

Abstract
During acute brain ischemia, α2-antiplasmin markedly enhances brain injury, blood brain barrier breakdown and matrix metalloproteinase-9 (MMP-9) expression. Although α2-antiplasmin inhibits fibrin thrombus-degradation, and MMP-9 is a collagen-degrading enzyme altering blood brain barrier, both have similar deleterious effects on the ischemic brain. We examined the hypothesis that MMP-9 is an essential downstream mediator of α2-antiplasmin’s deleterious effects during brain ischemia. Middle cerebral artery thromboembolic stroke was induced in a randomized, blinded fashion in mice with increased blood levels of α2-antiplasmin. There was a robust increase in MMP-9 expression (immunofluorescence) in the ischemic vs. the non-ischemic hemisphere of MMP-9+/+ but not MMP-9−/− mice, 24 h after stroke. Brain swelling and hemorrhage were significantly increased in the ischemic vs. the non-ischemic hemisphere of MMP-9+/+ mice. By comparison to MMP-9+/+ mice, the ischemic hemispheres of MMP-9−/− mice showed a ~6-fold reduction in brain swelling (p<0.001) and a ~9-fold reduction in brain hemorrhage. Brain infarction (p<0.0001) and TUNEL-positive cell death (p<0.001) were significantly diminished in the ischemic hemisphere of MMP-9−/− mice vs. MMP-9+/+ mice. Ischemic breakdown of the blood brain barrier and fibrin deposition were also significantly reduced in MMP-9−/− mice vs. MMP-9+/+ mice (p<0.05), as measured by quantitative immunofluorescence. We conclude that MMP-9 deficiency ablates many of the deleterious effects of high α2-antiplasmin levels, significantly reducing blood brain barrier breakdown, TUNEL-positive cell death, brain hemorrhage, swelling and infarction. This suggests that the two molecules may be in a shared pathway in which MMP-9 is essential downstream for the deleterious effects of α2-antiplasmin in ischemic stroke.
Keywords: α2-antiplasmin, matrix metalloproteinase-9, blood brain barrier, ischemic stroke, fibrinogen
Introduction
Ischemic stroke is a leading cause of death and disability worldwide and affects at least ~795,000 Americans each year (Writing Group et al., 2016). Most ischemic strokes are caused by arterial obstruction by fibrin-containing thrombi, which reduces cerebral blood flow and causes ischemic damage with blood brain barrier (BBB) breakdown, brain swelling, hemorrhage and cell death (Lo et al., 2003).
α2-Antiplasmin impairs the dissolution of fibrin thrombi by inhibiting plasmin, the enzyme that dissolves fibrin (Collen and Wiman, 1978; Reed et al., 1990; Lijnen et al., 1999; Reed et al., 2016; Singh et al., 2017). Higher blood levels of α2-antiplasmin may be associated with increased risk for ischemic stroke in humans (Suri et al., 2010; Reed et al., 2016). Blood levels of α2-antiplasmin have dose-related, deleterious effects on ischemic brain injury after experimental thromboembolic stroke (Reed et al., 2014). Higher α2-antiplasmin levels increase BBB breakdown, swelling, microvascular thrombosis and cell death (Reed et al., 2014). High blood levels of α2-antiplasmin oppose the therapeutic effects of treatment with recombinant tissue plasminogen activator (tPA) in ischemic stroke (Marti-Fabregas et al., 2005). However, the molecular mechanisms through which α2-antiplasmin modulates ischemic stroke outcomes are not fully understood.
There is a dose-dependent relationship between α2-antiplasmin blood levels and the expression of matrix metalloproteinase-9 (MMP-9) in experimental stroke, with high α2-antiplasmin blood levels linked to increased MMP-9 in the ischemic hemisphere (Reed et al., 2014). MMP-9 has been implicated in BBB breakdown, cerebral edema, infarction and hemorrhagic transformation during ischemic stroke (Asahi et al., 2000; Asahi et al., 2001; Turner and Sharp, 2016). Studies with MMP-9−/− mice indicate that MMP-9 increases infarct volumes, hemorrhagic transformation, cerebral edema, and functional deficits (Asahi et al., 2000; Asahi et al., 2001; Hu et al., 2009; Wang et al., 2009). Both α2-antiplasmin and MMP-9 enhance BBB leakage, brain swelling, hemorrhage, and neuronal cell death. The fact that MMP-9 and α2-antiplasmin have similar effects on the ischemic brain, suggests they may act through a common pathway. In this study we examined the hypothesis that MMP-9 is essential for the deleterious effects of α2-antiplasmin on the ischemic brain during experimental thromboembolic stroke in mice.
Experimental Procedures
Proteins and reagents
Reagents were purchased from the following sources: bovine thrombin (Sigma, St. Louis; MO); citrated frozen human plasma (Lampire Biological Laboratories; Everett, PA);125I-fibrinogen (Perkin-Elmer; Waltham, MA); human α2-antiplasmin (Athens Research & Technology, Inc.; Athens, GA), proteinase K (Invitrogen Corporation Carlsbad, CA); all the other reagents (Sigma, St. Louis, MO).
Thromboembolic Ischemic stroke
Animal studies were approved by the Institutional Animal Care and Use Committee, University of Tennessee Health Science Center, Memphis. Adult male and female MMP-9+/+ and MMP-9−/− mice on a C57Black/6J background (Jackson Labs, Bar Harbor, ME) of 9-15 weeks of age were used in a series of randomized and blinded experiments in which the operator and data analyst were blinded to the agent under study. Thromboembolic stroke was induced by injecting plasma clots made with pooled fresh frozen normal mouse plasma (with traces of I125-fibrinogen~ 5000 cpm) at the origin of left middle cerebral artery (MCA) essentially as we have described (Houng et al., 2014; Singh et al., 2016). Embolization was confirmed by ≥ 80% drop in hemispheric blood flow measured by a laser Doppler flow (LDF, ADInstruments, Oxford Optronics, UK) probe mounted on the skull. Mice were kept on continuous anesthesia during surgery and injected with 200 μl of 0.9% saline after surgery. After 24 h of thromboembolism, mice were euthanized and the blood was collected by cardiac puncture. To increase blood α2-antiplasmin levels, mice were infused with human α2-antiplasmin, (4.7 mg/kg) through jugular vein 45 min before inducing stroke (Reed et al., 2014). 4.7 mg/kg α2-antiplasmin represents a dose of approximately 1 μM based on an average 25-30 g mice weight with ~ 2 ml blood volume. Our recent studies show that this dose is sufficient to achieve physiological levels (~1 μM) when injected to α2-antiplasmin KO mice (Singh et al., 2017) and a 2 fold increase in WT C57Bl/6 mice (Reed et al., 2014). Human and murine α2-antiplasmin have equivalent binding and inhibition constants for cross species plasmin (Lijnen et al., 1994). So, based on large dose requirements of α2-antiplasmin for in vivo studies and availability of highly purified protein, human α2-antiplasmin was used in these studies. There was a 19 % mortality during 24 h stroke in different mice groups under study and the mice that died before the 24 h time period were excluded from analysis for ischemic brain injury.
Measurement of thrombus dissolution, brain Infarction, hemorrhage and swelling
Following whole body saline perfusion, the whole brain tissue was harvested and residual I125-fibrinogen radioactivity was immediately recorded in a gamma counter to calculate percent thrombus dissolution as we have previously described (Houng et al., 2014). The brain tissue was sectioned (2 mm) coronally to calculate the percent brain swelling, hemorrhage and infarction as we have described (Houng et al., 2014; Singh et al., 2016). Brain slices were immediately photographed digitally on both sides through a microscope before and after triphenyl tetrazolium chloride (TTC, 2%) staining. Quantitative analysis of digital images on both hemispheres and both faces of brain images were done using Image Pro Plus 6.2 (Media Cybernetics, Bethesda, MD). For brain swelling, the volumes of ischemic and contralateral hemispheres on both faces of all brain slices were measured and quantitated by using the formula: swelling percentage = 100 × (volume of the infarcted hemisphere – volume of the control hemisphere)/volume of the control hemisphere). For brain hemorrhage, the area of hemorrhage was measured on infarct and contralateral hemispheres on both sides of brain slices and quantitated by using the formula: the percent hemorrhage = 100 × (volume of hemorrhage in the infarcted hemisphere/volume of the control hemisphere). To determine the percent hemisphere infarction, the TTC-stained areas of the ischemic and non-ischemic hemispheres were measured on both faces of each brain slice. The percent infarction was calculated for each brain by the formula: infarct percentage = 100 × (VC-VL/VC), where VC = TTC-stained area in the control hemisphere × slice thickness, VL = TTC-stained area in the infarct hemisphere × slice thickness.
Tissue histology and Immunofluorescence staining
Brain tissues were fixed in 4% paraformaldehyde for 24 h and paraffin-embedded brain sections (5 μm) were used for immunofluorescence staining as described (Houng et al., 2014; Singh et al., 2016). Sections were deparaffinized (Safeclear II, Fisher Diagnostics, MI) and antigen retrieval was performed by heat-induced epitope retrieval at 98 °C in 10 mM sodium citrate buffer, pH 6.0 for 20 min or 20 μg/ml proteinase K solution (in 50 mM Tris, 2 mM EDTA pH 8.0) for 15 min at 37 °C under moist conditions. After blocking with 10% normal donkey serum for 1 h at room temperature, the sections were incubated with primary antibody in 2% normal donkey serum overnight at 4 °C followed by 45 min incubation with fluorophore-conjugated donkey secondary antibodies. The primary antibodies include rabbit anti-mouse type IV collagen (Karlan Research Products Corporation, Cottonwood, AZ, #ECM451), goat anti type IV collagen (SouthernBiotech Birmingham, Al, # 1340-01), goat anti-mouse albumin (Abcam, Cambridge, MA #ab19194), goat anti-mouse MMP-9 (R&D systems, #AF 909, Minneapolis, MN) and rabbit anti mouse fibrinogen (MyBioSource, San Diego, CA, #MBS315814). The secondary antibodies were Alexa Fluor® 555 donkey anti-goat and donkey anti rabbit, and Alexa Fluor® 488 donkey anti-goat and donkey anti rabbit (Life technologies, Eugene, OR). The slides were mounted with Vectashield hardset mounting media (Vector Laboratories, CA, #H-1500) containing DAPI for nuclei staining. TUNEL staining for ischemic cell death was performed with the In situ Cell Detection Kit (Roche Life Sciences # 11684795910) as per manual’s instructions.
Imaging and analysis
The slides were scanned by a blinded experimenter on Aperio FL (Vista, CA) the digital fluorescence slide scanner using constant excitation/exposure settings. All the slides were scanned under the same experimental conditions with a negative control on each slide. Images at different magnifications were captured with a scanner-compatible software, Imagescope using the same contrast/brightness/gain settings for all images in both groups under study. Quantification of the fluorescence area/intensity was performed in Image-Pro Plus, a powerful image analysis software with the images set to a constant color range in the histogram mode analysis. The same parameters were applied to all images in the different mice brains under study. BBB breakdown was determined by the leakage of albumin in to the brain by measuring the average area of albumin immunofluorescence in 8-10 representative images at 20X magnification (100 μm) from the ischemic and the non-ischemic hemisphere as we have described previously (Singh et al., 2016). MMP-9 expression was examined by total immunofluorescence intensity of MMP-9 in the ischemic vs non-ischemic hemisphere and representative images at 20X magnification images (100 μm) were used. Fibrinogen deposition in the brain was quantified by measuring average fibrin(ogen) positive area in 8-10 representative images at 20X magnification (100 μm) from the ischemic and the non-ischemic hemisphere. The total numbers of TUNEL positive cells were counted as total numbers of objects (green fluorescence) in 1 mm images in the whole ischemic hemisphere for both groups.
Statistical analysis
Graph Pad Prism 5.0 software (San Diego, CA) was used for statistical analysis. Data are represented as the mean ± standard error. Normally distributed data were analyzed by an unpaired Student’s t-test or a one-way ANOVA using the Neuman–Keuls correction. Non-parametric data were analyzed by a Mann–Whitney test. A two-tailed p<0.05 was considered statistically significant.
Results
MMP-9 alters swelling, hemorrhage and decreases cerebral infarction caused by increased α2-antiplasmin levels
Our previous studies show that higher α2-antiplasmin levels during thromboembolic stroke induced dose-dependent increases in MMP-9 expression (r=0.94, P<0.0001) in the ischemic hemisphere as early as 6 h, that were associated with increased gelatinase activity (Reed et al., 2014). To confirm this earlier observation, we examined mice with ~2-fold increased α2-antiplasmin blood levels, achieved by an intravenous administration of human α2-antiplasmin (4.7 mg/kg) immediately prior to experimental stroke (Reed et al., 2014). As expected, MMP-9−/− mice did not show evidence of MMP-9 immunofluorescence in the ischemic or non-ischemic hemisphere after 24 h MCA thromboembolism (Figure 1A and B). MMP-9 expression was increased in the ischemic hemisphere of MMP-9+/+ mice supplemented with α2-antiplasmin while the non-ischemic hemisphere showed negligible to no MMP-9-positive immunofluorescence (Figure 1C, D and E, p<0.001).
Figure 1. Effect of increased blood α2-antiplasmin levels on MMP-9 expression.
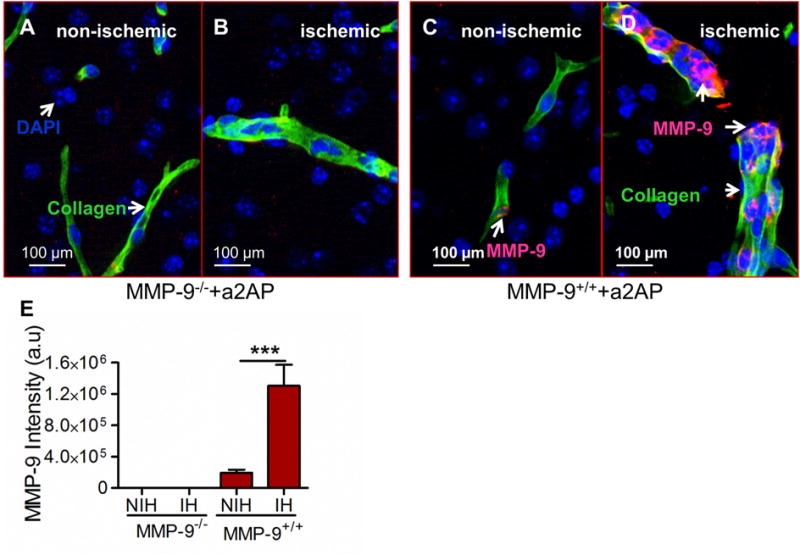
MMP-9+/+ and MMP-9−/− mice were intravenously supplemented with α2-antiplasmin prior to MCA thromboembolic stroke. Brain tissue was examined 24 h later. (A and B) Representative merged images (scale bar=100 μm) of the ischemic (IH) and non-ischemic (NIH) hemispheres after immunostaining for type IV collagen (green, blood vessels) and MMP-9 (red) in (A, B) MMP-9−/− mice and (C, D) MMP-9+/+ mice. Areas with both type IV collagen and MMP-9 stain yellow. MMP-9 expression (red color) inside type IV collagen (green, blood vessels) is indicated by white arrows. DAPI stained nuclei are blue in color. (E) Bar graph showing total MMP-9 intensity in arbitrary units (a.u.) in the ischemic hemisphere(IH) and non-ischemic hemisphere (NIH) of MMP-9+/+ mice. N= 4-6 ***p<0.001.
In MMP-9+/+ mice with high α2-antiplasmin levels, there was macroscopic evidence of brain swelling (Figure 2A) and hemorrhage (Figure 2B) in the ischemic hemisphere vs. the non-ischemic hemisphere. When compared to MMP-9+/+ mice, brain swelling was reduced ~6-fold in MMP-9−/− mice (Figure 2 A, D, p<0.001). Brain hemorrhage was also significantly reduced (~9-fold) in MMP-9−/− mice when compared to MMP-9+/+ mice (Figure 2B, E, p<0.01; Mann-Whitney test). Similarly, there was a 3-4-fold reduction in brain infarction in MMP-9−/− mice as compared with MMP-9+/+ mice (Figure 2 C, F, p<0.0001). The ischemic cell death was also determined by fluorescence TUNEL staining. The total number of TUNEL-positive cells was reduced by more than 3-fold (p<0.001) in the ischemic hemispheres of MMP-9−/− mice with increased α2-antiplasmin as compared to MMP-9+/+ mice with increased α2-antiplasmin levels (Figure 3 A-E). Further, brain infarct volumes were significantly reduced in MMP-9−/− mice with high α2-antiplasmin levels when compared to MMP-9+/+ mice with increased α2-antiplasmin levels (11.6±3.8% vs. 52.1±2.6%; p<0.001). In contrast, MMP-9−/− mice with α2-antiplasmin treatment had equivalent infarct volumes to MMP-9−/− mice with normal α2-antiplasmin levels (11.6±3.8% vs. 11.9±2.9%, P=NS). This indicates that MMP-9 is essential for much, if not all, of the deleterious effects of α2-antiplasmin on the ischemic brain.
Figure 2. Effect of increased blood α2-antiplasmin levels on brain swelling, hemorrhage and cerebral infarct in MMP-9+/+ vs MMP-9−/− mice.
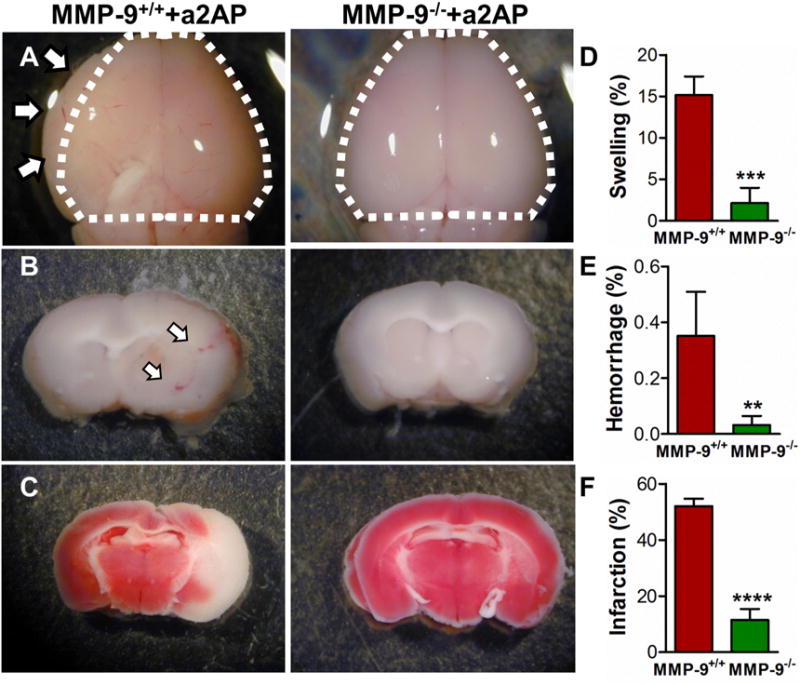
MMP-9+/+ and MMP-9−/− mice were intravenously supplemented with α2-antiplasmin prior to MCA thromboembolic stroke. Brain tissue was examined 24 h later. (A) Representative images of brain swelling in the ischemic hemisphere. To visualize the swelling, dotted lines outline the area of the contralateral non-ischemic hemisphere and arrows indicate visible brain swelling beyond that margin in the ischemic hemisphere. (B) Brain hemorrhage (red color; white arrows) in 2 mm brain sections before TTC staining. (C) Cerebral infarction (white) after TTC staining. The bar graphs show (D) quantitation of swelling (%), (E) hemorrhage (%) and (F) infarct volume (%) in the ischemic vs non-ischemic hemisphere. N=5-7 per group, **p<0.01***p<0.001, ****p<0.0001.
Figure 3. Effect of increased blood α2-antiplasmin levels on apoptotic/necrotic cell death in MMP-9+/+ vs MMP-9−/− mice.
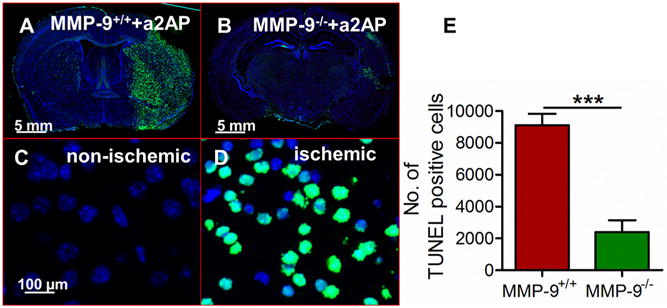
(A, B) Representative immunofluorescence images of whole brain showing apoptotic/necrotic cell death (green) by TUNEL staining in α2-antiplasmin supplemented MMP-9+/+ and MMP-9−/− mice. In low power images of brain slice, TUNEL-positive cell staining was largely restricted to the ischemic hemisphere (C, D) 20X magnification images (Scale bar =100 μm) showing TUNEL positive (green) cells and DAPI stained blue nuclei in the ischemic hemisphere vs. only blue nuclei in the non-ischemic hemisphere. (E) The bar graph shows total number of apoptotic/necrotic cells counted as number of objects in the ischemic hemisphere of MMP-9+/+ vs MMP-9−/− mice supplemented with exogenous α2-antiplasmin. N=4-6, ***p<0.001.
MMP-9 affects BBB breakdown caused by higher α2-antiplasmin
Previous studies have indicated that α2-antiplasmin contributes to BBB breakdown and MMP-9 is an important mediator of BBB breakdown (Asahi et al., 2001; Lakhan et al., 2013; Turner and Sharp, 2016). Therefore, we examined whether the effects of increased α2-antiplasmin on BBB breakdown are mediated through MMP-9. BBB breakdown was assessed by examining the leakage of albumin outside type IV collagen-stained blood vessels and quantitatively determined as the average albumin positive area in the ischemic and non-ischemic hemisphere as we have described previously (Reed et al., 2014; Singh et al., 2016). In MMP-9+/+ mice, the leakage of albumin outside of blood vessels in the ischemic hemisphere, was ~9-fold greater than observed in the non-ischemic control hemisphere (Figure 4A, B and E). In contrast, BBB breakdown in the ischemic hemisphere of MMP-9−/− mice was not significantly different from non-ischemic hemisphere (Figure 4C, D and E). When compared to MMP-9+/+ mice, in the ischemic hemisphere, the leakage of albumin outside collagen-stained blood vessels was diminished by 3-fold in MMP-9−/− mice, (p<0.001) (Figure 4B, D and E). Increased albumin leakage into the brain parenchymal tissue in MMP-9+/+ mice after α2-antiplasmin administration suggests that vascular permeability is increased in comparison to MMP-9−/− mice with α2-antiplasmin treatment.
Figure 4. Effect of increased bloodα2-antiplasmin levels on BBB breakdown in MMP-9−/− vs MMP-9+/+ mice.
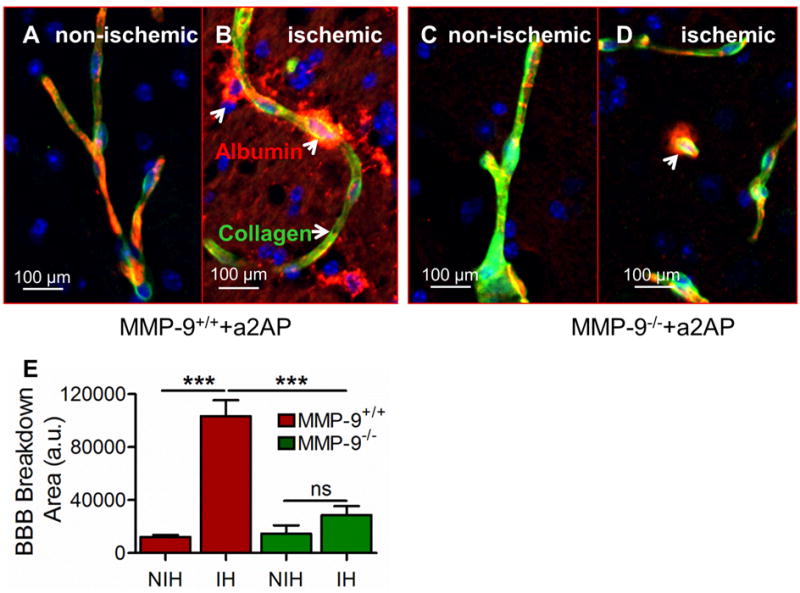
BBB breakdown was examined by leakage of albumin from intravascular to extravascular brain parenchymal tissue in MMP-9−/− and MMP-9+/+ mice supplemented with α2-antiplasmin. (A and B) Representative, merged 20X magnification (Scale bar = 100 μm) images of albumin leakage (red, arrows) outside blood vessels (type IV collagen; green) showing BBB breakdown (white arrows) in the ischemic vs. the non-ischemic hemisphere in MMP-9+/+ mice. Yellow color shows overlapping expression of collagen and albumin. (C and D) BBB breakdown in ischemic vs non-ischemic hemisphere in MMP-9−/− mice (E) The bar graph shows the average albumin positive area (arbitrary units, a.u.) in the ischemic hemisphere (IH) and the non-ischemic (NIH) hemisphere in MMP-9+/+ and MMP-9−/− mice, quantified in at least 8-10 representative images from each hemisphere. N=4-6, ***p<0.001, ns- non significant.
MMP-9 deficiency is associated with decreased fibrin(ogen) deposition caused by higher α2-antiplasmin levels in the brain
α2-antiplasmin increases brain microvascular thrombosis and plasminogen deficiency also increases microvascular thrombosis in the ischemic brain in a dose-dependent fashion in thromboembolic ischemic stroke (Reed et al., 2014; Singh et al., 2016). Fibrin is an important component of thrombo-inflammatory mechanisms in the brain causing ischemic brain injury. MMP-9+/+ mice supplemented with α2-antiplasmin, had increased fibrin(ogen) deposition in the ischemic hemisphere by comparison to the non-ischemic hemisphere (Figure 5A, B and E). Fibrin(ogen) staining was observed in the intravascular compartment (microvascular thrombosis) as well as in the brain parenchyma in the ischemic hemisphere (Figure 5B). In contrast, in MMP-9−/− mice supplemented with α2-antiplasmin, there was no apparent difference in fibrin(ogen) deposition in the ischemic vs. the non-ischemic hemisphere (Figure 5C, D and E). Total fibrin(ogen) deposition in the brain (intravascular and extravascular) was significantly less in MMP-9-deficient (MMP-9−/−) mice as compared to MMP-9+/+ mice after α2-antiplasmin supplementation (p<0.01) (Figure 5B, D and E). As expected MMP-9+/+ and MMP-9−/− had equivalent MCA thrombus dissolution (p=0.415) but there was an increased fibrin(ogen) deposition, emphasizing the potential contributions of secondary microvascular thrombosis and fibrin deposition to the ischemic brain injury.
Figure 5. Effects of MMP-9 deficiency on fibrin(ogen) deposition in the brain after stroke.
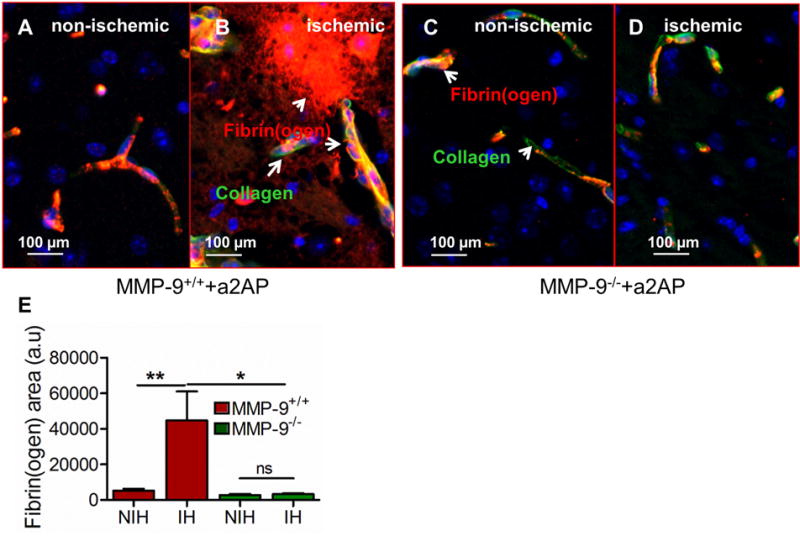
(A, B) Merged immunofluorescence images of fibrin(ogen) deposition (red, arrows), type IV collagen (green, blood vessels) or both (yellow) in the ischemic (A) vs non-ischemic hemisphere (B) of MMP-9+/+ mice with increased α2-antiplasmin. Blue color represents DAPI stained nuclei. (C, D) Representative (100 μm) images showing fibrinogen deposition in the ischemic (C) vs non-ischemic hemisphere (D) in MMP-9−/− mice with increased α2-antiplasmin. (E) Average area of fibrin(ogen) deposition in the ischemic (IH) vs the non- ischemic (NIH) hemisphere in 8-10 images from each hemisphere in MMP-9+/+ and MMP-9−/− mice. N=4-6, **p<0.01, *p<0.05, ns- non significant.
Discussion
α2-antiplasmin enhances ischemic brain injury, brain hemorrhage, swelling, BBB breakdown and TUNEL-positive cell death in experimental stroke (Reed et al., 2014). There is a dose-relationship relationship between blood levels of α2-antiplasmin and MMP-9 expression in the ischemic brain, suggesting that the harmful effects of α2-antiplasmin may be mediated through MMP-9 (Reed et al., 2014). Consistent with this hypothesis, we find that MMP-9 deficiency abrogates the deleterious effects of higher blood α2-antiplasmin levels, significantly reducing BBB breakdown, TUNEL-positive cell death, brain hemorrhage, swelling and infarction during experimental thromboembolic stroke. Taken together, these data show that MMP-9 appears to be essential for many of the harmful effects of increased α2-antiplasmin in ischemic stroke.
The fibrinolytic system is linked to MMP-9 expression-activation and BBB breakdown during ischemic stroke (Bardehle et al., 2015). Cerebral ischemia causes upregulation of tPA and the production of plasmin, which contributes to the activation of pro-MMP-9 and to BBB breakdown (Kaur et al., 2004). As a plasmin inhibitor, α2-antiplasmin might be expected to decrease MMP-9 activation and have protective effects on the BBB during ischemic injury. However, α2-antiplasmin increases MMP-9 expression in a dose-dependent fashion after thromboembolic stroke (Reed et al., 2014). Similarly, plasmin(ogen)-deficient mice also show increased MMP-9 expression and BBB breakdown after thromboembolic ischemic stroke (Singh et al., 2016). Higher α2-antiplasmin levels or plasminogen deficiency both decrease fibrinolysis and enhance microvascular thrombosis and fibrin(ogen) deposition in the brain (Reed et al., 2014; Singh et al., 2016). Even when fibrinolysis is suppressed by plasminogen deficiency, experimental prevention of intravascular fibrin thrombus formation significantly diminishes MMP-9 expression, ischemic brain injury and breakdown of the BBB (Singh et al., 2016). Previous studies indicate that MMP-9 binds to fibrin (Makowski and Ramsby, 1998) and vascular fibrin deposition enhances CD-18-mediated adherence of neutrophils, a major source of MMP-9 after ischemic stroke (Cooper et al., 1988; Gidday et al., 2005; Harris et al., 2005; Rosell et al., 2008; Wang et al., 2009). Increased deposition of fibrin(ogen) (by elevated α2-antiplasmin levels) in the microvasculature may locally concentrate tPA in the vessels and increase tPA’s direct activation of MMP-9. Increased fibrinogen in the micro vessels and in the brain parenchyma may also induce MMP-9 activation by other plasmin-independent mechanisms such as by neutrophils elastase (Ferry et al., 1997), cathepsin G (Wilson et al., 2009) or by other member of MMP family e.g. MMP-3 (Johnson et al., 2011) or MMP13 (Knauper et al., 1997), etc.
The mechanisms through which high blood levels of α2-antiplasmin promote MMP-9 expression are not well understood. There is no evidence for direct interactions between α2-antiplasmin and MMP-9. The C-terminal extension of α2-antiplasmin has been shown in vitro to interact with RGD motifs on human umbilical vein endothelial cells, (Thomas et al., 2007) but whether this interaction promotes MMP-9 expression during stroke is not known. Alternatively, the mechanistic link between α2-antiplasmin and MMP-9 may be indirect. As noted earlier, α2-antiplasmin enhances vascular fibrin deposition which may promote neutrophil retention and MMP-9 release. In aortic aneurysm, another disease associated with intravascular thrombus, blood levels of MMP-9 and α2-antiplasmin-plasmin complex are also elevated (Hellenthal et al., 2009b, a). Further studies will be necessary to examine the hypothesis that fibrin provides the key link between α2-antiplasmin and MMP-9-mediated effects in ischemic stroke. Although these data clearly show that deleterious effects of α2-antiplasmin on BBB breakdown and ischemic brain injury are mainly mediated through MMP-9, MMP-9 may act through other MMPs or other molecules to accomplish its effects. Previous studies have shown that genetic deficiency of MMP-9 and MMP-9 gene silencing in mice does not cause compensatory upregulation of MMP-2, another gelatinase, and MMP-2 levels do not increase after ischemic stroke (Asahi et al., 2000; Hu et al., 2009). However, further work will be necessary to determine whether α2-antiplasmin induces alterations of other members of MMP family including MMP-3 and MMP-13 during ischemic stroke. Moreover, plasmin-independent mechanisms of MMP-9 activation under reduced fibrinolysis conditions in vivo still remains a major question.
In summary, α2-antiplasmin increases brain MMP-9 levels during thromboembolic ischemic stroke in a dose-dependent fashion suggesting a mechanistic link. We show that MMP-9 is essential for many of the harmful effects of α2-antiplasmin in promoting BBB breakdown, swelling, hemorrhage and ischemic brain injury. While the mechanisms through which α2-antiplasmin enhances MMP-9 expression remain unproven, the current data suggest that the two molecules are linked through common fibrin-dependent, thrombo-inflammatory pathways.
Highlights.
High α2-antiplasmin levels are linked to ischemic stroke risk and severe experimental brain injury
α2-antiplasmin causes dose-related increases in matrix metalloproteinase-9 (MMP-9) in the ischemic brain
MMP-9 deficiency blocks α2-antiplasmin’s deleterious effects on brain infarction, swelling and hemorrhage
MMP-9 is essential for the α2-antiplasmin’s harmful effects on the ischemic brain in experimental stroke
Acknowledgments
We appreciate the technical efforts of Nelson Houng.
Funding This work was funded in part by NIH (NINDS NS089707 and by NHLBI HL092750 to GR) and American Heart Association postdoctoral fellowship (16POST31350010 Greater Southeast Affiliateto SS)
Abbreviations
- MMP-9
matrix metalloproteinase-9
- BBB
blood brain barrier
- a2AP
α2-antiplasmin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors’ contributions: SS performed experiments, analyzed data and wrote the manuscript. AH performed experiments and analyzed data. GR designed experiments, analyzed data and wrote the manuscript. All authors have read and approved the final article.
Declarations of Interests: G. L. Reed is a founder of Translational Sciences, Memphis. Satish Singh, Aiilyan K Houng - None
References
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardehle S, Rafalski VA, Akassoglou K. Breaking boundaries-coagulation and fibrinolysis at the neurovascular interface. Front Cell Neurosci. 2015;9:354. doi: 10.3389/fncel.2015.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collen D, Wiman B. Fast-acting plasmin inhibitor in human plasma. Blood. 1978;51:563–569. [PubMed] [Google Scholar]
- Cooper JA, Lo SK, Malik AB. Fibrin is a determinant of neutrophil sequestration in the lung. Circ Res. 1988;63:735–741. doi: 10.1161/01.res.63.4.735. [DOI] [PubMed] [Google Scholar]
- Ferry G, Lonchampt M, Pennel L, de Nanteuil G, Canet E, Tucker GC. Activation of MMP-9 by neutrophil elastase in an in vivo model of acute lung injury. FEBS letters. 1997;402:111–115. doi: 10.1016/s0014-5793(96)01508-6. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. American journal of physiology. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Harris AK, Ergul A, Kozak A, Machado LS, Johnson MH, Fagan SC. Effect of neutrophil depletion on gelatinase expression, edema formation and hemorrhagic transformation after focal ischemic stroke. BMC Neurosci. 2005;6:49. doi: 10.1186/1471-2202-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of abdominal aortic aneurysm progression. Part 2: inflammation. Nat Rev Cardiol. 2009a;6:543–552. doi: 10.1038/nrcardio.2009.102. [DOI] [PubMed] [Google Scholar]
- Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of AAA progression. Part 1: extracellular matrix degeneration. Nat Rev Cardiol. 2009b;6:464–474. doi: 10.1038/nrcardio.2009.80. [DOI] [PubMed] [Google Scholar]
- Houng AK, Wang D, Reed GL. Reversing the deleterious effects of alpha2-antiplasmin on tissue plasminogen activator therapy improves outcomes in experimental ischemic stroke. Exp Neurol. 2014;255:56–62. doi: 10.1016/j.expneurol.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Chen C, Yan J, Yang X, Shi X, Zhao J, Lei J, Yang L, Wang K, Chen L, Huang H, Han J, Zhang JH, Zhou C. Therapeutic application of gene silencing MMP-9 in a middle cerebral artery occlusion-induced focal ischemia rat model. Exp Neurol. 2009;216:35–46. doi: 10.1016/j.expneurol.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. 2011;31:e35–44. doi: 10.1161/ATVBAHA.111.225623. [DOI] [PubMed] [Google Scholar]
- Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator? J Cereb Blood Flow Metab. 2004;24:945–963. doi: 10.1097/01.WCB.0000137868.50767.E8. [DOI] [PubMed] [Google Scholar]
- Knauper V, Smith B, Lopez-Otin C, Murphy G. Activation of progelatinase B (proMMP-9) by active collagenase-3 (MMP-13) European journal of biochemistry. 1997;248:369–373. doi: 10.1111/j.1432-1033.1997.00369.x. [DOI] [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. doi: 10.3389/fneur.2013.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lijnen HR, van Hoef B, Beelen V, Collen D. Characterization of the murine plasma fibrinolytic system. European journal of biochemistry. 1994;224:863–871. doi: 10.1111/j.1432-1033.1994.00863.x. [DOI] [PubMed] [Google Scholar]
- Lijnen HR, Okada K, Matsuo O, Collen D, Dewerchin M. Alpha2-antiplasmin gene deficiency in mice is associated with enhanced fibrinolytic potential without overt bleeding. Blood. 1999;93:2274–2281. [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Makowski GS, Ramsby ML. Binding of latent matrix metalloproteinase 9 to fibrin: activation via a plasmin-dependent pathway. Inflammation. 1998;22:287–305. doi: 10.1023/a:1022300216202. [DOI] [PubMed] [Google Scholar]
- Marti-Fabregas J, Borrell M, Cocho D, Belvis R, Castellanos M, Montaner J, Pagonabarraga J, Aleu A, Molina-Porcel L, Diaz-Manera J, Bravo Y, Alvarez-Sabin J, Davalos A, Fontcuberta J, Marti-Vilalta JL. Hemostatic markers of recanalization in patients with ischemic stroke treated with rt-PA. Neurology. 2005;65:366–370. doi: 10.1212/01.wnl.0000171704.50395.ba. [DOI] [PubMed] [Google Scholar]
- Reed GL, Houng AK, Wang D. Microvascular thrombosis, fibrinolysis, ischemic injury, and death after cerebral thromboembolism are affected by levels of circulating alpha2-antiplasmin. Arterioscler Thromb Vasc Biol. 2014;34:2586–2593. doi: 10.1161/ATVBAHA.114.304530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed GL, Houng AK, Singh S, Wang D. alpha2-Antiplasmin: New Insights and Opportunities for Ischemic Stroke. Semin Thromb Hemost. 2016 doi: 10.1055/s-0036-1585077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed GL, 3rd, Matsueda GR, Haber E. Inhibition of clot-bound alpha 2-antiplasmin enhances in vivo thrombolysis. Circulation. 1990;82:164–168. doi: 10.1161/01.cir.82.1.164. [DOI] [PubMed] [Google Scholar]
- Rosell A, Cuadrado E, Ortega-Aznar A, Hernandez-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–1126. doi: 10.1161/STROKEAHA.107.500868. [DOI] [PubMed] [Google Scholar]
- Singh S, Houng A, Reed GL. Releasing the Brakes on the Fibrinolytic System in Pulmonary Emboli: Unique Effects of Plasminogen Activation and alpha2-Antiplasmin Inactivation. Circulation. 2017;135:1011–1020. doi: 10.1161/CIRCULATIONAHA.116.024421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Houng AK, Wang D, Reed GL. Physiologic variations in blood plasminogen levels affect outcomes after acute cerebral thromboembolism in mice: a pathophysiologic role for microvascular thrombosis. J Thromb Haemost. 2016;14:1822–1832. doi: 10.1111/jth.13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri MF, Yamagishi K, Aleksic N, Hannan PJ, Folsom AR. Novel hemostatic factor levels and risk of ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Cerebrovasc Dis. 2010;29:497–502. doi: 10.1159/000297966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas L, Moore NR, Miller S, Booth NA. The C-terminus of alpha2-antiplasmin interacts with endothelial cells. Br J Haematol. 2007;136:472–479. doi: 10.1111/j.1365-2141.2006.06452.x. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci. 2016;10:56. doi: 10.3389/fncel.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Guo Q, Hossain M, Fazio V, Zeynalov E, Janigro D, Mayberg MR, Namura S. Bone marrow-derived cells are the major source of MMP-9 contributing to blood-brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res. 2009;1294:183–192. doi: 10.1016/j.brainres.2009.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TJ, Nannuru KC, Singh RK. Cathepsin G-mediated activation of pro-matrix metalloproteinase 9 at the tumor-bone interface promotes transforming growth factor-beta signaling and bone destruction. Molecular cancer research: MCR. 2009;7:1224–1233. doi: 10.1158/1541-7786.MCR-09-0028. [DOI] [PubMed] [Google Scholar]
- Writing Group M et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]