

Abstract
The pediatric ocular cancer retinoblastoma is the only central nervous system tumor readily observed without specialized equipment: it can be seen by, and in, the naked eye. This accessibility enables unique imaging modalities. Here, we review this cancer for a neuroscience audience, highlighting these clinical and research imaging options, including fundus imaging, optical coherence tomography, ultrasound, and magnetic resonance imaging. We also discuss the subtype of retinoblastoma driven by the MYCN oncogene more commonly associated with neuroblastoma, and consider trilateral retinoblastoma, in which an intracranial tumor arises along with ocular tumors in patients with germline RB1 gene mutations. Retinoblastoma research and clinical care can offer insights applicable to CNS malignancies, and also benefit from approaches developed elsewhere in the CNS.
Keywords: pediatric cancer, optical coherence tomography, MYCN, cancer genetics, pineoblastoma, neuroimaging
Graphical Abstract
Retinoblastoma is a uniquely accessible CNS tumor. Imaging techniques for this cancer, the biology of its MYCN-driven form, and its “trilateral,” intracranial manifestation provide interesting contrasts and parallels with other CNS malignancies.
INTRODUCTION
The pediatric retinal tumor retinoblastoma is the most common eye cancer in children. It is unique amongst CNS tumors as it is visible through the eye without requiring invasive imaging. This provides an unparalleled opportunity to image tumors as they develop or respond to treatment. Moreover, retinoblastoma provides intriguing insights into nervous system disease in its trilateral and MYCN-driven forms.
In this review, we introduce retinoblastoma with a focus on these features. In the first part of the review, we describe the clinical, molecular and histological features of this tumor in the context of its treatment and prognosis. In the second section of the review, we focus on innovations in the imaging of retinoblastoma to highlight what this pediatric ocular cancer tells us about CNS malignancy. We follow this with a discussion of the aforementioned trilateral and MYCN-driven forms of retinoblastoma, with the goal of highlighting what retinoblastoma research can gain from studies of other CNS malignancies, and vice versa.
RETINOBLASTOMA OVERVIEW
Clinical vignette
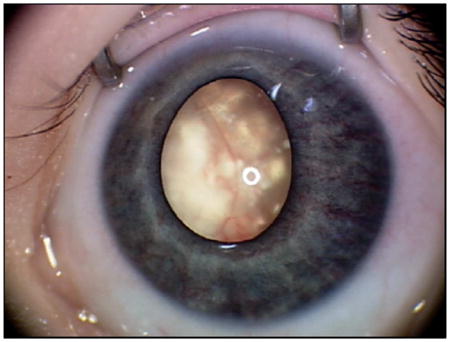
A mother in urban Canada is taking photographs of her child, when she notices a strange white reflex in the child’s eye (Fig. 1a). Unsure what it is, later that night she types “white eye in baby” into an Internet search engine. A list of results pops up, alerting her that a white pupil could be a sign of cancer. She immediately seeks attention at the nearest hospital, where the child is diagnosed with retinoblastoma.
Fig. 1.

Clinical features of retinoblastoma. (a) External RetcamTM image of retinoblastoma; calcified tumor is visible through the pupil as leukocoria. (b) RetcamTM fundus image revealing two tumors (arrowheads), * = optic nerve head. (c) Gross pathology of an enucleated eye. A large, calcified tumor is apparent on the left side of the globe (arrowhead). (d) Pupillary-optic nerve section of an eye enucleated for retinoblastoma as primary treatment. (e) Homer Wright rosettes (arrowhead) and packed cells; (f) Viable collars of cells around blood vessels, and abundant necroses between them; (g) Area of retinoma, with fleurettes (arrowhead). Scale bars = 100 μm.
Similarly, one evening a mother in rural India notices a white reflex in the eye of her child. Her baby does not seem bothered by it, so she does not worry about it too much. Later that month a community health worker arrives to check on the family. The mother asks about the odd white reflection in her baby’s eye. The health worker has just been trained on a new app that helps in the diagnosis of eye conditions. Using her smartphone, she takes a flash photo of the child’s eye, and uses the app’s database of eye conditions to compare her photograph. She realizes the photo she took looks similar to the database photo of retinoblastoma, so she refers the family to hospital.
Clinical features
These hypothetical cases illustrate typical presentations of retinoblastoma. The incidence of retinoblastoma is constant worldwide at 1 in every 16,000 live births (Dimaras et al. 2015; Dimaras et al. 2012). As such, countries with higher birth rates and populations observe the highest number of cases. The most common first signs of retinoblastoma are leukocoria, a white reflex visible through the pupil (Fig. 1a), and strabismus, misaligned eyes. Awareness of these early signs of eye cancer is crucial to good outcomes for affected children. However, where awareness of these signs is low, and access to healthcare is challenging, retinoblastoma is often diagnosed late. In this case, the most common sign at presentation may be proptosis, where tumor causes the eye to bulge from the orbit. Retinoblastoma can affect one or both eyes, and sometimes also the pineal, parasellar or suprasellar regions (trilateral retinoblastoma, discussed below).
Detection of retinoblastoma by lay people is possible because retinoblastoma is a visible tumor with distinct features. Several awareness efforts aim to improve public knowledge of the early visible signs of retinoblastoma. These include low-tech innovations, such as including images of leukocoria in Maternal Child Health booklets (Ministry of Health Kenya 2010) provided to mothers shortly after childbirth, to alert them of the signs of retinoblastoma. High-tech innovations include concepts like the Portable Eye Examination Kit (Peek), a smartphone application that allows users to recognize and detect eye conditions, including retinoblastoma (Bastawrous 2016; Bastawrous et al. 2015; Lodhia et al. 2016; Morjaria et al. 2017).
Medical diagnosis of retinoblastoma is based on the clinical features of the tumors visible in the eye upon dilation of the pupil (Dimaras et al. 2012) (Fig. 1b). This differs from diagnosis of other cancers, where histological confirmation via biopsy is usually required. Biopsy of retinoblastoma is not recommended, as it can induce seeding and extraocular spread along the needle tract (Karcioglu 2002).
Retinoblastoma is often highly calcified (Fig. 1c) (Bullock et al. 1977). Calcification is visible to the naked eye, and can also be detected by ultrasound or MRI (Rodjan et al. 2015) (see below). Calcification in retinoblastoma is ‘dystrophic’ (Eagle 2013), that is, it occurs in response to tissue damage or necrosis. It is more often observed in tumors of older children and advanced retinoblastoma (Levy et al. 2011).
Retinoma, the benign precursor of retinoblastoma, also displays calcification, along with several distinct clinical features; it is described clinically as a grey, translucent mass, often with chorioretinal atrophy and variation in the retinal pigment epithelium (Dimaras et al. 2009; Dimaras et al. 2008; Gallie et al. 1982b). Retinoma is rare, but can be sometimes observed in parents of children with retinoblastoma (Dimaras et al. 2008; Gallie et al. 1982a). Its rare occurrence suggests that most retinomas undergo malignant transformation to retinoblastoma.
Growing retinoblastoma tumors produce seeds that detach and adhere below the retina (subretinal seeds) or float into the vitreous (vitreous seeds). The appearance of seeds has been described as fine dust, spheres, or clouds, each with a progressively worse prognosis, respectively (Francis et al. 2015; Francis et al. 2016; Munier 2014). Large tumors can lift and detach the retina, and cause significant inflammation and necrosis, sometimes leading to phthisis bulbi (shrinkage of the eye) (Gallie et al. 1982b; Kashyap et al. 2011).
Tumor can spread beyond the eye by surpassing retinal boundaries, such as Bruch’s membrane, to invade the choroid and reach the blood supply. Another common route for tumor dissemination is via the optic nerve to reach the brain and cerebrospinal fluid (CSF) (Mallipatna et al. 2017).
Staging
The features of retinoblastoma at presentation provide data to accurately classify the disease in each eye, and consequently direct treatment. During the era when retinoblastoma was primarily treated by radiation, the Reese-Ellsworth Classification (Reese and Ellsworth 1963) system was used, separating tumor classes by their predicted response to radiation treatment. As systemic chemotherapy began to replace radiation as the most common form of treatment, the International Intraocular Retinoblastoma Classification (IIRC) (Murphree 2005) system was developed. This system is largely based on natural progression of disease from small tumors away from the macula (easily treatable with laser therapy) and larger, central tumors with variable degrees of seeding and retinal detachment (requiring chemotherapy in addition to focal therapy). Modifications to the original IIRC were later introduced by another group (Shields et al. 2006), making it difficult to compare across studies and centers (Novetsky et al. 2009). The critical differences between the classification systems have previously been reviewed (Dimaras et al. 2015). Other classification schemes include staging of extraocular retinoblastoma (Chantada et al. 2006) and staging of vitreous seeding (Francis et al. 2015; Francis et al. 2016; Munier 2014).
As with other cancers, retinoblastoma is also staged using the American Joint Commission on Cancer (AJCC)’s “Tumor Node Metastasis” (TNM) classification system. The most recent 8th edition of the TNM (Mallipatna et al. 2017) aims to provide a way forward for retinoblastoma classification given the above eye-based classification confusion. The Tumor stage is first applied per eye based on clinical features, and the patient’s score is based on the most advanced eye (Mallipatna et al. 2017). Radiology plays an important role in determining the Metastasis stage, particularly in delineating any involvement of the CNS. The 8th edition of the TNM also includes a new feature, the additional “H” or heritable trait, which denotes risk of multifocal tumors that can affect both eyes and brain (trilateral retinoblastoma). As yet, trilateral retinoblastoma is not staged by the TNM system. In fact, no other tumors of the CNS are staged by TNM, as tumor size is not a prognostic indicator, the brain and spinal cord do not have a lymphatic system, and metastases are rarely distant (Laws Jr. et al. 2017). Retinoblastoma is a unique CNS tumor in this respect, as tumor size is prognostic, it can affect preauricular, submandibular or cervical nodes, and distant metastases occur (Mallipatna et al. 2017).
Pathology
Retinoblastoma is a small round blue-cell tumor (Fig. 1d–e), and thus bears histologic similarity to other childhood nervous system tumors including neuroblastoma and medulloblastoma. Cells are primarily composed of large basophilic nuclei and little cytoplasm. The tumor is highly proliferative, displaying abundant mitoses and necrosis. Cells may take on a polygonal shape and mimic cobblestones when packed together (Tso 1980). Often, collars of viable cells can be observed around a blood vessel, surrounded by necrosing cells (Fig. 1f). Tumors display varying degrees of differentiation. Cellular differentiation is primarily in the form of Flexner-Wintersteiner rosettes and Homer Wright rosettes (Tso 1980) (Fig. 1e). Flexner-Wintersteiner rosettes are composed of an ‘empty’ lumen surrounded by columnar cells (Wippold and Perry 2006). Homer Wright rosettes consist of cells surrounding a central lumen made up of their processes (Wippold and Perry 2006). While each type is individually common in other neural tumors, the dual presence of Homer Wright and Flexner-Wintersteiner rosettes is pathognomonic for retinoblastoma.
Retinoma histopathology is distinct from retinoblastoma (Fig. 1g). This benign precursor is composed of sparsely spaced round cells, and does not contain rosettes. Instead, it is characterized by the presence of fleurettes, composed of clusters of cells with long, bulbous cytoplasmic processes, mimicking the fleur-de-lys (Dimaras et al. 2009; Dimaras et al. 2008). Retinoma is observed next to retinoblastoma in 16–20% of eyes enucleated for retinoblastoma (Dimaras et al. 2008; Eagle 2009).
Cellular differentiation has not been conclusively linked to prognosis, and thus does not form part of the histological evaluation of tumors. However, the degree of anaplasia evident in retinoblastoma has been shown to correlate with prognosis (Mendoza et al. 2015), but this has not yet become part of routine histological evaluation. The pathology TNM (pTNM) classification determines extent of tumor involvement of the optic nerve, choroid, sclera and anterior segment (Finger 2009; Gallie et al. 2016; Mallipatna et al. 2017). High risk of metastasis (pT3) is conveyed by >3 mm of tumor in the choroid (the vascular layer of the eye, under the retina), tumor extending beyond the lamina cribrosa of the optic nerve (the perforated or ‘mesh-like’ structure in the sclera through which the fibers of the optic nerve exit the eye), or significant invasion of the sclera. Extraocular disease (pT4) is marked by tumor at the cut end of the nerve, involvement of the episclera, or invasion of adjacent bone, eyelid, conjunctiva, or muscle (Mallipatna et al. 2017).
Genetics
Retinoblastoma is perhaps best known to cancer biologists as the inspiration for the “two-hit hypothesis” for oncogenesis, which states that loss of both alleles of a tumor suppressor gene are necessary to initiate cancer. Working in the pre-genomic era, Alfred Knudson noted an earlier age of onset for bilateral retinoblastoma than for unilateral (Knudson 1971). His mathematical analysis implied two rate-limiting steps were needed for unilateral retinoblastoma to develop, while only a single rate-limiting step was required for bilateral cases. Fifteen years later, the “retinoblastoma gene,” RB1, was cloned, confirming Knudson’s prediction (Friend et al. 1986; Fung et al. 1987; Lee et al. 1987). Unilateral patients (usually) have lost both wild-type alleles of RB1 in a susceptible retinal cell (two hits), leading to a single, unifocal tumor (Fig. 2). Conversely, all bilateral patients have a germline mutation in RB1, thus only require a second hit to initiate cancer, which (usually) happens in multiple cells, in both eyes. The situation is complicated somewhat by low-penetrance alleles, which can be mutated in the germline without promoting bilateral disease (Lohmann et al. 1994). Because of this, approximately 15% of unilateral cases have a heritable mutation.
Fig. 2.

Retinoblastoma genetics. The cancer can initiate with a germline RB1 mutation followed by a single somatic hit, or with two somatic mutations (M1 and M2). Loss of RB1 leads to the benign retinoma; further mutations (M3 to Mn) are required for malignancy. Retinoblastoma can also initiate by amplification of MYCN, with other genomic changes as yet unknown.
The irony is not lost on us that while being a visible tumor, retinoblastoma can rob vision from the children it affects. If patients grow up to have children of their own, they should learn if they have a heritable mutation that could affect their offspring. For all families with heritable retinoblastoma, the “visible feature” of retinoblastoma that allows early detection is knowledge of the RB1 mutation. Prenatal genetic detection coupled with early term delivery (37–38 weeks’ gestation) of the affected infant allows clinical teams to monitor the newborn’s retina for the earliest sign of tumor and treat it immediately (Soliman et al. 2016).
The RB1 gene encodes pRB, a key negative regulatory transcription factor governing the G1 to S phase cell cycle transition by blocking the interactions of DNA-binding E2F and DP transcription factors with DNA (Dick and Rubin 2013). In response to mitogen-stimulated cyclin-dependent kinase signaling, normal pRB is hyperphosphorylated, releasing it from E2F interactions, allowing these factors to activate transcription. pRB also has a growing spectrum of roles in differentiation, chromatin remodeling, genome stability, and apoptosis (Dimaras and Corson 2016; Velez-Cruz and Johnson 2017).
Over 1000 mutations in RB1 have been documented in retinoblastoma tumors, including missense and nonsense mutations, splicing mutations, frameshifts, micro- and macrodeletions, and promoter methylation (Lohmann and Gallie 2004). Despite this, a small fraction of unilateral cases lack identified RB1 mutations, so it was long postulated that an alternative mechanism of retinoblastomagenesis might be possible. As part of an international team, in 2013 we identified that 1.4% of unilateral retinoblastoma, lacking RB1 mutations, instead showed amplification of MYCN (Rushlow et al. 2013) (Fig. 2), a transcription factor-encoding gene more frequently associated with the pediatric peripheral nervous system tumor, neuroblastoma (Ruiz-Perez et al. 2017). This finding raises interesting questions about the parallels between retinoblastoma and other neuronal tumors; see below.
Even in retinoblastoma tumors with a defined RB1 mutation, further mutational events are needed for malignancy (Corson and Gallie 2007). Retinal cells homozygous for RB1 loss can form the benign tumor, retinoma, but develop other genomic changes in the progression to malignancy (Dimaras et al. 2008). The spectrum of changes is characterized by relatively few mutated genes, especially compared to common adult epithelial cancers. The transcriptional corepressor BCOR (BCL6 corepressor) is the most commonly mutated gene in retinoblastoma after RB1 (Zhang et al. 2012). However, retinoblastoma displays a distinct pattern of genomic gains and losses, notably 1q, 2p, and 6p gain, and 16q loss (Kooi et al. 2016b). Candidate progression genes with functional evidence in these regions include MDM4 and KIF14 on 1q, encoding the p53 regulator mouse double minute 4 and the oncogenic motor protein kinesin family member 14, respectively. MYCN is the most commonly amplified gene on 2p, while on 6p, the best-studied genes are the DNA-binding proto-oncogene DEK and the transcription factor E2F3. Genes commonly lost on 16q include CDH11, encoding the adhesion protein cadherin-11, and RBL2, encoding the retinoblastoma family member p130 (Kooi et al. 2016b; Theriault et al. 2014). In addition, aberrant methylation is seen in the retinoblastoma genome (Benavente and Dyer 2015), including of the oncogenic kinase SYK (spleen tyrosine kinase). As in other cancers, a variety of microRNAs are also dysregulated in retinoblastoma (Singh et al. 2016), including the miR-17~92 cluster and miR-106b~25 (Conkrite et al. 2011).
Treatment and prognosis
Treatment of retinoblastoma is complex and varied. Eyes containing tumors that display clinical features threatening to extend beyond the globe may be surgically removed (enucleation) and histologically assessed to determine risk of metastasis. Eye salvage may be attempted on less severely affected eyes (Dimaras et al. 2015; National Retinoblastoma Strategy 2009), with focal therapy (laser, cryotherapy) (Hamel et al. 2000), brachytherapy (Hernandez et al. 1993), and/or chemotherapy. Chemotherapy may be systemic, usually with a cocktail of vincristine, etoposide, and carboplatin (Chan et al. 1996; Gallie et al. 1996), intra-arterial (Yousef et al. 2016), and/or intravitreal (Munier et al. 2012a; Munier et al. 2012b)). Melphalan is the preferred drug for these latter modalities. External beam radiation is another effective treatment, however this is not preferred due to its added second cancer risks (Temming et al. 2016). Trilateral retinoblastoma treatment and outcomes are discussed in a later section.
Left untreated, retinoblastoma will grow and extend beyond the eye. Commonly, it invades the regional lymph nodes, bone, bone marrow, and the central nervous system. Extension into the CNS can be detected with radiologic imaging. Prognosis for CNS disease is much worse than for bone-marrow, as treatment with systemic chemotherapy is impeded by the blood-brain barrier (Mallipatna et al. 2017). Cytologic examination can detect tumor dissemination in CSF and bone marrow. Monitoring of CSF disease can be also done molecularly, by looking for tumor-specific (non-germline) RB1 mutations or post-RB1 loss genomic gains and losses (Bowles et al. 2007; Dimaras et al. 2010b; Racher et al. 2016), or tumor-derived GD2 or CRX mRNA expression (Laurent et al. 2010; Laurent et al. 2013; Torbidoni et al. 2015).
Individuals with a constitutional RB1 mutation have an increased risk of developing second primary tumors, like osteosarcoma, melanoma, lung or bladder cancer later in life (MacCarthy et al. 2013). Surveillance for second primary malignancies is not yet supported by strong evidence or broad consensus, though some suggest there may be a role for annual screening by whole body MRI (Friedman et al. 2014; Kamihara et al. 2017). More commonly, retinoblastoma survivors are educated on second primary tumor risks and instructed to be vigilant about potential signs of new malignancies (Kamihara et al. 2017).
Guidelines for treatment in high income (National Retinoblastoma Strategy 2009) and low-and-middle income countries (Kenyan National Retinoblastoma Strategy Group 2014) are similar, although they account for differences in availability of resources and stage of disease at presentation (i.e., low-and-middle-income countries see more cases of extraocular and metastatic disease). Patient outcomes are notably worse in low-and-middle income countries than in high (Dimaras et al. 2015), and further research is needed to address social determinants affecting access to quality care (Dimaras et al. 2010a).
IMAGING RETINOBLASTOMA
Clinical vignette
An adult survivor of bilateral retinoblastoma is pregnant with her first child. She undergoes prenatal genetic testing. Her fetus is found to carry her familial RB1 mutation and is therefore at risk of developing retinoblastoma. Shortly after birth, the infant’s eyes are visually examined, and the retina appears normal in each eye. Monthly examinations are scheduled, during which the ophthalmologist inspects each eye with the indirect ophthalmoscope, and an imaging specialist documents the fundus with digital images. At the second examination after birth, the ophthalmologist still does not see any tumors in the eyes. A clear fundus is documented in each eye using a high-resolution fundus camera. Next, the imaging specialist uses Optical Coherence Tomography (OCT) to image the eyes. Surprisingly, the OCT reveals a tiny lump within the inner nuclear layer of the retina in the left eye. Looking back at the fundus images, the ophthalmologist notes a slight haze in the exact spot picked up by OCT: it is a tiny tumor, originally missed on examination of the photographic images. The tumor is treated with focal therapy, preventing its growth and sparing vision (Based on similar cases previously reported; Rootman et al. 2013; Soliman et al. 2016).
Fundus imaging
Images of the fundus (interior surface of the eye), which become part of the medical record, facilitate consultation and collaboration, and precise tracking of tumor response or resistance to treatment. This kind of longitudinal, visual documentation is unique amongst CNS tumors. Before photographic imaging equipment was a standard part of ophthalmic care, retinal drawings (Wilson 1975) were used to map retinal topography, lesions, relevant landmarks and treatments applied, as observed through the indirect ophthalmoscope (Dvorak and Russell 2011). Retinal imaging has changed practice by providing a way to digitally document the eye, but retinal drawings still remain an important part of learning to understand the disease trajectory.
Digital retinal imaging via portable fundus camera has revolutionized clinical documentation of retinoblastoma (Fig. 1b, 3a, 3d), allowing comparison of pre- and post-treatment images (Romanowska Dixon and Morawski 2017) and facilitating consultation between centers (National Retinoblastoma Strategy 2009). The RetCamTM (Clarity Medical) captures high-resolution, wide-angle photographs, and with scleral depression, allows imaging of the retina up to the ora serrata (the anterior edge of the retina) (National Retinoblastoma Strategy 2009). Similarly, the ICON camera (Phoenix Technology Group), produces wide-field, high-contrast, high-resolution fundus images.
Fig. 3.

Retinoblastoma imaging. (a–b) RetcamTM (a) image of an eye reveals two large tumors, and ultrasound image (b) allows measurement of tumor height (yellow and green lines). (c) Axial T1 MRI post-gadolinium reveals tumor at back of eye (arrowhead). (d–f) Imaging of the eye of an infant with heritable retinoblastoma by RetcamTM (d) shows no visible tumor. OCT scan line shown in green/red. OCT imaging (e, en face; f, transverse section) reveals small tumor appearing to arise out of the inner nuclear layer of the retina (arrowhead). (g) Fundus photo (top) and OCT image (bottom) show a retinoblastoma-like tumor (arrowheads) arising in the TAg-RB mouse model. OCT scan line shown in red. Scale bars = 100 μm.
Unlike fundus drawings, which depict the entire retina in one image, fundus images are partial views of the entire retina and must be stitched together to get a whole-retina view. Fluorescein angiography with portable fundus camera imaging is also important to document vascularization, especially for follow-up of advanced tumors (Kim et al. 2014).
Ultrasound
The B-scan (10 MHz) 2D ultrasound is commonly used for ocular imaging, and has been a part of the diagnostic toolkit in ophthalmology for decades (Giglio and Sherman 1979; Kendall et al. 2015; Mrochuk 1990). It is able to distinguish between ocular structures such as lens, choroid, retina and sclera, and image and measure aberrations in the eye, such as tumors or retinal detachment. Importantly for retinoblastoma, B-scan ultrasound allows visualization of calcification, a key feature of these tumors, and thus can help confirm diagnosis (National Retinoblastoma Strategy 2009; Novotny and Krasny 1990) (Fig. 3b).
High frequency B-scan ultrasonography (20–50MHz), also known as ultrasound biomicroscopy (UBM), produces images with higher resolution than traditional B-scan (Pavlin et al. 1990). It first showed promise in the imaging of anterior segment tumors more consistently than B-scan (Pavlin et al. 1992; Reminick et al. 1998). For retinoblastoma, UBM has been shown to be essential for determining the proximity of the tumor to the anterior portion of eye (Vasquez et al. 2011). These are important diagnostic details that assist with accurate classification at presentation.
Furthermore, like other imaging modalities, ocular ultrasonography also allows monitoring of tumor regression as treatment progresses, primarily by comparing the size of the tumor at each examination to measurements taken at baseline. Unlike the use of intraoperative ultrasound for detection of residual disease in brain tumors (Bohringer et al. 2009), ocular ultrasound cannot currently distinguish between active and inactive retinoblastoma tumor.
Magnetic Resonance Imaging (MRI)
As with other CNS tumors, MRI can provide important diagnostic and prognostic information in retinoblastoma (Fig. 3c) and is vital for screening for trilateral retinoblastoma (see below). MRI can also aid in characterizing the tumor in cases where it is not visible, for instance if it is behind a cataract-containing lens.
Where available, MRI is often used as part of the workup of all patients with retinoblastoma, as it can provide vital information about high-risk pathological features prior to enucleation (de Graaf et al. 2012). These include: optic nerve invasion evidenced by thickening; massive choroidal invasion evidenced by thickening or irregularities, scleral invasion evidenced by signal-enhancing tissue beyond the choroid, and intracranial invasion into the suprasellar region (Razek and Elkhamary 2011). However, sensitivity for detecting these high-risk features varies considerably (de Jong et al. 2014a). High field-strength MRI can also confirm retinoblastoma diagnosis by corroborating the calcifications observed by ultrasound or fundus imaging (Galluzzi et al. 2009), and can detect vitreous seeds or anterior segment involvement (Razek and Elkhamary 2011). Again, these uses are particularly valuable when fundus imaging is not possible. Note that computerized tomography (CT) imaging is generally avoided in retinoblastoma due to the radiation risks, especially in patients with a germline RB1 mutation, already at increased risk for second primary tumors.
In intracranial tumors like gliomas, intraoperative MRI has assumed a pivotal role, to detect removal of diseased tissue during surgery (Hlavac et al. 2017). Intraoperative MRI is not necessary in retinoblastoma, as enucleation usually removes the entirety of the tumor, unless extraocular spread has been previously detected, or high risk features are subsequently discovered on pathology. However, one retinoblastoma case report describes the use of intraoperative MRI to track the delivery and tumor penetration of intra-arterial melphalan delivered in a cocktail with gadolinium (Materin et al. 2012).
Optical coherence tomography (OCT)
OCT, a non-invasive imaging technique based on differential near-infrared light reflection, offers a powerful tool for imaging retinoblastoma, with resolution exceeding that of ultrasound or MRI. Conceptually similar to ultrasound, spectral domain (SD) OCT can generate high resolution, cross-sectional images of tissues where light can penetrate (Fujimoto and Swanson 2016), so has found utility in dermatology, urology, and in particular, ophthalmology, where it is routinely used in adult clinics. It has also been used in the CNS: for ex vivo analysis of gliomas (Valdes et al. 2016) and medulloblastoma (Vuong et al. 2015), and is being trialed for intraoperative imaging of gliomas to detect residual tumor, as is also done via ultrasound and MRI (Bohringer et al. 2009).
Recently, with the advent of hand-held intraoperative OCT systems, OCT has become popular for pediatric ophthalmology as well, including retinoblastoma assessment (Do et al. 2017). Initial publications were case reports indicating the value of OCT for detecting cavitary retinoblastoma (Mashayekhi et al. 2005) and reasons for visual loss (such as retinal detachment) post-therapy (Shields et al. 2005). Subsequently, OCT showed utility for documenting response to therapy, spotting edge recurrence, and notably, detecting “invisible” tiny retinoblastoma (Fig. 3d–f) (Rootman et al. 2013), as well as assessing foveal anatomy, which impacts the likelihood of visual function post-therapy (Cao et al. 2014; Samara et al. 2015). One case report even documented optic nerve head disease, usually detected by MRI (see above) (Yousef et al. 2012). OCT’s ability to look “behind” small features such as vitreous seeds to the underlying tumor or normal retina can be valuable for assessing retinoblastoma comprehensively.
This growing body of work also contributed to an ongoing debate on the cell of origin of retinoblastoma, as some tiny tumors detected by OCT seem to originate in the inner nuclear layer (Rootman et al. 2013) (Fig. 3f), while others are more localized to the outer nuclear layer, suggestive of a cone precursor origin for at least some retinoblastoma (Berry et al. 2016), which is consistent with experimental data (Xu et al. 2014).
Although not yet in use for retinoblastoma, recently-developed OCT angiography holds considerable promise for documenting vascularization and blood flow in and around tumors (Gao et al. 2016). However, OCT does have limitations: it images the periphery of the retina poorly (although technology for widefield OCT is improving), and relies on transparent media: large tumors and/or cataracts limit its usefulness. Despite this, OCT for retinoblastoma is usually successful and can guide treatment decisions (Soliman et al. 2017b). With time, OCT will doubtless assume a greater role in the staging and management of retinoblastoma, taking true advantage of the accessibility of the eye to manage this cancer.
Imaging animal models
The wealth of imaging tools available for the eye has made longitudinal studies of retinoblastoma animal models possible, complementing more traditional histological analyses (Fig. 3g). The advent of small animal intraocular imagers enabled documentation of intraocular tumors in both transgenic and orthotopic xenograft models of the cancer. In particular, coupling fluorescence and brightfield imaging of GFP-expressing xenografts allowed qualitative assessment of tumor growth and (importantly) dissemination within the eye (Corson et al. 2014). Similarly, a number of preclinical therapeutic studies have included qualitative fundus and fluorescein angiography images (Brennan et al. 2011; McEvoy et al. 2011; Zhang et al. 2012). In the future, quantification of fluorescence should allow volumetric analysis of tumor growth, as is currently done using bioluminescence of luciferase-expressing xenografts (Corson et al. 2014; Ji et al. 2009; Laurie et al. 2005).
MRI has also been employed in preclinical treatment trials, and in one report was used for volumetric estimation of tumor (McEvoy et al. 2011). Ultrasound has also been used for animal models in limited cases (Brennan et al. 2011); one study showed fundus, ultrasound and MRI images of transgenic animals in a treatment trial, but did not use these images for assessing efficacy (Nemeth et al. 2011).
OCT has become a favored method for detecting lesions, especially in genetic models. The first work in this area was done with custom-built OCT apparatus, enabling detection of intraretinal tumors in the SV40 T-antigen driven mouse model of retinoblastoma (TAg-RB) (Ruggeri et al. 2007), and assessing their response to therapy (Cebulla et al. 2008). Even retinal tumors in utero could be detected through the uterine horn (Larina et al. 2012). TAg-RB tumors have also been documented by ultrasound (Foster and Brown 2012).
More recently, using a commercially-available small animal OCT system, we showed that this technique can detect the earliest signs of tumors in the TAg-RB model, corresponding to their first appearance by histology (Fig. 3g) (Wenzel et al. 2015), and we used this finding to show that overexpression of the oncogene Kif14 accelerates not just tumor growth, but tumor initiation in this model as well (O’Hare et al. 2016).
OCT has proven useful for xenografts: intravitreal xenografts could be monitored (Corson et al. 2014; Tschulakow et al. 2016) and subretinal xenografts could be rapidly assessed by a combination of OCT and funduscopy (Lemaitre et al. 2017). Of course, OCT assessment of morphology of xenografts is less clinically relevant than the morphology of tumors arising intraretinally as in genetic retinoblastoma models. However, the prospects for rapidly assessing genetic modifiers and/or therapeutic efficacy by OCT in both types of model are appealing, especially with the advent of methods to volumetrically assess lesions on OCT (Ruggeri et al. 2009; Sulaiman et al. 2015).
MYCN-AMPLIFIED RETINOBLASTOMA
Clinical vignette
A 1-month-old infant with no family history of retinoblastoma has a large retinoblastoma identified in one eye. On enucleation, no rosettes are noted, but the high-risk pathological feature of optic nerve invasion is present. Since early-onset, aggressive tumors are often indicative of a germline RB1 mutation, heritable disease is suspected, with the concomitant risk of tumors in the fellow eye and a need for lifelong surveillance and risk to offspring. But genetic analysis of the enucleated tumor fails to find any mutated RB1 alleles, despite >96% detection rate in specialized labs (Soliman et al. 2017a). However, further analysis of common copy number changes in retinoblastoma reveals that the oncogene MYCN is amplified to 53 copies in this tumor, and on subsequent re-review, the pathologist notices some neuroblastoma-like histological features. These findings strongly suggest MYCN-driven disease, which is not heritable, leaving this child at only population risk for future malignancies.
Discovery and characteristics
MYCN retinoblastoma was identified based on careful analysis of over 1000 tumor genotypes (Rushlow et al. 2013). Although MYCN amplification had been documented in occasional retinoblastoma since the MYCN gene was first identified (Gilbert et al. 1981), this large-scale study was needed to reveal that MYCN amplification is much more common amongst unilateral retinoblastoma without a detectable RB1 mutation. This finding has since been replicated in other cohorts (Ewens et al. 2017; Kooi et al. 2016a; McEvoy et al. 2014), but it is important to note that MYCN retinoblastoma is currently only identifiable after enucleation. Retrospective analysis of MYCN retinoblastoma revealed several further differentiating features (Fig. 4). Clinically, these tumors are amongst the very earliest-onset unilateral tumors, and often had high-risk pathological features, although there is not yet sufficient evidence to conclude that MYCN amplification has a dramatic effect on aggressiveness, as it does in neuroblastoma or medulloblastoma (Ruiz-Perez et al. 2017). Histopathologically, these tumors also differ from RB1−/− retinoblastoma. Comparatively, they show more rounded, undifferentiated cells, with prominent nucleoli (Fig. 4c), and lack the nuclear molding, differentiated rosettes, and extensive calcification commonly seen in retinoblastoma. Again, the histology of MYCN retinoblastoma is more similar to neuroblastoma than it is to RB1−/− retinoblastoma.
Fig. 4.
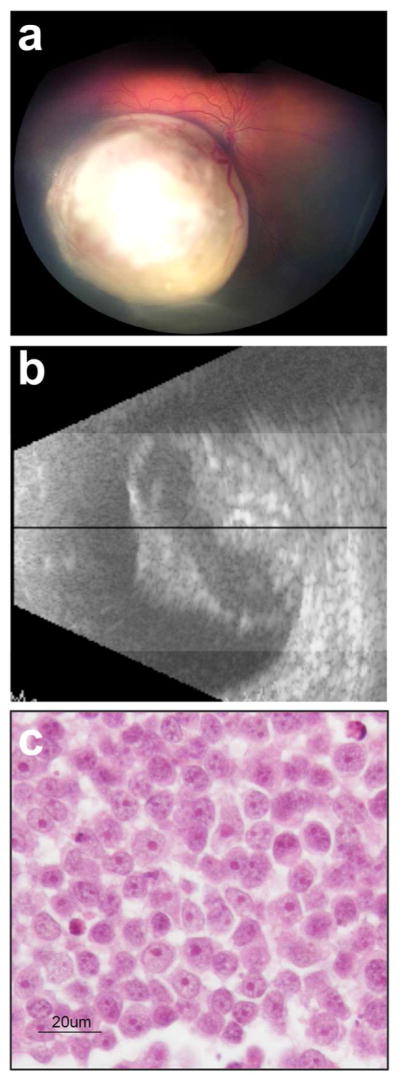
MYCN retinoblastoma. (a) Tumor appears retinoblastoma-like on fundus image. (b) Ultrasound reveals characteristic calcification. (c) Histopathology of tumor in enucleated eye reveals round nuclei with prominent large multiple nucleoli; contrast with RB1−/− retinoblastoma in Fig. 1e. Adapted with permission from (Rushlow et al. 2013).
Molecular features
Beyond the lack of RB1 mutations, MYCN retinoblastoma had fewer copy number alterations than RB1−/− tumors in one study (Rushlow et al. 2013), although this was not seen in a subsequent report (Ewens et al. 2017). MYCN retinoblastoma were also less likely to display the characteristic chromosomal gains and losses of retinoblastoma (1q and 6p gain, 16q loss), instead showing some variations in common with neuroblastoma (17q gain, 11q loss) (Rushlow et al. 2013).
MYCN retinoblastomas have functional, hyperphosphorylated pRB (Ewens et al. 2017), and high levels of N-Myc protein (Rushlow et al. 2013). The fact that pRB is expressed, yet inactivated by post-translational modification, strongly implies that genetic changes at the RB1 locus are not driving this disease. The SKP2-p27 axis leading to pRB phosphorylation was not activated in 3 of 4 MYCN retinoblastoma (Ewens et al. 2017), suggesting that this MYCN-activated pathway is not a key driver in MYCN retinoblastoma, unlike in neuroblastoma (Evans et al. 2015).
Interestingly, high retinal Mycn levels alone (driven by the Pax6α enhancer) could not initiate retinoblastoma-like tumors in mice, but could cooperate with Rb1 loss to form tumors (which normally require loss of another retinoblastoma family member such as p107) (Wu et al. 2017). This discovery also offered a caution toward MYCN-targeted therapy: when Mycn levels were reduced in this mouse model, tumors reoccurred with Mycn-independence (Wu et al. 2017). Further insight into how MYCN amplification leads to pRB inactivation by hyperphosphorylation and drives the human tumors is needed. Perhaps the Pax6α enhancer does not drive expression in the retinal cell type susceptible to Mycn-driven retinoblastoma; Mycn overexpression alone can promote cancer in other tissues, as observed with the tyrosine hydroxylase-driven Mycn neuroblastoma model (Weiss et al. 1997). In the absence of perfect mouse models, cell lines and patient-derived xenografts will be best suited for these experiments, but given the rarity of MYCN retinoblastoma, few are currently available.
Are MYCN-amplified ocular tumors retinoblastoma?
Despite the histopathological and genetic similarities with neuroblastoma, there is no reason to believe that “MYCN retinoblastoma” is not retinoblastoma: it apparently arises in a retinal neuronal precursor, and (based on a small number of tumors) has a gene expression profile not statistically distinct from that of a major group of retinoblastoma, characterized by low “photoreceptorness”, but high expression of RNA biogenesis and M phase genes (Kooi et al. 2015). Thus, to date it is unclear if MYCN retinoblastoma derives from a different retinal cell of origin than RB1−/− retinoblastoma, or if the cell of origin is the same, but the initiating MYCN amplification event pushes these cells into a distinct phenotype.
TRILATERAL RETINOBLASTOMA
Clinical vignette
A 6-month old presents with a 2-week history of leukocoria in one eye. Upon ophthalmic examination, the child is diagnosed with unilateral retinoblastoma. As part of routine work-up, the child undergoes CT of head and both orbits. The CT reveals an intracranial midline tumor. Cytologic examination of the CSF shows presence of malignant cells. The child undergoes treatment for trilateral retinoblastoma, which includes high-dose chemotherapy followed by autologous stem cell transplant. The child is alive and well 10 years post-treatment. (Based on a previously reported case; Dimaras et al. 2015; Dimaras et al. 2011).
A 4-month old presents to the emergency room with atypical eye movements and overgrowth syndrome. A CT scan reveals a suprasellar tumor (Fig. 5). The eyes are not examined at this time. The tumor is biopsied and shows a small blue cell tumor with abundant Flexner-Wintersteiner rosettes. The eyes are then examined, and reveal multifocal tumors in both eyes. Systemic and intrathecal chemotherapy for trilateral retinoblastoma is prescribed. During the placement of the intrathecal catheter, the treating physicians observe tumor seeding along the needle tract of the initial biopsy. The child’s tumor reoccurs and she dies. (Based on a previously reported case; (Dai et al. 2008; Dimaras et al. 2011).
Fig. 5.

Trilateral retinoblastoma. (a) Sagittal T1-weighted and (b) coronal T2-weighted fluid-attenuated inversion recovery (FLAIR) contrast MRI reveals suprasellar mass in a patient who also has bilateral retinoblastoma. (c) Histology of this mass reveals abundant Flexner-Wintersteiner and Homer Wright rosettes. Scale bar = 25 μm.
Origins and Pathology
Trilateral retinoblastoma, first described in 1977 (Jakobiec et al. 1977) and coined in 1980 (Bader et al. 1980), is the heritable form of retinoblastoma associated with an intracranial tumor. The intracranial tumors most commonly affect the pineal gland, as well as the suprasellar or intrasellar region of the brain (Fig. 5). Pineal tumors are generally smaller than non-pineal tumors in trilateral retinoblastoma (de Jong et al. 2014b). Benign pineal cysts have also been observed with retinoblastoma (Beck Popovic et al. 2006; Gupta et al. 2016; Karatza et al. 2006). In rare cases pineal cysts can transform into malignant tumors, suggesting they should be monitored over time (de Jong et al. 2016). Cystic normal pineal glands, however, are also regularly observed in children; their size is often comparable to solid and cystic pineoblastoma (Sirin et al. 2016). This makes it challenging to know which cystic lesions require follow-up, and which do not. A classification for pineal glands has been proposed to differentiate glands based on absence or presence of cysts, number of cysts, and enlargement (Type 0–4) (Sirin et al. 2016). The European Retinoblastoma Imaging Collaboration proposed guidelines for follow-up of cystic pineal lesions based on this classification: they recommend monitoring Type 3 and 4 lesions (multicystic with enlargement) by routine MRI for changes in size or morphology of the solid component of the lesion (Sirin et al. 2016). It is sometimes equally challenging to differentiate large solid pineal glands from pineoblastoma (Galluzzi et al. 2016).
Trilateral retinoblastoma often presents with leptomeningeal metastasis or CSF involvement, which is difficult to cure. The histological features of the intracranial tumor are identical to those of retinoblastoma (Fig. 5c). Because of its rarity, little work on the molecular genetics of this tumor has been reported (Li et al. 2005). Mouse models may yet offer some insights: 15%–27% of TAg-RB mice develop primitive neuroectodermal tumors, analogous to the parasellar and suprasellar tumors arising in patients with trilateral retinoblastoma (Marcus et al. 1991; O’Brien et al. 1990). The trilateral tumors in mice have histological features consistent with their human counterparts (Marcus et al. 1991). However, this murine retinoblastoma model does not develop pineoblastoma (Marcus et al. 1991), although other T-antigen-driven tumor models do (Marcus et al. 1996). Also, Rb1+/− mice with loss of p53 develop pineoblastomas, but these mice do not develop retinal tumors (Williams et al. 1994). Despite the existence of these models, no preclinical studies on trilateral retinoblastoma have yet been reported.
In addition to predisposition by RB1 mutation, pineoblastoma alone is associated with germline mutations in DICER1 (de Kock et al. 2014). In murine retinoblastoma models, Dicer1 is a haploinsufficient tumor suppressor (Lambertz et al. 2010). Further investigation is warranted into the development of pineoblastoma within and without the context of retinoblastoma.
Epidemiology
The incidence of trilateral retinoblastoma among patients with heritable retinoblastoma is 3.5% (de Jong et al. 2015). Prior systemic chemotherapy to treat retinoblastoma is associated with decreased risk of developing an intracranial tumor (Shields et al. 2001). Suprasellar or intrasellar tumors are diagnosed earlier than pineal, and often concurrently with the retinoblastoma (Kivela 1999). In contrast, 60% of pineal tumors are diagnosed within 1 year of initial retinoblastoma diagnosis, suggesting that routine screening by MRI could contribute to earlier diagnosis (de Jong et al. 2014b). Some centers screen by MRI every 6 months until 5 years of age (Kamihara et al. 2017), however it is unclear if this approach is warranted or effective (De Ioris et al. 2014).
Becoming treatable
Treatment for trilateral retinoblastoma generally involves chemotherapy delivered systemically and intrathecally via an Ommaya reservoir, followed by high-dose chemotherapy and autologous stem cell rescue (Dimaras et al. 2011). Surgical treatment has been attempted in some cases; however, complete resection is difficult due to the locations of the intracranial tumors (Dunkel et al. 2010). Craniospinal radiation, while effective, is not preferred because of its long-term developmental, neurocognitive and endocrine damage (Duffner et al. 1985; Mulhern et al. 1999). Indeed, the use of radiation in the treatment of trilateral retinoblastoma has declined in recent years (de Jong et al. 2014b).
Survival from trilateral retinoblastoma has improved over time, most notably with the introduction of high-dose chemotherapy and autologous stem cell transplant (de Jong et al. 2014b). Looking at outcomes before and after 1995, when chemotherapy changed treatment for retinoblastoma, the 5-year survival for pineal trilateral retinoblastoma has increased from 6% to 44%, and non-pineal from 0 to 57% (de Jong et al. 2014b; Kivela 1999). The presence of leptomeningeal metastases considerably reduces probability of survival (de Jong et al. 2014b).
CONCLUSIONS AND OUTLOOK
Although rarer than many other CNS cancers, retinoblastoma has offered fundamental cancer knowledge, and clinicians and researchers have taken advantage of its accessibility to carefully document and treat it using multiple modalities. We envisage several areas for future developments to improve our understanding and clinical management of this cancer.
Trilateral retinoblastoma
Our improving knowledge of the clinical behavior of intracranial tumors in retinoblastoma has not been matched by our understanding of their molecular features. It will be valuable to determine if there are molecular characteristics of certain heritable retinoblastomas that might predispose to formation of trilateral tumors. These might include RB1 genotype, DICER1 genotype, protein marker expression, features on high resolution imaging, or others. Trilateral retinoblastoma tumors (like their anatomical counterparts not associated with retinoblastoma) might benefit from targeted therapy, and each could benefit from discoveries in the other in this domain. The availability of mice that develop trilateral retinoblastoma-like tumors makes preclinical studies technically feasible, but the low frequency of intracranial tumor formation in these mice plus the need for histological diagnosis makes such studies challenging.
MYCN retinoblastoma
The study of MYCN retinoblastoma can gain much from the wealth of knowledge available for MYCN neuroblastoma (Huang and Weiss 2013). Despite caveats (Wu et al. 2017), the potential for targeted agents borrowed from neuroblastoma research to treat MYCN retinoblastoma is exciting. Currently, these would only be useful for metastatic disease as the MYCN genotype is only determined post-enucleation (recall that intraocular retinoblastoma is not biopsied due to risk of tumor dissemination). But a promising new ‘liquid biopsy’ technique could change this, as it allows for capture and molecular analysis of tumor DNA from the aqueous humor of the eye, which can be safely removed with techniques that minimize risk of tumor spread (Berry et al. 2017). This approach could potentially be used to identify MYCN tumors in situ. Moreover, if distinguishing characteristics of MYCN tumors could be discovered in OCT, fundus, ultrasound, or magnetic resonance imaging, or in circulating tumor cells or DNA, targeting the primary tumor would also be possible. Large, prospective studies will be needed to find these characteristics. Similarly, comparison of the molecular features (genomic, proteomic, and signaling changes) of MYCN retinoblastoma with MYCN neuroblastoma may reveal shared vulnerabilities or unique characteristics.
Deciphering the cell of origin of MYCN retinoblastoma is likewise important. One way to explore this would be to parallel the work that identified cone precursors as a likely origin of at least some RB1−/− retinoblastoma by knocking down RB1 in dissociated human fetal retinal cells and observing which cell types can proliferate (Xu et al. 2014). A similar experiment, overexpressing MYCN instead of removing RB1, could perhaps provide insight into retinal cells susceptible to proliferation after MYCN gain.
Imaging
Tumor imaging has revolutionized care of retinoblastoma, and could perhaps be further applied to research on other CNS tumors. The powerful combination of fundus imaging and OCT makes the prospect of xenografting other tumor types into the eye an appealing one, especially for neural tumors where intravitreal or subretinal injection would be more facile than intracranial xenografting. Although the intraocular microenvironment differs from the brain, it offers similarities (neuronal and glial neighboring cells, separation from the blood by a semi-permeable barrier) that subcutaneous xenografts lack. Such paratopic xenografts could be easily followed using the imaging modalities above, and will be an interesting area for further exploration.
Retinoblastoma may also learn from imaging in CNS tumors. Molecular imaging via positron emission tomography (PET) or single photon emission computed tomography (SPECT) is showing promise for brain tumors, especially using radiolabeled amino acid tracers (Brindle et al. 2017). But the rare research on these imaging modalities in retinoblastoma has shown little-to-no promise, offering no additional insight on tumor features or response to treatment (Moll et al. 2004). Further research including novel tracer development may be indicated. However, the need to keep radiation exposure low for heritable retinoblastoma cases must also be considered.
Large imaging studies might also reveal other imaging biomarkers that could help stratify retinoblastomas for improved prognostication and treatment decisions. Such work could impact identification of both trilateral-predisposed and MYCN retinoblastoma, as noted above. Further ability to look out for distinctive features in the eyes of retinoblastoma patients will continue to improve the outlook for this visible CNS tumor.
SIGNIFICANCE.
Retinoblastoma is the most common eye cancer in children. It arises in the developing retina, part of the central nervous system (CNS). Thus, retinoblastoma is a CNS cancer viewable from outside. Retinoblastoma is usually caused by mutations in the RB1 gene, but can also be caused by amplification of the neuroblastoma oncogene MYCN. Rarely, RB1 gene mutations cause a tumor within the brain as well as eye tumors. Retinoblastoma can be imaged by multiple means, including visually through the eye, by ultrasound, magnetic resonance, and optical coherence tomography. Together, these features make retinoblastoma a unique case study amongst CNS tumors.
Acknowledgments
Support: Work in the laboratory of TWC is supported by NIH/NEI R01EY025641, St. Baldrick’s Foundation, the Retina Research Foundation, and an unrestricted grant from Research to Prevent Blindness, Inc. Work in the laboratory of HD is supported by the Canadian Institutes for Health Research and Brandan’s Eye Research Foundation.
We thank Dr. Furqan Shaikh for comments on the draft manuscript. We thank Leslie MacKeen, Dr. Cynthia Hawkins and Dr. Brenda L. Gallie for providing images for the figures.
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest.
ROLE OF AUTHORS
Conceptualization: H.D., T.W.C.; Writing – Original Draft: H.D., T.W.C.; Writing – Review & Editing: H.D., T.W.C.; Visualization: H.D., T.W.C.
References
- Bader JL, Miller RW, Meadows AT, Zimmerman LE, Champion LA, Voute PA. Trilateral retinoblastoma. Lancet. 1980;2(8194):582–583. doi: 10.1016/s0140-6736(80)92009-7. [DOI] [PubMed] [Google Scholar]
- Bastawrous A. Increasing access to eye care ... there’s an app for that. Peek: smartphone technology for eye health. Int J Epidemiol. 2016;45(4):1040–1043. doi: 10.1093/ije/dyw086. [DOI] [PubMed] [Google Scholar]
- Bastawrous A, Rono HK, Livingstone IA, Weiss HA, Jordan S, Kuper H, Burton MJ. Development and validation of a smartphone-based visual acuity test (Peek Acuity) for clinical practice and community-based fieldwork. JAMA Ophthalmol. 2015;133(8):930–937. doi: 10.1001/jamaophthalmol.2015.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck Popovic M, Balmer A, Maeder P, Braganca T, Munier FL. Benign pineal cysts in children with bilateral retinoblastoma: a new variant of trilateral retinoblastoma? Pediatr Blood Cancer. 2006;46(7):755–761. doi: 10.1002/pbc.20464. [DOI] [PubMed] [Google Scholar]
- Benavente CA, Dyer MA. Genetics and epigenetics of human retinoblastoma. Annu Rev Pathol. 2015;10:547–562. doi: 10.1146/annurev-pathol-012414-040259. [DOI] [PubMed] [Google Scholar]
- Berry JL, Cobrinik D, Kim JW. Detection and intraretinal localization of an ‘invisible’ retinoblastoma using optical coherence tomography. Ocul Oncol Pathol. 2016;2(3):148–152. doi: 10.1159/000442167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry JL, Xu L, Murphree A, et al. Potential of aqueous humor as a surrogate tumor biopsy for retinoblastoma. JAMA Ophthalmol. 2017;135(11):1221–1230. doi: 10.1001/jamaophthalmol.2017.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohringer HJ, Lankenau E, Stellmacher F, Reusche E, Huttmann G, Giese A. Imaging of human brain tumor tissue by near-infrared laser coherence tomography. Acta Neurochir (Wien) 2009;151(5):507–517. doi: 10.1007/s00701-009-0248-y. discussion 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles E, Corson TW, Bayani J, Squire JA, Wong N, Lai PB, Gallie BL. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosomes Cancer. 2007;46(2):118–129. doi: 10.1002/gcc.20383. [DOI] [PubMed] [Google Scholar]
- Brennan RC, Federico S, Bradley C, Zhang J, Flores-Otero J, Wilson M, Stewart C, Zhu F, Guy K, Dyer MA. Targeting the p53 pathway in retinoblastoma with subconjunctival Nutlin-3a. Cancer Res. 2011;71(12):4205–4213. doi: 10.1158/0008-5472.CAN-11-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindle KM, Izquierdo-Garcia JL, Lewis DY, Mair RJ, Wright AJ. Brain tumor imaging. J Clin Oncol. 2017;35(21):2432–2438. doi: 10.1200/JCO.2017.72.7636. [DOI] [PubMed] [Google Scholar]
- Bullock JD, Campbell RJ, Waller RR. Calcification in retinoblastoma. Invest Ophthalmol Vis Sci. 1977;16(3):252–255. [PubMed] [Google Scholar]
- Cao C, Markovitz M, Ferenczy S, Shields CL. Hand-held spectral-domain optical coherence tomography of small macular retinoblastoma in infants before and after chemotherapy. J Pediatr Ophthalmol Strabismus. 2014;51(4):230–234. doi: 10.3928/01913913-20140603-01. [DOI] [PubMed] [Google Scholar]
- Cebulla CM, Jockovich ME, Boutrid H, Pina Y, Ruggeri M, Jiao S, Bhattacharya SK, Feuer WJ, Murray TG. Lack of effect of SU1498, an inhibitor of vascular endothelial growth factor receptor-2, in a transgenic murine model of retinoblastoma. Open Ophthalmol J. 2008;2:62–67. doi: 10.2174/1874364100802010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HS, DeBoer G, Thiessen JJ, Budning A, Kingston JE, O'Brien JM, Koren G, Giesbrecht E, Haddad G, Verjee Z, Hungerford JL, Ling V, Gallie BL. Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin Cancer Res. 1996;2(9):1499–1508. [PubMed] [Google Scholar]
- Chantada G, Doz F, Antoneli CB, Grundy R, Clare Stannard FF, Dunkel IJ, Grabowski E, Leal-Leal C, Rodriguez-Galindo C, Schvartzman E, Popovic MB, Kremens B, Meadows AT, Zucker JM. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47(6):801–805. doi: 10.1002/pbc.20606. [DOI] [PubMed] [Google Scholar]
- Conkrite K, Sundby M, Mukai S, Thomson JM, Mu D, Hammond SM, MacPherson D. miR-17~92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev. 2011;25(16):1734–1745. doi: 10.1101/gad.17027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46(7):617–634. doi: 10.1002/gcc.20457. [DOI] [PubMed] [Google Scholar]
- Corson TW, Samuels BC, Wenzel AA, Geary AJ, Riley AA, McCarthy BP, Hanenberg H, Bailey BJ, Rogers PI, Pollok KE, Rajashekhar G, Territo PR. Multimodality imaging methods for assessing retinoblastoma orthotopic xenograft growth and development. PLoS One. 2014;9(6):e99036. doi: 10.1371/journal.pone.0099036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Dimaras H, Heon E, Budning A, Doyle J, Halliday W, Drake J, Gallie BL, Chan HS. Trilateral retinoblastoma with pituitary-hypothalamic dysfunction. Ophthalmic Genet. 2008;29(3):120–125. doi: 10.1080/13816810802043678. [DOI] [PubMed] [Google Scholar]
- de Graaf P, Goricke S, Rodjan F, Galluzzi P, Maeder P, Castelijns JA, Brisse HJ European Retinoblastoma Imaging Collaboration. Guidelines for imaging retinoblastoma: imaging principles and MRI standardization. Pediatr Radiol. 2012;42(1):2–14. doi: 10.1007/s00247-011-2201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ioris MA, Valente P, Randisi F, Buzzonetti L, Carai A, Cozza R, Del Bufalo F, Romanzo A, Angioni A, Cacchione A, Bernardi B, Mastronuzzi A. Baseline central nervous system magnetic resonance imaging in early detection of trilateral retinoblastoma: pitfalls in the diagnosis of pineal gland lesions. Anticancer Res. 2014;34(12):7449–7454. [PubMed] [Google Scholar]
- de Jong MC, de Graaf P, Noij DP, Goricke S, Maeder P, Galluzzi P, Brisse HJ, Moll AC, Castelijns JA European Retinoblastoma Imaging Collaboration. Diagnostic performance of magnetic resonance imaging and computed tomography for advanced retinoblastoma: a systematic review and meta-analysis. Ophthalmology. 2014a;121(5):1109–1118. doi: 10.1016/j.ophtha.2013.11.021. [DOI] [PubMed] [Google Scholar]
- de Jong MC, Kors WA, de Graaf P, Castelijns JA, Kivela T, Moll AC. Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol. 2014b;15(10):1157–1167. doi: 10.1016/S1470-2045(14)70336-5. [DOI] [PubMed] [Google Scholar]
- de Jong MC, Kors WA, de Graaf P, Castelijns JA, Moll AC, Kivela T. The incidence of trilateral retinoblastoma: a systematic review and meta-analysis. Am J Ophthalmol. 2015;160(6):1116–1126. e1115. doi: 10.1016/j.ajo.2015.09.009. [DOI] [PubMed] [Google Scholar]
- de Jong MC, Moll AC, Goricke S, van der Valk P, Kors WA, Castelijns JA, de Graaf P. From a suspicious cystic pineal gland to pineoblastoma in a patient with familial unilateral retinoblastoma. Ophthalmic Genet. 2016;37(1):116–118. doi: 10.3109/13816810.2014.929717. [DOI] [PubMed] [Google Scholar]
- de Kock L, Sabbaghian N, Druker H, Weber E, Hamel N, Miller S, Choong CS, Gottardo NG, Kees UR, Rednam SP, van Hest LP, Jongmans MC, Jhangiani S, Lupski JR, Zacharin M, Bouron-Dal Soglio D, Huang A, Priest JR, Perry A, Mueller S, Albrecht S, Malkin D, Grundy RG, Foulkes WD. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol. 2014;128(4):583–595. doi: 10.1007/s00401-014-1318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14(5):297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaras H, Corson TW. The molecular genetics of retinoblastoma. Rev Cell Biol Mol Med. 2016;2:105–142. [Google Scholar]
- Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, Abramson DH, Shields CL, Chantada GL, Njuguna F, Gallie BL. Retinoblastoma. Nat Rev Dis Primers. 2015;1:15021. doi: 10.1038/nrdp.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaras H, Dimba EA, Gallie BL. Challenging the global retinoblastoma survival disparity through a collaborative research effort. Br J Ophthalmol. 2010a;94(11):1415–1416. doi: 10.1136/bjo.2009.174136. [DOI] [PubMed] [Google Scholar]
- Dimaras H, Heon E, Doyle J, Strahlendorf C, Paton KE, Halliday W, Babyn P, Gallie BL, Chan HS. Multifaceted chemotherapy for trilateral retinoblastoma. Arch Ophthalmol. 2011;129(3):362–365. doi: 10.1001/archophthalmol.2011.17. [DOI] [PubMed] [Google Scholar]
- Dimaras H, Khetan V, Halliday W, Heon E, Chan HS, Gallie BL. Retinoma underlying retinoblastoma revealed after tumor response to 1 cycle of chemotherapy. Arch Ophthalmol. 2009;127(8):1066–1068. doi: 10.1001/archophthalmol.2009.178. [DOI] [PubMed] [Google Scholar]
- Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, Marrano P, Corson TW, Eagle RC, Jr, Squire JA, Gallie BL. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17(10):1363–1372. doi: 10.1093/hmg/ddn024. [DOI] [PubMed] [Google Scholar]
- Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS, Gallie BL. Retinoblastoma. Lancet. 2012;379(9824):1436–1446. doi: 10.1016/S0140-6736(11)61137-9. [DOI] [PubMed] [Google Scholar]
- Dimaras H, Rushlow D, Halliday W, Doyle JJ, Babyn P, Abella EM, Williams J, Heon E, Gallie BL, Chan HS. Using RB1 mutations to assess minimal residual disease in metastatic retinoblastoma. Transl Res. 2010b;156(2):91–97. doi: 10.1016/j.trsl.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Do JL, Do B, Berry JL. Optical coherence tomography for diagnosis and management of retinoblastoma. Adv Ophthalmol Optom. 2017;2(1):101–118. [Google Scholar]
- Duffner PK, Cohen ME, Thomas PR, Lansky SB. The long-term effects of cranial irradiation on the central nervous system. Cancer. 1985;56(7 Suppl):1841–1846. doi: 10.1002/1097-0142(19851001)56:7+<1841::aid-cncr2820561325>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Dunkel IJ, Jubran RF, Gururangan S, Chantada GL, Finlay JL, Goldman S, Khakoo Y, O’Brien JM, Orjuela M, Rodriguez-Galindo C, Souweidane MM, Abramson DH. Trilateral retinoblastoma: potentially curable with intensive chemotherapy. Pediatr Blood Cancer. 2010;54(3):384–387. doi: 10.1002/pbc.22336. [DOI] [PubMed] [Google Scholar]
- Dvorak L, Russell SR. Retinal drawing: a lost art of medicine. Perm J. 2011;15(3):74–75. doi: 10.7812/tpp/11-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle RC., Jr High-risk features and tumor differentiation in retinoblastoma: a retrospective histopathologic study. Arch Pathol Lab Med. 2009;133(8):1203–1209. doi: 10.5858/133.8.1203. [DOI] [PubMed] [Google Scholar]
- Eagle RC., Jr The pathology of ocular cancer. Eye. 2013;27(2):128–136. doi: 10.1038/eye.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans L, Chen L, Milazzo G, Gherardi S, Perini G, Willmore E, Newell DR, Tweddle DA. SKP2 is a direct transcriptional target of MYCN and a potential therapeutic target in neuroblastoma. Cancer Lett. 2015;363(1):37–45. doi: 10.1016/j.canlet.2015.03.044. [DOI] [PubMed] [Google Scholar]
- Ewens KG, Bhatti TR, Moran KA, Richards-Yutz J, Shields CL, Eagle RC, Ganguly A. Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med. 2017;6(3):619–630. doi: 10.1002/cam4.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger PT. The 7th Edition AJCC Staging System for Eye Cancer an international language for ophthalmic oncology. Arch Pathol Lab Med. 2009;133(8):1197–1198. doi: 10.5858/133.8.1197. [DOI] [PubMed] [Google Scholar]
- Foster FS, Brown AS. Microultrasound and its application to longitudinal studies of mouse eye development and disease. Cold Spring Harb Protoc. 2012;2012(4):494–503. doi: 10.1101/pdb.prot068544. [DOI] [PubMed] [Google Scholar]
- Francis JH, Abramson DH, Gaillard MC, Marr BP, Beck-Popovic M, Munier FL. The classification of vitreous seeds in retinoblastoma and response to intravitreal melphalan. Ophthalmology. 2015;122(6):1173–1179. doi: 10.1016/j.ophtha.2015.01.017. [DOI] [PubMed] [Google Scholar]
- Francis JH, Marr BP, Abramson DH. Classification of vitreous seeds in retinoblastoma: Correlations with patient, tumor, and treatment characteristics. Ophthalmology. 2016;123(7):1601–1605. doi: 10.1016/j.ophtha.2016.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DN, Lis E, Sklar CA, Oeffinger KC, Reppucci M, Fleischut MH, Francis JH, Marr B, Abramson DH, Dunkel IJ. Whole-body magnetic resonance hereditary retinoblastoma: a pilot study. Pediatr Blood Cancer. 2014;61(8):1440–1444. doi: 10.1002/pbc.24835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323(6089):643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- Fujimoto J, Swanson E. The development, commercialization, and impact of optical coherence tomography. Invest Ophthalmol Vis Sci. 2016;57(9):OCT1–OCT13. doi: 10.1167/iovs.16-19963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung YK, Murphree AL, T’Ang A, Qian J, Hinrichs SH, Benedict WF. Structural evidence for the authenticity of the human retinoblastoma gene. Science. 1987;236(4809):1657–1661. doi: 10.1126/science.2885916. [DOI] [PubMed] [Google Scholar]
- Gallie BL, Budning A, DeBoer G, Thiessen JJ, Koren G, Verjee Z, Ling V, Chan HS. Chemotherapy with focal therapy can cure intraocular retinoblastoma without radiotherapy. Arch Ophthalmol. 1996;114(11):1321–1328. doi: 10.1001/archopht.1996.01100140521001. [DOI] [PubMed] [Google Scholar]
- Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma: spontaneous regression of retinoblastoma or benign manifestation of the mutation? Br J Cancer. 1982a;45(4):513–521. doi: 10.1038/bjc.1982.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallie BL, Mallipatna A, Finger P, Zhao J, Kivela T, Chantada G, Lau W, Ramirez-Ortiz MA, Catala J, Yousef Y, Ushakova T, Yarovoy A, Wilson M, Khetan V, Walsh J, Blair P, Renner L, Teekappanava N. International survey of staging for retinoblastoma provides evidence for the 2016 8th Edition AJCC TNMH retinoblastoma cancer staging. Pediatr Blood Cancer. 2016;63:S21–S21. [Google Scholar]
- Gallie BL, Phillips RA, Ellsworth RM, Abramson DH. Significance of retinoma and phthisis bulbi for retinoblastoma. Ophthalmology. 1982b;89(12):1393–1399. doi: 10.1016/s0161-6420(82)34622-9. [DOI] [PubMed] [Google Scholar]
- Galluzzi P, de Jong MC, Sirin S, Maeder P, Piu P, Cerase A, Monti L, Brisse HJ, Castelijns JA, de Graaf P, Goericke SL European Retinoblastoma Imaging Collaboration. MRI-based assessment of the pineal gland in a large population of children aged 0–5 years and comparison with pineoblastoma: part I, the solid gland. Neuroradiology. 2016;58(7):705–712. doi: 10.1007/s00234-016-1684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi P, Hadjistilianou T, Cerase A, De Francesco S, Toti P, Venturi C. Is CT still useful in the study protocol of retinoblastoma? AJNR Am J Neuroradiol. 2009;30(9):1760–1765. doi: 10.3174/ajnr.A1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SS, Jia Y, Zhang M, Su JP, Liu G, Hwang TS, Bailey ST, Huang D. Optical coherence tomography angiography. Invest Ophthalmol Vis Sci. 2016;57(9):OCT27–36. doi: 10.1167/iovs.15-19043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio E, Sherman J. Ophthalmic ultrasound as a diagnostic tool. J Am Optom Assoc. 1979;50(1):73–78. [PubMed] [Google Scholar]
- Gilbert F, Balaban G, Breg WR, Gallie B, Reid T, Nichols W. Homogeneously staining region in a retinoblastoma cell line: relevance to tumor initiation and progression. J Natl Cancer Inst. 1981;67(2):301–306. [PubMed] [Google Scholar]
- Gupta AK, Jones M, Prelog K, Bui J, Zhu J, Ng A, Dalla-Pozza L. Pineal cysts-A benign association with familial retinoblastoma. Pediatr Hematol Oncol. 2016;33(6):408–414. doi: 10.1080/08880018.2016.1225326. [DOI] [PubMed] [Google Scholar]
- Hamel P, Heon E, Gallie BL, Budning AS. Focal therapy in the management of retinoblastoma: when to start and when to stop. J AAPOS. 2000;4(6):334–337. doi: 10.1067/mpa.2000.107902. [DOI] [PubMed] [Google Scholar]
- Hernandez JC, Brady LW, Shields CL, Shields JA, DePotter P. Conservative treatment of retinoblastoma. The use of plaque brachytherapy. Am J Clin Oncol. 1993;16(5):397–401. doi: 10.1097/00000421-199310000-00005. [DOI] [PubMed] [Google Scholar]
- Hlavac M, Wirtz CR, Halatsch ME. Intraoperative magnetic resonance imaging. HNO. 2017;65(1):25–29. doi: 10.1007/s00106-016-0240-9. [DOI] [PubMed] [Google Scholar]
- Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013;3(10):a014415. doi: 10.1101/cshperspect.a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobiec FA, Tso MO, Zimmerman LE, Danis P. Retinoblastoma and intracranial malignancy. Cancer. 1977;39(5):2048–2058. doi: 10.1002/1097-0142(197705)39:5<2048::aid-cncr2820390522>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Ji X, Cheng L, Wei F, Li H, Wang M, Tian Y, Chen X, Wang Y, Wolf F, Li C, Huang Q. Noninvasive visualization of retinoblastoma growth and metastasis via bioluminescence imaging. Invest Ophthalmol Vis Sci. 2009;50(12):5544–5551. doi: 10.1167/iovs.08-3258. [DOI] [PubMed] [Google Scholar]
- Kamihara J, Bourdeaut F, Foulkes WD, Molenaar JJ, Mosse YP, Nakagawara A, Parareda A, Scollon SR, Schneider KW, Skalet AH, States LJ, Walsh MF, Diller LR, Brodeur GM. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin Cancer Res. 2017;23(13):e98–e106. doi: 10.1158/1078-0432.CCR-17-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatza EC, Shields CL, Flanders AE, Gonzalez ME, Shields JA. Pineal cyst simulating pinealoblastoma in 11 children with retinoblastoma. Arch Ophthalmol. 2006;124(4):595–597. doi: 10.1001/archopht.124.4.595. [DOI] [PubMed] [Google Scholar]
- Karcioglu ZA. Fine needle aspiration biopsy (FNAB) for retinoblastoma. Retina. 2002;22(6):707–710. doi: 10.1097/00006982-200212000-00004. [DOI] [PubMed] [Google Scholar]
- Kashyap S, Meel R, Pushker N, Sen S, Bakhshi S, Bajaj MS, Chawla B, Sethi S, Ghose S, Chandra M. Phthisis bulbi in retinoblastoma. Clin Exp Ophthalmol. 2011;39(2):105–110. doi: 10.1111/j.1442-9071.2010.02426.x. [DOI] [PubMed] [Google Scholar]
- Kendall CJ, Prager TC, Cheng H, Gombos D, Tang RA, Schiffman JS. Diagnostic ophthalmic ultrasound for radiologists. Neuroimaging Clin N Am. 2015;25(3):327–365. doi: 10.1016/j.nic.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Kenyan National Retinoblastoma Strategy Group. Retinoblastoma Best Practice Guidelines. 2014. [Google Scholar]
- Kim JW, Ngai LK, Sadda S, Murakami Y, Lee DK, Murphree AL. Retcam fluorescein angiography findings in eyes with advanced retinoblastoma. Br J Ophthalmol. 2014;98(12):1666–1671. doi: 10.1136/bjophthalmol-2014-305180. [DOI] [PubMed] [Google Scholar]
- Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17(6):1829–1837. doi: 10.1200/JCO.1999.17.6.1829. [DOI] [PubMed] [Google Scholar]
- Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooi IE, Mol BM, Massink MP, Ameziane N, Meijers-Heijboer H, Dommering CJ, van Mil SE, de Vries Y, van der Hout AH, Kaspers GJ, Moll AC, Te Riele H, Cloos J, Dorsman JC. Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep. 2016a;6:25264. doi: 10.1038/srep25264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooi IE, Mol BM, Massink MP, de Jong MC, de Graaf P, van der Valk P, Meijers-Heijboer H, Kaspers GJ, Moll AC, Te Riele H, Cloos J, Dorsman JC. A meta-analysis of retinoblastoma copy numbers refines the list of possible driver genes involved in tumor progression. PLoS One. 2016b;11(4):e0153323. doi: 10.1371/journal.pone.0153323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooi IE, Mol BM, Moll AC, van der Valk P, de Jong MC, de Graaf P, van Mil SE, Schouten-van Meeteren AY, Meijers-Heijboer H, Kaspers GL, Te Riele H, Cloos J, Dorsman JC. Loss of photoreceptorness and gain of genomic alterations in retinoblastoma reveal tumor progression. EBioMedicine. 2015;2(7):660–670. doi: 10.1016/j.ebiom.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertz I, Nittner D, Mestdagh P, Denecker G, Vandesompele J, Dyer MA, Marine JC. Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ. 2010;17(4):633–641. doi: 10.1038/cdd.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larina IV, Syed SH, Sudheendran N, Overbeek PA, Dickinson ME, Larin KV. Optical coherence tomography for live phenotypic analysis of embryonic ocular structures in mouse models. J Biomed Optics. 2012;17(8):081410–081411. doi: 10.1117/1.JBO.17.8.081410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent VE, Otero LL, Vazquez V, Camarero S, Gabri MR, Labraga M, De Davila MT, Chantada GL, Alonso DF. Optimization of molecular detection of GD2 synthase mRNA in retinoblastoma. Mol Med Rep. 2010;3(2):253–259. doi: 10.3892/mmr_00000248. [DOI] [PubMed] [Google Scholar]
- Laurent VE, Sampor C, Solernou V, Rossi J, Gabri M, Eandi-Eberle S, de Davila MT, Alonso DF, Chantada GL. Detection of minimally disseminated disease in the cerebrospinal fluid of children with high-risk retinoblastoma by reverse transcriptase-polymerase chain reaction for GD2 synthase mRNA. Eur J Cancer. 2013;49(13):2892–2899. doi: 10.1016/j.ejca.2013.04.021. [DOI] [PubMed] [Google Scholar]
- Laurie NA, Gray JK, Zhang J, Leggas M, Relling M, Egorin M, Stewart C, Dyer MA. Topotecan combination chemotherapy in two new rodent models of retinoblastoma. Clin Cancer Res. 2005;11(20):7569–7578. doi: 10.1158/1078-0432.CCR-05-0849. [DOI] [PubMed] [Google Scholar]
- Laws ER, Jr, Curran WJ, Jr, Bondy ML, Brat DJ, Brem H, Chang SM, Colen RR, Lopes MB, Louis DN, Prados MD, Schiff D, Vallerand TM, Wen PY, Werner-Wasik M. Brain and Spinal Cord. In: Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR, Sullivan DC, Jessup JM, Brierley JD, Gaspar LE, Schilsky RL, Balch CM, Winchester DP, Asare EA, Madera M, Gress DM, Meyer LR, editors. AJCC Cancer Staging Manual. 8. New York: Springer International Publishing; 2017. [Google Scholar]
- Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235(4794):1394–1399. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- Lemaitre S, Poyer F, Marco S, Freneaux P, Doz F, Aerts I, Desjardins L, Cassoux N, Thomas CD. Looking for the most suitable orthotopic retinoblastoma mouse model in order to characterize the tumoral development. Invest Ophthalmol Vis Sci. 2017;58(7):3055–3064. doi: 10.1167/iovs.17-21760. [DOI] [PubMed] [Google Scholar]
- Levy J, Frenkel S, Baras M, Neufeld M, Pe’er J. Calcification in retinoblastoma: histopathologic findings and statistical analysis of 302 cases. Br J Ophthalmol. 2011;95(8):1145–1150. doi: 10.1136/bjo.2010.193961. [DOI] [PubMed] [Google Scholar]
- Li MH, Bouffet E, Hawkins CE, Squire JA, Huang A. Molecular genetics of supratentorial primitive neuroectodermal tumors and pineoblastoma. Neurosurg Focus. 2005;19(5):E3. doi: 10.3171/foc.2005.19.5.4. [DOI] [PubMed] [Google Scholar]
- Lodhia V, Karanja S, Lees S, Bastawrous A. Acceptability, usability, and views on deployment of Peek, a mobile phone mhealth intervention for eye care in Kenya: Qualitative study. JMIR Mhealth Uhealth. 2016;4(2):e30. doi: 10.2196/mhealth.4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94(4):349–354. doi: 10.1007/BF00201591. [DOI] [PubMed] [Google Scholar]
- Lohmann DR, Gallie BL. Retinoblastoma: revisiting the model prototype of inherited cancer. Am J Med Genet. 2004;129C(1):23–28. doi: 10.1002/ajmg.c.30024. [DOI] [PubMed] [Google Scholar]
- MacCarthy A, Bayne AM, Brownbill PA, Bunch KJ, Diggens NL, Draper GJ, Hawkins MM, Jenkinson HC, Kingston JE, Stiller CA, Vincent TJ, Murphy MF. Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951–2004. Br J Cancer. 2013;108(12):2455–2463. doi: 10.1038/bjc.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallipatna AC, Gallie BL, Chevez-Barrios P, Lumbroso-Le Rouic L, Chantada GL, Brisse HJ, Doz F, Munier FL, Albert DM, Català-Mora J, Desjardins LG, Suzuki S, Carroll WL, Coupland SE, Finger PT. Retinoblastoma. In: Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR, Sullivan DC, Jessup JM, Brierley JD, Gaspar LE, Schilsky RL, Balch CM, Winchester DP, Asare EA, Madera M, Gress DM, Meyer LR, editors. AJCC Cancer Staging Manual. 8. New York: Springer International Publishing; 2017. [Google Scholar]
- Marcus DM, Carpenter JL, O’Brien JM, Kivela T, Brauner E, Tarkkanen A, Virtanen I, Albert DM. Primitive neuroectodermal tumor of the midbrain in a murine model of retinoblastoma. Invest Ophthalmol Vis Sci. 1991;32(2):293–301. [PubMed] [Google Scholar]
- Marcus DM, Lasudry JG, Carpenter JL, Windle J, Howes KA, al-Ubaidi MR, Baehr W, Overbeek PA, Font RL, Albert DM. Trilateral tumors in four different lines of transgenic mice expressing SV40 T-antigen. Invest Ophthalmol Vis Sci. 1996;37(2):392–396. [PubMed] [Google Scholar]
- Mashayekhi A, Shields CL, Eagle RC, Jr, Shields JA. Cavitary changes in retinoblastoma: relationship to chemoresistance. Ophthalmology. 2005;112(6):1145–1150. doi: 10.1016/j.ophtha.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Materin MA, Kuzmik GA, Jubinsky PT, Minja FJ, Asnes JD, Bulsara KR. Verification of supraselective drug delivery for retinoblastoma using intra-arterial gadolinium. BMJ Case Rep. 2012 doi: 10.1136/bcr-2012-010508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy J, Flores-Otero J, Zhang J, Nemeth K, Brennan R, Bradley C, Krafcik F, Rodriguez-Galindo C, Wilson M, Xiong S, Lozano G, Sage J, Fu L, Louhibi L, Trimarchi J, Pani A, Smeyne R, Johnson D, Dyer MA. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell. 2011;20(2):260–275. doi: 10.1016/j.ccr.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy J, Nagahawatte P, Finkelstein D, Richards-Yutz J, Valentine M, Ma J, Mullighan C, Song G, Chen X, Wilson M, Brennan R, Pounds S, Becksfort J, Huether R, Lu C, Fulton RS, Fulton LL, Hong X, Dooling DJ, Ochoa K, Mardis ER, Wilson RK, Easton J, Zhang J, Downing JR, Ganguly A, Dyer MA. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget. 2014;5(2):438–450. doi: 10.18632/oncotarget.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza PR, Specht CS, Hubbard GB, Wells JR, Lynn MJ, Zhang Q, Kong J, Grossniklaus HE. Histopathologic grading of anaplasia in retinoblastoma. Am J Ophthalmol. 2015;159(4):764–776. doi: 10.1016/j.ajo.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ministry of Health Kenya. Mother & Child Health Booklet. 2010. [Google Scholar]
- Moll AC, Hoekstra OS, Imhof SM, Comans EF, Schouten-van Meeteren AY, van der Valk P, Boers M. Fluorine-18 fluorodeoxyglucose positron emission tomography (PET) to detect vital retinoblastoma in the eye: preliminary experience. Ophthalmic Genet. 2004;25(1):31–35. doi: 10.1076/opge.25.1.31.29001. [DOI] [PubMed] [Google Scholar]
- Morjaria P, Bastawrous A, Murthy GVS, Evans J, Gilbert C. Effectiveness of a novel mobile health education intervention (Peek) on spectacle wear among children in India: study protocol for a randomized controlled trial. Trials. 2017;18(1):168. doi: 10.1186/s13063-017-1888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrochuk J. Introduction to diagnostic ophthalmic ultrasound for nurses in ophthalmology. J Ophthalmic Nurs Technol. 1990;9(6):234–239. [PubMed] [Google Scholar]
- Mulhern RK, Reddick WE, Palmer SL, Glass JO, Elkin TD, Kun LE, Taylor J, Langston J, Gajjar A. Neurocognitive deficits in medulloblastoma survivors and white matter loss. Ann Neurol. 1999;46(6):834–841. doi: 10.1002/1531-8249(199912)46:6<834::aid-ana5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Munier FL. Classification and management of seeds in retinoblastoma. Ellsworth Lecture Ghent August 24th 2013. Ophthalmic Genet. 2014;35(4):193–207. doi: 10.3109/13816810.2014.973045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munier FL, Gaillard MC, Balmer A, Soliman S, Podilsky G, Moulin AP, Beck-Popovic M. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: from prohibition to conditional indications. Br J Ophthalmol. 2012a;96(8):1078–1083. doi: 10.1136/bjophthalmol-2011-301450. [DOI] [PubMed] [Google Scholar]
- Munier FL, Soliman S, Moulin AP, Gaillard MC, Balmer A, Beck-Popovic M. Profiling safety of intravitreal injections for retinoblastoma using an anti-reflux procedure and sterilisation of the needle track. Br J Ophthalmol. 2012b;96(8):1084–1087. doi: 10.1136/bjophthalmol-2011-301016. [DOI] [PubMed] [Google Scholar]
- Murphree AL. Intraocular retinoblastoma: the case for a new group classification. Ophthalmol Clin North Am. 2005;18(1):41–53. viii. doi: 10.1016/j.ohc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- National Retinoblastoma Strategy. National Retinoblastoma Strategy Canadian Guidelines for Care: Strategie therapeutique du retinoblastome guide clinique canadien. Can J Ophthalmol. 2009;44(Suppl 2):S1–88. doi: 10.3129/i09-194. [DOI] [PubMed] [Google Scholar]
- Nemeth KM, Federico S, Carcaboso AM, Shen Y, Schaiquevich P, Zhang J, Egorin M, Stewart C, Dyer MA. Subconjunctival carboplatin and systemic topotecan treatment in preclinical models of retinoblastoma. Cancer. 2011;117(2):421–434. doi: 10.1002/cncr.25574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novetsky DE, Abramson DH, Kim JW, Dunkel IJ. Published international classification of retinoblastoma (ICRB) definitions contain inconsistencies--an analysis of impact. Ophthalmic Genet. 2009;30(1):40–44. doi: 10.1080/13816810802452168. [DOI] [PubMed] [Google Scholar]
- Novotny A, Krasny J. Diagnosis of retinoblastoma using ultrasound. Cas Lek Cesk. 1990;129(12):364–365. [PubMed] [Google Scholar]
- O’Brien JM, Marcus DM, Bernards R, Carpenter JL, Windle JJ, Mellon P, Albert DM. A transgenic mouse model for trilateral retinoblastoma. Arch Ophthalmol. 1990;108(8):1145–1151. doi: 10.1001/archopht.1990.01070100101043. [DOI] [PubMed] [Google Scholar]
- O’Hare M, Shadmand M, Sulaiman RS, Sishtla K, Sakisaka T, Corson TW. Kif14 overexpression accelerates murine retinoblastoma development. Int J Cancer. 2016;139(8):1752–1758. doi: 10.1002/ijc.30221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlin CJ, McWhae JA, McGowan HD, Foster FS. Ultrasound biomicroscopy of anterior segment tumors. Ophthalmology. 1992;99(8):1220–1228. doi: 10.1016/s0161-6420(92)31820-2. [DOI] [PubMed] [Google Scholar]
- Pavlin CJ, Sherar MD, Foster FS. Subsurface ultrasound microscopic imaging of the intact eye. Ophthalmology. 1990;97(2):244–250. doi: 10.1016/s0161-6420(90)32598-8. [DOI] [PubMed] [Google Scholar]
- Racher H, Soliman S, Argiropoulos B, Chan HS, Gallie BL, Perrier R, Matevski D, Rushlow D, Piovesan B, Shaikh F, MacDonald H, Corson TW. Molecular analysis distinguishes metastatic disease from second cancers in patients with retinoblastoma. Cancer Genet. 2016;209(7–8):359–363. doi: 10.1016/j.cancergen.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razek AA, Elkhamary S. MRI of retinoblastoma. Br J Radiol. 2011;84(1005):775–784. doi: 10.1259/bjr/32022497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese AB, Ellsworth RM. The evaluation and current concept of retinoblastoma therapy. Trans Am Acad Ophthalmol Otolaryngol. 1963;67:164–172. [PubMed] [Google Scholar]
- Reminick LR, Finger PT, Ritch R, Weiss S, Ishikawa H. Ultrasound biomicroscopy in the diagnosis and management of anterior segment tumors. J Am Optom Assoc. 1998;69(9):575–582. [PubMed] [Google Scholar]
- Rodjan F, de Graaf P, van der Valk P, Hadjistilianou T, Cerase A, Toti P, de Jong MC, Moll AC, Castelijns JA, Galluzzi P European Retinoblastoma Imaging Collaboration. Detection of calcifications in retinoblastoma using gradient-echo MR imaging sequences: comparative study between in vivo MR imaging and ex vivo high-resolution CT. AJNR Am J Neuroradiol. 2015;36(2):355–360. doi: 10.3174/ajnr.A4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanowska Dixon B, Morawski K. Usefulness of Ret-Cam imaging in diagnosis, treatment and monitoring of retinoblastoma. Acta Ophthalmol. 2017;95 doi: 10.1111/j.1755-3768.2017.1110S1079. [DOI] [Google Scholar]
- Rootman DB, Gonzalez E, Mallipatna A, Vandenhoven C, Hampton L, Dimaras H, Chan HS, Gallie BL, Heon E. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: clinical and morphologic considerations. Br J Ophthalmol. 2013;97(1):59–65. doi: 10.1136/bjophthalmol-2012-302133. [DOI] [PubMed] [Google Scholar]
- Ruggeri M, Tsechpenakis G, Jiao S, Jockovich ME, Cebulla C, Hernandez E, Murray TG, Puliafito CA. Retinal tumor imaging and volume quantification in mouse model using spectral-domain optical coherence tomography. Opt Express. 2009;17(5):4074–4083. doi: 10.1364/oe.17.004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri M, Wehbe H, Jiao S, Gregori G, Jockovich ME, Hackam A, Duan Y, Puliafito CA. In vivo three-dimensional high-resolution imaging of rodent retina with spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2007;48(4):1808–1814. doi: 10.1167/iovs.06-0815. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez MV, Henley AB, Arsenian-Henriksson M. The MYCN protein in health and disease. Genes (Basel) 2017;8(4) doi: 10.3390/genes8040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Theriault BL, Prigoda-Lee NL, Spencer C, Dimaras H, Corson TW, Pang R, Massey C, Godbout R, Jiang Z, Zacksenhaus E, Paton K, Moll AC, Houdayer C, Raizis A, Halliday W, Lam WL, Boutros PC, Lohmann D, Dorsman JC, Gallie BL. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14(4):327–334. doi: 10.1016/S1470-2045(13)70045-7. [DOI] [PubMed] [Google Scholar]
- Samara WA, Pointdujour-Lim R, Say EA, Shields CL. Foveal microanatomy documented by SD-OCT following treatment of advanced retinoblastoma. J AAPOS. 2015;19(4):368–372. doi: 10.1016/j.jaapos.2015.02.019. [DOI] [PubMed] [Google Scholar]
- Shields CL, Mashayekhi A, Au AK, Czyz C, Leahey A, Meadows AT, Shields JA. The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology. 2006;113(12):2276–2280. doi: 10.1016/j.ophtha.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Shields CL, Materin MA, Shields JA. Restoration of foveal anatomy and function following chemoreduction for bilateral advanced retinoblastoma with total retinal detachment. Arch Ophthalmol. 2005;123(11):1610–1612. doi: 10.1001/archopht.123.11.1610. [DOI] [PubMed] [Google Scholar]
- Shields CL, Meadows AT, Shields JA, Carvalho C, Smith AF. Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma) Arch Ophthalmol. 2001;119(9):1269–1272. doi: 10.1001/archopht.119.9.1269. [DOI] [PubMed] [Google Scholar]
- Singh U, Malik MA, Goswami S, Shukla S, Kaur J. Epigenetic regulation of human retinoblastoma. Tumour Biol. 2016;37(11):14427–14441. doi: 10.1007/s13277-016-5308-3. [DOI] [PubMed] [Google Scholar]
- Sirin S, de Jong MC, Galluzzi P, Maeder P, Brisse HJ, Castelijns JA, de Graaf P, Goericke SL European Retinoblastoma Imaging Collaboration. MRI-based assessment of the pineal gland in a large population of children aged 0–5 years and comparison with pineoblastoma: part II, the cystic gland. Neuroradiology. 2016;58(7):713–721. doi: 10.1007/s00234-016-1683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman SE, Dimaras H, Khetan V, Gardiner JA, Chan HS, Heon E, Gallie BL. Prenatal versus postnatal screening for familial retinoblastoma. Ophthalmology. 2016;123(12):2610–2617. doi: 10.1016/j.ophtha.2016.08.027. [DOI] [PubMed] [Google Scholar]
- Soliman SE, Racher H, Zhang C, MacDonald H, Gallie BL. Genetics and molecular diagnostics in retinoblastoma--an update. Asia Pac J Ophthalmol. 2017a;6(2):197–207. doi: 10.22608/APO.201711. [DOI] [PubMed] [Google Scholar]
- Soliman SE, VandenHoven C, MacKeen LD, Heon E, Gallie BL. Optical coherence tomography-guided decisions in retinoblastoma management. Ophthalmology. 2017b;124(6):859–872. doi: 10.1016/j.ophtha.2017.01.052. [DOI] [PubMed] [Google Scholar]
- Sulaiman RS, Quigley J, Qi X, O’Hare MN, Grant MB, Boulton ME, Corson TW. A simple optical coherence tomography quantification method for choroidal neovascularization. J Ocul Pharmacol Ther. 2015;31(8):447–454. doi: 10.1089/jop.2015.0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temming P, Arendt M, Viehmann A, Eisele L, Le Guin CH, Schundeln MM, Biewald E, Mausert J, Wieland R, Bornfeld N, Sauerwein W, Eggert A, Lohmann DR, Jockel KH. How eye-preserving therapy affects long-term overall survival in heritable retinoblastoma survivors. J Clin Oncol. 2016;34(26):3183–3188. doi: 10.1200/JCO.2015.65.4012. [DOI] [PubMed] [Google Scholar]
- Theriault BL, Dimaras H, Gallie BL, Corson TW. The genomic landscape of retinoblastoma: a review. Clin Experiment Ophthalmol. 2014;42(1):33–52. doi: 10.1111/ceo.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbidoni AV, Laurent VE, Sampor C, Ottaviani D, Vazquez V, Gabri MR, Rossi J, de Davila MT, Alonso C, Alonso DF, Chantada GL. Association of cone-rod homeobox transcription factor messenger RNA with pediatric metastatic retinoblastoma. JAMA Ophthalmol. 2015;133(7):805–812. doi: 10.1001/jamaophthalmol.2015.0900. [DOI] [PubMed] [Google Scholar]
- Tschulakow AV, Schraermeyer U, Rodemann HP, Julien-Schraermeyer S. Establishment of a novel retinoblastoma (Rb) nude mouse model by intravitreal injection of human Rb Y79 cells - comparison of in vivo analysis versus histological follow up. Biol Open. 2016;5(11):1625–1630. doi: 10.1242/bio.019976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso MO. Clues to the cells of origin in retinoblastoma. Int Ophthalmol Clin. 1980;20(2):191–210. [PubMed] [Google Scholar]
- Valdes PA, Roberts DW, Lu FK, Golby A. Optical technologies for intraoperative neurosurgical guidance. Neurosurg Focus. 2016;40(3):E8. doi: 10.3171/2015.12.FOCUS15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez LM, Giuliari GP, Halliday W, Pavlin CJ, Gallie BL, Heon E. Ultrasound biomicroscopy in the management of retinoblastoma. Eye. 2011;25(2):141–147. doi: 10.1038/eye.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez-Cruz R, Johnson DG. The retinoblastoma (RB) tumor suppressor: Pushing back against genome instability on multiple fronts. Int J Mol Sci. 2017;18(8):1776. doi: 10.3390/ijms18081776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong B, Skowron P, Kiehl TR, Kyan M, Garzia L, Sun C, Taylor MD, Yang VX. Measuring the optical characteristics of medulloblastoma with optical coherence tomography. Biomed Opt Express. 2015;6(4):1487–1501. doi: 10.1364/BOE.6.001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel AA, O’Hare MN, Shadmand M, Corson TW. Optical coherence tomography enables imaging of tumor initiation in the TAg-RB mouse model of retinoblastoma. Mol Vis. 2015;21:515–522. [PMC free article] [PubMed] [Google Scholar]
- Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994;7(4):480–484. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- Wilson RS. Retinal drawing chart. Ophthalmic Surg. 1975;6(1):17–21. [PubMed] [Google Scholar]
- Wippold FJ, 2nd, Perry A. Neuropathology for the neuroradiologist: rosettes and pseudorosettes. AJNR Am J Neuroradiol. 2006;27(3):488–492. [PMC free article] [PubMed] [Google Scholar]
- Wu N, Jia D, Bates B, Basom R, Eberhart CG, MacPherson D. A mouse model of MYCN-driven retinoblastoma reveals MYCN-independent tumor reemergence. J Clin Invest. 2017;127(3):888–898. doi: 10.1172/JCI88508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XL, Singh HP, Wang L, Qi DL, Poulos BK, Abramson DH, Jhanwar SC, Cobrinik D. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature. 2014;514(7522):385–388. doi: 10.1038/nature13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef YA, Shroff M, Halliday W, Gallie BL, Heon E. Detection of optic nerve disease in retinoblastoma by use of spectral domain optical coherence tomography. J AAPOS. 2012;16(5):481–483. doi: 10.1016/j.jaapos.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Yousef YA, Soliman SE, Astudillo PP, Durairaj P, Dimaras H, Chan HS, Heon E, Gallie BL, Shaikh F. Intra-arterial chemotherapy for retinoblastoma: a systematic review. JAMA Ophthalmol. 2016;134(5):584–591. doi: 10.1001/jamaophthalmol.2016.0244. [DOI] [PubMed] [Google Scholar]
- Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, Brennan R, Rusch M, Manning AL, Ma J, Easton J, Shurtleff S, Mullighan C, Pounds S, Mukatira S, Gupta P, Neale G, Zhao D, Lu C, Fulton RS, Fulton LL, Hong X, Dooling DJ, Ochoa K, Naeve C, Dyson NJ, Mardis ER, Bahrami A, Ellison D, Wilson RK, Downing JR, Dyer MA. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481(7381):329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]