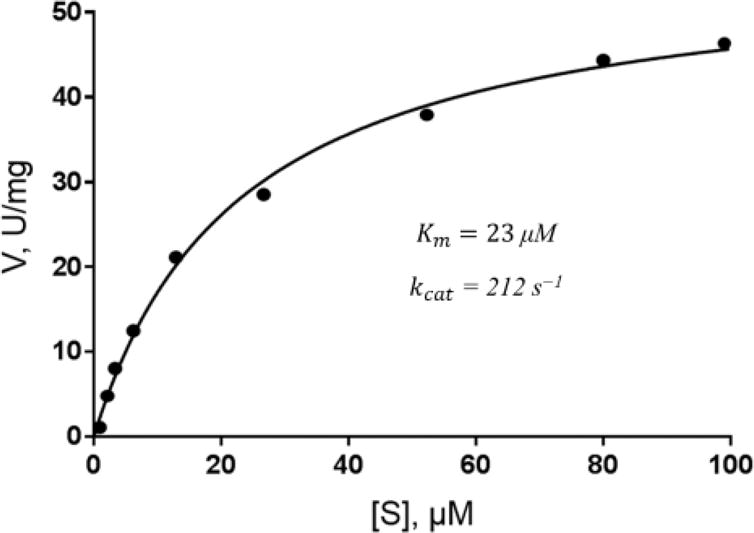
Steady-state kinetics for the hydrolysis of nitrocefin by VIM-2.
Table 1.
Single dose inhibition assay against VIM2
| Compound | % Inhibition at 50 μM |
|---|---|
| L-Captopril | 98 |
| Clavulanate | 15 |
| Tazobactam | 25 |
| SAHA | 29 |
| 1 | 12 |
| 2a (zinc salt) | 15 |
| 2b (sodium salt) | 93 |
| 3 | 98 |
| 4 | 18 |
| 5 | 34 |
The IC50 and inhibitory constant, Ki, for the four compounds with the highest single dose VIM2 inhibition were determined and are shown in Table 2. The determined IC50 for L-captopril was 6.6 μM and was comparable to that in an earlier report.[4] The Ki for L-captopril was 630 nM, corresponding to a ligand efficiency (LE) of 0.51. For compound 2b (sodium salt), the determined IC50 and Ki values were 908 nM and 217 nM, respectively, resulting in a remarkable LE of 1.15 and is likely the highest ever determined for a reported MBLi. For compound 3, which has been shown to be a selective HDAC inhibitor in our earlier study [10], the determined IC50 and Ki values were 270 nM and 13 nM, respectively, which corresponds to a 0.99 LE. Incorporation of a single amino acid with phenyl sidechain, 5, significantly raises its Ki by 576-fold from 0.013 nM to 7.5 μM. Given the observed data from the biochemical assays, the 20-fold enhanced potency by the addition of the carboxylic acid group adjacent to the N1 position of compound 2b is likely due to its electronic effect on 1,2-HPT zinc binding affinity, a perturbation to the 1,2-HPT pKa, and its interaction with nearby residues within the active site. Removal of the carboxylic acid from the adjacent N1 position of compound 3 or its separation by three atom spacer through amino acid addition could explain the significant of loss of inhibition potency between 4 and 5.
Table 2.
Inhibitory activity against VIM2
| Compound | N | Ki (μM) | IC50 (μM) | LE |
|---|---|---|---|---|
| L-Captopril | 14 | 0.63 | 6.6 (4.4) | 0.61 |
| 2b | 8 | 0.217 | 0.908 | 1.15 |
| 3 | 11 | 0.013 | 0.27 | 0.99 |
| 5 | 21 | 7.5 | 67.9 | 0.34 |
Ligand efficiency (LE) = -1.38 log (Ki) / N where N = number of heavy atoms.
IC50 in parenthesis reported from ref. 3.
Numerous structural studies of VIM2 have been carried out to examine the exact mechanism of ligand binding for various well-establish MBLi’s.[4, 18] To better understand the mechanism of MBL binding, we examined the previously solved X-ray structures complexes of VIM2. As shown in Figure 3A, in the absence of ligands, two water molecules (W1 and W2) with W1 acting as a bridging ligand between the Zn1 and Zn2 ions. 1 is tetra-coordinated to H114, H116, H179 and W1, while Zn2 is penta-coordinated to D118, S198, H240, W1, and W2. The carboxylates of formic acid displaces W2 from Zn2 while the thiolate ion of D-captopril replaces W1 as the bridging ligand (Figure 3B). D-captopril also participates in hydrogen bonding to the amide hydrogen of N210 sidechain and forms a direct salt bridge with R205 (Figure 3C). Cross examination with other available MBLs (PDB: 1DD6, 3VQZ, 2QDT, 2FU9) with bound ligands consistently showed thiolate as the preferred bridging ligand over carboxylate when both are present. Given the fact that pyrithione can undergo resonance to form a thiolate ion, the expected mode of binding involves thiolate ion as the bridging atom between the two zinc ions (Figure 3D).
Figure 3.

VIM2 active site (A) without ligand (4NQ2), with (B) formic acid (4BZ3), (C) D-captopril (4C1E) and (D) model compound 3. The chelated waters are labeled as W1 and W2.
To improve our understanding of potential mode of binding for 3, molecular modeling was carried out using Schrodinger modeling suites[19]. Docking with Glide into metalloenzymes did not reliably reproduce the crystallographically-observed mode of binding with observed sulfur atoms acting as a bridging ligand between the two zinc ions (unpublished). Due to the presence of the charged carboxylate group, the dominant pose involved chelation of the carboxylate to the zinc ions. As such, comparative modeling based on established structurally-determined mode of binding was carried out to yield the most consistent model to corroborate with our biochemical data (Figure 3D). The model of 3 binding to VIM2 was developed through overlaying of the chelating S and O atoms from 1,2-HPT moiety and its carboxylate group to the two crystallographic water sites (W1 and W2) and D-captopril’s carboxylate group. Such model would preserve the commonly observed mode of thiolate ligand binding with the sulfur acting as the chelating atom for bridging the two zinc ions for VIM2 binding. This model also consistently replaced the other chelating water atom, W2, and preserved the salt bridge interaction with R205. As D-captopril’s salt bridge interaction with Lys does not yield nanomolar inhibition in IMP1 and BcII,REF this mode of binding would corroborate with the observed nanomolar inhibitory activity of D-captopril that is not achieved in BcII and IMP1. The removal of the carboxylic acid would lead to the loss of the salt bridge interaction to R205, which would explain the 20-fold reduction in Ki for compound 2. Furthermore, this potential mode of binding will likely restrict the placement of the amino acid group at the D-captopril carboxylate site leading to significant loss in binding affinity to the active site as observed in 5.
We further demonstrated the clinical relevance of compound 3 by its ability to restore amoxicillin efficacy against VIM2-expressing E. coli. Clavulanate was included as control. As shown in Table 3, the growth of wild type E. coli can be effectively inhibited by 50 μM of amoxicillin regardless of the presence of the MBLi. The transformation and expression of VIM2 MBL lead to amoxicillin resistance in E. coli, rendering the treatment with amoxicillin alone and the amoxicillin – clavulanic acid combination therapy ineffective. Compound 2, which has a Ki of 217 nM, exhibits potent antibacterial activity against both E. coli strains with over 89% growth inhibition activity on its own, making it unsuitable as a β-lactamase inhibitor for combination antibacterial therapy. For L-captopril and 3, both compounds exhibit only moderate antibacterial activity on their own against both wild-type and VIM2-expressing E coli. As nanomolar inhibitors, both compounds effectively inhibited VIM2 and restored amoxicillin efficacy with over 90% synergistic growth inhibition against VIM2-expressing E. coli.
Table 3.
Cell viability assay
| Compound | E. coli | VIM2 expressing E. coli | ||
|---|---|---|---|---|
| Amox (−) | Amox (+) | Amox (−) | Amox (+) | |
| Amox | - | 3.6 | - | 77 |
| Clav | 86 | 2.9 | 93 | 57 |
| SAHA | 83 | 3.6 | 79 | 60 |
| L-Captopril | 86 | 5.0 | 87 | 9.3 |
| 2b | 5.3 | 1.9 | 11 | 2.1 |
| 3 | 77 | 3.6 | 61 | 3.2 |
The data are reported as relative percentage growth compared to untreated cells.
Since β-lactamase inhibitors serve primarily as a β-lactam antibiotic re-activating agents, β-lactamase inhibitors commonly do not exhibit sufficient antibacterial activity on their own. To evaluate its therapeutic potential, it is important to examine its efficacy in the presence of a β-lactam antibiotic. Here, we evaluated the effective β-lactamase inhibitor concentration in which a fixed concentration of antibiotic regains 50% of its growth inhibitory activities (EC50*). Such approach would avoid the immediate need to determine an optimal combination index necessary for future formulation studies and allow the direct comparison between two potential β-lactamase inhibitor candidates. As the growth of the wild-type E. coli could be effectively inhibited at over 96% by amoxicillin alone at 50 μM, the EC50* for 3 and L-captopril was determined under the same conditions against VIM-2 expressing E. coli. The determined EC50* was 0.110 μM for 3 and 1.7 μM for L-captopril (Table 4), which corresponds to a 2- to 10-fold change from its observed Ki.
Table 4.
Cell bioactivities and plasma stability
| Compound | CC50 (μM) | EC50* (μM) | TI | Human Plasma t1/2 (mins) | Mouse Plasma t1/2 (mins) |
|---|---|---|---|---|---|
| L-Captopril | - | 1.7 | - | - | - |
| 2b | 0.014 | - | - | - | - |
| 3 | 97 | 0.110 | 880 | 11.7 | 12.7 |
t1/2 – Half-life.
EC50* was determined in the presence of 50 μM amoxicillin.
Therapeutic index (TI) = CC50/50.
To determine the overall therapeutic index of these two compounds, we further assessed their cytotoxicities against human embryonic kidney HEK 293 cell lines by MTT assay as described earlier.[15] The CC50 of 3 was reported earlier.[15] Compound 2 was included to compare the pharmacological difference for the addition of the carboxylic acid group at the 6− position of 2. As 1,2HPT (2) is known as a non-selective, zinc-chelating agent with wide spectrum antimicrobial activities which was also observed here against both E. coli strains, our earlier studies have demonstrated compound 3 is a selective HDAC inhibitor with nearly 5000-fold range selectivity among all eleven HDACs and may not possess the same therapeutic profile as the parent compound 2. As shown in Table 4, the determined CC50 for 2 and 3 after 72 hrs treatment against HEK293 cells was 0.014 μM and 97 μM. This modest modification resulted in an unexpected and remarkable 6900-fold change in cytotoxicity that further supports 3 as a selective inhibitor that does not exhibit any reasonable high affinity for other biologically important zinc enzymes, a major concern in rational drug design. The determined TI for 3 was 880 UNITS, giving it a promising starting therapeutic window for further development.
To further explore the therapeutic potential of 1-hydroxypyridine-2-thione-6-carboxylic acid, 3, its stability in mouse and human plasma was also assessed. While the half-life for both compounds was determined to be quite short in mouse plasma, the half-life of 3 in human plasma was determined to be 11.7 and 12.7 mins, respectively (Table 4), suggesting further pharmacokinetics optimization is necessary.
Lastly, in an effort to address the mode of action of 1-hydroxypyridine-2-thione-6-carboxylic acid, 3, equilibrium dialysis experiments were conducted. Since compound 3 is known to coordinate metal ions, it was not clear whether the mode of inhibition was removal of the Zn(II) or simple binding to the Zn(II). Therefore, we incubated VIM-2 with increasing concentrations of compounds 2b and 3 (Figure 4) and used equilibrium dialysis and metal analysis to evaluate the metal content in the resulting enzyme samples. L-Captopril and EDTA were used as controls because of their contrasting modes of inhibition. EDTA, a well-known metal scavenger, inhibits all MBLs through a chelation mechanism.[doi:10.1128/AAC.01009-09] Conversely, L-captopril has been shown to be a competitive inhibitor of several MBLs.REF In alignment with the ICP-AES results, the metal content of VIM-2 is relatively unaffected by increasing concentrations of L-captopril, as opposed to the inverse relationship apparent with EDTA. (needs to be expanded on after EDTA data is collected)
Figure 4.
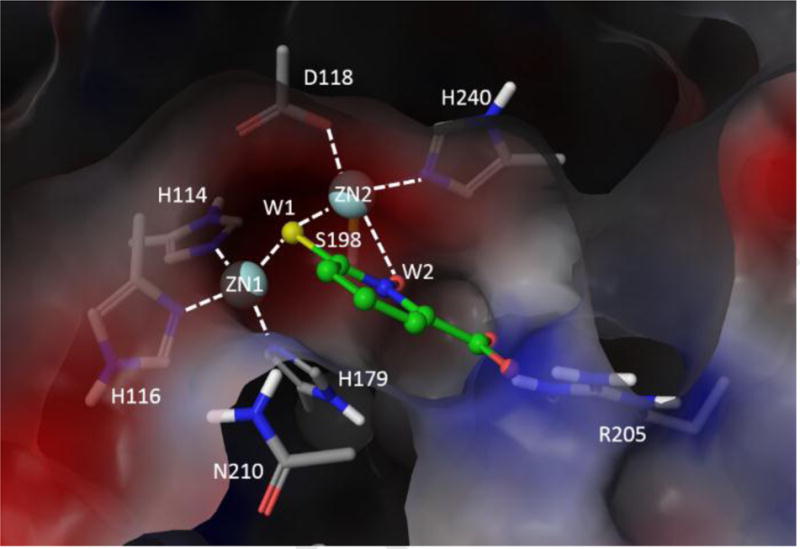
Top view of VIM2 binding site with compound 3 with its sulfur and oxygen atom chelating to ZN1 and ZN2 at the W1 and W2 water sites.
Conclusion
Herein we report the inhibition activity of three representative classes of zinc specific chelators as potential MBL inhibitors, namely hydroxamic acid, cyclic hydroxamic acid, and pyrithione (1,2HPT). The study demonstrated 1-hydroxypyridine-2(1H)-thiones-6-carboxylic acid, 3, as a potent nanomolar inhibitor of VIM2 with a Ki of 13 nM that corresponds to a remarkable 0.99 ligand efficiency. There was a 6900-fold change in cytotoxicity from its parent compound, 2, observed through the addition of a carboxylic acid group at the 6th position of 1,2-HPT that suggested limited off target liability against other cellular zinc enzymes. The mode of binding for 3 was further assessed by molecular docking, which corroborates with our biochemical activities. Binding of compound 3 involves the sulfur acting as a bridging ligand between the zinc ions, while its carboxylate group forms a salt bridge interaction to R205. This work is the first study of 1,2-HPT analogues as a potent and a novel MBLi and should serve as a lead candidate for further development.
Experimental Procedure
Materials and methods
Materials
L-captopril, SAHA, and pyrithione salts (2) were commercially purchased from Sigma Aldrich. Compounds 3-5 were described in our earlier studies.[10, 15] Compound 1 was from our in-house unpublished library and was included as a comparison compound to hydroxamic acid and 1,2-HPT. For the synthesis of 1, all chemicals were purchased from commercial suppliers and used as received unless otherwise stated. Flash silica gel chromatography was performed using standard commercial source (40-60 μm mesh). Inert reactions were carried out under nitrogen atmosphere (balloon), 1H NMR spectra were recorded at ambient temperature on a 300 MHz Varian FT-NMR instrument.
Synthesis
Methyl 6-chloronicotinate (7)
To a cooled (0 °C) solution of 6-chloronicotinic acid 6 (200 mg, 1.29 mmol) in CH2Cl2 (5 mL), 2 drops of DMF and oxalyl chloride (0.13 mL, 1.52 mmol) were added drop wise. The reaction mixture was stirred at room temperature for 16 h, and the volatiles were removed under reduced pressure. The residue was cooled to 0 °C and dissolved in CH2Cl2 (5 mL), methanol (1 mL) and heated to 40 oC (monitored by TLC). After 15 min, the solvents were removed under vacuum and diluted with ether (HOW MUCH?). The organic layer was washed with water and brine solution. The organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure, to afford methyl 6-chloronicotinate 7 (200 mg, 91%) as off-white solid; 1H NMR (400 MHz, CDCl3): δ 9.00 (s, 1H), 8.26 (d, J = 8.4Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 3.96 (s, 3H).
Methyl 6- chloropyridine-3-carboxylate-1-oxide (8)
To a suspension of methyl 6-chloronicotinate 7 (200 mg, 1.14 mmol)) and urea hydrogen peroxide (230 mg, 2.44 mmol) in CH3CN at 0 °C, trifluoro acetic anhydride (0.35 mL, 2.33 mmol) was added drop wise and stirred at room temperature for 16 h. The reaction was monitored with TLC; volatiles were concentrated under reduced pressure. The residue was partitioned between EtOAc (10 mL) and saturated sodium hydrogen sulfite (10 mL). The aqueous layer was extracted with EtOAc (2 × 10mL). The combined organic extracts were washed with brine solution, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford crude methyl-6-chloropyridine-3-carboxylate-1-oxide 8, which was carried forward to the next step without any further purification; 1H NMR (500MHz, DMSO-d6): δ 8.73 (s, 1H), 7.95 (d, J = 8.5Hz, 1H), 7.76 (d, J = 8.5Hz, 1H), 3.89 (s, 3H).
Methyl-1-hydroxy-6-oxo-1,6-dihydropyridine-3-carboxylate (9)
To the crude methyl-6-chloropyridine-3-carboxylate-1-oxide 8 (50 mg, 0.26 mmol) in CH3CN (2 mL), trifluoro acetic anhydride (2 mL) was added dropwise and stirred at room temperature. The reaction was monitored with TLC. After 1h, the volatiles were removed under reduced pressure. Solid sodium bicarbonate (100 mg), MeOH (10 mL) was added to the residue, and the suspension was filtered. The filtrate was concentrated under reduced pressure to provide the crude methyl-1-hydroxy-6-oxo-1,6-dihydropyridine-3-carboxylate 9, which was taken to the next step without any further purification. 1H NMR (400MHz, CD3OD): δ 8.56 (s, 1H), 7.87(d, J = 8.8Hz, 4H), 6.64 (d, J = 8.8 Hz, 4H), 3.85 (s, 3H).
Methyl 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-3-carboxylate (10)
To a stirred solution of methyl-1-hydroxy-6-oxo-1,6-dihydropyridine-3-carboxylate 9 (800 mg, 5.26 mmol), K2CO3 (2.1 gm, 15.6 mmol) in DMF (2 mL) at 0 °C, benzyl bromide (0.7 5mL, 6.3 mmol) was added drop wise and stirred at room temperature for 16 h. The reaction mixture was diluted with cold water (15 mL) and extracted with EtOAc (3 × 5 mL). The combined organic extracts were washed with water and brine solution, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified with silica gel (60-120 mesh) column chromatography (20% EtOAc-hexane) to afford methyl 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-3-carboxylate S5 (410 mg, 38.6 % after 3 steps) as a solid. 1H NMR (500 MHz, CD3OD): δ 8.22 (s, 1H), 7.9 (d, J = 9.5 Hz, 1H), 7.47-7.45 (m, 2H), 7.39-7.31 (m, 3H), 6.67(d, J = 9.5 Hz, 4H) 5.28 (s, 2H), 3.79 (s, 3H).
1-(Benzyloxy)-6-oxo-1,6-dihydropyridine-3-carboxylic acid (11)
To compound 5 (3.2 gm, 12.35 mmol) dissolved in MeOH (30 mL) at 0 °C, 1 N NaOH (25 mL, 24.7 mmol) was added and stirred at room temperature for 16 h. The volatiles were concentrated under reduced pressure to give the residue, which was acidified with 8 N HCl during which a solid was precipitated, filtered, washed with water, and dried under vacuum to afford 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-3-carboxylic acid 11 (2.08 gm, 66 %) as a solid. 1H NMR (400 MHz, CD3OD): δ 8.18 (s, 1H), 7.91 (d, J = 9.6 Hz, 1H), 7.47-7.45 (m, 2H), 7.40-7.38 (m, 3H), 6.67(d, J = 9.6 Hz, 4H) 5.29 (s, 2H).
Methyl 2-(1-(benzyloxy)-6-oxo-1, 6-dihydropyridine-3-carboxamido)-2-phenylacetate (13)
A mixture of 1-(benzyloxy)-6-oxo-1, 6-dihydropyridine-3-carboxylic acid 11 (1.6 g, 6.53 mmol) (R)-methyl 2-amino-2-phenylacetate hydrochloride 12 (1.3 g, 6.53 mmol), EDC.HCl (1.5 g, 7.82 mmol), HOBt (1.05 g, 7.83 mmol), and DIPEA (2.5 mL, 15.06 mmol) in DMF (10 mL) was stirred at room temperature for 6 h. The mixture was diluted with water (30 mL) and extracted with EtOAc (3 × 20mL), and combined organic extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure to give the crude residue. The residue was purified by column chromatography on silica gel (60-120) with 2% MeOH/CH2Cl2 as an eluent to yield methyl 2-(1-(benzyloxy)-6–oxo-1,6-dihydropyridine-3-carboxamido)-2-phenylacetate 13 (1.5 g, 62.5 %) as a solid. 1H NMR (400 MHz, CDCl3): δ 7.83 (s, 1H), 7.63 (d, J = 6.8Hz, 1H), 7.43-7.31 (m, 10H), 6.67-6.65 (m, 2H), 5.62 (d, J = 9.6Hz, 1H), 5.27(s,2H), 3.75 (s, 3H).
2-(1-(benzyloxy)-6-oxo-1,6-dihydropyridine-3-carboxamido)-2-phenylacetic acid (14)
To a stirred solution of methyl 2-(1-(benzyloxy)-6-oxo-1,6-dihydropyridine-3-carboxamido)-2-phenylacetate 13 (500 mg, 1.27 mmol) in THF: MeOH (1:1, 5 mL), 5% LiOH (5 mL,w/v) solution was added slowly and stirred at room temperature for 16 h, solvent was concentrated under reduced pressure. The residue was cooled to 0 °C and acidified with 1N HCl and extracted with 10% MeOH in CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with saturated brine solution, dried over Na2SO4 and concentrated to obtain 2-(1-(benzyloxy)-6-oxo-1, 6-dihydropyridine-3-carboxamido)-2-phenylacetic acid 14 (230 mg, 48%) as an off-white fluffy solid. 1H NMR (400 MHz, CD3OD): δ 8.33 (s, 1H), 7.94 (d, J = 6.8Hz, 1H), 7.48-7.32 (m, 11H), 6.68 (d, J = 9.6Hz, 1H), 5.53 (s, 1H), 5.27 (s, 2H).
2-(1-hydroxy-6-oxo-1, 6-dihydropyridine-3-carboxamido)-2-phenylacetic acid (1)
To 2-(1-(benzyloxy)-6-oxo-1,6-dihydropyridine-3-carboxamido)-2-phenylacetic acid 14 (3 g, 7.93 mmol) dissolved in 1,4-dioxan (300 mL), 10% Pd/C (300 mg) was added portion wise and stirred under hydrogen atmosphere for 8 h. The reaction was monitored with TLC. The catalyst was filtered through Celite bed and concentrated. The crude residue was purified using chiral HPLC, to obtain 1 (racemic, 700 mg, 23%) as an off-white solid. 1H NMR (400 MHz, CD3OD): δ 8.57 (s, 1H), 7.99 (d, J = 6.8Hz, 1H), 7.51-7.49 (m, 2H), 7.43-7.37 (m,3H), 6.66 (d, J =9.6 Hz, 1H), 5.64 (s, 3H).
Protein Expression and Purification
For our biochemical assay, the blaVIM2 gene, from a clinical strain of P. aeruginosa, was expressed using the pET24a (+) vector [20]. The pET24a-VIM-2 plasmid was transformed into competent BL21(DE3) E. coli cells. The cells were plated onto an LB-agar plate with kanamycin (25 μg/mL) and incubated overnight at 37 °C. A single colony was used to inoculate 50 mL of LB, containing 25 μg/mL kanamycin, and the culture was shaken overnight at 37 °C. From the overnight culture, 10 mL were transferred to 4 × 1L LB medium containing 25 μg/mL kanamycin. The cultures were grown at 37 °C until the optical density (OD600nm) reached 0.6-0.8, at which point protein production was induced with IPTG (0.5 mM) and ZnCl2 (100 μM). The temperature was reduced to 20 °C, and the cells were shaken for an additional 18 h. The cultures were harvested by centrifugation (8,000xg) for 10 min at 4 °C. The resulting pellets were re-suspended with 25 mL of 50 mM HEPES, pH 7.5, containing 500 mM NaCl (buffer B). The cells were lysed with three passes through a French Press. The lysate was centrifuged (15,000xg) for 30 min at 4 °C. The supernatant was dialyzed against 2L of 50 mM HEPES, pH 7.5 (buffer A), for 4 h. Buffer A was used to equilibrate a 25 mL Q-Sepharose column using an FPLC. The sample was loaded onto the column, and proteins were eluted with a linear gradient 0-500 mM NaCl with buffer B. Fractions containing VIM2, determined by SDS-PAGE, were pooled and concentrated to 2-3 mL in an Amicon ultraconcentrator equipped with a YM-10 membrane. Further purification was conducted with a Sephacryl S-200 gel filtration column using 50 mM HEPES, pH 7.5, containing 150 mM NaCl. Fractions containing pure VIM2 were pooled, and metal analyses were performed.[18]
Biochemical Assay
Nitrocefin (Cayman, CAS 41906-86-9) was used as the chromogenic substrate for all biochemical assay. The enzymatic activity of purified VIM2 was determined spectrophotometrically (Spectramax-M5-reader) at room temperature in 50 mM potassium phosphate buffer, pH 7.0. The rate of product formation was monitored based on the λmax=486 nm absorbance taken at 10 s intervals for 30 mins. The Km and kcat values were determined from 10 different concentrations of nitrocefin ranging from 0.001 to 100 μM with at least four independent initial-velocity measurements and fitted by nonlinear regression using Michaelis-Menten Enzyme kinetics with Graphpad Prism 6.
Single Dose Enzymatic Inhibition Assay
To identify potential MBL inhibitors, the relative change in the formation of hydrolyzed nitrocefin between treated and untreated VIM2 was determined as percentage inhibition. VIM-2 (5 nM) was pre-incubated for 10 mins with 50 μM of each compound, followed by the addition of 10 μM nitrocefin. The relative change in the absorbance at 486 nm after 30 mins was evaluated as percentage inhibition.
Dose Response Enzymatic Inhibition Assay and Inhibition Constant
Each inhibitor was pre-incubated at concentrations from 0.001 to 50 μM with 5 nM VIM-2 for 5 mins at room temperature in detergent buffer before addition of 10 μM nitrocefin. The rate of product formation was monitored based on the absorbance at 486 nm taken at 10s intervals for 30 mins. The relative change in absorbance was evaluated as percentage inhibition, and the IC50 was determined by fitting the data to a sigmoidal dose-response curve. The enzyme inhibition constant (Ki) was derived from initial-velocity measurements by nonlinear regression using competitive-inhibition enzyme kinetics using Graphpad Prism 6.
Bacteria Cell culture
Both the native and transformed cells lines have been reported previously [21]. The cells were cultured on 0.8 g/100 ml nutrient agar plate at pH 7.0 and 37 °C. The bacterial growth medium was diluted in nutrient broth (NB) to a concentration absorbance of 1.5 at OD600nm and incubated overnight in 10 ml capped culture tubes with shaking. An overnight culture of the bacterial strain was sub-cultured to an optical density of 0.06 at OD600nm into the NB medium and was then seeded at max 200 μl into the wells of a 96 well microtiter plate. Samples were then incubated at 37 °C and shaken at 200 rpm for 18 h. The absorbance was measured on an ELIZA plate reader at 600 nm and analyzed with the Gen5TM software suite (version 1.08).
Half Maximal Effective Concentration (EC50)
The bacterial culture was prepared as described above. The diluted subculture bacteria in NB medium was then set up to a final volume of 200 μl in clear flat-bottom 96-well plates containing ten different concentrations of each tested compound ranging from 0.001 μM to 50 μM. The mixing of the bacterial culture plate were then incubated in a 37 °C stationary shaken incubator at 200 rpm for 18 h before measuring their optical density at 600 nm. The EC50 values were obtained by fitting the data to a sigmoidal dose-response equation using Graphpad Prism 6.
Human cell line culture
Human embryonic kidney cell line (HEK293) was grown in DMEM (Dulbecco’s modifications of Eagle’s medium with L-glutamine & 4.5 g/L glucose) supplemented with fetal bovine serum 100 units/ml of penicillin G and 0.1 mg/ml of streptomycin sulfate in a humidified atmosphere of a 5% CO2 at 37 °C.
Cell Proliferation Assays
Cell proliferations were measured by counting viable cells by using the 3(4,5dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, St. Louis, MO) colorimetric dye-reduction assay. RPMI-1640 Gibco® (10% FBS, 2 mM L-glutamine, 1 mM pyruvate, 1% penicillin/streptomycin) growth medium was used, and cells were seeded at a concentration of 1× 104 cells/well in 200 μl culture medium and incubated at 37 °C in 5% CO2 incubator. After 72 hrs, 10 μl of MTT (5 mg/ml) dye was added to each well, and the plates were incubated for 4 hours at 37 °C in 5% CO2 incubator. After centrifuge with 1,500 rpm/10 mins, the supernatant was removed, 200 μl of dimethyl sulfoxide (DMSO) was added, and the plates were gently shaken to solubilize the formed formazan for 30 min. The absorbance was measured using a micro-plate reader at wavelength 590 nm. The CC50 values were obtained by fitting the data to a sigmoidal dose-response equation using Graphpad Prism 6.
Molecular Modeling
All modeling was performed using the Schrodinger modeling package.[19] The modeling study was based on the X-ray crystallographic structures of VIM2 (PDB code: 4NQ2, 4BZ3, 4C1E). All missing sidechains and hydrogen atoms were added with standard protein preparation protocols at physiological pH, followed by energy minimization using OPLS-AA 2005 force field with implicit solvent to optimize all hydrogen-bonding networks.
Plasma Stability Study
Each tested compound was spiked into pooled mouse or human plasma at a final concentration of 10 μM (0.1% DMSO) and incubated at 37 °C. The incubations were performed in triplicate. At different time points, 40 μL of incubation mixture were removed and immediately quenched by addition of 120 μL of acetonitrile containing appropriate internal standard and 0.5% formic acid. The quenched samples were centrifuged at 14,000 rpm for 5 min at 4 °C. The supernatants were injected into LC-MS/MS for analysis. The electrospray mass spectrum showed two prominent peaks that indicated that these compounds exist predominantly as dimers, but are at constant equilibrium to one another. Plasma stability was evaluated by monitoring the disappearance of both monomer and dimer of each compound over a certain time period. The peak area ratios of the analyte versus internal standard were used to calculate the remaining percentage at each time point. The natural logarithm of remaining percentage is plotted against time and the gradient of the line is used to determine the half-life t1/2 in plasma.
Equilibrium Dialysis – ICP-AES
VIM-2 was diluted to 6 μM with 50 mM HEPES, pH 7.5, containing 150 mM NaCl. The diluted protein solution was used to make several 5 mL aliquots. L-Captopril, EDTA, 2b, and 3 were each added to two aliquots. One aliquot contained one molar equivalent of inhibitor (6 μM), with respect to enzyme concentration, and the second aliquot contained two molar equivalents (12 μM). One aliquot received no inhibitor. Each aliquot was then incubated for 4 h at 4 °C. Following incubation, each aliquot was dialyzed versus 500 mL of 50 mM HEPES, pH 7.5, for 4 h at 4 °C. The aliquots were analyzed using ICP-AES, as previously described REF. The emission wavelengths used were 213.856, 238.892, 259.940, 231.604, and 324.754 nm for Zn, Co, Fe, Ni, and Cu, respectively.
Acknowledgments
We thank the Center for Drug Design and Department of Experimental and Clinical Pharmacology for the research support. We thank the University of Minnesota Supercomputing Institute for providing all the necessary computational resources.
References
- 1.Drawz SM, Papp-Wallace KM, Bonomo RA. New beta-Lactamase Inhibitors: a Therapeutic Renaissance in an MDR World. Antimicrob Agents Ch. 2014;58:1835–1846. doi: 10.1128/AAC.00826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palzkill T. Metallo-beta-lactamase structure and function. Ann Ny Acad Sci. 2013;1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinz U, Bauer R, Wommer S, Meyer-Klaucke W, Papamichaels C, Bateson J, Adolph HW. Coordination geometries of metal ions in d- or l-captopril-inhibited metallo-beta-lactamases. J Biol Chem. 2003;278:20659–20666. doi: 10.1074/jbc.M212581200. [DOI] [PubMed] [Google Scholar]
- 4.Brem J, van Berkel SS, Zollman D, Lee SY, Gileadi O, McHugh PJ, Walsh TR, McDonough MA, Schofield CJ. Structural Basis of Metallo-beta-Lactamase Inhibition by Captopril Stereoisomers. Antimicrob Agents Chemother. 2016;60:142–150. doi: 10.1128/AAC.01335-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou XH, Li Wan Po A. Stability and in vitro absorption of captopril, enalapril and lisinopril across the rat intestine. Biochem Pharmacol. 1994;47:1121–1126. doi: 10.1016/0006-2952(94)90382-4. [DOI] [PubMed] [Google Scholar]
- 6.Jacobsen FE, Lewis JA, Cohen SM. The design of inhibitors for medicinally relevant metalloproteins. Chemmedchem. 2007;2:152–171. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 7.Yao JZ, Liang HQ, Liao SX. Studies on the active Constituents of Polyalthia nemoralis A. et DC. Acta Pharmaceutica Sinica. 1994;29:845–850. [PubMed] [Google Scholar]
- 8.Marcheselli M, Azzoni P, Mauri M. Novel antifouling agent-zinc pyrithione: Stress induction and genotoxicity to the marine mussel Mytilus galloprovincialis. Aquat Toxicol. 2011;102:39–47. doi: 10.1016/j.aquatox.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 9.Tailler M, Senovilla L, Lainey E, Thepot S, Metivier D, Sebert M, Baud V, Billot K, Fenaux P, Galluzzi L, Boehrer S, Kroemer G, Kepp O. Antineoplastic activity of ouabain and pyrithione zinc in acute myeloid leukemia. Oncogene. 2012;31:3536–3546. doi: 10.1038/onc.2011.521. [DOI] [PubMed] [Google Scholar]
- 10.Muthyala R, Shin WS, Xie JS, Sham YY. Discovery of 1-hydroxypyridine-2-thiones as selective histone deacetylase inhibitors and their potential application for treating leukemia. Bioorganic & medicinal chemistry letters. 2015;25:4320–4324. doi: 10.1016/j.bmcl.2015.07.065. [DOI] [PubMed] [Google Scholar]
- 11.Han GY, Xu BX, Wang XP, Liu MZ, Xu XY, Meng LN, Chen ZL, Zhu DY. Study on the Active Principle of Polyalthia nemoralis: I. The Isolation and Identification of Natural Zinc Compound. Acta Chimica Sinica. 1981;39:433–437. [Google Scholar]
- 12.Qiu M, Chen Y, Chu Y, Song SW, Yang N, Gao J, Wu ZW. Zinc ionophores pyrithione inhibits herpes simplex virus replication through interfering with proteasome function and NF-kappa B activation. Antivir Res. 2013;100:44–53. doi: 10.1016/j.antiviral.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Krenn BM, Gaudernak E, Holzer B, Lanke K, Van Kuppeveld FJM, Seipelt J. Antiviral Activity of the Zinc Ionophores Pyrithione and Hinokitiol against Picornavirus Infections. J Virol. 2009;83:58–64. doi: 10.1128/JVI.01543-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz J, Mizoguchi H, Bacon R. Comparative evaluation of antidandruff clinical efficacy of a potentiated zinc pyrithione shampoo and a zinc pyrithione/climbazole combination formula. J Am Acad Dermatol. 2013;68:Ab46–Ab46. [Google Scholar]
- 15.Muthyala R, Rastogi N, Shin WS, Peterson ML, Sham YY. Cell permeable vanX inhibitors as vancomycin re-sensitizing agents. Bioorganic & medicinal chemistry letters. 2014;24:2535–2538. doi: 10.1016/j.bmcl.2014.03.097. [DOI] [PubMed] [Google Scholar]
- 16.Ando M, Sato N, Nagase T, Nagai K, Ishikawa S, Takahashi H, Ohtake N, Ito J, Hirayama M, Mitobe Y, Iwaasa H, Gomori A, Matsushita H, Tadano K, Fujino N, Tanaka S, Ohe T, Ishihara A, Kanatani A, Fukami T. Discovery of pyridone-containing imidazolines as potent and selective inhibitors of neuropeptide Y Y5 receptor. Bioorgan Med Chem. 2009;17:6106–6122. doi: 10.1016/j.bmc.2009.05.069. [DOI] [PubMed] [Google Scholar]
- 17.Marchiaro P, Tomatis PE, Mussi MA, Pasteran F, Viale AM, Limansky AS, Vila AJ. Biochemical characterization of metallo-beta-lactamase VIM-11 from a Pseudomonas aeruginosa clinical strain. Antimicrob Agents Ch. 2008;52:2250–2252. doi: 10.1128/AAC.01025-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aitha M, Marts AR, Bergstrom A, Moller AJ, Moritz L, Tumer L, Nix JC, Bonomo RA, Page RC, Tierney DL, Crowder MW. Biochemical, Mechanistic, and Spectroscopic Characterization of Metallo-beta-lactamase VIM-2. Biochemistry-Us. 2014;53:7321–7331. doi: 10.1021/bi500916y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.N.Y. Schrodinger LLC, NY, Maestro v9.7,Bioluminate v1.2, Canvas v1.9, Epik v2.7, Glide v6.2, LigPrep v2.9, MacromModel v10.3, Prime v3.5, Schrodinger LLC, New York, NY, in, 2014
- 20.Borgianni L, Vandenameele J, Matagne A, Bini L, Bonomo RA, Frere JM, Rossolini GM, Docquier JD. Mutational Analysis of VIM-2 Reveals an Essential Determinant for Metallo-beta-Lactamase Stability and Folding. Antimicrob Agents Ch. 2010;54:3197–3204. doi: 10.1128/AAC.01336-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mojica MF, Mahler SG, Bethel CR, Taracila MA, Kosmopoulou M, Papp-Wallace KM, Llarrull LI, Wilson BM, Marshall SH, Wallace CJ, Villegas MV, Harris ME, Vila AJ, Spencee J, Bonomo RA. Exploring the Role of Residue 228 in Substrate and Inhibitor Recognition by VIM Metallo-beta-lactamases. Biochemistry-Us. 2015;54:3183–3196. doi: 10.1021/acs.biochem.5b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]