

Abstract
Hearing loss is the most common sensorineural disorder, affecting over 5% of the population worldwide. Its most frequent cause is the loss of hair cells (HCs), the mechanosensory receptors of the cochlea. HCs transduce incoming sounds into electrical signals that activate auditory neurons, which in turn send this information to the brain. Although some spontaneous HC regeneration has been observed in neonatal mammals, the very small pool of putative progenitor cells that have been identified in the adult mammalian cochlea is not able to replace the damaged HCs, making any hearing impairment permanent. To date, guided differentiation of human cells to HC-like cells has only been achieved using either embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs). However, use of such cell types suffers from a number of important disadvantages, such as the risk of tumourigenicity if transplanted into the host´s tissue. We have obtained cells expressing hair cell markers from cultures of human fibroblasts by overexpression of GFI1, Pou4f3 and ATOH1 (GPA), three genes that are known to play a critical role in the development of HCs. Immunocytochemical, qPCR and RNAseq analyses demonstrate the expression of genes typically expressed by HCs in the transdifferentiated cells. Our protocol represents a much faster approach than the methods applied to ESCs and iPSCs and validates the combination of GPA as a set of genes whose activation leads to the direct conversion of human somatic cells towards the hair cell lineage. Our observations are expected to contribute to the development of future therapies aimed at the regeneration of the auditory organ and the restoration of hearing.
Introduction
Hearing loss is the most prevalent sensorineural deficit in humans, most frequently caused by damage of hair cells (HCs). These are mechanoreceptor cells in the cochlear part of the inner ear and responsible for transducing the information arriving in the form of incoming sound waves to the auditory neurons that connect to the brain. Although a small number of inner ear progenitor cells have been identified in neonatal animals that allow for a certain degree of repair following damage, these appear to be dormant in older individuals, which could explain the observed lack of HC regeneration [1,2]. Approaches that could thus be envisaged towards the restoration of hearing are either the in vivo transdifferentiation into HCs of other cell types present in the inner ear (e.g. supporting cells) or transplantation of HC-like cells that have been derived from distinct tissue sources [2,3,4]. HC-like cells have been obtained from embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) [5,6,7,8,9,10,11,12,13]. Although the studies where these cells have been more thoroughly characterized have identified properties of vestibular rather than cochlear HCs [9,14], they set the basis for obtaining their auditory counterparts.
A promising alternative to the ESCs or iPSCs may be the use of patient-derived somatic cells that, although fully differentiated, can be changed into the desired cell fate while bypassing the full reprogramming process typically undergone by iPSCs [15,16,17,18,19]. This strategy has already resulted in the successful transdifferentiation of fibroblasts, hepatocytes, astrocytes and various differentiated blood cell types into other cell lineages (e.g. cardiomyocytes, neurons, macrophages) [20,21,22,23,24,25,26]. For reprogramming, two main routes have been followed, either the induction of epigenetic changes, or the direct conversion of somatic cells into sought-for lineages through the forced expression of lineage-determining factors [16,18,20,26,27,28,29,30]. Epigenetic alterations have been shown to occur following exposure of the cells to chromatin modifiers such as demethylating agents and histone deacetylase inhibitors, or overexpression of transcription factors such as Sox-2 [31] and Oct-4 [24,32]. This results in the transient expression of sets of genes that are associated with a variety of cell lineages. Subsequent culture of these cells under conditions known to drive the emergence of the cell type of interest in vivo will then promote their differentiation. On the other hand, overexpression of transcription factors associated with a given lineage is thought to result in the direct conversion of the donor cells, through the recruitment of downstream genes and the activation of the corresponding signalling cascades [22,28,29]. Importantly, this approach has already been successfully applied in vivo [2,23,26,33,34].
In the present study, we have adopted a direct conversion approach in order to obtain cells expressing HC markers from cultures of human fibroblasts (hFIBs). In order to do so, and based on the publication by Costa et al. [7], we overexpressed GFI1, Pou4f3 and ATOH1 (hereafter referred to as GPA) coding sequences in human fibroblast cell cultures. This led to the differentiation of cells expressing HC markers, and our work thus validates the potential of the GPA combination to convert fibroblasts of human origin into cells expressing hair cell markers. To our knowledge, this is the first time that conversion of human somatic cells towards the hair cell lineage is reported.
Materials and methods
Culture of hFIBs
hFIB cultures were established from skin biopsies from human donors from whom written consent had been obtained, as described in [35]. The project was approved by the clinical research ethic committee of the University of Valladolid (reference: 2012/15). The cells were routinely passaged 1:3 and grown in standard human fibroblast medium (hFib Med), consisting of Dulbecco´s Modified Eagle Medium (DMEM; Life Technologies, Eugene, OR, USA) supplemented with 1x GlutamaxTM-I CTSTM (Gibco, Life technologies, Grand Island, NY, USA) and 1x MEM non-essential amino acid solution (NEAA; Merck, Darmstadt, Germany), Penicillin (100U/ml)-Streptomycin (100 μg/ml, Life technologies, Grand Island, NY, USA) and 10% foetal bovine serum (FBS; Gibco). Infection of the cultures was carried out at passage numbers 4–8.
DNA constructs and preparation of lentiviral particles
The OCT4 sequence was removed from the lentiviral plasmid pSIN-EF2-OCT4-Pur (Addgene, Cambridge, MA; plasmid 16579, deposited by James Thomson) as an SpeI-EcoRI fragment and the remaining DNA vector was used to prepare the constructs pSIN-EF2-Pou4f3-Pur and pSIN-EF2-GFI1-Pur. The former was built as follows: a 1,7kb SalI-EcoRI fragment containing the mouse Pou4f3 ORF (1kb) and a 0,7kb 3´ UTR genomic sequence was excised from pRK5KS-Brn3c (kindly provided by Mengqing Xiang; Piscataway, NJ, USA) and cloned into a pCDNA3 vector; this sequence was subsequently subcloned as an XbaI-EcoRI fragment into the SpeI/EcoRI-digested pSIN-EF2-Pur vector. In order to build the pSIN-EF2-GFI1-Pur construct, the sequence coding for GFI1 was amplified from the human cDNA clone SC126131 (OriGene Technologies, Rockville, MD, USA) using PCR primers SpeI-GFI1-F (5´-AACTAGTGCCGGACCACCATGC-3´) and ERI-GFI1-R (5´- GGAATTCATTTGAGCCCATGCTG-3´), carrying an SpeI and an EcoRI sequence, respectively, and Platinum Taq (Invitrogen, Carlsbad, CA, USA); amplification conditions were: 1 cycle at 95° for 5 minutes; 30 cycles at 94°C for 30 seconds, 53°C for 45 seconds, and 72°C for 2 minutes; 1 cycle at 72°C for 10 minutes. The PCR fragment was purified and its sequence confirmed, and subsequently digested with SpeI and EcoRI and cloned into the SpeI/EcoRI-digested pSIN-EF2-Pur vector. The pLV-ATOH1-IRES-eGFP construct was obtained by excising a BamHI-XhoI fragment containing the ATOH1 coding sequence from the pRV-IRES-MATH1 (provided by Micaela Gallozzi) and subcloning it into the lentiviral vector pLV-IRES-eGFP, kindly provided by Carlos Vicario Abejón (Madrid, Spain).
Although pSIN-EF2-(Pou4f3 or GFI1)-Pur and pLV-ATOH1-IRES-eGFP belong to the third-generation of lentiviral transfer vectors [36,37], pSIN-EF2-Pou4f3-Pur and pSIN-EF2-GFI1-Pur viral particles were obtained by co-transfecting either of these DNAs with the second generation packaging plasmid psPAX-2 (Addgene plasmid 12260) and the envelope plasmid pMD2.G (Addgene plasmid 12259) into 293FT cells (Invitrogen), using LipofectamineTM 2000 (ThermoFisher Scientific, Waltham, MA, USA) and according to manufacturer’s instructions. When preparing pLV-ATOH1-IRES-eGFP lentiviral stocks, the pLV-ATOH1-IRES-eGFP construct was co-transfected with the plasmids pMDLg, pRSV and pMD2.C. The former two are third-generation packaging plasmids and the latter is an envelope plasmid [36,37,38]. Viral supernatants were collected at 48 and 72 hours post-transfection, pooled and concentrated by ultracentrifugation.
Titering of the pSIN-EF2-(Pou4f3 or GFI1)-Pur viral stocks was carried out by incubating HeLa cells (plated the previous day at 2x105 cells/well on 6-well dishes in DMEM medium supplemented with 1x GlutamaxTM-I CTSTM, 1x NEAA, penicillin/streptomycin, 1mM sodium pyruvate (Sigma, Merck), and 10% FBS) with serial dilutions (0, 10−2, 10−3, 10−4, 10−5 and 10−6) of each viral stock, in the presence of 8μg/ml polybrene (Sigma, Merck). Cultures were changed to fresh growth medium the following day and 24 hours later they were exposed to 1μg/ml puromycin (Sigma, Merck). Following 12–14 days in puromycin selection, the cultures were washed with 1x phosphate buffered saline (PBS; ThermoFisher Scientific, Waltham, MA, USA) and stained for 10 minutes in a solution of 1% crystal violet prepared in 10% ethanol. The cultures were thereafter gently washed with 1x PBS and the number of blue-stained colonies was counted in the 10−5 and 10−6 wells. Lentiviral titer (transforming units per ml (TU/ml)) was calculated as (Number of colonies/volume the cells were transduced in (ml)) x Dilution factor. Viral titers were 2,3–4,7 x 107 and 4,8–6 x 106 TU/ml for pSIN-EF2-Pou4f3-Pur and pSIN-EF2-GFI1-Pur stocks, respectively. Titers of pLV-ATOH1-IRES-eGFP viral preparations were measured by analysing the percentage of GFP-expressing cells in HeLa cultures that had been transduced with the virus 72 hours earlier, following the same protocol as that for the pSIN-LVs. This was carried out by flow cytometric analysis using a Gallios Flow Cytometer (Beckman Coulter, Brea, CA, US); GFP was detected with a blue solid-state laser (488nm) and a 525 BP 40 filter. pLV-ATOH1-IRES-eGFP titers were calculated as [(%GFP(+) cells in well x total No of cells infected)/Volume the cells were transduced in] x Dilution factor, and were in the range of 2,7–6 x 107 TU/ml.
Lentiviral transduction of hFIB cultures and culture of GPA hFIBs
2,2x104 cells were plated on each well in 6-well dishes, in hFib Med. Cultures were infected four days later, at a MOI of 8, 7 and 1,8 with pLV-ATOH1-IRES-eGFP, pSIN-EF2-Pou4f3-Pur, and pSIN-EF2-GFI1-Pur lentiviral preparations, respectively, in fresh hFib Med with 8μg/ml polybrene added. The medium was changed to fresh hFib Med 24 hours later. These cultures are hereafter referred to as GPA hFIBs. Transduction efficiencies performed in triplicate samples were 46,72 ± 20,88%, 76,49 ± 11.01%, and 85,93 ± 2,62% for the pLVATOH1-IRES-eGFP, the pSIN-EF2Pou4f3-Pur and the pSIN-EF2-GFI1 viruses, respectively.
Following removal of the transduction medium, some cultures were maintained in hFib Med for 12–15 days; others were grown in serum-free medium (SFM), consisting of DMEM:F12 (Gibco), 1x Glutamax, 1x N-2 Supplement (Gibco), and 2x B27 Supplement (Gibco), containing 20ng/ml recombinant human rhEGF (ImmunoTools, Friesoythe, Germany) and 1μM retinoic acid (RA) (Merck). No antibiotic was added to the culture media during differentiation (except puromycin when preparing samples for RNAseq analyses (see below)) and cells were passaged once onto wells and coverslips coated with 0,1% gelatin (Millipore, Merck).
Preparation of GPA hFIB samples for RNAseq analysis
Four days after lentiviral transduction, cultures grown in SFM containing 20ng/ml EGF and 1μM RA were changed to fresh medium containing 1,5μg/ml puromycin (Sigma, Merck) and maintained in selection for an additional 8 days. RNA was then extracted from these GPA hFIB cultures using the RNeasy Micro kit (QIAGEN, Hilden, Germany), introducing the DNAse treatment step and according to manufacturer´s instructions. RNA libraries were prepared for sequencing using standard Illumina protocols. Mapping of the reads to the GRCh38 reference genome was performed with Star (version 2.5.2) [39]. Generation of count tables and differential expression was done by parsing Star output with edgeR (version 3.2.3) [40]. Gene ontology was performed using PANTHER [41]. RNAseq data have been deposited at GEO with accession number GSE108905.
Quantitative reverse transcription polymerase chain reaction (qPCR)
Isolation of total RNA from cell cultures was performed using TRIzol® Reagent (Invitrogen), following the manufacturer’s protocol. RNA samples were quantified on a Spectrophotometer ND-1000 (NanoDrop, Thermo Fisher Scientific). One to two micrograms of total RNA was used to synthesize cDNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Life Technologies). cDNA samples were amplified on a LightCycler® 480 II (Roche Molecular Diagnostics, Pleasanton, CA, USA) using SYBR® Green PCR Master Mix (Life Technologies). The thermocycling conditions consisted of an initial denaturation step of 10 minutes at 95°C, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 min. Primers for each of the genes studied in this work were: GAPDH, ACACCCACTCCTCCACCTTTG and CATACCAGGAAATGAGCTTGACAA; MYOSIN VIIA, CCCATTCTGGAAGCATTTGG and ACTTTCCGAAACGGCTTGAG; POU4F3, TTCTCCAGTCTGCACTCTGG and TGCTCATGGTATGGTAGGTGG; ESPIN [11], CAGAGTGCAGGACAAAGACAA and GCAGCGTAGTGGATAGGCAG, and murine Pou4f3, TTTGATGAGAGCCTGCTGGC and TGCTCATGGTATGGTAGGTGG. Data were analysed using the Software version LCS480 1.5.0.39. Relative levels of mRNA expression were calculated according to the 2-ΔΔCt method, using GAPDH as the housekeeping gene [42].
Immunocytochemistry
Immunocytochemical analyses were carried out on cultures grown on glass coverslips coated with 0,1% gelatin (Millipore, Merck). Cells were fixed at room temperature in 4% paraformaldehyde (Merck) for 10–15 minutes. Blocking of non-specific binding sites and cell permeabilization were carried out by adding PBT1 solution (PBS containing 0,1% Triton X-100 (Sigma, Merck), 1% BSA (Sigma, Merck) and 5% FBS) for 15 minutes prior to the addition of the primary antibody; this was diluted in PBT1 and left to incubate for 1 hour at room temperature. Primary antibodies used in this work were: polyclonal rabbit antibody against MYOSIN VIIA (1:500; Proteus BioSciences, Inc., Ramona, CA, USA), mouse monoclonal antibody against POU4F3 (1:50; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, US), polyclonal goat antibody against GFI1 (1:50; R&D Systems, Minneapolis, MN, US), polyclonal goat antibody against ANNEXIN A4 (1:50; R&D Systems, Bio-Techne, Minneapolis, MN, USA) and polyclonal rabbit antibody against ESPIN (1:1000; kind gift from Professor James Hudspeth, New York, NY, USA). Following incubation with the primary antibody, the secondary antibody was added, diluted in PBT2 solution (PBS containing 0.1% Triton X-100 and 1% BSA). Secondary antibodies used were Alexa Fluor® 488-conjugated goat anti-mouse IgG and Alexa Fluor® 568-conjugated goat anti-rabbit IgG, and Alexa Fluor® 647-conjugated donkey anti-goat IgG (Invitrogen, Molecular Probes, Eugene, OR, USA). After 1 hour at room temperature, the cultures were mounted on glass slides using Vectashield mounting medium containing DAPI (4’, 6’-diamidino-2-phenylindole) (Vector Laboratories, Inc., Burlingame, CA, USA) to counterstain nuclear DNA. Samples were viewed under an Eclipse 90i fluorescence microscope (Nikon Instruments Europe B.V., Amsterdam, Netherlands). Confocal images of the GPA hFIB cultures were obtained with a Leica DMI 6000B microscope with a TCS SP5 X confocal system and a WLL laser controlled by LAS AF software (Leica, Spain).
Statistics
During the course of this work, a total of 12 independent experiments were performed using hFIB Med and another 12 using RA&EGF-containing SFM; each experiment consisted of a control culture kept in the corresponding medium all throughout the course of the experiment and a culture grown in the same medium but to which the GPA viral combination had been added. Each culture was passaged once during the length of the experiment and replated as duplicate cultures that were ultimately pooled together in order to extract RNA for qPCR analysis (for these analyses, each sample was loaded onto duplicate wells into the qPCR plate, with a deviation between duplicates of 0,1–0,3 Ct units), as well as on sets of 2–4 duplicate coverslips that were used for immunocytochemical analyses. At least two duplicate cultures were stained in parallel with each antibody. Data are presented as the mean ± SD; the Student´s t-test was used to analyse the significance of any differences between control and treatment groups and also between cultures grown in hFIB Med and those maintained in RA/EGF. Values of p<0,05 were considered to be statistically significant.
Results
Effects of GPA overexpression in hFIB cultures
A direct conversion approach was followed [16,17,30], based on the over-expression of GFI1, Pou4f3 and ATOH1 (GPA), three genes whose co-expression has been shown to result in the emergence of HC-like cells in cultures of mESCs [7]. Forced expression of GPA was achieved through infection with lentiviral particles carrying the coding sequences of each of these genes. Overexpression of the three genes was confirmed by qPCR and their coexpression in the transduced cells was analyzed by immunocytochemistry (see Fig 1 and methods). Individual transduction rates were 46,72 ± 20,88%, 76,49 ± 11.01%, and 85,93 ± 2,62% for the LVATOH1-IRES-GFP, the LVPou4f3 and the LVGFI1 viruses, respectively, with 72,81 ± 6,14% of the GFP-positive cells simultaneously expressing both the Pou4f3 and GFI1 proteins (Fig 1B).
Fig 1. Overexpression of the GPA genes in hFIB cultures.

(A) Significantly increased mRNA expression of the genes was observed compared to non-infected control cultures by qPCR analyses. Asterisks: p-value<0,05. (B) GPA protein expression in hFIB cultures simultaneously transduced with pSIN-EF2-Pou4f3-Pur, pSIN-EF2-GFI1-Pur and pLV-ATOH1-IRES-eGFP lentiviruses. Immunostaining of GPA hFIB cultures indicated that a high rate of the GFP(+) cells also expressed the Pou4f3 and the GFI1 proteins. Scale bar: 50μm.
In a first set of experiments hFIBs were grown in standard fibroblast medium (hFIB Med) following infection. No apparent morphological changes were observed after two weeks (Fig 2A and 2B). To assess conversion towards the HC lineage we examined the expression of the HC markers MYOSIN VIIA and ESPIN, typically used for this purpose, together with endogenous expression of POU4F3 via qPCR. This analysis revealed a significant increase in the mRNA levels of all three genes in the GPA hFIB cultures (Fig 3A). Unexpectedly, however, the high levels of MYOSIN VIIA mRNA were accompanied by a very weak induction of MYOSIN VIIA protein in a small percentage of the transduced cells, as revealed by immunocytochemistry (Fig 3B). Only 13,29±3,53% of the GFP-positive cells expressed MYOSIN VIIA protein (Fig 4).
Fig 2. Cultures of hFIBs.
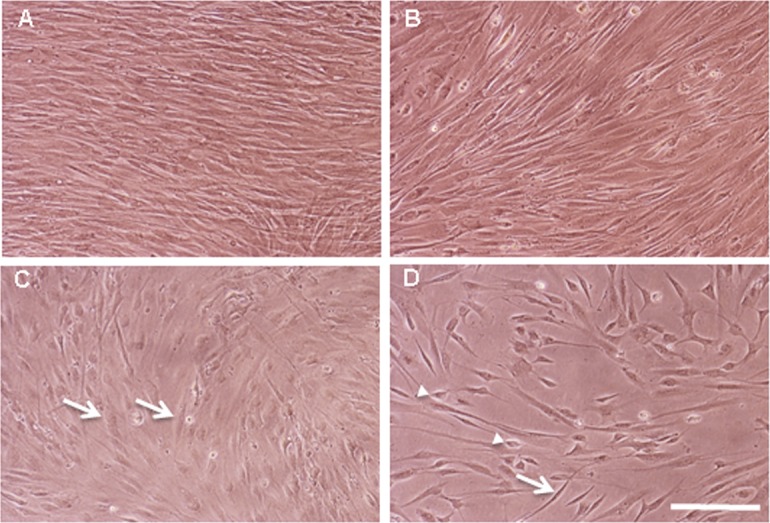
Photomicrographs of living hFIB cultures grown in hFIB Med (A and B) or in RA&EGF-containing SFM (C and D) for two weeks. (A and C) Non-infected control cultures; (B and D) GPA hFIBs transduced with lentiviral particles carrying the GFI1, Pou4f3 and ATOH1 ORFs. Non-infected control cultures (A) and GPA transduced hFIBs (B) grown in hFIB Med showed densely packed fusiform cells, while in the presence of RA&EGF-containing SFM (C) a lower cell density was observed with cells that appeared more flattened out (arrows). (D) GPA transduced hFIB cultures grown in RA&EGF-containing SFM showed a further reduction in cell density and the presence of birefringent elongated (arrow) and smaller polygonal cells (arrowheads). Scale bar: 70μm.
Fig 3. Induction of HC genes in GPA hFIBs grown in hFIB Med.

(A) Relative mRNA expression of the endogenous genes MYOSIN VIIA, POU4F3 and ESPIN, as measured by qPCR analysis. (B) Induction of MYOSIN VIIA protein expression in transduced (GFP-positive) hFIB cells. Note that due to the very high increase in mRNA levels in GPA-infected samples the low levels of mRNAs detected in control samples was barely visualized in the graph in (A). Asterisks: p-value<0.05; scale bar: 20μm.
Fig 4. Quantification of the percentage of GFP- and Myosin VIIA colabelled cells.

GPA hFIB cultures grown for two weeks in hFIB Med (green), and GPA hFIB cultures grown for two weeks in RA&EGF (red), following transduction. Asterisk: p value<0.005.
Culture of GPA transduced hFIBs in the presence of retinoic acid and EGF promotes the induction of HC genes and MYOSIN VIIA protein expression
Previous studies have demonstrated that EGF and retinoic acid (RA) improve the differentiation of HC progenitors and the induction of Myosin VIIA protein [43,44,45]. Furthermore, Costa et al. [7] observed that addition of RA to GPA-expressing mESC cultures enhanced the efficiency of their programming protocol. With this in mind, we conducted a second set of experiments where GPA hFIBs were grown following infection in serum-free medium (SFM) containing both EGF and RA (Fig 5). Compared to GPA hFIB cultures maintained in hFIB Med, RA and EGF induced cells with increased birefringence (Fig 2C and 2D). These cultures also tended to contain a high proportion of GFP-positive cells (56,36±2,9% versus 39,51±7,00%, respectively) (Fig 4), although this difference was not statistically significant (p>0,06).
Fig 5. Induction of HC genes in GPA hFIBs grown for two weeks in RA&EGF.

(A) Induction of MYOSIN VIIA protein expression visualized by immunocytochemistry. (B) Relative mRNA expression of the endogenous genes MYOSIN VIIA, POU4F3 and ESPIN, as measured by qPCR analysis. (C) Induction of ANNEXIN A4 and ESPIN protein expression visualized by immunocytochemistry. Note that due to the very high increase in mRNA levels in GPA-infected samples the low levels of mRNAs detected in RA&EGF control samples were barely visualized in the graph in (B). Asterisks: p-value<0.05; scale bar: 20μm.
Immunocytochemistry revealed that a larger percentage of GFP-positive cells now expressed MYOSIN VIIA protein in cultures treated with RA and EGF compared to cells maintained in hFIB Med (50,95±9,87% in RA&EGF vs. 13,29±3,53% in hFIB Med; p<0,05) (compare Figs 5A with 3B). This was accompanied by very high levels of MYOSIN VIIA mRNA, well above those observed in control experiments, and those of GPA hFIBs grown in hFIB Med (p<0,005; Figs 5B and 3A). Compared to non-infected controls (grown either in hFib Med or in RA&EGF) expression of endogenous POU4F3 and ESPIN mRNAs was also high in these experiments (Figs 3A and 5B). Additional immunocytochemical analysis was conducted in order to search for ANNEXIN A4 and ESPIN expression in the transduced cultures, both markers being typical of HCs [9]. Expression of ANNEXIN A4 occurred in 9,44±0,12% of the GFP-positive cells (Fig 5C). Despite the marked increase in ESPIN mRNA levels indicated by qPCR analysis (see above) we were unable to detect any polarized membrane projections that would indicate the formation of stereocilia, but rather a homogenous staining in a subset of the transduced cells (Fig 5C).
In order to characterize the transcriptional profile of GPA hFIB cultures grown in RA&EGF, we carried out RNAseq analysis (see Methods). RNAseq from GPA hFIB cultures grown in RA&EGF was compared to that from non-treated control hFIBs cells (S1 and S2 Tables). Gene ontology analysis revealed an enrichment of genes related to HC development and differentiation, next to genes involved in neuronal differentiation in RA&EGF hFIBs (Fig 6A and 6B). This included genes involved in HC functions such as mechanoreception and sensory transduction. RNAseq analysis confirmed the upregulation of the hair cell markers MYO VIIA and ESPIN, next to MYO15A/B, USH1C and CDH23 amongst others (S3 Table). Inner ear developmental genes that were up-regulated included SOX9, BMP2/4, TGFB2, FGF9, NTN1, EPHB2 or PROX1 (S3 Table). Additionally, 94 genes that belong to the core group of HC-specific genes isolated from P1 mouse Atoh1-GFP-sorted hair populations were found to be upregulated in the transdifferentiated hFIBs [46]. Finally, consistent with the conversion of GPA-treated cultures, genes associated with cell cycle and cell division were found to be repressed (Fig 6C). Altogether these data indicate that the protocol based on GPA and RA/EGF treatment is able to initiate the conversion of human fibroblasts towards the HC lineage.
Fig 6. Gene ontology analysis.
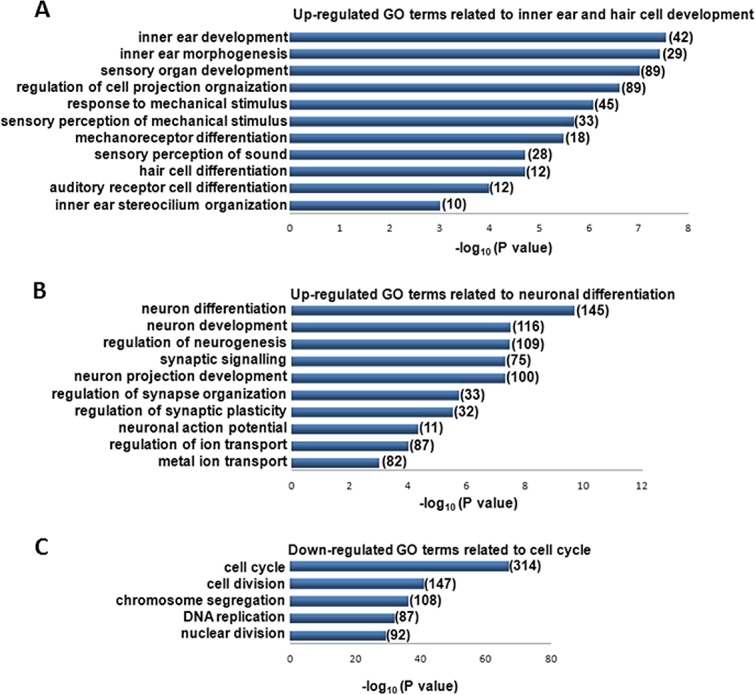
Performed using the PANTHER functional annotation tool for genes significantly up- or downregulated (fold change >2, P<0.01) in fibroblasts transduced with GPA and differentiated towards the HC lineage relative to undifferentiated fibroblasts. The number of upregulated genes included within each gene ontology functional term is shown.
Discussion
According to the latest estimates (March 2018) by the World Health Organization (WHO), disabling hearing loss affects 466 million people worldwide, and about one third of people over 65 years of age suffer from it (http://www.who.int/en/news-room/fact-sheets/detail/deafness-and-hearing-loss). It is the most prevalent sensorineural disorder in humans and its incidence is on the rise. The most frequent cause of hearing deficits is the loss of HCs, the mechanosensory receptors residing in the cochlea. There are just 15 thousand HCs in each cochlea, as compared to the millions of photoreceptors or olfactory receptors, and when they die there is no spontaneous regeneration. Although a very small number of supporting cells may behave as putative progenitor cells in post-natal mice, they do not seem to become active following damage to the adult mammalian cochlea and missing HCs are not replaced by new ones [1,2,29,47].
A large source of HCs and otic progenitors is necessary in order to provide a platform where to carry out extensive analyses on the molecular mechanisms governing the proliferation of otic progenitors, their differentiation and HC survival. The final aim of the research in the field is to identify a set of transcription factors, miRNAs or small molecule combinations that induce new HCs in vivo, to establish cultures of otic progenitors or HC-like cells amenable to analyses and to use them in future regenerative therapies. Accordingly, over the last few years, several groups reported the expansion of otic progenitors [6,48] and the differentiation of ESCs and iPSCs, of human and murine origin, to HC-like cells [6,7,8,9,10,11,49]. This is the first step in order to identify transcription factors and signalling pathways that can lead to regeneration in the inner ear, and thus the restoration of hearing.
Transplantation of progenitors or HC-like cells derived from exogenous cell types may be the only available option in some instances. For instance, when the lost HCs are replaced by a flat epithelium constituted by supporting cells that have become refractory to transdifferentiation, or when a genetic mutation is the underlying cause of the existing hearing deficits [49,50,51]. With respect to the latter, recent progress in iPSC and CRISPR technologies has rendered the in vitro correction of such mutations and the subsequent differentiation of the genetically corrected cells into HC-like cells a feasible option [49,52,53]. Nonetheless, the use of human ESCs or iPSCs as sources for HC-like cells raises a series of concerns. Next to ethical issues and the possibility of immune rejection, there is also the risk that ESCs or iPSCs that remain undifferentiated in the transplant may result in the development of teratomas [54]. In addition, despite the clear advantages posed by the advent of iPSC technology [15,19,49,52], such as the possibility of obtaining patient-derived HC-like cells that would not encounter any immune rejection response, obtaining true iPSC clones is costly and requires specific expertise [19], something that limits its widespread application.
It is in this setting that transdifferentiation of human somatic cells to HC-like cells may become an interesting alternative worthwhile to explore. In this work, we adopted a direct conversion approach whereby we obtained transdifferentiated cells from hFIB cultures that expressed a battery of HC genes, as evidenced by qPCR immunochemistry and RNAseq analysis. This was achieved through the over-expression of three transcription factors, ATOH1, POU4F3 and GFI1, the former being considered a master regulatory gene in the HC lineage [55,56], while the latter two also play critical roles in HC survival [51,57,58,59]. Transient expression of this combination of genes in mESC cultures induces a very fast (4 days) conversion of over 50% of the ES cells into HC-like cells [7]. This is in contrast to the much longer duration of the protocols that have met with success when differentiating hESCs or hiPSCs [6,8,9,11]. In our experimental set-up we confirmed the expression of MYOSIN VIIA protein and MYOSIN VIIA, POU4F3 and ESPIN mRNAs after 12 days of infection. Furthermore, preliminary experiments indicated that expression of these markers requires an even shorter time span, since high levels of MYOSIN VIIA protein expression are already observed at 7 days post-infection (data not shown). On the other hand, preliminary analysis also indicates that a co-expression of ATOH1 and GFI1 alone is already able to induce some MYOSIN VII A protein expression, while this is not the case when any of the factors is expressed by its own.
Despite transduction rates of above 50%, accompanied by the expression of MYOSIN VIIA protein in about half of the transduced GFP-positive cells, and the induction of other HC genes such as POU4F3, ESPIN, and ANNEXIN A4, our protocol faces a series of important limitations. Firstly, although our results provide evidence that conversion towards the hair cell lineage has been initiated, it is unclear if true reprogramming of the hFIB cells is taking place. This would require the confirmation of a stable phenotype of the transdifferentiated cells in the absence of the exogenous expression of GPA. Furthermore, each of the coding sequences for GFI1, Pou4f3 and ATOH1, were cloned into separate lentiviral vectors. As a consequence, and although qPCR data and immunocytochemistry demonstrated the overexpression of each of the different transgenes, actual expression levels of the three transcription factors may vary from cell to cell, leading to differences in the extent of conversion within individual cells. Additionally, the use of lentiviral vectors conveys the integration of exogenous DNA sequences into the host genome, and the concomitant risk of insertional mutagenesis. This also ensures long-term expression of the transgene, while transdifferentiation to stable cellular phenotypes has often been correlated to a loss of transgene expression and the induction of the endogenous counterparts [26,49]. ATOH1 expression, although necessary for the appearance of HCs in the inner ear, needs to be downregulated during HC maturation and its overexpression has been reported to result in cell death [60]. This effect, together with the absence of hair cell stereocilia, that could indicate further differentiation of the transduced cells [61], might be caused by the sustained expression of ATOH1. On the other hand, it is possible that our culture system lacks the appropriate conditions to sustain the survival and maturation towards transdifferentiating cells. Modification of these conditions might lead to better yields of more mature HC-like cells in our cultures [5,9].
Soon after transduction, cell density of GPA hFIB cultures grown in RA&EGF is reduced, and the observation of the cultured cells suggested a halt in cell proliferation. RNAseq analysis confirmed a downregulation of cell cycle genes very much like that of GPA-expressing mESCs [7]. Cell cycle arrest associated with the conversion of GPA hFIBs into post-mitotic cells may thus hinder the extensive characterization of these cells. For example, isolation of individual cell clones where to carry out more extensive analyses was not feasible, and whole transduced cultures had to be used for this work.
Several modifications will be potentially useful to improve our current protocols. For instance, inducible systems carrying the three ORFs together [7,30] and the use of non-integrative vectors may allow tight control of the expression of the transgenes, avoid the risk of insertional mutagenesis and, eventually, long-term expression of the transgenes [62,63]. Given the short period of time required for the conversion process, and the unsuitability of maintaining high levels of expression of the ATOH1 gene, a transient overexpression of the GPA genes should suffice. In this regard, adenoviruses, as well as integrase-defective lentiviral vectors have already yielded successful outcomes [62,64,65,66]. Furthermore, there are other ways to guide cellular transdifferentiation, such as the introduction of DNA sequences via non-viral methods [67,68], or the use of proteins, microRNAs, RNAs or combinations of small molecules [17,69]. In order to improve cell survival and differentiation, several reports have shown that 3-dimensional culture settings promote self-organization into tissue-like structures and favour the emergence of mature phenotypes [5,8,9,14,70,71].
Further work may also improve the limited yield of differentiated cells obtained. Differentiation into proliferating otic progenitors would represent a clear advantage over the current model, both regarding the availability of cells for in vitro analysis and, more importantly, as an abundant source of cells for future regenerative therapies. Regarding this point, it has already been shown for other lineages that it is possible to obtain progenitor cells by the overexpression of key cell fate determination genes together with pluripotency genes [72,73,74].
In summary, our data validate the potential of the GPA combination to transdifferentiate human somatic cells into cells expressing hair cell markers. Direct conversion of somatic cells into another cell type reaches higher efficiencies when the starting and the sought-for cell populations present closer epigenomes [26]. Therefore, the ground is also set for in vivo studies to overexpress the GPA combination in the cochlea, where cell types such as the supporting cells are more closely lineage related to HCs, and where transdifferentiation should occur under the optimal environmental conditions. Successful in vivo transdifferentiation of cochlear cell types towards the hair cell fate following GPA overexpression in mature animals would lend support to its application as a possible avenue towards future regenerative therapies.
Supporting information
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank James Hudspeth for providing ESPIN antibodies, Carlos Vicario-Abejón for pLV-IRES-eGFP, Mengqing Xiang for pRK5KS-mBrn3c, Kiril Schimmang Alonso for assisting in the gene ontology analysis, and Cristina Sánchez and Alvaro Martín for technical assistance.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The project was funded by Junta de Castilla y Leon (Project VA024U16, Feder 2014-2020), Fundacion La Marato (Project 201227-30-31), Red de Terapia Celular and Red de medicina regenerativa y terapia celular de Castilla y Leon. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Shi F, Kempfle JS, Edge AS (2012) Wnt-responsive Lgr5-expressing stem cells are hair cell progenitors in the cochlea. Journal of Neuroscience 32: 9639–9648. doi: 10.1523/JNEUROSCI.1064-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walters BJ, Coak E, Dearman J, Bailey G, Yamashita T, Kuo B, et al. (2017) In Vivo Interplay between p27Kip1, GATA3, ATOH1, and POU4F3 Converts Non-sensory Cells to Hair Cells in Adult Mice. Cell Reports 19: 307–320. doi: 10.1016/j.celrep.2017.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conde de Felipe MM, Feijoo Redondo A, Garcia-Sancho J, Schimmang T, Duran Alonso MB (2011) Cell- and gene-therapy approaches to inner ear repair. Histology & Histopathology 26: 923–940. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z, Fang J, Dearman J, Zhang L, Zuo J (2014) In vivo generation of immature inner hair cells in neonatal mouse cochleae by ectopic Atoh1 expression. PLoS ONE [Electronic Resource] 9: e89377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abboud N, Fontbonne A, Watabe I, Tonetto A, Brezun JM, Feron F, et al. (2017) Culture conditions have an impact on the maturation of traceable, transplantable mouse embryonic stem cell-derived otic progenitor cells. J Tissue Eng Regen Med 11: 2629–2642. doi: 10.1002/term.2163 [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Jongkamonwiwat N, Abbas L, Eshtan SJ, Johnson SL, Kuhn S, et al. (2012) Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature 490: 278–282. doi: 10.1038/nature11415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa A, Sanchez-Guardado L, Juniat S, Gale JE, Daudet N, Henrique D (2015) Generation of sensory hair cells by genetic programming with a combination of transcription factors. Development 142: 1948–1959. doi: 10.1242/dev.119149 [DOI] [PubMed] [Google Scholar]
- 8.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E (2013) Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500: 217–221. doi: 10.1038/nature12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koehler KR, Nie J, Longworth-Mills E, Liu XP, Lee J, Holt JR, et al. (2017) Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nature Biotechnology 35: 583–589. doi: 10.1038/nbt.3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oshima K, Shin K, Diensthuber M, Peng AW, Ricci AJ, Heller S (2010) Mechanosensitive hair cell-like cells from embryonic and induced pluripotent stem cells. Cell 141: 704–716. doi: 10.1016/j.cell.2010.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ronaghi M, Nasr M, Ealy M, Durruthy-Durruthy R, Waldhaus J, Diaz GH, et al. (2014) Inner ear hair cell-like cells from human embryonic stem cells. Stem Cells & Development 23: 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouji Y, Ishizaka S, Nakamura-Uchiyama F, Wanaka A, Yoshikawa M (2013) Induction of inner ear hair cell-like cells from Math1-transfected mouse ES cells. Cell Death Dis 4: e700 doi: 10.1038/cddis.2013.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohnishi H, Skerleva D, Kitajiri S, Sakamoto T, Yamamoto N, Ito J, et al. (2015) Limited hair cell induction from human induced pluripotent stem cells using a simple stepwise method. Neurosci Lett 599: 49–54. doi: 10.1016/j.neulet.2015.05.032 [DOI] [PubMed] [Google Scholar]
- 14.Liu XP, Koehler KR, Mikosz AM, Hashino E, Holt JR (2016) Functional development of mechanosensitive hair cells in stem cell-derived organoids parallels native vestibular hair cells. Nature communications 7: 11508 doi: 10.1038/ncomms11508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eguizabal C, Montserrat N, Veiga A, Izpisua Belmonte JC (2013) Dedifferentiation, transdifferentiation, and reprogramming: future directions in regenerative medicine. Seminars in Reproductive Medicine 31: 82–94. doi: 10.1055/s-0032-1331802 [DOI] [PubMed] [Google Scholar]
- 16.Graf T, Enver T (2009) Forcing cells to change lineages. Nature 462: 587–594. doi: 10.1038/nature08533 [DOI] [PubMed] [Google Scholar]
- 17.Kelaini S, Cochrane A, Margariti A (2014) Direct reprogramming of adult cells: avoiding the pluripotent state. Stem Cells Cloning 7: 19–29. doi: 10.2147/SCCAA.S38006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ladewig J, Koch P, Brustle O (2013) Leveling Waddington: the emergence of direct programming and the loss of cell fate hierarchies. Nature Reviews Molecular Cell Biology 14: 225–236. [DOI] [PubMed] [Google Scholar]
- 19.Vierbuchen T, Wernig M (2012) Molecular roadblocks for cellular reprogramming. Molecular Cell 47: 827–838. doi: 10.1016/j.molcel.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis RL, Weintraub H, Lassar AB (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51: 987–1000. [DOI] [PubMed] [Google Scholar]
- 21.Marro S, Pang ZP, Yang N, Tsai MC, Qu K, Chang HY, et al. (2011) Direct lineage conversion of terminally differentiated hepatocytes to functional neurons. Cell Stem Cell 9: 374–382. doi: 10.1016/j.stem.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, et al. (2011) Induction of human neuronal cells by defined transcription factors. Nature 476: 220–223. doi: 10.1038/nature10202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, et al. (2012) Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485: 599–604. doi: 10.1038/nature11139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szabo E, Rampalli S, Risueno RM, Schnerch A, Mitchell R, Fiebig-Comyn A, et al. (2010) Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 468: 521–526. doi: 10.1038/nature09591 [DOI] [PubMed] [Google Scholar]
- 25.Xie H, Ye M, Feng R, Graf T (2004) Stepwise reprogramming of B cells into macrophages. Cell 117: 663–676. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA (2008) In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455: 627–632. doi: 10.1038/nature07314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Yu H, Zhang Y, Li W, Lu N, Ni W, et al. (2013) Cotransfection of Pax2 and Math1 promote in situ cochlear hair cell regeneration after neomycin insult. Sci Rep 3: 2996 doi: 10.1038/srep02996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. (2010) Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142: 375–386. doi: 10.1016/j.cell.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo BR, Baldwin EM, Layman WS, Taketo MM, Zuo J (2015) In Vivo Cochlear Hair Cell Generation and Survival by Coactivation of beta-Catenin and Atoh1. Journal of Neuroscience 35: 10786–10798. doi: 10.1523/JNEUROSCI.0967-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes JH, O'Shea KS, Wys NL, Velkey JM, Prieskorn DM, Wesolowski K, et al. (2008) Glutamatergic neuronal differentiation of mouse embryonic stem cells after transient expression of neurogenin 1 and treatment with BDNF and GDNF: in vitro and in vivo studies. Journal of Neuroscience 28: 12622–12631. doi: 10.1523/JNEUROSCI.0563-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ring KL, Tong LM, Balestra ME, Javier R, Andrews-Zwilling Y, Li G, et al. (2012) Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 11: 100–109. doi: 10.1016/j.stem.2012.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell RR, Szabo E, Benoit YD, Case DT, Mechael R, Alamilla J, et al. (2014) Activation of neural cell fate programs toward direct conversion of adult human fibroblasts into tri-potent neural progenitors using OCT-4. Stem Cells & Development 23: 1937–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gubbels SP, Woessner DW, Mitchell JC, Ricci AJ, Brigande JV (2008) Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455: 537–541. doi: 10.1038/nature07265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, et al. (2012) In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485: 593–598. doi: 10.1038/nature11044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vangipuram M, Ting D, Kim S, Diaz R, Schule B (2013) Skin punch biopsy explant culture for derivation of primary human fibroblasts. J Vis Exp: e3779 doi: 10.3791/3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakuma T, Barry MA, Ikeda Y (2012) Lentiviral vectors: basic to translational. Biochemical Journal 443: 603–618. doi: 10.1042/BJ20120146 [DOI] [PubMed] [Google Scholar]
- 37.Shearer RF, Saunders DN (2015) Experimental design for stable genetic manipulation in mammalian cell lines: lentivirus and alternatives. Genes to Cells 20: 1–10. doi: 10.1111/gtc.12183 [DOI] [PubMed] [Google Scholar]
- 38.Pluta K, Kacprzak MM (2009) Use of HIV as a gene transfer vector. Acta Biochimica Polonica 56: 531–595. [PubMed] [Google Scholar]
- 39.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mi H, Muruganujan A, Thomas PD (2013) PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41: D377–386. doi: 10.1093/nar/gks1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duran Alonso MB, Feijoo-Redondo A, Conde de Felipe M, Carnicero E, Garcia AS, Garcia-Sancho J, et al. (2012) Generation of inner ear sensory cells from bone marrow-derived human mesenchymal stem cells. Regenerative Medicine 7: 769–783. doi: 10.2217/rme.12.65 [DOI] [PubMed] [Google Scholar]
- 43.Doetzlhofer A, White PM, Johnson JE, Segil N, Groves AK (2004) In vitro growth and differentiation of mammalian sensory hair cell progenitors: a requirement for EGF and periotic mesenchyme. Developmental Biology 272: 432–447. doi: 10.1016/j.ydbio.2004.05.013 [DOI] [PubMed] [Google Scholar]
- 44.Helyer R, Cacciabue-Rivolta D, Davies D, Rivolta MN, Kros CJ, Holley MC (2007) A model for mammalian cochlear hair cell differentiation in vitro: effects of retinoic acid on cytoskeletal proteins and potassium conductances. European Journal of Neuroscience 25: 957–973. doi: 10.1111/j.1460-9568.2007.05338.x [DOI] [PubMed] [Google Scholar]
- 45.Kelley MW, Xu XM, Wagner MA, Warchol ME, Corwin JT (1993) The developing organ of Corti contains retinoic acid and forms supernumerary hair cells in response to exogenous retinoic acid in culture. Development 119: 1041–1053. [DOI] [PubMed] [Google Scholar]
- 46.Cai T, Jen HI, Kang H, Klisch TJ, Zoghbi HY, Groves AK (2015) Characterization of the transcriptome of nascent hair cells and identification of direct targets of the Atoh1 transcription factor. Journal of Neuroscience 35: 5870–5883. doi: 10.1523/JNEUROSCI.5083-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chai R, Xia A, Wang T, Jan TA, Hayashi T, Bermingham-McDonogh O, et al. (2011) Dynamic expression of Lgr5, a Wnt target gene, in the developing and mature mouse cochlea. Jaro 12: 455–469. doi: 10.1007/s10162-011-0267-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McLean WJ, Yin X, Lu L, Lenz DR, McLean D, Langer R, et al. (2017) Clonal Expansion of Lgr5-Positive Cells from Mammalian Cochlea and High-Purity Generation of Sensory Hair Cells. Cell Reports 18: 1917–1929. doi: 10.1016/j.celrep.2017.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang ZH, Chen JR, Zheng J, Shi HS, Ding J, Qian XD, et al. (2016) Genetic Correction of Induced Pluripotent Stem Cells From a Deaf Patient With MYO7A Mutation Results in Morphologic and Functional Recovery of the Derived Hair Cell-Like Cells. Stem Cells Transl Med 5: 561–571. doi: 10.5966/sctm.2015-0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Izumikawa M, Batts SA, Miyazawa T, Swiderski DL, Raphael Y (2008) Response of the flat cochlear epithelium to forced expression of Atoh1. Hear Res 240: 52–56. doi: 10.1016/j.heares.2008.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, et al. (1998) Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279: 1950–1954. [DOI] [PubMed] [Google Scholar]
- 52.Chen JR, Tang ZH, Zheng J, Shi HS, Ding J, Qian XD, et al. (2016) Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death Differ 23: 1347–1357. doi: 10.1038/cdd.2016.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, et al. (2018) Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553: 217–221. doi: 10.1038/nature25164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nishimura K, Nakagawa T, Sakamoto T, Ito J (2012) Fates of murine pluripotent stem cell-derived neural progenitors following transplantation into mouse cochleae. Cell Transplantation 21: 763–771. doi: 10.3727/096368911X623907 [DOI] [PubMed] [Google Scholar]
- 55.Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, et al. (1999) Math1: an essential gene for the generation of inner ear hair cells. Science 284: 1837–1841. [DOI] [PubMed] [Google Scholar]
- 56.Chonko KT, Jahan I, Stone J, Wright MC, Fujiyama T, Hoshino M, et al. (2013) Atoh1 directs hair cell differentiation and survival in the late embryonic mouse inner ear. Developmental Biology 381: 401–410. doi: 10.1016/j.ydbio.2013.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hertzano R, Montcouquiol M, Rashi-Elkeles S, Elkon R, Yucel R, Frankel WN, et al. (2004) Transcription profiling of inner ears from Pou4f3(ddl/ddl) identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum Mol Genet 13: 2143–2153. doi: 10.1093/hmg/ddh218 [DOI] [PubMed] [Google Scholar]
- 58.Wallis D, Hamblen M, Zhou Y, Venken KJ, Schumacher A, Grimes HL, et al. (2003) The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 130: 221–232. [DOI] [PubMed] [Google Scholar]
- 59.Xiang M, Gan L, Li D, Chen ZY, Zhou L, O'Malley BW Jr., et al. (1997) Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proceedings of the National Academy of Sciences of the United States of America 94: 9445–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Z, Dearman JA, Cox BC, Walters BJ, Zhang L, Ayrault O, et al. (2012) Age-dependent in vivo conversion of mouse cochlear pillar and Deiters' cells to immature hair cells by Atoh1 ectopic expression. Journal of Neuroscience 32: 6600–6610. doi: 10.1523/JNEUROSCI.0818-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang SM, Chen W, Guo WW, Jia S, Sun JH, Liu HZ, et al. (2012) Regeneration of stereocilia of hair cells by forced Atoh1 expression in the adult mammalian cochlea. PLoS ONE [Electronic Resource] 7: e46355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K (2008) Induced pluripotent stem cells generated without viral integration. Science 322: 945–949. doi: 10.1126/science.1162494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, et al. (2009) Human induced pluripotent stem cells free of vector and transgene sequences. Science 324: 797–801. doi: 10.1126/science.1172482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banasik MB, McCray PB Jr. (2010) Integrase-defective lentiviral vectors: progress and applications. Gene Therapy 17: 150–157. doi: 10.1038/gt.2009.135 [DOI] [PubMed] [Google Scholar]
- 65.Horio F, Sakurai H, Ohsawa Y, Nakano S, Matsukura M, Fujii I (2017) Functional validation and expression analysis of myotubes converted from skin fibroblasts using a simple direct reprogramming strategy. eNeurologicalSci 6: 9–15. doi: 10.1016/j.ensci.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pay SL, Qi X, Willard JF, Godoy J, Sankhavaram K, Horton R, et al. (2017) Improving the Transduction of Bone Marrow-Derived Cells with an Integrase-Defective Lentiviral Vector. Hum Gene Ther Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang JH, Tsai PH, Wang KY, Wei YT, Chiou SH, Mou CY (2018) Generation of Functional Dopaminergic Neurons from Reprogramming Fibroblasts by Nonviral-based Mesoporous Silica Nanoparticles. Sci Rep 8: 11 doi: 10.1038/s41598-017-18324-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gonzalez F, Barragan Monasterio M, Tiscornia G, Montserrat Pulido N, Vassena R, Batlle Morera L, et al. (2009) Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proceedings of the National Academy of Sciences of the United States of America 106: 8918–8922. doi: 10.1073/pnas.0901471106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fu Y, Huang C, Xu X, Gu H, Ye Y, Jiang C, et al. (2015) Direct reprogramming of mouse fibroblasts into cardiomyocytes with chemical cocktails. Cell Res 25: 1013–1024. doi: 10.1038/cr.2015.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DeJonge RE, Liu XP, Deig CR, Heller S, Koehler KR, Hashino E (2016) Modulation of Wnt Signaling Enhances Inner Ear Organoid Development in 3D Culture. PLoS ONE [Electronic Resource] 11: e0162508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perny M, Ting CC, Kleinlogel S, Senn P, Roccio M (2017) Generation of Otic Sensory Neurons from Mouse Embryonic Stem Cells in 3D Culture. Front Cell Neurosci 11: 409 doi: 10.3389/fncel.2017.00409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S, et al. (2011) Direct reprogramming of mouse fibroblasts to neural progenitors. Proceedings of the National Academy of Sciences of the United States of America 108: 7838–7843. doi: 10.1073/pnas.1103113108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Serrano F, Garcia-Bravo M, Blazquez M, Torres J, Castell JV, Segovia JC, et al. (2016) Silencing of hepatic fate-conversion factors induce tumorigenesis in reprogrammed hepatic progenitor-like cells. Stem Cell Res Ther 7: 96 doi: 10.1186/s13287-016-0349-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thier M, Worsdorfer P, Lakes YB, Gorris R, Herms S, Opitz T, et al. (2012) Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell 10: 473–479. doi: 10.1016/j.stem.2012.03.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.