

Abstract
The present study aimed to investigate the anti-arthritic effect of physcion 8-O-β-glucopyranoside (POGD) and its possible mechanisms. The anti-proliferative effects of POGD on MH7A cells were detected using a CCK-8 assay, and the release of pro-inflammatory cytokines, interleukin (IL)-1β, IL-6, IL-8, IL-12 and IL-17A, were determined by ELISA. A type II collagen-induced arthritis (CIA) rat model was established to evaluate the anti-arthritic effect of POGD in vivo. The paw volumes, arthritis indices and serum levels of tumor necrosis factor (TNF)-α, IL-1β, IL-6, IL-8, IL-17A were determined by ELISA. The mRNA expression levels of matrix metalloproteinase (MMP)-2, MMP-3, MMP-9, vascular endothelial growth factor and cyclooxygenase-2 were determined by reverse transcription-quantitative polymerase chain reaction analysis, and the expression levels of transforming growth factor (TGF)-β1, small mothers against decapentaplegic (Smad)4, Smad7, c-Jun N-terminal kinase (JNK), phosphorylated (p-)JNK, p-P38, P38, p-extracellular signal-regulated kinase (ERK)1/2, ERK1/2, nuclear factor (NF)-κB p65 in the nucleus (N), cytosolic NF-κB p65 (C), and inhibitor of NF-κB (IκB) were determined by western blot analysis. The results indicated that POGD significantly inhibited MH7A cell growth. POGD markedly inhibited paw swelling and the arthritis indices of the CIA rats, and POGD may also inhibit the release of pro-inflammatory cytokines. Furthermore, POGD downregulated the expression levels of TGF-β1, Smad4, NF-κB p65 (N), p38, p-p38, p-ERK1/2, JNK, p-JNK, TGF-β1, Smad4, p-JNK, JNK, p-P38, P38, p-ERK1/2, ERK1/2 and NF-κB p65 (N), and upregulated the Smad7, NF-κB p65 (C) and IκB in TNF-α induced MH7A cells. In conclusion, the results suggested that POGD is a promising potential anti-inflammatory drug, and that POGD may decrease the expression of pro-inflammatory cytokines and mediators via inhibiting the TGF-β/NF-κB/mitogen-activated protein kinase pathways.
Keywords: physcion 8-O-β-glucopyranoside, anti-proliferative activity, anti-inflammatory effects, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease, which causes chronic inflammation of the joints and other areas of the body (1). The damaged of tissues in the body by its own immune system are termed autoimmune diseases (2). RA is characterized by inflammation of the tissue around the joints, and this disease can also cause inflammation and injury in other organs in the body (3). Chronic inflammation of RA can cause permanent joint destruction and deformity, which begins with the proliferation of synovial macrophages and fibroblasts (4). The treatment of RA involves a combination of patient education, rest and exercise, joint protection, medications and occasionally surgery (5). For pharmacologic treatment, non-steroidal anti-inflammatory drugs are commonly used to reduce joint pain. Disease-modifying anti-rheumatic drugs, including methotrexate and sulfasalazine, are initiated to slow disease progression as early as possible. In addition, novel drugs, including transforming growth factor-α (TNF-α) inhibitors, interleukin-6 (IL-6) inhibitors and Janus kinase (JAK) inhibitors have been introduced (6). However, there is no cure for RA. Early RA treatment leads to an improved prognosis. The fibroblast-like synoviocytes (FLS) is the most prominent cell type in the rheumatoid joint and is central role to the propagation of RA (7). It has been reported that the FLS is directly responsible for cartilage destruction, and drives inflammation and autoimmunity (7). Therefore, treatment designed to inhibit the proliferation of RA-FLS may provide a reasonably efficient approach for curing RA.
Polygonum cuspidatum, also known as Reynoutria japonica Houtt and Huzhang in China, is a perennial herb plant belonging to the family Polygonaceae (8). Polygonum cuspidatum is widely distributed in southern China and Japan, and is a traditional and well-known Chinese medicinal herb (8). It has long been used in folk medicine for the treatment of inflammation, infection, jaundice, skin burns and hyper-lipemia (8,9). As demonstrated by extensive reports, the root of Polygonum cuspidatum has wide pharmacological effects, including anti-shock, anti-inflammatory, antioxidant, anticancer and hepatoprotective effects (10–14). Currently, >67 compounds have been isolated and identified from this plant, including quinones, stilbenes, flavonoids, counmarins and ligans (8). Physcion 8-O-β-glucopyranoside (POGD) is an anthraquinonesone (Fig. 1) isolated from the root of Polygonum cuspidatum. It has been reported that POGD has significant anti-proliferative activity on A549 cell lines by inducing cell cycle arrest and apoptosis (9). The aim of the present study was to increase current understanding of the anti-proliferative and anti-inflammatory potency of POGD in MH7A RA-derived fibroblast-like synoviocytes.
Figure 1.
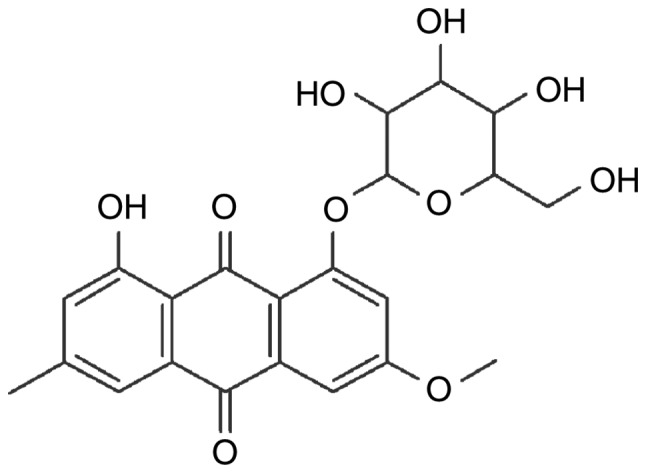
Chemical structure of physcion 8-O-β-glucopyranoside.
Materials and methods
Herbal medicines
Polygonum cuspidatum was purchased from the traditional Chinese Medicine Store of Shandong Province (Shandong, China). A voucher specimen of P. cuspidatum was deposited at the Department of Pharmacy, Shandong Zibo Central Hospital (Shandong, China).
Cells and culture
The MH7A RA-derived fibroblast-like synoviocyte cell line was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The MH7A cells were cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS), 1% penicillin and 1% streptomycin in a 5% CO2 humidified atmosphere at 37°C.
Chemicals and reagents
Interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-10 (IL-10), interleukin-12 (IL-12) and interleukin-17A (IL-17A) enzyme-linked immunosorbent assay (ELISA) kits were purchased from Invitrogen; Thermo Fisher Scientific, Inc. (Waltham, MA USA); the Dulbecco's modified Eagle's medium (DMEM), FBS and trypsinase were from Gibco; Thermo Fisher Scientific, Inc. The Cell Counting Kit-8, BCA protein assay reagent and goat-anti-rabbit/rat horseradish-peroxidase-conjugated (HRP) secondary antibodies were purchased from Beyotime Institute of Biotechnology (Haimen, Jiangshu province, China); the tumor necrosis factor-α (TNF-α), dimethyl sulfoxide (DMSO), dexamethasone, complete Freund's adjuvant (CFA) and incomplete Freund's adjuvant (IFA) were purchased from Sigma (Shanghai, China); EMD Millipore (Billerica, MA, USA); TGF-β1 (cat. no. sc-130348), small mothers against decapentaplegic (Smad)4 (cat. no. sc-7154), Smad7 (cat. no. sc-11392), Histone H1 (cat. no. sc-8030) and β-actin (cat. no. sc-47778) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); c-Jun N-terminal kinase (JNK; cat. no. ab179461), phosphorylated [(p-)JNK (cat. no. ab124956)], p-P38 (cat. no. ab4822), P38 (cat. no. ab170099), p-extracellular signal-regulated kinase (ERK)1/2 (cat. no. ab223500), ERK1/2 (cat. no. ab17942), nuclear factor (NF)-κB p65 (cat. no. ab32536), and inhibitor of NF-κB (IκB; cat. no. ab32518) antibodies were purchased from Abcam (Cambridge, MA, USA). Chicken type II collagen (CII) was purchased from Xi'an Herb King Biotechnology Co., Ltd. (Xi'an, Shanxi, China). All other reagents used were of analytically pure grade.
Isolation and preparation of POGD
The preparation of POGD was performed according to a previously described method with minor modification (9). Briefly, the comminuted herbal medicines were extracted with 8X 75% aqueous ethanol by reflux three times. The total extracts were evaporated under a vacuum to remove the ethanol. The residual extract solution was then orderly extracted with ethyl acetate and n-butanol, and the n-butanol fraction was evaporated under a vacuum to obtain the dried n-butanol extracts. Subsequently, the n-butanol extracts were isolated by silica gel (200-300 mesh) column chromatography eluting with chloroform: Methanol (20:1, 15:1, 10:1, 7:1, 5:1, 2:1 and 1:1). Based on the thin layer chromatography assay, the similar fractions of the sub-fractions of n-butanol extracts were combined to obtain six sub-fractions (Fra. 1-Fra. 6). Subsequently, using a series of repeated silica gel column chromatography and Sephadex LH-20 chromatography, the target compound was isolated from fractions 4.
Identification of the POGD
The POGD was identified based on the 1H NMR and 13C NMR experiments, and the spectral data were as follows: 1H-NMR (600 MHz, DMSO-d6) δ: 11.32 (1H, s, 1-OH), 7.42 (1H, d, J=1.6 Hz, H-4), 7.25 (1H, d, J=2.8 Hz, H-5), 7.12 (1H, d, J=2.8 Hz, H-7), 6.98 (H, s, H-2), 5.09 (1H, d, J=7.4 Hz, H-1′), 3.14 (3H, s, 6-OCH3), 2.37 (3H, s, 3-CH3); 13C-NMR (150 MHz, DMSO-d6) δ: 187.9 (C-9), 183.6 (C-10), 165.7 (C-6), 163.2 (C-1), 162.6 (C-8), 148.3 (C-3), 138.0 (C-10a), 133.5 (C-4a), 125.6 (C-2), 120.7 (C-4), 115.9 (C-8a), 114.7 (C-9a), 109.8 (C-7), 109.8 (C-5), 102.3 (C-1′), 78.8 (C-3′), 77.9 (C-5′), 74.7 (C-2′), 70.9 (C-4′), 62.1 (C-6′), 50.1 (6-OCH3), 22.9 (3-CH3). These data were similar to the previously reported data (9).
Cell viability assay using Cell-Counting Kit-8 (CCK-8)
The effects of POGD on cellular viability were determined using CCK-8 according to the manufacturer's protocol. The MH7A cells (5×103 cells/well) were seeded in 96-well plates and incubated with different concentrations of POGD (4, 8, 16, 32, 64, 128 and 256 μg/ml) for 12, 24, 36, 48 or 72 h. The CCK-8 solution was then added to each well and incubated for another 1 h in the incubator. Finally, the optical density (OD) value was read at 450 nm using a 96-well plate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The results were calculated as a percentage of the DMSO-treated control cells.
Determination of cell cytokine levels by ELISA
The MH7A cells (5×105 cells/well) were treated with TNF-α (10 ng/ml) for 12 h at 37°C in a 5% CO2 humidified atmosphere prior to the assay. The cell supernatant was removed via pippetting and cells were washed twice with PBS. The cells were then exposed to increased concentrations of POGD (8, 16 and 32 μg/ml) for another 24 h. Following treatment, the cell supernatant was collected, and the levels of IL-1β, IL-6, IL-8, IL-10, IL-12, and IL-17 cytokines were determined using ELISA assay kits according to the manufacturer's protocol (R&D Systems, Inc., Minneapolis, MN, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis of the mRNA expression levels of matrix metalloproteinase (MMP)-2, MMP-3, MMP-9, cyclooxygenase-2 (COX-2) and vascular endothelial growth factor (VEGF) in MH7A cells
The effects of POGD on the mRNA expression levels of MMP-2, MMP-3, MMP-9, COX-2 and VEGF were determined by RT-qPCR analysis. The MH7A cells (3×105 cells/well) were seeded in 6-well plates and incubated with either increasing concentrations of POGD (15, 30 and 60 μg/ml) or vehicle (DMSO) in the presence of 10 ng/ml TNF-α for 72 h at 37°C. Subsequently, the cells were collected and the total RNA was extracted using an RNAiso Plus kit (Takara Biotechnology Co., Ltd., Dalian, China). Total RNA was then used to synthesize cDNAs of MMP-2, MMP-3, MMP-9, COX-2, VEGF and β-actin using PrimeScript™ RT reagent kits (Takara Biotechnology Co., Ltd.). The synthesized cDNAs were amplified using SYBR Green mixture on a CFX96 Touch Real-Time PCR Detection system (Bio-Rad Laboratories, Inc.). Each 10 μl reaction mixture contained 5 μl of SYBR Green mixture, 1 μl of 2 μM forward and reverse primer mixture (Table I), 1 μl of cDNA, and 3 μl of ddH2O. The PCR conditions were 95°C for 10 min, followed by 39 cycles of 95°C for 15 sec and 58°C for 60 sec. The primers used for RT-qPCR analysis are shown in Table I. The relative mRNA expression levels of TNF-α, IL-6 and IL-1β were measured using the 2−ΔΔ Cq relative quantitative analysis method (15) and all samples were analyzed in triplicate.
Table I.
Primers used for reverse transcription-quantitative polymerase chain reaction analysis.
| Author, year | Gene | Primer sequence (5′-3′) | Product size (bp) | (Refs.) |
|---|---|---|---|---|
| Pu et al, 2016 | MMP-2 | Forward: TGGCAAGTACGGCTTCTGTC | 179 | (16) |
| Reverse: TTCTTGTCGCGGTCGTAGTC | ||||
| Tran et al, 2005 | MMP-3 | Forward: CAGGCTTTCCCAAGCAAATA | 129 | (17) |
| Reverse: TTGCATTTGGGTCAAACTCC | ||||
| Pu et al, 2016 | MMP-9 | Forward: TGCGCTACCACCTCGAACTT | 200 | (16) |
| Reverse: GATGCCATTGACGTCGTCCT | ||||
| Ospelt et al, 2008 | COX-2 | Forward: TTCAAATGAGATTGTGGGAAAT | 305 | (18) |
| Reverse: AGATCATCTCTGCCTGAGTATCTT | ||||
| Pu et al, 2016 | VEGF | Forward: CGGCGAAGAGAAGAGACACA | 196 | (16) |
| Reverse: GGAGGAAGGTCAACCACTCA | ||||
| Pu et al, 2016 | GAPDH | Forward: GAGTCAACGGATTTGGTCGT | 185 | (16) |
| Reverse: GACAAGCTTCCCGTTCTCAG |
MMP-2, matrix metalloproteinase 2; MMP-3, matrix metalloproteinase 3; MMP-9, matrix metalloproteinase 9; COX-2, cyclooxygenase-2; VEGF, vascular endothelial growth factor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Western blot analysis
The effects of POGD on the protein expression levels of TGF-β1, Smad4, Smad2, Smad7, ERK1/2, P-ERK1/2, JNK, P-JNK, P-38, P-P38, NF-KB p65 (N), NF-KB p65 (C) and IKB were measured in MH7A cells using western blot analysis. Following treatment with either grade concentrations of POGD (15, 30 and 60 μg/ml) or vehicle (DMSO) in the presence of 10 ng/ml TNF-α for 72 h, the cells were collected and total protein was extracted using western blot & IP cell lysis buffer (Sangon Biotech Co., Ltd., Shanghai, China). Subsequently, the concentration of total protein was determined using BCA protein assay reagent (Sangon Biotech Co., Ltd.). Total protein (40 μg) in each sample was separated using SDS-PAGE (12% gel) and blotted onto a polyvinylidene difluoride filter membrane (PVDF). Subsequently, the protein on the PVDF was probed with anti-TGF-β1 (1:3,000), Smad4 (1:1,000), Smad2 (1:1,000), Smad7 (1:1,000), ERK1/2 (1:1,000), P-ERK1/2 (1:1,000), JNK (1:1,000), P-JNK (1:1,000), P-38 (1:1,000), P-P38 (1:1,000), NF-KB p65 (1:500), IκB (1:500), β-actin (1:3,000) and Histone H1 (1:1,000) antibodies at 4°C for 12 h, followed by incubation with corresponding HRP-conjugated secondary antibodies for 2 h at 37°C. Finally, the immunoreactive bands were visualized with ECL-detecting reagents and the OD values were analyzed using ImageJ software (version 1.48; National Institutes of Health, Bethesda, MD, USA). To normalize for protein loading, the expression levels of TGF-β1, Smad4, Smad2, Smad7, ERK1/2, P-ERK1/2, JNK, P-JNK, P-38, P-P38, NF-KB p65 (N), NF-KB p65 (C) and IKB were determined as a relative value to that of β-actin, and the expression level of p65 (N) was determined as a relative value to that of Histone H1.
Determination of the anti-arthritic effects of POGD in vivo
60 rats weighing 220±20 g with 30 male and 30 female were purchased from the Shanghai Laboratory Animal Centre (Shanghai, China). Each animal was housed under standard conditions (21±1°C, 50-10% relative humidity, 12 h light/dark cycle) and had free access to food and water. A total of 60 rats were randomly divided into the following six groups (n=10): i) normal, healthy rats treated with saline (10 ml/kg); ii) control, collagen-induced arthritis (CIA) rats treated with saline (10 ml/kg); iii) dexamethasone, CIA rats treated with dexamethasone (positive drug, 0.05 mg/kg); iv) POGD (20 mg/kg), CIA rats treated with POGD at a dose of 20 mg/kg; v) POGD (40 mg/kg), CIA rats treated with POGD at a dose of 40 mg/kg; and vi) POGD (60 mg/kg), CIA rats treated with POGD at a dose of 60 mg/kg. The experimental protocols were approved by the Animal Care and Use Committee of Dezhou People's Hospital (Shandong, China).
To evaluate the anti-arthritic effects of POGD in vivo, a CIA animal model was established as previously reported with minor modifications (16). In brief, CII in 0.1 mM acetic acid (4 mg/ml) was emulsified with an equal volume of CFA. The rats were initially immunized by a subcutaneous injection of CII emulsion at the base of the tail (100 μl/rat). After 14 days, the CII emulsion with an equal volume of IFA (100 μl/rat) was administered via subcutaneous injection at the same location, and the rats were administered orally with saline, dexamethasone and POGD for 21 days. During the experiment, the paw volumes and arthritic indices of the rats were measured every 3 days. The arthritic indices were recorded using the following ordinal scale: 0, unaffected, 1, one type of joint affected, 2, two types of joint affected, 3, three types of joint affected, 4, three types of joint affected plus maximal erythema and swelling. At the end of the experiment, blood samples were collected using an orbital blood sampling protocol and centrifuged for 15 min (1,800 × g, 4°C). The serum levels of TNF-α, IL-1β, IL-6, IL-8 and IL-17A were determined using commercial ELISA kits according to the manufacturer's protocols.
Statistical analysis
All results are expressed as the mean ± standard deviation of three independent experiments performed in triplicate. Statistical analyses were performed using the SPSS 19.0 software package (IBM SPSS, Armonk, NY, USA) and one-way analysis of variance with Dunnett's test was used to compare the means between two groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of POGD on MH7A cell viability
The effect of POGD on MH7A cell viability was determined using CCK-8. As shown in Fig. 2A, POGD exerted marked inhibitory effects on MH7A cell growth in a concentration-dependent manner with IC50 values of 49.76 μg/ml. In addition, POGD inhibited MH7A cell proliferation in time-dependent manner in 72 h (Fig. 2B).
Figure 2.
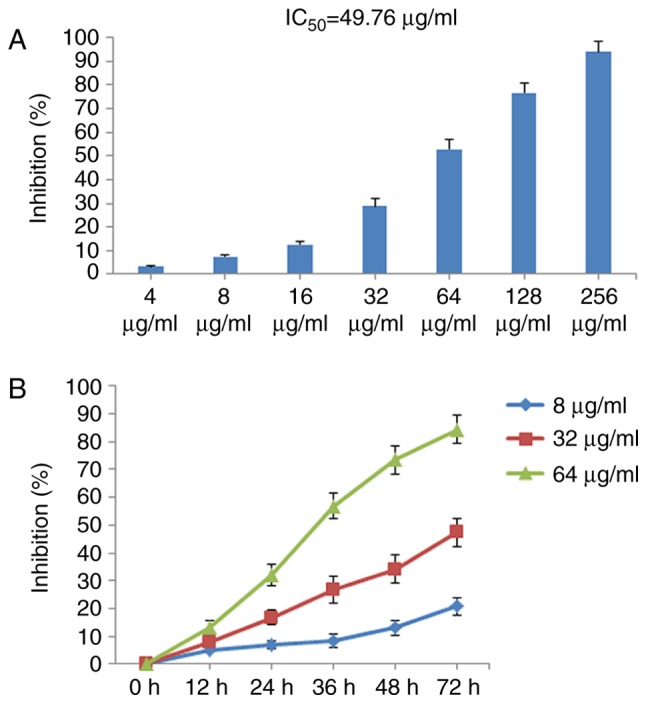
Inhibitory effects of POGD on the proliferation of MH7A cells. (A) Cells were treated with POGD (4, 8, 16, 32, 64, 128 and 256 μg/ml) for 36 h; a CCK-8 assay was performed to determine the cell percentage proliferation inhibition (n=4), and IC50 value of POGD was calculated. (B) Cells were treated with POGD (8, 32 and 64 μg/ml) for 12, 24, 36, 48 and 72 h, respectively; a CCK-8 assay was performed to determine the percentage cell proliferation inhibition (n=4). POGD, physcion 8-O-β-glucopyranoside; CCK-8, Cell Counting Kit-8.
Effects of POGD on levels of IL-1β, IL-6, IL-8, IL-10, IL-12 and IL-17A in TNF-α-stimulated MH7A cells
To verify the anti-inflammatory effects of POGD in MH7A cells, the levels of inflammatory cytokines IL-1β, IL-6, IL-8, IL-10, IL-12 and IL-17A were determined by ELISA in TNF-α-induced MH7A cells. As shown in Fig. 3, with the exception of IL-10, the levels of these inflammatory cytokines were significantly reduced, compared with those in the TNF-α group (P<0.01, by POGD at concentrations of 8, 16 and 32 μg/ml.
Figure 3.

Inhibitory effects of POGD on levels of IL-1β, IL-6, IL-8, IL-10, IL-12 and IL-17A in TNF-α-stimulated MH7A cells. Cells were pretreated with TNF-α (10 ng/ml) for 12 h, following which the cells were exposed to POGD (8, 16 and 32 μg/ml) for another 24 h; enzyme-linked immunosorbent assays were performed to determine the levels of cytokines in cell supernatants (n=4). **P<0.01, vs. control (TNF-α group). POGD, physcion 8-O-β-glucopyranoside; IL, interleukin; TNF-α, tumor necrosis factor-α.
Effects of POGD on mRNA expression levels of MMP-2, MMP-3, MMP-9, VEGF and COX-2 in TNF-α-stimulated MH7A cells
In the TNF-α-induced MH7A cells, the mRNA expression levels of MMP-2, MMP-3, MMP-9, VEGF and COX-2 were determined using RT-qPCR analysis. Following treatment with POGD for 24 h, the mRNA expression levels of MMP-2, MMP-3, MMP-9, VEGF and COX-2 were significantly decreased, compared with those in the TNF-α group (P<0.01; Fig. 4).
Figure 4.

Inhibitory effects of POGD on mRNA expression levels of MMP-2, MMP-3, MMP-9, VEGF and COX-2 in TNF-α-stimulated MH7A cells. Cells were pretreated with TNF-α (10 ng/ml) for 12 h, following which the cells were exposed to POGD (8, 16 and 32 μg/ml) for another 24 h; reverse transcription-quantitative polymerase chain reaction assays were performed to determine the mRNA expression levels of MMP-2, MMP-3, MMP-9, VEGF and COX-2 (n=4). **P<0.01, vs. control (TNF-α group). POGD, physcion 8-O-β-glucopyranoside; MMP, matrix metalloproteinase; VEGF, vascular endothelial factor; COX-2, cyclooxygenase 2; TNF-α, tumor necrosis factor-α.
Effects of POGD on expression levels of TGF-β1, Smad4 and Smad7 in TNF-α-stimulated MH7A cells
Based on the above results, POGD not only inhibited the expression levels of pro-inflammatory cytokines IL-1β, IL-6, IL-8, IL-12 and IL-17A in the TNF-α-stimulated MH7A cells, but also decreased the mRNA expression levels of MMP-2, MMP-3, MMP-9, VEGF and COX-2. To further investigate the possible mechanisms underlying these changes, the protein expression levels of TGF-β1, Smad4 and Smad7 were measured in the TNF-α-stimulated MH7A cells using western blot analysis. As shown in Fig. 5, compared with the TNF-α group, the protein expression levels of TGF-β1 and Smad4 were markedly down-regulated in the POGD groups, whereas the protein expression of Smad7 was significantly upregulated in the TNF-α-induced MH7A cells.
Figure 5.

Regulatory effects of POGD on the expression of TGF-β1, Smad4 and Smad7 in TNF-α-stimulated MH7A cells. Cells were pretreated with TNF-α (10 ng/ml) for 12 h, following which the cells were exposed to POGD (8, 16 and 32 μg/ml) for another 24 h; western blot assays were performed to determine the expressions of TGF-β, Smad4 and Smad7 (n=4). **P<0.01, vs. control (TNF-α group). POGD, physcion 8-O-β-glucopyranoside; TGF-β1, transforming growth factor-β1; Smad, small mothers against decapentaplegic; TNF-α, tumor necrosis factor-α.
Effects of POGD on expression levels of ERK1/2, P-ERK1/2, JNK, P-JNK, P-38, P-P38 in TNF-α-stimulated MH7A cells
The present study also examined the protein expression levels of ERK1/2, P-ERK1/2, JNK, P-JNK, P-38 and P-P38 in the TNF-α-stimulated and POGD-treated MH7A cells using western blot analysis. As shown in Fig. 6, POGD significantly inhibited the protein expression levels of P-JNK, JNK, P-P38, P38, P-ERK1/2 and ERK1/2 in the TNF-α-induced MH7A cells (P<0.01).
Figure 6.

Regulatory effects of POGD on the expression of p-JNK, JNK, p-P38, P38, p-ERK1/2 and ERK1/2 in TNF-α-stimulated MH7A cells. Cells were pretreated with TNF-α (10 ng/ml) for 12 h, following which the cells were exposed to POGD (8, 16 and 32 μg/ml) for another 24 h; western blot assays were performed to determine the expression levels of p-JNK, JNK, p-P38, P38, p-ERK1/2 and ERK1/2 (n=4). **P<0.01, vs. control (TNF-α group). POGD, physcion 8-O-β-glucopyranoside; TNF-α, tumor necrosis factor-α; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; p-, phosphorylated.
Effects of POGD on expression levels of NF-κB p65 (N), NF-κB p65 (C) and IκB in TNF-α-stimulated MH7A cells
The present study also examined the effects of POGD on the expression levels of NF-κB p65 (N), NF-κB p65 (C) and IκB using western blot analysis in TNF-α-stimulated MH7A cells. Following treatment with POGD for 24 h, the protein expression level of NF-κB p65 (N) was significantly reduced, whereas the protein expression levels of NF-κB p65 (C) and IκB were significantly increased (P<0.01), as shown in Fig. 7.
Figure 7.

Regulatory effects of POGD on the expression of NF-κB and IκB in TNF-α-stimulated MH7A cells. Cells were pretreated with TNF-α (10 ng/ml) for 12 h, following which the cells were exposed to POGD (8, 16 and 32 μg/ml) for another 24 h; western blotting assays were performed to determine the expression levels of NF-κB and IκB (n=4). **P<0.01, vs. control (TNF-α group). POGD, physcion 8-O-β-glucopyranoside; TNF-α, tumor necrosis factor-α; NF-κB, nuclear factor-κB; (N), nuclear; (C), cytoplasmic; IκB, inhibitor of NF-κB.
POGD decreases paw swelling and arthritis indices in CIA rats
A CIA rat model was established to evaluate the therapeutic effects of POGD in rats with RA. As shown in Fig. 8, RA symptoms were observed when the experimental rats were immunized twice with CII, including paw swelling and higher arthritis indices (P<0.01), compared with the normal rats. These symptoms were significantly improved following treatment with dexamethasone. The results indicated that, from the 6th day following initial dexamethasone administration, paw swelling was decreased in the CIA rats, compared with that in the control group (P<0.01; Fig. 8A). Additionally, the arthritis indices in the dexamethasone (0.05 mg/kg)-treated CIA rats were significantly decreased, compared with those in the control group (P<0.01; Fig. 8B). Similar to the results from dexamethasone treatment, POGD significantly decreased paw swelling when administered at a dose of 60 mg/kg (P<0.01), and moderately decreased paw swelling at doses of 20 and 40 mg/kg. In addition, at POGD doses of 20, 40 and 60 mg/kg, the arthritis indices were significantly reduced.
Figure 8.
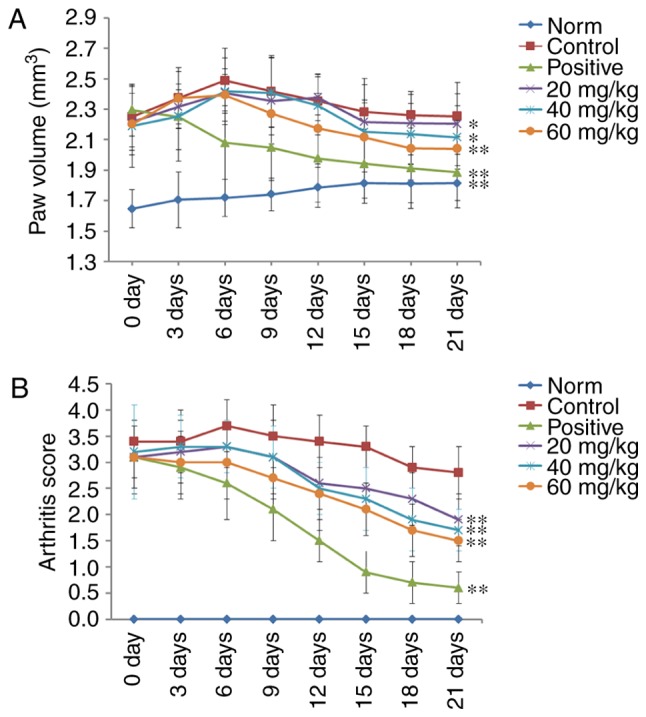
Effects of POGD on paw volume and arthritis score changes in CIA rats. Following establishment of the CIA rat model, rats were orally administered with saline, dexamethasone or different doses of POGD. During the experiment, the (A) paw volumes (mm3) and (B) arthritis indices of the rats were measured every 3 days. Data are presented as the mean ± standard deviation (n=10). *P<0.05, and **P<0.01, compared with the control group. POGD, physcion 8-O-β-glucopyranoside; CIA, type II collagen-induced arthritis.
POGD decreases the release of TNF-α, IL-1β, IL-6, IL-8 and IL-17A into the serum of CIA rats
As shown in Fig. 9, there was an increase in the levels of TNF-α, IL-1β, IL-6, IL-8 and IL-17A in the CIA rats (P<0.01), compared with those in the normal rats. Of note, daily treatment of the CIA rats with POGD at a dose of 20, 40 or 60 mg/kg significantly decreased the release of these regulatory cytokines into the serum (P<0.01), compared with the release observed in the control CIA rats. Similar results were observed in the dexamethasone treatment group.
Figure 9.

Effects of POGD on the release of TNF-α, IL-1β, IL-6, IL-8 and IL-17A into the serum of CIA rats. Following establishment of the CIA rat model, rats were orally administered with saline, dexamethasone or different doses of POGD. Following 30 days of treatment, blood samples were collected, and the serum levels of TNF-α, IL-1β, IL-6, IL-8 and IL-17A were determined using enzyme-linked immunosorbent assays. *P<0.05 and **P<0.01, compared with the control group. POGD, physcion 8-O-β-glucopyranoside; TNF-α, tumor necrosis factor-α; IL, interleukin; CIA, type II collagen-induced arthritis; CIA, type II collagen-induced arthritis.
Discussion
In the present study, the anti-proliferative and anti-inflammatory activities of POGD against MH7A] RA-derived fibroblast-like synoviocytes and CIA rats were analyzed. POGD significantly inhibited MH7A cell growth, and decreased the levels of inflammatory cytokines IL-1β, IL-6, IL-8, IL-12 and IL-17A following TNF-α stimulation. In addition, the mRNA expression levels of MMP-2, MMP-3, MMP-9, VEGF and COX-2 were markedly decreased, compared with those in the TNF-α group. POGD also significantly decreased the protein expression levels of TGF-β1, Smad4, P-JNK, JNK, P-P38, P38, P-ERK1/2, ERK1/2 and NF-κB p65 (N) in the TNF-α-induced MH7A cells, but increased the protein expression levels of Smad7, NF-κB p65 (C) and IκB. Furthermore, POGD inhibited paw swelling, arthritis indices and the release of pro-inflammatory cytokines in CIA rats.
The expansion of synovial cells is one of the main pathological findings in the inflamed synovium of patients with RA (17). The invasion of synovial hyperplasia into cartilage and bones causes direct joint damage and chronic inflammation in situ (18). In the hyperplastic synovium, RA-FLS is the main cell type and tumor-like expansion is the characteristic feature. It has been reported that RA-FLS always causes aggressive invasion into the cartilage (7,19). Therefore, the anti-proliferative effect on MH7A cells is considered to be an efficient therapeutic method in the treatment of RA. The inhibition of RA-FLS also reduced the production of proinflammatory cytokines. In the present study, POGD exhibited potent anti-proliferative and anti-inflammatory activities against MH7A cells in vitro. These effects are important for attenuating synovial hyperplasia, inflammation and bone degeneration in joints.
It is known that the CIA model is the most commonly used autoimmune model of RA, which induces immunological and pathological features similar to those in the RA disease observed in humans (20). In the present study, a CIA rat model was successfully established, which was used to evaluate the anti-arthritic activity of POGD. The resulting data demonstrated that treatment with POGD decreased paw swelling and arthritis indices in the CIA rats, indicating that POGD may have potential therapeutic effects in vivo.
TNF-α is a proinflammatory cytokine and has been reported to be important in contributing to the development and progression of human RA (21). It has been reported that TNF-α can induce lymphocytes, macrophages and synovial fibroblasts to release proinflammatory cytokines (22). In the present study, it was found that the levels of inflammatory cytokines IL-1β, IL-6, IL-8, IL-10, IL-12 and IL-17 were increased in TNF-α-stimulated MH7A cells, which indicated that the inflammatory cell model of human RA had been successfully established. The data obtained in the present study demonstrated that POGD significantly inhibited the expression levels of these cytokines (IL-1β, IL-6, IL-8, IL-12 and IL-17), suggesting that POGD exerted anti-inflammatory activities against the TNF-α-induced MH7A cells. In addition, the inhibition ratio of POGD (32 μg/ml) against MH7A cells was <30% at 36 h and <20% at 24 h (Fig. 2B). Therefore, it was hypothesized that the POGD-induced inhibition of inflammatory factor release was not caused by the anti-proliferative activity of the MH7A cells.
Previous studies have reported that cytokines, including TNF-α and IL-1β, induce the expression of MMPs and COX-2 (23,24). The production of MMPs is largely responsible for joint destruction, involving cartilage, bone and tendons (25). COX-2, a proinflammatory enzyme, is also important in the inflammatory reaction, and is upregulated in inflammatory sites and tumor tissues (26). COX-2 catalyzes the conversion of arachidonic acid into prostaglandins, in particular prostaglandin E2 (PGE2) (27). PGE2 is an important pro-inflammatory mediator involved in inflammatory diseases. In the present study, POGD significantly decreased the mRNA expression levels of MMP-2, MMP-3, MMP-9 and COX-2 in the MH7A cells treated with TNF-α, which indicated that POGD inhibited the targeted joint destruction mediated by MMPs and decreased the release of proinflammatory cytokines by reducing the expression of COX-2 in RA.
Angiogenesis is considered to be an essential process in the development of arthritis (28). Expression of VEGF has been demonstrated in the synovium and synovial fluids, where it can cause vascular permeability and angiogenesis in arthritic joints (29). Cytokines, particularly TNF-α, IL-1β and TGF-β1, can stimulate the release of VEGF, which may contribute significantly to angiogenesis in the synovium (30,31). Additionally, in RA, cartilage and bone matrix are degraded, and extracellular matrix (ECM) proteins, which act as cellular activators, are liberated (32). TGF-β may be important in cartilage repair, as it can induce collagen synthesis to result in synovial hyperplasia. In the present study, POGD inhibited the expression of VEGF, TGF-β1 and Smad4, and increased the expression of Smad7, which indicated that the inhibitory effect of POGD on the expression of VEGF was mediated by the TGF-β1 pathway.
Mitogen-activated protein kinases (MAPKs) have been reported to be involved in cell proliferation and the inflammatory response (33,34). Regulating MAPK kinases offer potential benefits for treating tumor and inflammatory diseases. As ERK and JNK can inhibit the expression of c-Jun and activator protein-1, which regulate the gene transcription of pro-inflammatory cytokines, and inhibiting ERK and JNK is beneficial in slowing the progression of RA (35). In the present study, POGD treatment downregulated the expression of p-JNK, JNK, p-ERK1/2 and ERK1/2 in the MH7A cells, which led to the inhibition of proinflammatory cytokine secretion. In addition, the decreased cytokine levels contributed to the anti-proliferative activity against MH7A cells. The modulation of p38 is considered to be key in the inhibitory properties of POGD against MH7A cells, with the upregulation of p38 being essential for the anti-proliferative activity.
The NF-κB signaling pathway is also pivotal in inflammatory and immune responses (36). Transcription factors of NF-κB are key coordinators of innate immunity and inflammation (37). p65, an NF-κB subunit, is generally localized to the cytoplasm by its inhibitor IκB (38). As IκB dissociates from NF-κB, NF-κB p65 can be translocated to the nucleus and can exert its transcriptional activity, including regulating the expression of inflammatory enzyme COX-2, and cytokines IL-1β, IL-6, IL-8, IL-12 and IL-17A (39). In the present study, the protein expression of nuclear p65 was significantly decreased by POGD, whereas the protein levels of IκB and cytosolic p65 were significantly increased, suggesting that the anti-inflammatory effect of POGD may be associated with a reduction in NF-κB protein by increasing the expression of its inhibitor IκB.
In conclusion, the present study demonstrated that POGD exhibited significant anti-proliferative and anti-inflammatory activities in vitro and in vivo. Furthermore, the possible mechanisms may involve reducing the release of pro-inflammatory cytokines, including IL-1β, IL-6, IL-8, IL-12 and IL-17A, and decreasing the expression of MMP-2, MMP-3, MMP-9, VEGF and COX-2 by inhibiting the TGF-β, NF-κB and MAPK signaling pathways.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
QG and SW contributed to the design of the study. QG, QW, SW, HQ, QZ, XIA LIU and XS contributed to the acquisition, collection and assembly of the data. QG and QW contributed to the statistical analysis. QG and SW wrote the main manuscript text. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The experimental protocols were approved by the Animal Care and Use Committee of Dezhou People's Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fry J, Moulds A, Strube G, Gambrill E. Rheumatic diseases. In: The family good health guide. Springer. 1982:157–164. [Google Scholar]
- 3.Collison J. Experimental arthritis: Do you want to treat arthritis? IDO2. Nat Rev Rheumatol. 2017;13:196–197. doi: 10.1038/nrrheum.2017.33. [DOI] [PubMed] [Google Scholar]
- 4.Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46:183–196. doi: 10.1016/j.immuni.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calabrese LH, Calabrese C, Kirchner E. The 2015 American college of rheumatology guideline for the treatment of rheumatoid arthritis should include new standards for hepatitis B screening: Comment on the article by Singh et al. Arthritis Care Res. 2016;68:723–724. doi: 10.1002/acr.22865. [DOI] [PubMed] [Google Scholar]
- 6.Singh JA, Saag KG, Bridges SL, Jr, Akl EA, Bannuru RR, Sullivan MC, Vaysbrot E, McNaughton C, Osani M, Shmerling RH, et al. 2015 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68:1–26. doi: 10.1002/art.39480. [DOI] [PubMed] [Google Scholar]
- 7.Mor A, Abramson SB, Pillinger MH. The fibroblast-like synovial cell in rheumatoid arthritis: A key player in inflammation and joint destruction. Clin Immunol. 2005;115:118–128. doi: 10.1016/j.clim.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Peng W, Qin R, Li X, Zhou H. Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb.et Zucc.: A review. J Ethnopharmacol. 2013;148:729–745. doi: 10.1016/j.jep.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Xie QC, Yang YP. Anti-proliferative of physcion 8-O-β-glucopyranoside isolated from Rumex japonicus Houtt. on A549 cell lines via inducing apoptosis and cell cycle arrest. BMC Complement Altern Med. 2014;14:377. doi: 10.1186/1472-6882-14-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao KS, Jin C, Huang X, Liu J, Yan WS, Huang Q, Kan W. The mechanism of Polydatin in shock treatment. Clin Hemorheol Microcirc. 2003;29:211–217. [PubMed] [Google Scholar]
- 11.Bralley EE, Greenspan P, Hargrove JL, Wicker L, Hartle DK. Topical anti-inflammatory activity of Polygonum cuspidatum extract in the TPA model of mouse ear inflammation. J Inflamm. 2008;5:1. doi: 10.1186/1476-9255-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu CY, Chan YP, Chang J. Antioxidant activity of extract from Polygonum cuspidatum. Biol Res. 2007;40:13–21. doi: 10.4067/S0716-97602007000100002. [DOI] [PubMed] [Google Scholar]
- 13.Kimura Y, Okuda H. Resveratrol isolated from Polygonum cuspidatum root prevents tumor growth and metastasis to lung and tumor-induced neovascularization in Lewis lung carcinoma-bearing mice. J Nutri. 2001;131:1844–1849. doi: 10.1093/jn/131.6.1844. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H, Yu CH, Jiang YP, Peng C, He K, Tang JY, Xin HL. Protective effects of polydatin from Polygonum cuspidatum against carbon tetrachloride-induced liver injury in mice. PLoS One. 2012;7:e46574. doi: 10.1371/journal.pone.0046574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC T method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Pu J, Fang FF, Li XQ, Shu ZH, Jiang YP, Han T, Peng W, Zheng CJ. Matrine exerts a strong anti-arthritic effect on type II collagen-induced arthritis in rats by inhibiting inflammatory responses. Int J Mol Sci. 2016;17:E1410. doi: 10.3390/ijms17091410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran CN, Lundy SK, Fox DA. Synovial biology and T cells in rheumatoid arthritis. Pathophysiology. 2005;12:183–189. doi: 10.1016/j.pathophys.2005.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ospelt C, Gay S. The role of resident synovial cells in destructive arthritis. Best Prac Res Clin Rheumatol. 2008;22:239–252. doi: 10.1016/j.berh.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Zuo J, Xia Y, Li X, Ou-Yang Z, Chen JW. Selective modulation of MAPKs contribute to the anti-proliferative and anti-inflammatory activities of 17-dihydroxy-3,4-dimethoxyxanthone in rheumatoid arthritis-derived fibroblast-like synoviocyte MH7A cells. J Ethnopharmacol. 2015;168:248–254. doi: 10.1016/j.jep.2015.03.069. [DOI] [PubMed] [Google Scholar]
- 20.Myers LK, Rosloniec EF, Cremer MA, Kang AH. Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 1997;61:1861–1878. doi: 10.1016/S0024-3205(97)00480-3. [DOI] [PubMed] [Google Scholar]
- 21.Giles JT, Bartlett SJ, Gelber AC, Nanda S, Fontaine K, Ruffing V, Bathon JM. Tumor necrosis factor inhibitor therapy and risk of serious postoperative orthopedic infection in rheumatoid arthritis. Arthritis Rheum. 2006;55:333–337. doi: 10.1002/art.21841. [DOI] [PubMed] [Google Scholar]
- 22.Umar S, Hedaya O, Singh AK, Ahmed S. Thymoquinone inhibits TNF-α-induced inflammation and cell adhesion in rheumatoid arthritis synovial fibroblasts by ASK1 regulation. Toxicol Appl Pharmacol. 2015;287:299–305. doi: 10.1016/j.taap.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lianxu C, Hongti J, Changlong Y. NF-kappaBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and TNF-alpha-induced chon-drocytes. Osteoarthritis Cartilage. 2006;14:367–376. doi: 10.1016/j.joca.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Tian J, Chen JW, Gao JS, Li L, Xie X. Resveratrol inhibits TNF-α-induced IL-1β, MMP-3 production in human rheumatoid arthritis fibroblast-like synoviocytes via modulation of PI3k inase/Akt pathway. Rheumatol Int. 2013;33:1829–1835. doi: 10.1007/s00296-012-2657-0. [DOI] [PubMed] [Google Scholar]
- 25.Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: Role in arthritis. Front Biosci. 2006;11:529–543. doi: 10.2741/1817. [DOI] [PubMed] [Google Scholar]
- 26.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: How hot is the link. Biochem Pharmacol. 2006;72:1605–1621. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 27.Hawkey CJ. COX-2 inhibitors. Lancet. 1999;353:307–314. doi: 10.1016/S0140-6736(98)12154-2. [DOI] [PubMed] [Google Scholar]
- 28.Koch A. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003;62(Suppl 2):ii60–ii67. doi: 10.1136/ard.62.suppl_2.ii60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagashima M, Yoshino S, Ishiwata T, Asano G. Role of vascular endothelial growth factor in angiogenesis of rheumatoid arthritis. J Rheumatol. 1995;22:1624–1630. [PubMed] [Google Scholar]
- 30.Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor α and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. 1998;41:1258–1265. doi: 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 31.Cheon H, YU SJ, Yoo DH, Chae IJ, Song GG, Sohn J. Increased expression of pro-inflammatory cytokines and metalloproteinase-1 by TGF-beta1 in synovial fibroblasts from rheumatoid arthritis and normal individuals. Clin Exp Immunol. 2002;127:547–552. doi: 10.1046/j.1365-2249.2002.01785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldring S. Pathogenesis of bone and cartilage destruction in rheumatoid arthritis. Rheumatology. 2003;42(Suppl 2):ii11–ii16. doi: 10.1093/rheumatology/keg327. [DOI] [PubMed] [Google Scholar]
- 33.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–261. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- 34.Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy-from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. 2005;1754:253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 35.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 36.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Ann Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 38.Beg AA, Baldwin AS., Jr The I kappa B proteins: Multifunctional regulators of Rel/NF-kappa B transcription factors. Genes Dev. 1993;7:2064–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 39.Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kappaB by directly targeting p65. J Biol Chem. 1998;273:33508–33516. doi: 10.1074/jbc.273.50.33508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.