

Abstract
Schwann cell (SC) transplantation has been comprehensively studied as a strategy for spinal cord injury (SCI) repair. SCs are neuroprotective and promote axon regeneration and myelination. Nonetheless, substantial SC death occurs post-implantation, which limits therapeutic efficacy. The use of extracellular matrix (ECM)-derived matrices, such as Matrigel, supports transplanted SC survival and axon growth, resulting in improved motor function. Because appropriate matrices are needed for clinical translation, we test here the use of an acellular injectable peripheral nerve (iPN) matrix. Implantation of SCs in iPN into a contusion lesion did not alter immune cell infiltration compared to injury only controls. iPN implants were larger and contained twice as many SC-myelinated axons as Matrigel grafts. SC/iPN animals performed as well as the SC/Matrigel group in the BBB locomotor test, and made fewer errors on the grid walk at 4 weeks, equalizing at 8 weeks. The fact that this clinically relevant iPN matrix is immunologically tolerated and supports SC survival and axon growth within the graft offers a highly translational possibility for improving efficacy of SC treatment after SCI. To our knowledge, it is the first time that an injectable PN matrix is being evaluated to improve the efficacy of cell SC transplantation in SCI repair.
Keywords: Schwann cells, injectable peripheral nerve, decellularization, spinal cord injury, transplantation, axon growth
1. Introduction
Following an insult to the mammalian spinal cord, a complex cascade of reactions contributes to an aggravated expansion of the initial injury, including the formation of fluidfilled cavities that partly contribute to the regenerative failure and cause extreme functional deficits [1]. Cell transplantation strategies following spinal cord injury (SCI) rely on reducing cavitation following damage, re-bridging the injured tissue and creating more favorable conditions for axonal regeneration [2]. Among the prime candidates for repair are Schwann cells (SCs), the myelinating glia of the peripheral nervous system. SCs can be obtained from SCI patient nerve, purified and expanded in culture, and autologously transplanted into the lesioned spinal cord [3]. SCs have been extensively studied and found to be neuroprotective, reduce cavitation, promote axon regeneration and myelination, and modestly improve hindlimb movements in rat SCI models [4–6]. These findings contributed to FDA-approved clinical trials evaluating the safety and efficacy of SC therapy in sub-acute and chronic SCI subjects (www.clinicaltrials.gov NCT01739023; NCT02354625) [7–9].
After transplantation, however, SC survival rates are considerably low which limits the extent of their therapeutic action. Previous studies have shown that after a contusion injury in rats, only approximately 20% of the SCs survived when transplanted in culture medium [10–12]. The harsh injury environment, where oxidative stress, inflammation and immune responses occur, undoubtedly contributes to these low survival levels. In addition, the SC detachment from extracellular matrix (ECM) in the culture dish before transplantation, contributes to significant cell death through an apoptotic process designated anoikis [13]. The vehicle in which SCs are transplanted is thus very important and affects the treatment efficacy. Patel et al. [14] compared different ECM-derived in situ gelling formulations with culture medium as vehicles for SC implantation into a SCI site. Transplanting SCs in these matrices, particularly Matrigel, enhanced SC survival, axon growth, re-vascularization and functional recovery compared to SC suspension. Matrigel is not clinically relevant, however, as it is derived from a mouse tumor and its composition varies from batch to batch [15]. A supportive and clinically acceptable matrix is needed for human SC implantation.
We hypothesize that peripheral nerve (PN) tissue, the natural environment of SCs, could provide an excellent support scaffold for SC transplantation after SCI. Decellularization technologies allow the removal of cellular components of source tissues that can elicit an immune response, while preserving the ECM components, thus providing attractive biomaterials for tissue engineering [16]. Implantation of decellularized biomaterials does not require immunosuppression, and the chemical intrinsic cues for cell function and guidance are maintained [17]. Schmidt et al. [18] have developed an optimized acellular PN graft that has been shown to elicit similar functional recovery to an autograft in a rat sciatic nerve injury model. A similar human-derived scaffold is now FDAapproved for use in patients with PN discontinuities. An injectable formulation of acellular PN, recently established, mimics the composition of native nerve ECM, gels at physiological temperature and possesses mechanical properties similar to rat neural tissue [19]. We here investigate the potential of injectable PN (iPN) matrix to support transplanted SC survival and axon growth following SCI. Accordingly, we transplanted SCs in iPN and compared this to Matrigel, in a rat model of thoracic contusion. We evaluated the subacute immune response, graft volume, axon ingrowth and SC myelination, and locomotor function 8 weeks post-transplantation. SCs transplanted in iPN promoted twice as much axonal regeneration and equivalent locomotor recovery, compared to Matrigel, raising exciting possibilities that can directly translate to future SCI clinical trial strategies.
2. Materials and Methods
2.1. Injectable acellular peripheral nerve matrix preparation
Decellularization of rat sciatic nerve was conducted according to methods previously developed in the Schmidt laboratory [20]. Briefly, sciatic nerves were aseptically harvested from Sprague Dawley rats and the epineurium was removed. The nerves were then stored in 1x phosphate buffer saline (PBS) at −20°C until processing. Nerves were washed with double distilled water and detergent, with buffer washes (100 mM sodium/ 50 mM phosphate buffer) performed between detergent washes. The detergent used was composed of sulfobetaine-10 (SB-10) in 50 mM sodium/10 mM phosphate buffer and 0.14% Triton X-200/ 0.6 mM sulfobetaine-16 (SB-16) in 50 mM sodium/100 mM phosphate. The nerve samples were then rinsed in 50 mM sodium/ 10 mM phosphate buffer and incubated in 200 μl of chondroitinase ABC (0.2 U/ml, Sigma- Aldrich, USA) at 37°C for 16 hours. The tissue was then washed three times in 1x PBS for 3 hours each. Samples were lyophilized and stored at −20°C until used for digestion. To create the injectable hydrogel, lyophilized nerves were aseptically weighed and minced into 1–2 mm segments using microscissors. The segments were collected and placed into a scintillation vial containing a magnetic stir bar. Samples were digested in 1 mg/ml pepsin in 0.01 M hydrochloric acid at a concentration of 20 mg ECM/ml solution on a stir plate for 64 hours at room temperature. At the end of the digestion, the homogenous digest was neutralized to a pH of 7.4 using 1 M sodium hydroxide in 10x PBS. The pregel solution was either stored at 4°C for a few days or at −20°C for up to one month.
2.2. Schwann cell isolation
Purified populations of SCs were obtained from the sciatic nerves of adult female Fischer rats (Envigo, USA) as described previously [21]. Briefly, after removal of the epineurium, the nerves were cut into 3 mm-long segments that were placed into culture dishes and covered with Dulbecco Modified Eagle Medium (DMEM, Invitrogen, USA) containing 10% fetal bovine serum (FBS, Hyclone, USA). The segments were transferred to new dishes weekly and the medium was refreshed twice a week. After 2 weeks, the explants were treated with dispase (Roche, Switzerland) and collagenase (Worthington, USA), dissociated and resuspended in culture medium with mitogens: pituitary extract (20 μg/ml; Biomedical Technologies, USA), forskolin (2 μΜ; Sigma-Aldrich), and heregulin (2.5 nM; Genentech, USA). Treatment with Thy-1 and rabbit complement (MP Biomedicals, USA) was performed to eliminate remaining fibroblasts. The cells were grown to confluence and passaged three times (P3) prior to in vitro studies or in vivo transplantation. Their purity for grafting was determined to be 95–98% [22].
2.3. Lentiviral vector preparation and transduction of Schwann cells
Lentiviral vectors encoding enhanced green fluorescent protein (GFP) were used to transduce the SCs, so as to enable in vivo tracking. Lentiviral vector preparation was performed as described previously [23]. The lentiviral particles were produced by the Miami Project Viral Vector Core. Cultured 293T cells were used for transfecting the plasmids and viral harvesting. The viral titer was determined by p24 ELISA assay (PerkinElmer, USA) for quantifying p24 core protein concentrations. Purified viral vector stocks were stored at −80°C until SC infection. The vectors were used to transduce SCs at passage one. An optimal multiplicity of infection of 30 was designated by highest transduction efficiency (>95% of GFP-positive cells) with no obvious signs of toxicity [24]. The transduced cells were harvested and stored in liquid nitrogen, and later thawed and expanded as needed.
2.4. Cell culture experiments
The iPN scaffold was evaluated for the ability to support viable and metabolically active SCs. For these experiments 1×105 SCs were added to either the pre-gel iPN or Matrigel solution equaling a total of 30 μl volume (n=8). The gel solutions were incubated at 37°C to initiate thermal gelation and then placed into a 48-well plate along with 400 μl of fresh culture medium with mitogens (as detailed above). The cells were incubated at 37°C and 5% CO2 for 10 days. Metabolic activity was assessed using the alamarBlue assay (Thermo Scientific, USA) according to the manufacturer’s instructions. In short, 40 μl of alamarBlue reagent was added to each well containing the gels/medium and allowed to incubate for 2 hours. Then 200 μl of liquid was removed and placed into a 96-well plate. This was repeated for each sample. The remaining supernatant was removed and fresh medium was placed onto each gel. The 96-well plate was then read using a Biotek Synergy HT Microplate Reader (Biotek, USA). The data were recorded and the percent reduction of the alamarBlue reagent (indicator of metabolic activity) was calculated as per the manufacturer’s instructions. Viability was assessed using a live/dead assay (Molecular Probes, USA). Briefly, this assay involved incubating the gels in 4 μΜ calcium AM/2 μΜ ethidium homodimer solution. After 30 minutes of incubation, the solution was removed and fresh medium was placed back onto the gels, and protected from light. Confocal microscopy (Zeiss LSM710 Confocal, Carl Zeiss Microimaging, USA) was used to assess live (green), and dead (red) cells. Zen 2012 Image Processing software (Carl Zeiss Microimaging) was programmed to appropriately auto-segment adjacent cells and to define cells based on pixel size and color. Based on these settings, the number of live and dead cells per frame of confocal micrographs was counted and the relative viability calculated.
2.5. Animal experiments
Female adult Fischer rats (160–180 g; Envigo, USA) were housed according to the National Institutes of Health (NIH) and the United States Department of Agriculture guidelines. The Institutional Animal Care and Use Committee of the University of Miami approved all animal procedures. Rats were anesthetized with an intraperitoneal injection of ketamine (25 mg/kg; Vedco Inc., USA) and xylazine (5 mg/kg; Lloyd Laboratories, USA). Adequate anesthesia was determined by monitoring the corneal reflex and hindlimb withdrawal to painful stimuli. During surgery, the rats were kept on heating pads. The back of the animal was shaved and aseptically prepared with chlorhexidine solution before surgery. Lacrilube opthalamic ointment (Allergen Pharmaceuticals, USA) was applied to the eyes to prevent drying. Animals were randomly assigned to the experimental groups at the time of transplantation. Rats were housed two per cage at a temperature of 24°C with ad libitum access to water and food. The animals were euthanized at 3, 14 days, or 8 weeks post-SC transplantation. Figure 1 summarizes the experimental timeline of this study.
Figure 1.
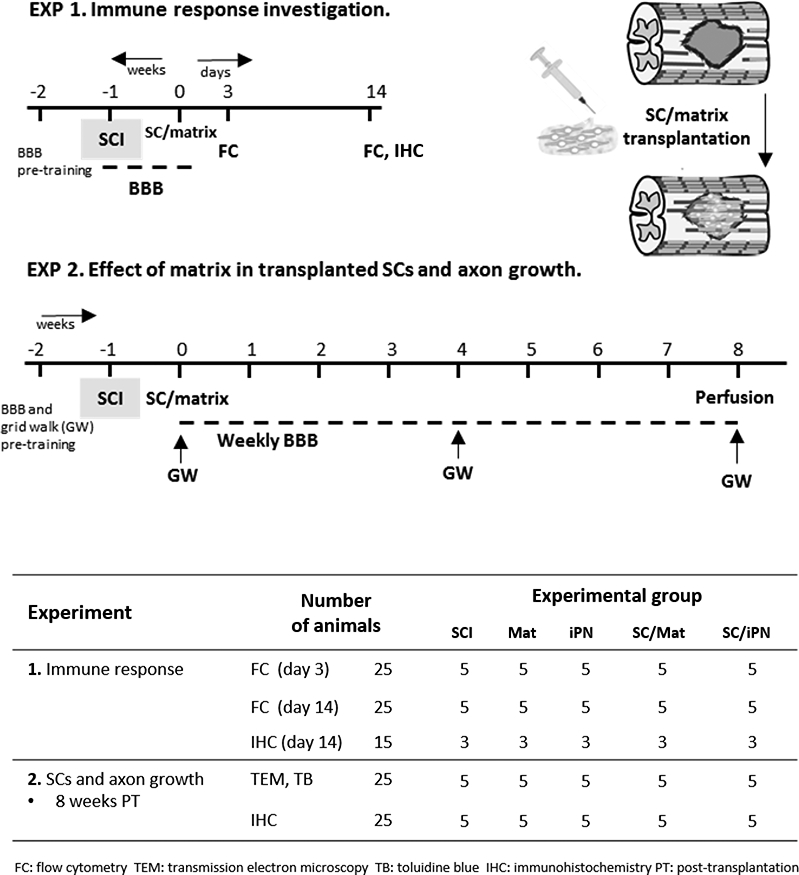
Summary of experimental design. Two independent experiments were conducted to investigate: (1) the host immune response to Matrigel and iPN, with and without SC grafts; and (2) long-term transplanted SC survival and axon growth in iPN or Matrigel.
2.5.1. Moderate thoracic contusion injury
A dorsal laminectomy was performed at thoracic vertebra T8, exposing the surface of the spinal cord without disrupting the dura. A moderate contusion injury on the dorsal surface was created using the Infinite Horizon (IH) Impactor (Precision Systems and Instrumentation, USA) set at a force of 200 kdynes. Animals were excluded when displaying abnormal contusion hits as defined by the force/displacement graph of the IH Impactor. The muscles and skin were sutured and closed with clips, respectively. The rats were allowed to recover in their cages with ad libitum access to food and water. Postoperative treatment and care included administration of Gentamicin (5 mg/kg, APP Pharmaceuticals, USA) to prevent infection, Buprenex (0.3 mg/kg, Reckitt Benckiser, UK) to reduce pain and Lactated Ringer’s to prevent dehydration. Bladders were emptied manually twice a day until bladder function returned.
2.5.2. Schwann cell transplantation
One week post-injury, the rats were anesthetized and the injury site re-exposed. Before implantation, DMEM/F12 medium was removed from the cell suspension and GFP-SCs were re-suspended in the respective matrix to be tested (iPN or Matrigel, 1:1 of matrix to SCs). GFP-SCs, Matrigel and iPN were maintained on ice until injection. A pulled pipette placed in a Hamilton syringe containing 6 μL of 2×106 GFP SCs was inserted into the spinal cord at a depth of 1 mm at the lesion epicenter and injected with a microinjector at 2 μl/min (World Precision Instruments, USA). The needle was kept in place for another 6 minutes after injection to prevent cell leakage upon withdrawal. The muscles and skin were sutured and closed with clips, respectively. Immediately after surgery, rats received postoperative care, as aforementioned.
2.6. Flow cytometry
Spinals cords (n=5/ group) and spleens (for single color controls) were harvested at 3 and 14 days post-transplantation. Spinal cord segments centered in the injury site were mechanically dissociated and tissue homogenates were obtained using the neural tissue dissociation kit (Miltenyi Biotec, Germany), and then passed through 40 μm cell strainers. Myelin was removed using myelin-specific magnetic beads and columns, according to the manufacturer’s protocol (Miltenyi Biotec). Non-labelled cells were used for immunolabeling. A near infrared viability dye (ThermoFisher Scientific, USA) allowed to determine cell viability prior to staining and fixation. Nonspecific staining was prevented with an anti-CD16/32 (FcR block, BD Pharmingen, USA) prior to adding the following surface marker antibody conjugates: V450 anti-rat CD11b (BD Biosciences, USA); Alexa Fluor 647 anti-rat CD45 (BioLegend, USA); and PE anti-rat CD3 (BD Biosciences). All incubations were performed at 4°C for 30 minutes. Cells were then fixed with 1% paraformaldehyde (PFA) in PBS containing 10% FBS. Control cell suspensions (single color and fluorescence minus one) were prepared for setting compensation. Cells were analyzed using a CytoFlex cytometer (Beckman Coulter, USA) and FACSDiva software (BD Biosciences).
2.7. Behavior testing
All locomotor assessments were performed by two examiners blinded to the group allocation. Open-field locomotion was evaluated using the BBB locomotor rating scale [25]. The hindlimb movement was scored from 0 (no observable locomotor movements) to 21 points (normal locomotor movements). Testing was performed weekly from 1 to 9 weeks after injury. In addition, a BBB subscore was obtained to provide a separate assessment of hindlimb and tail positioning. All animals in the present study scored 21 for the BBB score and 13 for the BBB subscore during baseline assessment. Animals were also scored 1 day and 7 days post-SCI to confirm the lesion severity. The animals scored 0.9 ± 0.5 at day 1 and 5.5 ± 0.7 at day 7 post-SCI. The ability to precisely control and place the hindlimbs was tested on an evenly spaced horizontal grid (grid walk test) at 4 and 8 weeks post-transplantation. The number of errors in foot placement (footslips) was counted on a metal grid runway elevated circa 30 cm from the ground.
2.8. Immunocytochemistry
At 14 days and 8 weeks after transplantation, animals were transcardially perfused with ice-cold saline solution, followed by 4% PFA (0.1 M PBS, pH 7.4). The spinal cords were removed and post-fixed overnight at 4°C, and then cryo-protected in 30% sucrose/ PBS at 4°C. The T7-T9 spinal cord segments centered at the injury site were embedded in gelatin. Serial sagittal cryostat sections (20 μm thick) were mounted on slides. Sections were incubated in a blocking solution of 10% normal goat serum with 0.1% Triton X-100 for 1 hour at room temperature. They were next incubated in the following antibodies overnight at 4°C: anti-GFP (1:500, Aves, USA) for SC identification, anti-CD11b (1:700, NovusBio, USA) or anti-lba1 (1:500, Abcam, USA) for microglia/macrophages identification, anti-CD3 (1:300, BioRad, USA) for T cells, anti-CD45 (1:300, BioRad) for leukocytes, anti-GFAP (1:1000, Dako, Denmark) to visualize astrocytes, and anti-RT-97 (1:3, hybridoma, ATCC, USA) for neurofilament staining. The sections were then incubated with fluorescent secondary antibodies: goat anti-chicken IgG Alexa Fluor 488, goat anti-rabbit IgG Alexa Fluor 594 or goat anti-mouse IgG Alexa Fluor 660 (1:200; Molecular Probes, USA) with 100 μΜ Floechst dye (Sigma-Aldrich) for 1 hour at room temperature. Sections were cover-slipped with Immumount mounting medium (Thermo Scientific) and maintained at 4°C until imaged in an Olympus FluoView™ FV1000 confocal laser scanning microscope (Olympus, USA).
2.9. Electron microscopy processing
Following perfusion, a 1 mm transverse slice from the injury/graft center was removed and fixed in glutaraldehyde and post-fixed in osmium tetroxide [26]. In brief, the tissue was dehydrated in a series of graded alcohol solutions, and embedded in Epon Alraldite resin (Electron Microscopy Sciences, USA) [27]. The 1 μm semi-thin coronal plastic sections were stained with Richardson’s toluidine blue stain for nuclear and cytoplasmic detail [28]. Thin sections were stained in 4% uranyl acetate and lead citrate and examined in a Philips CM-10 transmission electron microscope.
2.10. Stereological quantification
SC graft volume was analyzed in the serial sagittal sections using unbiased computer-assisted microscopy and Stereoinvestigator 10 software (MBF Bioscience, USA). The SC graft in each section (20 μm thickness, 140 μm intervals) was delineated using GFP fluorescence to determine the SC graft area. This area was automatically calculated and the graft volume determined using the equation: V=Σ (transplant area × section thickness × number of sections in each sampling interval). The graft area in each section was measured blindly. SC-myelinated axons were counted in semi-thin sections at 63× and the total numbers obtained using Stereoinvestigator 10 (MBF Bioscience). Using the optical fractionator sampling design (counting frame area, 25 × 25 μm; grid size, 75 × 75 μm), the graft epicenter was systematically sampled. These sections allow central myelin to be distinguished morphologically from peripheral myelin [27, 29]. The analysis was performed by two examiners blinded to the animal groups. Estimated total numbers of unmyelinated axons were determined by systematic random sampling of thin sections in the electron microscope to obtain the ratio of unmyelinated to myelinated axons in each condition [27]. The ratio of unmyelinated: myelinated axons in the Matrigel samples was estimated to be 6.7 : 1, whereas in the iPN samples it was 5.7 : 1.
2.11. Statistical analysis
All statistical analyses were performed using the GraphPad Prism v5.0 (GraphPad Software Inc., USA) or STATISTICA (Dell Inc, USA). All data are presented as mean ± SEM. Significant differences between the experimental groups were assessed using the parametric Student’s t test for in vitro studies, and chronic in vivo experiments. Differences of p < 0.05 were considered to be statistically significant. To analyze temporal differences in functional recovery, one-way analysis of variance (ANOVA) with Tukey’s post-hoc test were used.
3. Results
3.1. Quantification of SC viability in iPN and Matrigel in vitro
In vitro data demonstrated that the iPN scaffold was not cytotoxic to the seeded SCs. SCs were able to grow abundantly in both iPN and Matrigel, and in both conditions they were evenly distributed. SCs seeded in Matrigel exhibited a bipolar morphology (Figure 2A), whereas in iPN displayed a rounded phenotype (Figure 2B). Despite the unusual morphological appearance, SCs cultured in iPN maintained robust viability that was significantly higher (90.0 ± 1.7%) than the cells cultured in Matrigel (80.1 ± 2.0%) (Figure 2C). Three times more dead cells were found in Matrigel than in iPN based on a live/ dead assay (Figure 2D). Taken together, these numbers revealed that iPN is an adequate matrix that allows SC growth and activity. Furthemore, iPN significantly reduced cultured SC death when compared to Matrigel. IPN, however, is not an optimal matrix for SC process extension in vitro.
Figure 2.
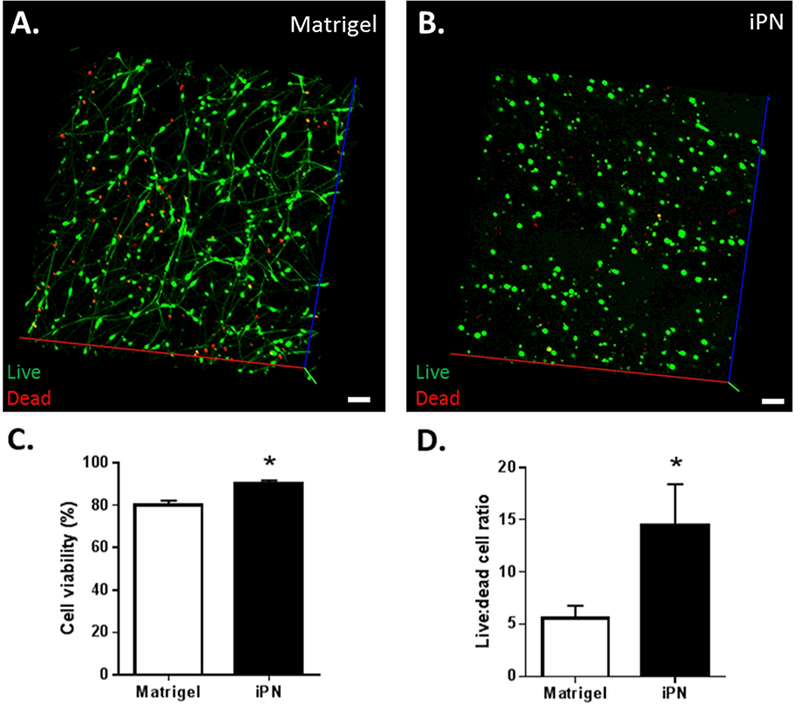
Confocal z-stack showing SCs seeded in Matrigel (A) or iPN (B) in vitro. Bar = 100 μm. Graphs depicting viability (C) and ratio of live/ dead SCs, when cultured in Matrigel or iPN (D) are shown. Results expressed as mean ± SEM (n=8). * p<0.05
3.2. Assessment of subacute cellular inflammation
Changes in cellular inflammation were investigated following matrix or SC/matrix injection into the injured spinal cord. Flow cytometry allowed quantification of immune cells at 3 and 14 days post-injection, after efficient myelin removal from the tissue. Several immune cell sub-populations were successfully identified and quantified, such as: microglia (CD11b+CD45low), macrophages (CD11b+CD45high), leukocytes (CD11b− CD45+), and T cells (CD3+) (Figures 3 and 4). Equivalent microglia and macrophage numbers were found in all groups, particularly at 14 days post-transplantation (Figure 3). However, at this same time point, there was a trend of increased number of leukocytes (Figure 3B) and T cells (Figure 4B) in Matrigel groups. Groups containing Matrigel, either with or without SCs, presented approximately three times more T cells and leukocytes than the control SCI group (Figures 3B and 4B). These differences however were not statistically significant due to some variability found within the Matrigel groups. Interestingly, the iPN groups displayed consistent leukocyte and T cell numbers identical to the control group. The signal from the transplanted cells (GFP+CD3-) was also visible in the groups receiving SC transplants. In the future, we will optimize efficient SC separation from spinal cord tissue to allow quantification of proliferation and survival of transplanted cells using this method.
Figure 3.
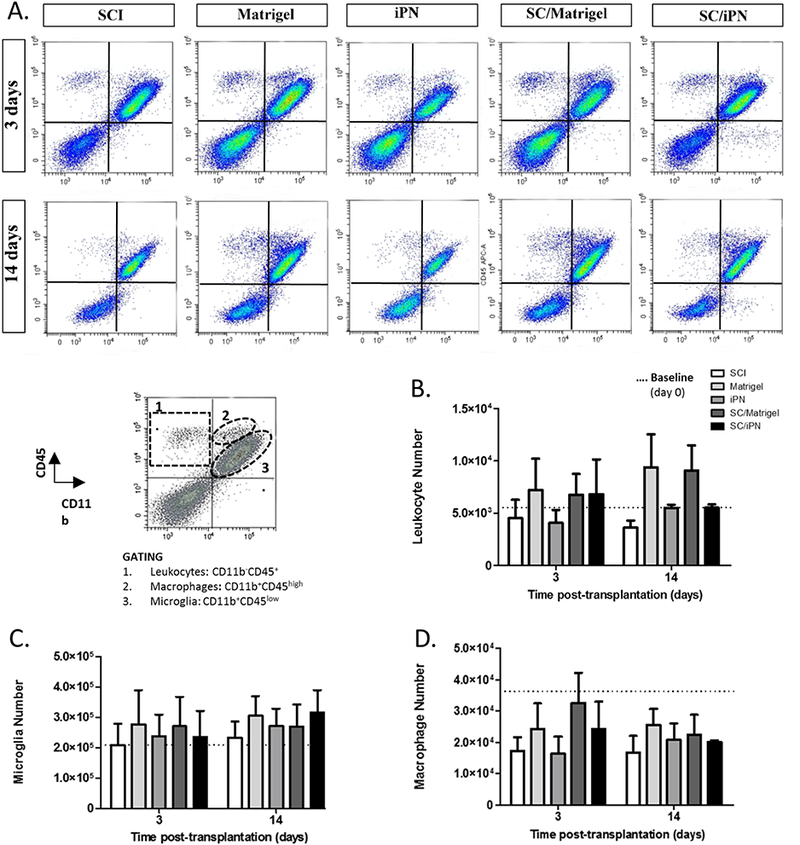
A. Representative images of flow cytometry analysis of subacute changes in immune cell number after SC transplantation and/or iPN or Matrigel injection, at 3 and 14 days post-transplantation. B-D. Quantification of the total number of leukocytes (B), microglia (C), and macrophages (D). Baseline levels measured before transplantation are represented by a dashed line. Results expressed as mean ± SEM.
Figure 4.
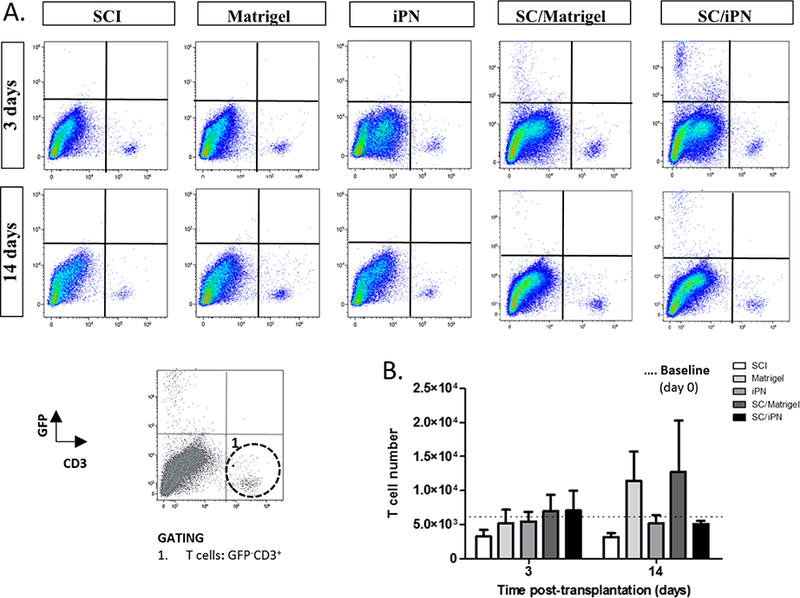
A. Representative images of flow cytometry analysis of changes in T cell number after SC transplantation and/or iPN or Matrigel injection, at 3 and 14 days posttransplantation. B. Quantification of the total number of T cells in the rat spinal cord 3 and 14 days post-transplantation. Results expressed as mean ± SEM.
Histological observation of sagittal sections of injured spinal cords at 14 days posttransplantation allowed visualization of the spatial distribution of microglia/macrophages, leukocytes and T cells (Figure 5). All cell types were seen in the tissue containing SC grafts in iPN or Matrigel. In both SC/Matrigel and SC/iPN groups, abundant macrophage/microglia were seen within the SC grafts. A higher density of microglia/macrophages was noted around the SC/Matrigel grafts, compared to SC/iPN. On the other hand, leukocytes and T cells were observed mostly surrounding the transplant area. Larger areas of intensely immunostained leukocytes and T cells were noted in the Matrigel groups, compared to iPN. Also, a border seemingly preventing direct contact of T cells and leukocytes with the graft was seen in Matrigel groups. This region was absent in the iPN groups. Grafts contained 3 times more SCs in iPN groups than Matrigel at 14 days post-transplantation (Figure 5B).
Figure 5.
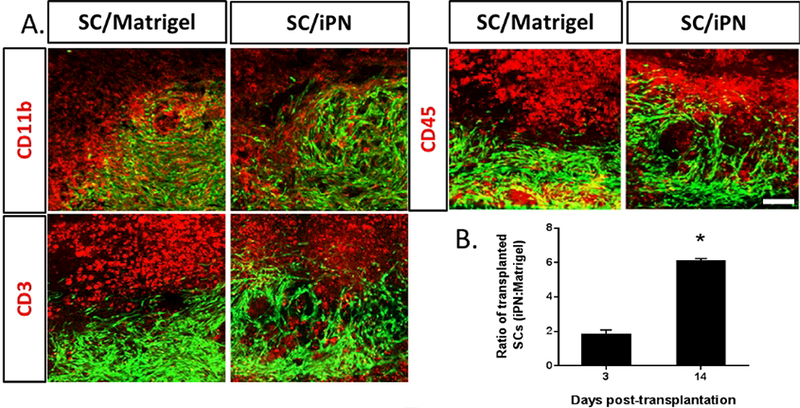
A. Fluorescent micrographs of sagittal spinal cord sections at 14 days posttransplantation to evaluate immune cells. SCs (green) transplanted either in Matrigel or iPN were stained for microglia/ macrophages (CD11b), T cells (CD3) and leukocytes (CD45), in red. Bar = 100 μm. B. Ratio of transplanted SCs in iPN: transplanted SCs in Matrigel at 14 days are shown. Results expressed as mean ± SEM. * p<0.05
3.3. Transplanted SC morphology and distribution in the lesion site
As expected, at 8 weeks the grafted SCs occupied most of the contusion cavity outlined by GFAP immunoreactive astrocytes (Figure 6A). Transplanted SCs could be seen as a continuous bridge from rostral to caudal and did not significantly migrate out of the lesion area, either in Matrigel or iPN. The grafts were very dense in both groups, not revealing any apparent evidence of the matrix in which SCs were implanted at the light microscope level. This suggests that the matrices were largely degraded during this period. Transplanted GFP-SCs exhibited the typical spindle shape and long processes in both groups (Figure 6Ai,ii). SCs aligned in a characteristic swirling pattern, which was discernable in sagittal sections of both iPN and Matrigel grafts. Also in both groups, astrocytes penetrated the graft into more central areas close to the lesion epicenter.
Figure 6.
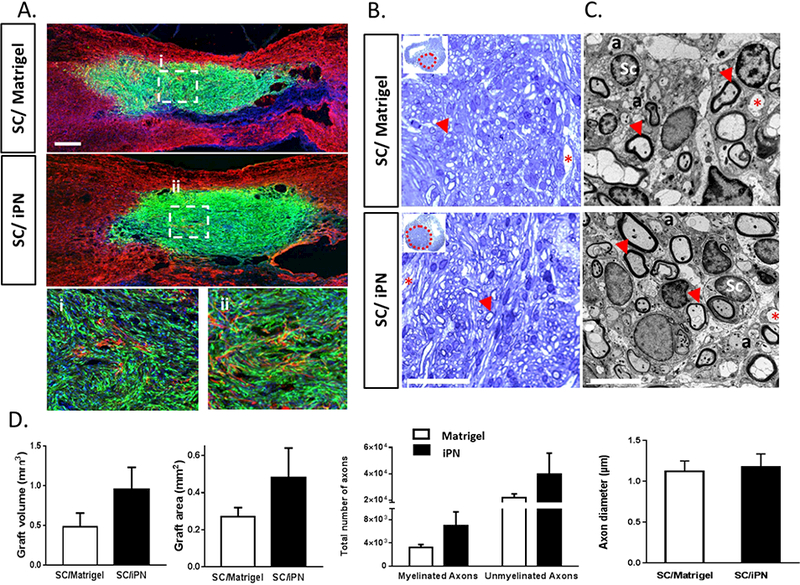
A. Confocal micrographs of sagittal spinal cord sections at 8 weeks posttransplantation of SCs in either Matrigel or iPN. Transplanted SCs (GFP, in green) and astrocytes (GFAP, in red) are shown. Cell nuclei are also visible (Hoechst, in blue). Bar = 500μm. Higher magnifications of the central areas of SC grafts in Matrigel (i) or iPN (ii) are shown. B. Bright field micrographs of SCs in either Matrigel or iPN in semi-thin, toluidine blue-stained coronal sections of spinal cord at 8 weeks post-transplantation. SCs were transplanted either in Matrigel or iPN. Graft areas are delineated by a dashed red line. Arrows indicate myelinated axons; asterisks indicates blood vessels. Bar = 50μm C. Transmission electron micrographs of the epicenter of the grafts. Legend: Sc: Schwann cell nucleus; a: unmyelinated axon. Arrows indicate myelinated axons; asterisks indicate blood vessels. Bar = 5μm. D. Quantification of graft volumes and areas, and number and diameter of axons in the graft. Results expressed as mean ± SEM.
3.4. SC graft volume quantification at 8 weeks post-transplantation
Stereological quantification of the graft volumes at 8 weeks post-transplantation revealed that these were two times larger in the animals that received SCs in iPN (0.95 ± 0.28 mm3), compared to the Matrigel group (0.48 ± 0.17 mm3) (Figure 6D). Although there was a 197% increase in the graft volume in the iPN group, compared to Matrigel, it was not statistically significant due to some within group variation. Earlier reports support that Matrigel per se induces a 256% increase in SC number, compared to medium suspension [14]. Similar quantification of areas from coronal sections confirmed the results obtained from the sagittal sections. Implants in the SC/iPN group were estimated to be 0.48 ± 0.16 mm2 compared while the SC/Matrigel group was 0.27 ± 0.05 mm2. There was a 177% increase in the iPN groups, which was not statistical significant (Figure 6D).
3.5. Analysis of axonal growth in SC transplants
Evaluation of semi-thin, toluidine blue-stained coronal sections of the SC graft/lesion epicenter demonstrated the presence of dense grafts with a plethora of SCmyelinated axons in both groups (Figure 6B). Stereological quantification of axon numbers allowed the examination of differences between the experimental groups. Electron microscopic observation of thin sections allowed the distinction of myelinating and non-myelinating axons (Figure 6C). As seen in Figure 6D, estimation of the number of axons within the implant revealed twice as many SC-myelinated axons in the SC/iPN group (6,927 ± 2,429) as in the SC/Matrigel group (3,259 ± 475). A 212% increase in the number of axons occurred in SC/iPN grafts. Quantification of non-myelinating axons showed the same trend, with a 181% increase in iPN (39,518 ± 16,116), compared to Matrigel (21,813 ± 2,861). No differences were observed in the myelinated axon diameters in both conditions (Figure 6D). Neurofilament immunostaining in sagittal sections of injured spinal cord 8 weeks post-transplantation confirmed the abundant presence of axons within SC grafts in iPN and Matrigel (Figure 7).
Figure 7.
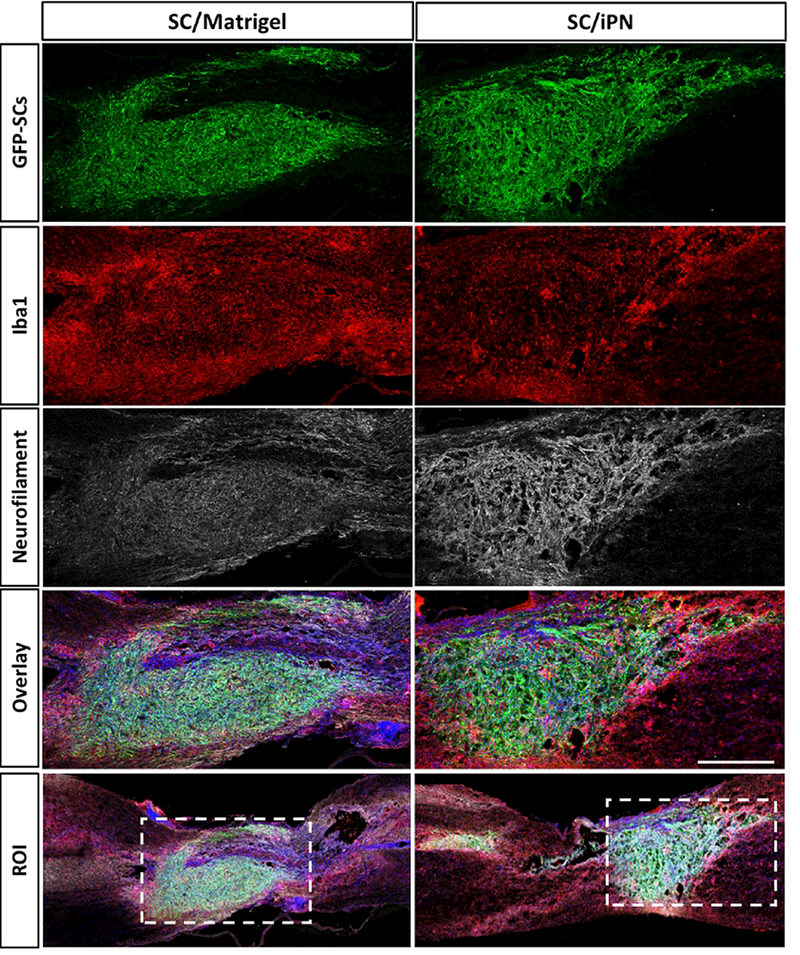
Confocal micrographs of sagittal spinal cord sections at 8 weeks posttransplantation. SCs (GFP, green) were transplanted in either Matrigel or iPN. The sections were immunostained for microglia/macrophage (Iba1, red) and neurofilamentpositive axon (NF, white) identification. Bar = 500 μm.
3.6. Immune response at 8 weeks post-transplantation
Immune responses were investigated by visualizing the presence of macrophage/microglia in and around the graft by immunostaining for the panmacrophage marker Iba1. Fluorescence images of sagittal sections revealed widespread macrophages/microglia inside and around the grafts (Figure 7). Most notably, the spinal cords that received SC/Matrigel grafts appeared to display a more intense border of immunostained macrophage and microglia around the graft, indicating a possibly more robust and long-lasting cellular inflammatory response.
3.7. SCs transplanted in iPN lead to similar locomotor recovery as Matrigel
Comparable trends in the locomotor performance in the open field, as measured by the BBB score [25], were observed between SC/iPN and SC/Matrigel groups during the 8 weeks following transplantation (Figure 8). Complementary analysis of BBB subscores indicated a faster recovery in hindpaw positioning in the SC/iPN group, particularly from 3 to 6 weeks post-transplantation. Evaluation of hindpaw placement on the grid-walk test revealed better performance of the SC/iPN group at 4 weeks posttransplantation (6.1 ± 1.1%), when compared to Matrigel (8.6 ± 1.8%). The number of footslip errors is equalized in the 2 groups at 8 weeks post-transplantation (5.0 ± 1.7%, in the Matrigel/SC group; 3.5 ± 1.6%, in the iPN/SC group).
Figure 8.
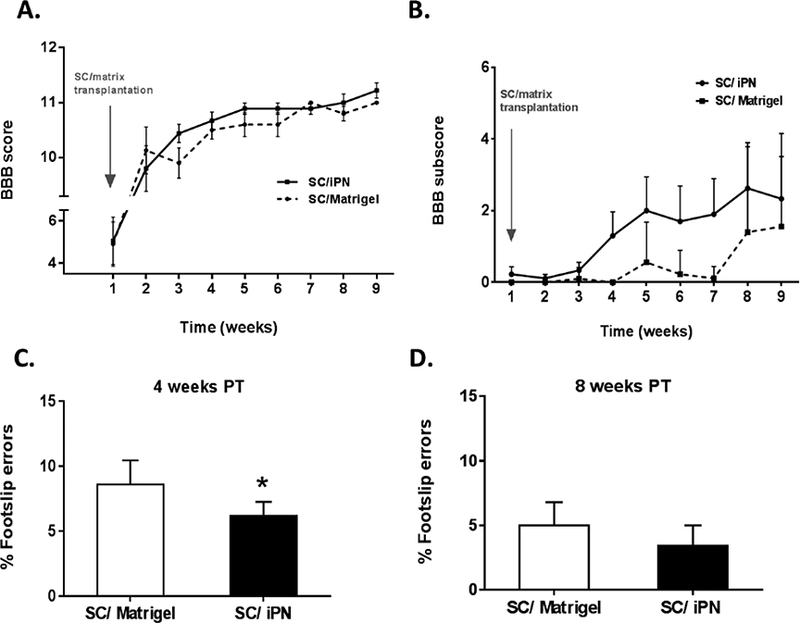
A, B. BBB scores (A) and subscores (B) of SC/iPN and SC/Matrigel implanted animals. C, D. Number of errors on the grid walk in the SC/iPN and SC/Matrigel implanted animals, at 4 and 8 weeks post-transplantation. Results expressed as mean ± SEM. *p<0.05.
4. Discussion
The current study demonstrates that iPN is an appropriate injectable biomaterial to support transplanted SC survival and axon growth into a SCI site. Moreover, injection of iPN into a lesion is immunologically tolerated and supports as much locomotor recovery as Matrigel, making this matrix an attractive possibility for future translational studies. SC transplantation in Matrigel has been compared to other matrices as well as with SC suspension and shown superior SC survival, axon growth and locomotor recovery. We here show that iPN equally supports graft volumes and their capacity to sustain axon growth within the transplant after SCI.
Matrigel is a basement membrane extract that enhances cell spreading and extension in vitro [38, 39]. It is a thermoresponsive hydrogel primarily containing laminin, collagen IV, and entactin, and also variable amounts of several growth factors [30]. Despite these attractive properties, however, its derivation from a mouse sarcoma and variable composition limit its use in clinical applications [31]. Nonetheless, it has been widely used as a matrix to support cell survival in vitro, and also in vivo [14, 32–34]. IPN is also a biologically derived material that can equally originate some compositional variablility. Nonetheless, during the processing of the biomaterial we have taken measures to prevent batch-to-batch variability. Peripheral nerve tissues were isolated from a single species of rat matched for same age and weight. We have previously observed that animal age was the principal effector of variability before and after decellularization (unpublished data). Based on our characterization of this matrix, the variability in ECM components, such as collagens and glycosaminoglycans, was not appreciable in different preparations of iPN [19].
Our data confirm that Matrigel has excellent properties for culturing SCs. After 10 days of culture, SCs displayed the typical bipolar morphology in Matrigel, whereas in iPN SCs did not extend their processes suggesting that iPN does not possess optimal properties for in vitro culturing. We believe these dissimilarities in vitro are caused mainly by the differences in protein and growth factor content between Matrigel and iPN. The increased content of adhesive basement membrane proteins in Matrigel is, therefore, a plausible reason for the differences in cell morphology in vitro. Despite the morphological variance, SC viability was higher in iPN scaffolds, indicating that SCs survive and are viable when cultured in iPN.
The composition of the ECM plays a key role in cell behavior [36, 37]. Biologically active factors like nerve growth factor, fibroblast growth factor (FGF), platelet-derived growth factor, insulin growth factor-1 (IGF-1) and heparin sulfate proteoglycans (HSPG) are released from Matrigel and can interact with SCs [35]. Both FGF and IGF-1 encourage SC growth and proliferation [34–36], and HSPG are known mediators of SC spreading on laminin-based surfaces [37]. To generate iPN, peripheral nerves undergo chemical and then enzymatic/ acidic digestion. Chemical decellularization has been shown to variably remove many growth factors from tissues [36]. For example, Reing et al. demonstrated that chemical decellularization of porcine dermis contained no detectable vascular endothelial growth factor and transforming growth factor beta1 while eliminating at least 60% of FGF compared to fresh dermis [37]. The ensuing enzymatic and acidic treatments may also facilitate removal and/or denaturation of growth factors [38, 39]. Although we have not explicitly tested for this in iPN, we predict similar results. Preliminary proteomic analysis of decellularized nerve indicated that these scaffolds are primarily composed of structural proteins. Moreover, iPN contains native ECM proteins, such as laminin and collagen, in lower and more physiologically-relevant concentrations than in Matrigel, which is more relevant for in vivo applications [19].
An important factor to consider when envisioning applications of decellularized matrices is the tissue specificity of the matrix source. While it is legitimate to assume that a homologous substrate will be the most favorable in clinical applications, this is a current subject of discussion [40]. A recent study comparing the neuroregenerative potential of CNS and non-CNS derived ECM hydrogels in SCI reported both materials to be equally beneficial in supporting tissue repair [41]. Different ECM hydrogels have been tested for SCI with encouraging results, independently of cell transplantation, by filling lesion cavities and also by immunomodulatory properties [42]. However, it is widely recognized that due to the complexity of the post-injury environment combination of therapies will be necessary for effective restoration of function. Cell transplantation strategies have been intensively studied and are believed to be one of the necessary components to a successful strategy in SCI repair [43].
In the current study we have chosen to investigate the use of iPN to support transplanted SC survival. PN is the native SC environment and our choice of source tissue because decellularized PN scaffolds have been widely characterized and have recently advanced to clinical applications in PN injury [44]. IPN possesses approximate chemical and mechanical properties of PN upon thermal gelation in physiological conditions. Injection of iPN hydrogels into a SCI site favored a regenerative macrophage phenotype subacutely, while no long-term functional benefits or deficits were reported [19]. The hydrogel was present in the first 2 weeks after injection, but not visible at 8 weeks indicating degradation of the material. A similar hydrogel was injected after SCI and the authors reported that the matrix was relatively degraded in as little as 2 weeks postinjection [41]. Both of these studies injected the hydrogel alone, and the addition of cells in our current study could alter degradation. Nonetheless, neither Matrigel nor iPN were detected in our tissue preparations at 8 weeks, indicating possibly similar degradation profiles.
Clinical application of biomaterials is dependent upon the host response to the material implantation. The host response is, in fact, the primary determinant of clinical success in most applications, as it affects the performance and integrity of the material in vivo. Understanding the kinetics of cellular inflammation after implantation of biomaterials into injury sites is essential for the development of effective therapeutic interventions. Neurotraumatic events induce the initiation of inflammation cascades and activate systemic immunity, with the potential to aggravate pathology or trigger reparative responses [45]. After SCI, inflammation is a multi-phasic response and, while not yet completely understood, acute neuroinflammation has been shown to be a determining factor in the extent of functional recovery [46].
In the fields of SCI research and also of tissue engineering, evaluation of immune cell infiltration is typically attained using histological analyses. Accurate quantification by means of histological evaluation can be challenging because early after injury the tissue is very fragile. Other factors such as sampling strategy, variability in the distribution of cells, tissue heterogeneity, and histological processing might also influence the final estimation of cell numbers. It is also a time consuming procedure. Hence, to evade these circumstances we quantified cellular inflammation using flow cytometry analysis [47]. The use of flow cytometry to assess the primary immune cell types in the injured spinal cord was recently described and validated [47, 48]. We investigated the inflammatory response at two time points in the subacute phase response (3 and 14 days post-implantation). Flow cytometry analysis allowed accurate comparison of SC implantation in different matrices providing insights into the dynamics of inflammation in response to the presence of different matrices. Consistent with previous reports, microglia and macrophages were the most abundant immune cells present in the injured tissue, followed by leukocytes and T cells.
Microglia, the immune resident cells of the CNS, are activated after injury, proliferating and peaking in number by 7 days after SCI [48]. They are among the first to respond to SCI by producing pro-inflammatory cytokines, nitric oxide and reactive oxygen species. Studies modulating this early microglia response have shown reduced secondary tissue damage and improved functional recovery [49, 50]. These cells can, however, adopt different phenotypes that have contrasting roles on triggering further damage (M1 cells) or contributing to spinal cord repair (M2 cells) [51]. Previous reports have shown that acellular scaffolds, including iPN, promote immune cell bias toward M2 regenerative phenotypes [19, 52]. In future studies it will be important to discriminate the contribution of each of these phenotypes in tissue repair after SC transplantation in biomaterials, such as the one we are proposing.
Whereas microglia and macrophage numbers equalized in all experimental groups by 14 days post-transplantation, leukocytes and T cells tended to accumulate in the groups containing Matrigel, with or without SCs. The role of leukocytes acutely after injury has been mainly associated with a progressive destructive process that exacerbates damage by releasing proteases, reactive oxygen species and pro-inflammatory cytokines [53]. In the long term, however, alternative reparative roles have been suggested for leukocytes. T cells are present in very low numbers in the healthy spinal cord, but increase in the first week post-injury [45]. There is evidence that T cells have roles in both injury progression and repair processes, depending on their phenotype and the microenvironment. Although usually present in lower numbers than infiltrating macrophages or activated microglia, they can be responsible for greater tissue damage [54]. Confirmation of their function after SCI was obtained from T cell inhibition studies that resulted in improved recovery from SCI [55, 56]. A thorough understanding of the function of these cells in the subacute phase and their impact on damage/ repair will be critical for a complete interpretation of the data, and future work will focus on investigating the basis of these differences. Nonetheless, iPN had no deleterious effect on the overall subacute immune response, as all iPN experimental groups resembled the SCI control group at 14 days post-implantation. The fact that Sprague Dawley-derived iPN does not elicit an immune response when implanted in Fischer rats is very promising in terms of clinical applications. In future studies, it will be important to use a different species as source of iPN and evaluate the host response to the biomaterial.
Previous studies from the Bunge group and others have shown that SCs transplanted in culture medium suffer a dramatic 80% loss in the first 3 weeks following transplantation [10, 11, 57]. The causes of this significant cell death may include the removal of SCs from their culture substratum (termed anoikis), as well as the hostile inflammatory environment of the injury site. Improving the survival rates of SCs in this critical phase may enhance their recognized neuroprotective effects [3]. Our data reveal that suspending SCs in iPN led to a more than threefold increase compared to the Matrigel group 14 days after transplantation. Both matrices contain laminin and collagen to support effective cellular adhesion and spreading, and also regulate SC survival and proliferation [58, 59]. More important, unlike Matrigel, iPN has potential to be translated and used in clinical applications.
Other strategies have been explored to enhance survival of transplanted SCs and improve the extent of anatomical and functional restoration following contusive SCI. Neurotrophin-transduced SCs have been shown to significantly improve graft survival and behavioral recovery [24, 60–62]. Delayed SC transplantation in a subacute phase by itself [63], or in conjunction with immunosuppression also resulted in enhanced survival and improved anatomical outcomes [12]. Hydrogels have the potential to be tuned to have chemically-defined composition, mechanical properties of soft tissues, and to be injectable [64]. Patel et al. [14], who compared different in situ gelling matrices over standard SC-medium suspensions, found that SCs implanted in Matrigel exhibited better survival and axon ingrowth, resulting in enhanced gross locomotor function. Whereas Matrigel boosts SC transplantation efficacy in SCI repair, it is not clinically applicable. Therefore, we investigated the potential of iPN to support chronic survival of SC implants, comparing it to Matrigel.
IPN is here shown to act as a supportive substrate for SC grafts, possibly because of SC adherence properties provided by the preserved nerve ECM proteins. Additionally, iPN in combination with SCs also proved to be supportive of axon regeneration. Axon growth within iPN implants likely contributes to larger grafts, as axons are known to express mitogenic factors that induce SC proliferation [65]. A plethora of SC-myelinated axons was seen in the dense SC/iPN grafts that filled the lesion cavity, with no distinct glial scar between the graft and the host tissue. The SC/iPN grafts integrated into the host tissue without observable toxicity, which would have been reflected in tissue loss and cavity formation.
To our knowledge, it is the first time that an injectable PN matrix is being evaluated to improve the efficacy of cell transplantation in SCI repair. Drawing from the clinical success of acellular nerve grafts, an injectable acellular nerve graft may offer an effective, clinically relevant treatment for SCI. Clinical trials at the Miami Project are testing the safety of autologous human SCs transplanted into SCI sites. It is tempting to consider the possibility, after removal of the SCs from nerve biopsy, of treating the remaining tissue to serve as an autologous matrix to support survival of the transplanted human SCs.
5. Conclusions
Matrices to support SC grafts have been recently explored and shown to improve SC transplantation efficacy after SCI. Nonetheless, no clinically relevant ECM matrix that could be used in human subjects has been reported to date. Our results with iPN revealed this material to be highly supportive for SC survival and myelination, axon growth, and locomotor recovery. Additionally, implantation of this matrix does not aggravate immune responses in the lesion site. As both SC transplantation in SCI and acellular nerve scaffold implantation in PN injury are FDA-approved methods, this work raises an exciting possibility that can directly translate to future SCI clinical trials.
Acknowledgements
This work was supported by the National Institutes of Health [NIH NS 09923 (MBB)], The Miami Project to Cure Paralysis (MPCP) at the University of Miami, The Buoniconti Fund and the Craig Neilsen Foundation [222456 (CES)]. The authors would like to acknowledge: Yelena Pressman for preparing SCs for transplantation; the MPCP Virus Core for the virus production; the Animal Facility Core at the MPCP for assisting in animal care; Margaret Bates and Vania Almeida for TEM imaging and sample processing; Dr. Melissa Carballosa-Gautam for help in confocal imaging; Jason Wagner for cell counting; and Drs. Oliver Umland and Roberta Brambilla for assistance with flow cytometry. SRC would also like to acknowledge the Spinal Cord Injury Training Program at Ohio State University.
Footnotes
Author contributions SRC,YL, RAW, CES and MBB designed the experiments; SRC, YL, MWM and RCC performed the experimental studies and acquired data; SRC, MWM and RAW analyzed data and performed statistical analysis; SRC prepared the manuscript; CES and MBB edited and reviewed the manuscript.
Disclosures The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowland JW, et al. , Current status of acute spinal cord injury pathophysiology and emerging therapies: promise on the horizon. Neurosurgical Focus, 2008. 25(5): p. E2. [DOI] [PubMed] [Google Scholar]
- 2.Tetzlaff W, et al. , A Systematic Review of Cellular Transplantation Therapies for Spinal Cord Injury. Journal of Neurotrauma, 2011. 28(8): p. 1611–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunge MB, Efficacy of Schwann cell transplantation for spinal cord repair is improved with combinatorial strategies. The Journal of Physiology, 2016. 594(13): p. 3533–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bunge MB and Wood PM, Chapter 32 - Realizing the maximum potential of Schwann cells to promote recovery from spinal cord injury, in Handbook of Clinical Neurology, Joost V John WM, 2012, Elsevier; p. 523–540. [DOI] [PubMed] [Google Scholar]
- 5.Kanno H, et al. , Schwann cell transplantation for spinal cord injury repair: its significant therapeutic potential and prospectus, in Reviews in the Neurosciences. 2015. p. 121. [DOI] [PubMed] [Google Scholar]
- 6.Fortun J, Hill CE, and Bunge MB, Combinatorial Strategies with Schwann Cell Transplantation to Improve Repair of the Injured Spinal Cord. Neuroscience letters, 2009. 456(3): p. 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guest J, Santamaria AJ, and Benavides FD, Clinical translation of autologous Schwann cell transplantation for the treatment of spinal cord injury. Current Opinion in Organ Transplantation, 2013. 18(6): p. 682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson KD, et al. , Safety of Autologous Human Schwann Cell Transplantation in Subacute Thoracic Spinal Cord Injury. J Neurotrauma, 2017. [DOI] [PubMed] [Google Scholar]
- 9.Bunge MB, et al. , From transplanting Schwann cells in experimental rat spinal cord injury to their transplantation into human injured spinal cord in clinical trials. Prog Brain Res, 2017. 231: p. 107–133. [DOI] [PubMed] [Google Scholar]
- 10.Hill CE, et al. , Early necrosis and apoptosis of Schwann cells transplanted into the injured rat spinal cord. European Journal of Neuroscience, 2007. 26(6): p. 1433–1445. [DOI] [PubMed] [Google Scholar]
- 11.Pearse DD, et al. , Transplantation of Schwann cells and/or olfactory ensheathing glia into the contused spinal cord: Survival, migration, axon association, and functional recovery. Glia, 2007. 55(9): p. 976–1000. [DOI] [PubMed] [Google Scholar]
- 12.Hill CE, et al. , Labeled Schwann cell transplantation: Cell loss, host Schwann cell replacement, and strategies to enhance survival. Glia, 2006. 53(3): p. 338–343. [DOI] [PubMed] [Google Scholar]
- 13.Koda M, et al. , Brain-derived neurotrophic factor suppresses anoikis-induced death of Schwann cells. Neuroscience Letters, 2008. 444(2): p. 143–147. [DOI] [PubMed] [Google Scholar]
- 14.Patel V, et al. , Suspension matrices for improved Schwann-cell survival after implantation into the injured rat spinal cord. Journal of Neurotrauma, 2010. 27(5): p. 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benton G, et al. , Matrigel: From discovery and ECM mimicry to assays and models for cancer research. Advanced Drug Delivery Reviews, 2014. 79–80: p. 3–18. [DOI] [PubMed] [Google Scholar]
- 16.Crapo PM, Gilbert TW, and Badylak SF, An overview of tissue and whole organ decellularization processes. Biomaterials, 2011. 32(12): p. 3233–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoshiba T, et al. , Decellularized matrices for tissue engineering. Expert Opinion on Biological Therapy, 2010. 10(12): p. 1717–1728. [DOI] [PubMed] [Google Scholar]
- 18.Nagao RJ, et al. , Functional characterization of optimized acellular peripheral nerve graft in a rat sciatic nerve injury model. Neurological Research, 2011. 33(6): p. 600–608. [DOI] [PubMed] [Google Scholar]
- 19.Cornelison RC, et al. , Injectable hydrogels of optimized acellular nerve for injection in the injured spinal cord. Biomed Mater, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudson TW, Liu SY, and Schmidt CE, Engineering an improved acellular nerve graft via optimized chemical processing. Tissue Eng, 2004. 10(9–10): p. 1346–58. [DOI] [PubMed] [Google Scholar]
- 21.Morrissey TK, Kleitman N, and Bunge RP, Isolation and functional characterization of Schwann cells derived from adult peripheral nerve. The Journal of neuroscience, 1991. 11(8): p. 2433–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takami T, et al. , Schwann cell but not olfactory ensheathing glia transplants Improve hindlimb locomotor performance In the moderately contused adult rat thoracic spinal cord. J Neurosci, 2002. 22(15): p. 6670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Follenzi A, et al. , Gene transfer by lentiviral vectors Is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat Genet, 2000. 25(2): p. 217–22. [DOI] [PubMed] [Google Scholar]
- 24.Kanno H, et al. , Combination of engineered Schwann cell grafts to secrete neurotrophin and chondroitinase promotes axonal regeneration and locomotion after spinal cord Injury. J Neurosci, 2014. 34(5): p. 1838–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basso DM, Beattie MS, and Bresnahan JC, A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma, 1995. 12(1): p. 1–21. [DOI] [PubMed] [Google Scholar]
- 26.Xu XM, et al. , Axonal regeneration Into Schwann cell-seeded guidance channels grafted Into transected adult rat spinal cord. Journal of Comparative Neurology, 1995. 351(1): p. 145–160. [DOI] [PubMed] [Google Scholar]
- 27.Bates ML, Puzis R, and Bunge MB, Preparation of spinal cord injured tissue for light and electron microscopy including preparation for immunostaining. Animal Models of Movement Disorders: Volume II, 2011: p. 381–399. [Google Scholar]
- 28.Murray PD, et al. , Spontaneous remyelination following extensive demyelination Is associated with Improved neurological function In a viral model of multiple sclerosis. Brain, 2001. 124(7): p. 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunge MB, et al. , Characterization of photochemically induced spinal cord Injury In the rat by light and electron microscopy. Experimental neurology, 1994. 127(1): p. 76–93. [DOI] [PubMed] [Google Scholar]
- 30.Vukicevic S, et al. , Identification of multiple active growth factors In basement membrane Matrigel suggests caution In Interpretation of cellular activity related to extracellular matrix components. Exp Cell Res, 1992. 202(1): p. 1–8. [DOI] [PubMed] [Google Scholar]
- 31.Kleinman HK and Martin GR, Matrigel: Basement membrane matrix with biological activity. Seminars in Cancer Biology, 2005. 15(5): p. 378–386. [DOI] [PubMed] [Google Scholar]
- 32.Akhtar N, Dickerson EB, and Auerbach R, The sponge/Matrigel angiogenesis assay. Angiogenesis, 2002. 5(1): p. 75–80. [DOI] [PubMed] [Google Scholar]
- 33.Benton G, et al. , Multiple uses of basement membrane-like matrix (BME/Matrigel) In vitro and in vivo with cancer cells. Int J Cancer, 2011. 128(8): p. 1751–7. [DOI] [PubMed] [Google Scholar]
- 34.Williams RR, et al. , Permissive Schwann Cell Graft/Spinal Cord Interfaces for Axon Regeneration. Cell transplantation, 2015. 24(1): p. 115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes CS, Postovit LM, and Lajoie GA, Matrigel: A complex protein mixture required for optimal growth of cell culture. PROTEOMICS, 2010. 10(9): p. 1886–1890. [DOI] [PubMed] [Google Scholar]
- 36.Caralt M, et al. , Optimization and critical evaluation of decellularization strategies to develop renal extracellular matrix scaffolds as biological templates for organ engineering and transplantation. American journal of transplantation, 2015. 15(1): p. 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reing JE, et al. , The effects of processing methods upon mechanical and biologic properties of porcine dermal extracellular matrix scaffolds. Biomaterials, 2010. 31(33): p. 8626–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chi Y. h., et al. , Thermodynamic characterization of the human acidic fibroblast growth factor: Evidence for cold denaturation. Biochemistry, 2001. 40(25): p. 7746–7753. [DOI] [PubMed] [Google Scholar]
- 39.Playford RJ, et al. , Epidermal growth factor Is digested to smaller, less active forms In acidic gastric juice. Gastroenterology, 1995. 108(1): p. 92–101. [DOI] [PubMed] [Google Scholar]
- 40.Londono R and Badylak SF, Biologic scaffolds for regenerative medicine: mechanisms of In vivo remodeling. Annals of biomedical engineering, 2015. 43(3): p. 577–592. [DOI] [PubMed] [Google Scholar]
- 41.Tukmachev D, et al. , Injectable extracellular matrix hydrogels as scaffolds for spinal cord injury repair. Tissue Engineering Part A, 2016. 22(3–4): p. 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Führmann T, Anandakumaran PN, and Shoichet MS, Combinatorial Therapies After Spinal Cord Injury: How Can Biomaterials Help? Advanced healthcare materials, 2017. [DOI] [PubMed] [Google Scholar]
- 43.Assinck P, et al. , Cell transplantation therapy for spinal cord Injury. Nat Neurosci, 2017. 20(05): p. 637–647. [DOI] [PubMed] [Google Scholar]
- 44.Spearman BS, et al. , Tissue-Engineered Peripheral Nerve Interfaces. Advanced Functional Materials, 2018. 28(12): p. 1701713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donnelly DJ and Popovich PG, Inflammation and Its role In neuroprotection, axonal regeneration and functional recovery after spinal cord Injury. Experimental neurology, 2008. 209(2): p. 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okada S, The pathophysiological role of acute Inflammation after spinal cord Injury. Inflammation and Regeneration, 2016. 36(1): p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen HX, Beck KD, and Anderson AJ, Quantitative assessment of Immune cells In the Injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. JoVE (Journal of Visualized Experiments), 2011(50): p. e2698–e2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beck KD, et al. , Quantitative analysis of cellular Inflammation after traumatic spinal cord Injury: evidence for a multiphasic Inflammatory response In the acute to chronic environment. Brain, 2010: p. awp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stirling DP, et al. , Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and Improves functional outcome after spinal cord Injury. J Neurosci, 2004. 24(9): p. 2182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerqueira SR, et al. , Microglia Response and In Vivo Therapeutic Potential of Methylprednisolone-Loaded Dendrimer Nanoparticles In Spinal Cord Injury. Small, 2013. 9(5): p. 738–749. [DOI] [PubMed] [Google Scholar]
- 51.David S and Kroner A, Repertoire of microglial and macrophage responses after spinal cord injury. Nature Reviews Neuroscience, 2011. 12(7): p. 388–399. [DOI] [PubMed] [Google Scholar]
- 52.Brown BN, et al. , Macrophage phenotype and remodeling outcomes In response to biologic scaffolds with and without a cellular component. Biomaterials, 2009. 30(8): p. 1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taoka Y and Okajima K, Role of leukocytes In spinal cord Injury In rats. J Neurotrauma, 2000. 17(3): p. 219–29. [DOI] [PubMed] [Google Scholar]
- 54.Trivedi A, Olivas AD, and Noble-Haeusslein LJ, Inflammation and Spinal Cord Injury: Infiltrating Leukocytes as Determinants of Injury and Repair Processes. Clinical neuroscience research, 2006. 6(5): p. 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fleming JC, et al. , Alpha4beta1 integrin blockade after spinal cord Injury decreases damage and Improves neurological function. Exp Neurol, 2008. 214(2): p. 147–59. [DOI] [PubMed] [Google Scholar]
- 56.Lu HZ, et al. , Cyclosporin A Increases recovery after spinal cord Injury but does not Improve myelination by oligodendrocyte progenitor cell transplantation. BMC Neurosci, 2010. 11: p. 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barakat DJ, et al. , Survival, Integration, and Axon Growth Support of Glia Transplanted Into the Chronically Contused Spinal Cord. Cell Transplantation, 2005. 14(4): p. 225–240. [DOI] [PubMed] [Google Scholar]
- 58.Armstrong SJ, et al. , ECM molecules mediate both Schwann cell proliferation and activation to enhance neurIte outgrowth. Tissue engineering, 2007. 13(12): p. 2863–2870. [DOI] [PubMed] [Google Scholar]
- 59.Chernousov MA, et al. , Regulation of Schwann cell function by the extracellular matrix. Glia, 2008. 56(14): p. 1498–1507. [DOI] [PubMed] [Google Scholar]
- 60.Enomoto Μ, Bunge MB, and Tsoulfas P, A multifunctional neurotrophin with reduced affinity to p75 NTR enhances transplanted Schwann cell survival and axon growth after spinal cord Injury. Experimental neurology, 2013. 248: p. 170–182. [DOI] [PubMed] [Google Scholar]
- 61.Golden KL, et al. , Transduced Schwann cells promote axon growth and myelination after spinal cord injury. Experimental neurology, 2007. 207(2): p. 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tuszynski MH, et al. , Grafts of genetically modified Schwann cells to the spinal cord: survival, axon growth, and myelination. Cell Transplantation, 1998. 7(2): p. 187–196. [DOI] [PubMed] [Google Scholar]
- 63.Barbour HR, et al. , Tissue sparing, behavioral recovery, supraspinal axonal sparing/regeneration following sub-acute glial transplantation in a model of spinal cord contusion. BMC Neuroscience, 2013. 14(1): p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alvarado-Velez M, Pai SB, and Bellamkonda RV, Hydrogels as carriers for stem cell transplantation. IEEE Trans Biomed Eng, 2014. 61(5): p. 1474–81. [DOI] [PubMed] [Google Scholar]
- 65.Wood PM and Bunge RP, Evidence that sensory axons are mitogenic for Schwann cells. 1975. [DOI] [PubMed] [Google Scholar]