

Abstract
Purpose
CHARGE syndrome is an autosomal dominant, multiple congenital anomaly condition characterized by vision and hearing loss, congenital heart disease, and malformations of craniofacial and other structures. Pathogenic variants in CHD7, encoding ATP-dependent Chromodomain Helicase DNA binding protein 7, are present in the majority of affected individuals. However, no causal variant can be found in 5-30% (depending on the cohort) of individuals with a clinical diagnosis of CHARGE syndrome.
Methods
We performed whole exome sequencing (WES) on 28 families from which at least one individual presented with features highly suggestive of CHARGE syndrome.
Results
Pathogenic variants in CHD7 were present in 15 of 28 individuals (53.6%), whereas four(14.3%) individuals had pathogenic variants in other genes (RERE, KMT2D, EP300, or PUF60). A variant of uncertain clinical significance in KDM6A was identified in one (3.5%) individual. The remaining eight (28.6%)individuals were not found to have pathogenic variants by WES.
Conclusion
These results demonstrate that the phenotypic features of CHARGE syndrome overlap with multiple other rare single-gene syndromes. Additionally, they implicate a shared molecular pathology that disrupts epigenetic regulation of multiple-organ development.
Keywords: CHARGE, Genetics, Exome, Oligogenicity, Chromatin
Introduction
Epigenomic regulation is essential for human development. De novo pathogenic variants in genes that encode chromatin effectors are important for developmental transcriptional plasticity and make important genetic contributions to developmental disorders. CHARGE syndrome (MIM: 214800) is an autosomal dominant multiple congenital anomaly condition. Individuals with CHARGE syndrome present with a combination of distinct findings such as coloboma of the eye, choanal atresia, cleft palate, vestibular abnormalities, cranial nerve and other brain anomalies, and characteristic dysmorphology particularly of the ear and face1.It occurs in about 1 in 10,000 births worldwide2. CHARGE syndrome can be caused by heterozygous pathogenic variants in CHD7, encoding Chromodomain Helicase DNA binding protein 7, an ATP-dependent chromatin remodeler3. However, a pathogenic variant in CHD7 cannot be found in up to 30% of individuals with clinical features of CHARGE syndrome, suggesting other genes or pathologies may be involved in a subset of cases.
CHARGE syndrome is one of several multi-organ developmental disorders attributed to pathogenic variants in genes encoding chromatin remodelers of the CHD family. Despite similarities in protein domain structure between CHD family members, individual CHD genes are associated with clinically distinct syndromes. Pathogenic variants in CHD4 are implicated in Sifram-Hitz-Weiss syndrome (MIM: 617159), and CHD8 is an important genetic risk factor for a subtype of autism spectrum disorders (MIM: 615032). Pathogenic variants in CHD genes are characterized by variable expressivity and reduced penetrance. The same pathogenic CHD7 variant within a single family can present with disparate features, ranging from multiple organ system involvement to mild developmental delay or isolated clefting and hearing loss4.While there is minimal clinical overlap between individual CHD related disorders, a number of syndromes that mimic CHARGE have been described, including 22q11 Deletion syndrome (MIM: 188400), Kabuki syndrome (MIM: 147920, 300867), Renal-coloboma syndrome (MIM: 610536), and Mandibulofacial dysostosis (MIM: 610536)5-8. This clinical variability highlights the importance of genetic testing in diagnosis of developmental syndromes.
Within this clinical diversity of developmental disorders, a major unanswered question is whether pathogenic variants in genes other than CHD7 might explain the genetic etiology in individuals not previously diagnosed with CHD7 pathogenic variants. We performed WES for a cohort of 28 individuals with clinical features of CHARGE who had no previous history of genetic testing or tested negative for pathogenic variants, deletions or duplications in CHD7.WES revealed pathogenic variants in genes previously shown to cause other developmental disorders, including Rubinstein-Taybi syndrome (EP300), Kabuki syndrome (KMT2D),Verheji syndrome (PUF60, MIM: 615583), and RERE-associated developmental disorder (RERE, MIM: 616975). These results illustrate a higher degree of oligogenicity than previously recognized for individuals with clinical CHARGE features, suggesting common underlying molecular biological mechanisms that contribute to development of affected organs and tissues.
Materials and Methods
Subject recruitment and sample collection
The study protocol was approved by the Institutional Review Board for the Protection of Human Subjects at The University of Michigan Medical School. Individuals with clinical features of CHARGE and their family members were recruited using standard practices at the 12th International CHARGE Syndrome Foundation conference (Chicago, Illinois, 2015), the Pediatrics Genetics clinic at The University of Michigan C.S. Mott Children's Hospital, and external healthcare facilities. We included individuals with features of CHARGE syndrome for which a genetic etiology had not been identified and excluded individuals with features of CHARGE syndrome with a previously identified CHD7 pathogenic variant. Study participation involved informed written consent, a clinical history questionnaire, and either a saliva or peripheral blood sample. DNA was extracted per the manufacturer's instructions from saliva samples using DNA Genotek Prep-IT L2P (catalog no. PT-L2P) or blood samples using Qiagen DNeasy Blood & Tissue Kit (catalog no. 69504). DNA quantification was employed using Life Technologies Quant-iT PicoGreen dsDNA Assay Kit (catalog no. P7589) with a microplate reader to obtain 3 μg of DNA for WES.
Exome sequencing
DNA samples were submitted to the University of Washington Center for Mendelian Genomics (UW-CMG) where library construction, WES and analysis were performed. Briefly, sequencing libraries were generated for each DNA sample in an automated, 96-well plate format (Perkin-Elmer Janus II). Sample libraries were constructed from 1 μg of genomic DNA which underwent a series of shotgun library construction steps including: acoustic sonication (Covaris), end-repair, A-tailing ligation of unique sequencing adaptors, and PCR amplification. Sample libraries were hybridized to the Nimblegen SeqCap EZ v2.0 target (∼36.5 Mb) in multiplex for a period of 72 hrs. Captured DNA was then purified, PCR amplified, and normalized for sequencing.
Captured DNA was sequenced on Illumina HiSeq machines using paired-end sequencing. Raw sequence data in FASTQ format were aligned to the human genome reference hg19 using the BWA algorithm for the generation of BAM files9. The quality of each sample was assessed for coverage (80% of sequenced target with ≥20× coverage and 90% of target with ≥8× coverage) and Ts/Tv ratios. Additionally, samples were quality controlled to confirm sex using PLINK (v1.90b2m) software, and estimations of kinship were corroborated with pedigrees using KINGv1.4.0 software.
Variant detection, interpretation and validation
Variants and indels were detected and genotyped using HaplotypeCaller from GATK with hard filtering parameters. A multi-sample VCF was generated for all samples following GATK best practices10.The multi-sample VCF was annotated using Variant Effect Predictor (VEP) tool v83 within GEMINI (http://uswest.ensembl.org/info/docs/tools/vep/index.html). Copy number variants were detected from exome sequence data using the program CoNIFER11.
GEMINI v0.19.1 was used to filter variants and indels detected in affected individuals12. Variants were excluded if allele frequency in reference populations (ExAC, NHLBI Exome Sequencing Project (ESP) 6500, or 1000 Genomes) exceeded 0.005 or exceeded 0.05 in the UW-CMG internal database. Variant impact determined by VEP was also utilized to exclude variants and indels with an impact severity of “low” (ex. intergenic, intronic, synonymous, 5′UTR, and 3′ UTR variants). Variant prioritization was based on the following parameters: variant impact, presence in a disease-associated gene, conservation of impacted nucleotide by GERP, or predicted deleteriousness (CADD > 15)13; 14. Interpretation of detected CNVs was based on size, frequency in the UW-CMG internal database, and overlap with known disease-associated regions. In addition, CNVs were filtered based on phenotypic overlap according to the ACMG guidelines for postnatal CNV calling15.
Sanger sequencing was used to validate rare, protein-altering variants. Variant segregation was also used to confirm inheritance patterns when family member samples were available.
Results
Cohort
We assembled a cohort of 28 affected individuals and their parents (Table 1). Probands ranged in age from 18 months to 40 years old, with 14 probands being <18 years of age. The majority of probands (26/28) presented with two or more major clinical CHARGE criteria, achieving a diagnosis of CHARGE as defined by Hale et al.16. Probands also exhibited minor clinical features according to the diagnostic criteria (Table 1 and Supplemental Table 1). Nineteen of the families in the cohort were trios, one a duo and the remaining eight were proband only. A previous history of negative CHD7 genetic testing was reported for 12 probands with the remaining 16 probands reporting no prior history of clinical genetic testing at the level of CHD7 or WES.
Table 1. Cohort Demographics and Clinical Features.
| Cohort | |
| Total individuals | 67 |
| Number of probands | 28 |
| Trios (proband and parents) | 19 |
| Duo (proband and mother) | 1 |
| Singletons | 8 |
| Proband Demographics | |
| Age | 18m – 40yrs |
| Males | 13 (46.4%) |
| Females | 15 (53.6%) |
| Proband History of CHD7 Genetic Testing | |
| Negative | 12 (42.9%) |
| Not previously tested | 16 (57.1%) |
| Proband Major Clinical Features | |
| Coloboma | 26/28 (92.9%) |
| Choanal Atresia or Cleft Palate | 19/28 (67.9%) |
| Abnormal External, Middle, or Inner Ears | 19/28 (67.9%) |
| Proband Minor Clinical Features | |
| Cranial Nerve Dysfunction (including hearing loss) | 20/28 (71.4%) |
| Dysphagia/feeding difficulties | 8/28 (28.6%) |
| Structural brain abnormalities | 3/28 (10.7%) |
| Developmental delay, intellectual disability or autism spectrum disorder | 22/28 (78.6%) |
| Hypothalamo-hypophyseal dysfunction and genital anomalies | 13/28 (46.4%) |
| Heart or esophageal malformations | 20/28 (71.4%) |
| Renal anomalies or skeletal/limb anomalies | 12/28 (42.9%) |
CHD7
WES and analysis were conducted at the UW-CMG. Fifteen probands, one of whom had a previous history of negative clinical CHD7 genetic testing, were found to have pathogenic variants in CHD7 [GenBank: NM_017780] by WES(Table 2). Of these 15 CHD7 variants, 11 were designated as de novo because the variant was absent in parental samples and 1 variant was inherited from a clinically affected mother. For three probands, parental DNA was unavailable for segregation analysis. The majority of CHD7 variants (13) were classified as either a stop-gained or frameshift, consistent with the previously described allelic spectrum of CHD7 variants that cause CHARGE syndrome17. One CHD7 splicing variant and one missense variant were also detected. Seven of the fifteen CHD7 variants were described in 1000 Genomes with an association with CHARGE syndrome by The Human Gene Mutation Database or ClinVar. Otherwise, all CHD7 variants were absent in ExAC, 1000 Genomes, and NHLBI ESP Exome Variant Server. CNV analysis of WES data did not reveal pathogenic changes (Supp. Table 3).
Table 2. Probands, CHARGE major diagnostic features, gene variants, and pathogenicity evidence.
| Proband | CHARGE Major Criteria | Gene | Genomic Position |
cDNA variant | Protein alteration | CADD Score |
GERP Score |
Inheritance | Status | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coloboma | Choanal atresia or cleft palate |
Abnormal external, middle or inner ears |
Pathogenic CHD7 variant |
|||||||||
| 502 | + | + | + | + | CHD7 | chr8: 61,761,664 | c.5355G>A | p.Trp1785* | 48 | 5.87 | de novo | known |
| 503 | + | + | - | + | CHD7 | chr8: 61,655,332 | c.1342_1343delAG | p.Arg448Glufs*126 | N/A | 5.539 | de novo | novel |
| 504 | + | + | + | + | CHD7 | chr8: 61,754,595 | c.4835delA | p.Asn1612Ilefs*28 | N/A | 3.85 | unknown | novel |
| 505 | + | + | + | + | CHD7 | chr8: 61,773,655 | c.7802dupA | p.Tyr2601* | N/A | 5.73 | de novo | novela |
| 506 | + | - | + | + | CHD7 | chr8: 61,736,406 | c.3209delT | p.Val1070Glyfs*2 | N/A | 5.69 | de novo | novela |
| 507 | + | + | - | + | CHD7 | chr8: 61,742,962 | c.3606_3616dupAGAAACTATTA | p.Ile1206Lysfs*9 | N/A | 5.47 | de novo | novela |
| 508 | + | + | + | + | CHD7 | chr8: 61,757,970 | c.5210+2T>C | 22.6 | 5.48 | de novo | novelb | |
| 509 | + | - | - | + | CHD7 | chr8: 61,714,150 | c.2440C>T | p.Gln814* | 44 | 6.17 | inherited | known |
| 510 | + | + | + | + | CHD7 | chr8: 61,750,761 | c.4480C>T | p.Arg1494* | 48 | 4.42 | unknown | known |
| 511 | + | - | - | + | CHD7 | chr8: 61,763,056 | c.5409T>G | p.Tyr1803* | 49 | -1.18 | de novo | novel |
| 512 | + | - | + | + | CHD7 | chr8: 61,654,916 | c.925C>T | p.Gln309* | 40 | 5.56 | unknown | known |
| 513 | + | + | + | + | CHD7 | chr8: 61,765,241 | c.6079C>T | p.Arg2027* | 49 | 3.64 | de novo | known |
| 514 | + | - | + | + | CHD7 | chr8: 61,734,662 | c.2915A>G | p.Gln972Arg | 24.7 | 5.53 | de novo | novel |
| 515 | + | + | + | + | CHD7 | chr8: 61,736,402 | c.3205C>T | p.Arg1069* | 45 | 5.69 | de novo | known |
| 516 | + | + | + | + | CHD7 | chr8: 61,735,210 | c.3106C>T | p.Arg1036* | 45 | 5.53 | de novo | known |
| 517 | + | + | + | - | RERE | chr1: 8,418,276 | c.4313_4318dupTCCACC | p.Leu1438_His1439dup | N/A | 5.61 | de novo | known |
| 518 | + | - | + | - | KMT2D | chr12: 49,431,536 | c.9602dupT | p.Ser3202Glufs*13 | N/A | -0.809 | unknown | novel |
| 519 | + | + | + | - | KDM6A | chrX: 44,820,553 | c.250A>G | p.Ile84Val | 17.16 | 5.66 | X-linked recessive | novel |
| 520 | - | + | + | - | EP300 | chr22: 41,553,171 | c.3262-2A>G | 24.4 | 5.7 | de novo | novel | |
| 521 | + | - | - | - | PUF60 | chr8:144,900,664 | c.389G>A | p.Arg130His | 25.9 | 5.18 | de novo | novel |
Known or novel status of CHD7 variants were defined according to presence in the CHD7 database at molgenis51.gcc.rug.nl on June 29, 2017. Known CHD7 variants were also observed in 1000 Genomes with an association to CHARGE syndrome by the Human Gene Mutation Database or ClinVar. Abbreviations are as follows: known, identified in database and defined as pathogenic;
novel, known frameshifts observed at this protein position and defined as pathogenic;
novel, a splice site mutation is observed one nucleotide away at c.5210+3A>G and defined as pathogenic; N/A, not applicable.
RERE
Proband 517, a female at the age of 8 years old at the time of study recruitment, presented with a medical history consistent with CHARGE syndrome. Notably, she exhibited bilateral choanal atresia, right inferior iris coloboma, abnormal external ears, progressive sensorineural hearing loss and bilateral cochlear dysplasia identified by temporal bone CT. Additionally, proband 517 was also noted to exhibit growth retardation, short stature, microcephaly, developmental delay, and truncus arteriosus repaired surgically at one-month of age. Consistent with the results of her previous CMA and CHD7 genetic testing, proband 517 was not found to harbour a pathogenic variant in CHD7 or CNV by WES (Supplemental Table 2). Rather, a de novo in-frame duplication (c.4313_4318dupTCCACC [p.Leu1438_His1439dup] [GenBank: NM_001042681]; hg19 chr1: 8,418,276) in the RE (Arginine-glutamic acid) dipeptide repeats gene RERE was identified. This in-frame duplication occurs in a histidine rich region of RERE (UniProt: QP2R6), and is absent in publically available databases ExAC, 1000 Genomes, and NHLBI ESP Exome Variant Server. The RERE variant detected for proband 517 is the same duplication identified for male subject S2 in Fregeau et al. who exhibits similar features to proband 517 including choanal atresia, coloboma, growth retardation, short stature, developmental delay, and microcephaly. Such phenotypic overlap suggests that this variant contributes to the clinical findings of proband 517 18.
KMT2D and KDM6A
Variants in KMT2D and KDM6A, genes that underlie Kabuki syndrome, were identified in our cohort. Proband 518, a 16-year-old female, had clinical features of CHARGE syndrome (Table 2). Specifically, her medical history was notable for external ear abnormalities, mild hearing loss in the right ear, moderate to severe hearing loss in the left ear, long palpebral fissures, everted lashes, and persistent fingertip pads. Multiple ophthalmologic abnormalities were also noted, including bilateral chorioretinal colobomas, strabismus, and nystagmus. In addition, proband 518 exhibits cardiac and renal abnormalities, smal stature, precocious puberty, developmental delay, and skeletal related problems including kyphosis and scoliosis. However, previous genetic testing did not identify a pathogenic variant in CHD7. WES reconfirmed previous negative CHD7 genetic testing results and identified a single nucleotide duplication in KMT2D (c.9602dupT [p.Ser3202Glufs*13] [GenBank:NM_003482]; hg19 chr12: 49,431,536) (Supplemental Table 2). This duplication is predicted to result in a frameshift, generation of a premature stop codon in the central portion of KMT2D (UniProt: O14686), and nonsense mediated decay. The c.9602dupT variant in KMT2D is absent from the ExAC, NHLBI ESP Exome Variant Server and 1000 Genomes databases, and resemble previously described KMT2D variants ascribed to Kabuki syndrome19.
Proband 519, a male just under 18 months of age at the time of recruitment, had clinical findings consistent with CHARGE syndrome including bilateral choanal atresia, bilateral iris coloboma and asymmetric microphthalmia, low external ear placement on the left side with normal hearing, but no CHD7 pathogenic variant. A family history of unilateral choanal atresia in a maternal great-grandfather was also reported. WES confirmed absence of a pathogenic CHD7 variant (Supplemental Table 2). A hemizygous missense variant in KDM6A (c.250A>G [p.Ile84Val] [GenBank: NM_021140]; hg19 chrX: 44,820,553) was identified just N-terminal to the first tetratricopeptide motif (UniProt: O15550). A GERP conservation score of 5.660 and a CADD score of 17.16, along with the variant's absence from publically available databases ExAC, 1000 Genomes and NHLBI ESP Exome Variant Server, suggest deleteriousness. The inheritance of the KDM6A variant from a self-reported unaffected mother is unexpected as carrier mothers are reported to exhibit mild features of Kabuki syndrome, but this may reflect favorable skewing of X-inactivation19. Consequently, we classify the p.Ile84Val variant observed in proband 519 as a variant of uncertain clinical significance. Further experimental studies including X-inactivation profiling, and a clinical evaluation of the mother of proband 519 would be required to change the variant classification.
EP300
Proband 520 was an 11-year-old male with a clinical diagnosis of CHARGE syndrome with a normal chromosomal microarray and CHD7 sequencing. Significant findings included right-sided choanal atresia, sensorineural hearing loss, external ear abnormalities, dysphagia, developmental delay, growth deficiency, a ventricular septal defect and patent ductus arteriosus with spontaneous closure. He was born prematurely with polyhydramnios. Facial features included level palpebral fissures, a short nose with anteverted nares, an underbite with a symmetric facial grimace, and left-sided esotropia. He was found to have a de novo splice acceptor variant in EP300 3′ to exon 18 (c.3262-2A>G [Genbank: NM_001429]; hg19 chr22: 41,553,171). Pathogenic variants in EP300 are a minor cause of Rubinstein-Taybi syndrome (RTS) (OMIM: 180849)20. The c.3262-2A nucleotide encompasses the AG splice acceptor site, which is well-conserved as described by a GERP score of 5.700. The transition to G is predicted to impact splicing (UniProt: Q09472). The c.3262-2A>G EP300 splice acceptor variant was not observed in ExAC, 1000 Genomes, and NHLBI ESP Exome Variant Server databases. Moreover, the pathogenic role of the c.3262-2A>G variant is supported by the similarity between proband 520 and descriptions of RTS, and that RTS EP300 variants are located throughout the gene.
PUF60
Proband 521 was a male recruited into our CHARGE cohort at 22 years of age with a clinical history of global developmental delay, irido-retinal colobomas, mild hearing loss, small stature, genital abnormalities, and a ventricular septal defect corrected surgically. Surgical history was also notable for multiple back surgeries. Consistent with his previous history of normal CHD7 genetic testing, WES did not identify a pathogenic variant in CHD7 or a CNV (Supplemental Table 2). Instead, a de novo missense variant in the RNA recognition motif of poly-U-binding splicing factor 60 (PUF60) (c.389G>A [p.Arg130His] [GenBank: NM_078480]; hg19 chr8: 144,900,664) was identified. PUF60 encodes a protein involved in pre-mRNA splicing21. Heterozygous loss of PUF60 is predicted to contribute to the clinical features of recurrent microdeletions of 8q24.3, also called Verheij syndrome (OMIM: 615583)22, characterized by a constellation of features including microcephaly, craniofacial dysmorphisms, ocular colobomata, developmental delay, short stature, and skeletal, cardiac and renal abnormalities. Conservation of the PUF60 c.389G>A variant based on GERP score, pathogenicity predicted by CADD, and absence of the missense variant in the online databases ExAC, 1000 Genomes, and NHLBI ESP Exome Variant Server all provide evidence to support the pathogenicity of this variant (Table 2).
FLNA
Proband 522 was a 10-year-old male with clinical features suggestive of CHARGE syndrome including bilateral retinal colobomas, bilateral cleft lip and palate, bilateral hypoplastic vestibular apparatus and semicircular canals, and abnormal external ears. He also has a history of hearing loss, resolved patent ductus arteriosus, developmental delay, dysphagia, left hydronephrosis, hiatal hernia, genital anomalies, and normal brain imaging. Consistent with his previous genetic workup, proband 522 was not found to have variants impacting CHD7 nor CNVs by WES. Instead, he was found to have a missense variant (c.2309A>G [p.Asn770Ser] [GenBank: NM_001456]; hg19 chrX: 153,591,124) in the rod-domain repeat 6 (UniProt: P21333-2) of filamin A (FLNA) that was inherited from his unaffected mother. Variants in FLNA often cluster in the calponin homology domain 2 and the rod-domain repeat 10, and are associated with several X-linked disorders together termed otopalatodigital spectrum disorders including Otopalatodigital syndrome types I and II (OPD, MIM: 311300 and 304120), Melnick-Needles syndrome (MIM: 309350), and Frontometaphyseal dysplasia type I (MIM: 305620)23.Some clinical features of proband 522 overlap with features of FLNA-associated conditions, specifically OPD1, including hearing loss and bilateral cleft lip/palate. However, he had only mild syndactyly of the left second and third toes, which is less severe than the skeletal or limb abnormalities typically observed in FLNA-related disorders. Important in our analysis was additional genetic testing of an unaffected male sibling, who was found to be positive for the c.2309A>G FLNA variant. This information provided support for a variant classification of likely benign according to ACMG guidelines.
Discussion
We report results of WES of a cohort of 28 individuals with clinical features of CHARGE syndrome. Pathogenic variants in CHD7 were discovered in 15/28 (53.6%)individuals and 4/28 (14.3%) individuals had pathogenic variants in four genes other than CHD7, including RERE, KMT2D, EP300, and PUF60.Our results suggest that careful phenotype-genotype analysis in the setting of comprehensive sequencing studies can uncover new relationships between genes and phenotypes, and sheds light on the underlying molecular pathology contributing to CHARGE syndrome and related disorders.
Our findings broaden the spectrum of clinical features associated with pathogenic variants in genes such as EP300. RTS is characterized by intellectual disability, growth delay, and distinctive craniofacial and digital dysmorphisms24. CHARGE can typically be differentiated from RTS by the presence of semicircular canal dysgenesis, choanal atresia, facial asymmetry,gondal hypoplasia, and syndrome-specific dysmorphology. While hearing loss has been reported in individuals with EP300-associated RTS, the choanal atresia remains unique to proband 52025. It is possible that choanal atresia is a low penetrance phenotype of EP300-associated RTS, but remains seldom detected due to the limited number of individuals with EP300 variants to date. This illustrates how the unbiased use of WES can broaden the phenotypic spectrum associated with known disease genes to uncover novel phenotypic overlap between developmental disorders. Alternatively, dual molecular diagnoses may account for his unique RTS presentation. Although WES did not uncover any additional variants suggestive of this alternative, the possibility remains that proband 520 possesses an undiscovered genetic or non-genetic etiology.
The substantial clinical overlap between RERE-associated developmental disorder and CHARGE syndrome raises the possibility of classifying RERE as a genetic cause of CHARGE syndrome. Heterozygous pathogenic variants in RERE have been reported in 10 individuals with complex and variable phenotypes that overlap with those observed in CHARGE syndrome, including structural brain malformations, intellectual disability, seizures, coloboma, sensorineural hearing loss, choanal atresia, cardiac malformations, and gastroesophageal reflux disease 18. Clinical evaluation of individuals with RERE pathogenic variants shows that two of the ten individuals meet clinical criteria for CHARGE syndrome 17; 18. Inner ear dysplasia and hypogonadotropic hypogonadism are not reported in RERE-associated developmental disorder and the craniofacial features differ from those commonly observed in CHARGE syndrome (square shaped facies, auricular anomalies, facial asymmetry and seventh nerve palsies). These differences suggest that pathogenic variants in RERE and CHD7 are the genetic etiologies of distinct disorders.
Our results show that complex phenotypes presenting as CHARGE syndrome may be caused by pathogenic variants in CHD7 or other genes associated with other syndromes. The high degree of phenotypic overlap among these disorders raises the question of whether CHARGE syndrome should be considered as a single gene disorder associated only with pathogenic variants in CHD7 or an oligogenic disorder26. Isolated features of CHARGE, such as congenital heart disease and congenital hypogonadotropic hypogonadism, can occur due to pathogenic variants in multiple individual genes, and are therefore considered to be oligogenic27. Careful attention to both clinical diagnosis and molecular results is sure to continue shedding light on this important question.
Phenotypic overlap between CHARGE and Kabuki syndromes is well described, and may reflect direct interactions between CHD7, KMT2D and KDM6A or shared effects on DNA methylation28; 29. Furthermore, overlapping features with Rubinstein-Taybi, Verheij and RERE-associated syndromes may also indicate shared molecular pathology between proteins encoded by CHD7, KMT2D, KDM6A,EP300, PUF60 and RERE, the genetic findings in this study21; 30-34. Such functional synergy is supported by experimental observations from primary cells derived from individuals with CHARGE syndrome and animal models. RERE is a co-regulator of retinoic acid (RA) signaling and forms a complex with NR2F2 (nuclear receptor subfamily 2, group F, member 2), P300 and retinoic acid receptors (RARs/RXRs), which is recruited to the retinoic acid response element (RARE) of retinoic acid transcriptional targets, such as FGF8 (Figure 1) 32.Activation of RAREs by RA promotes an exchange of repressive histone modification (H3K27Me3)in favor of activating ones (H3K27Ac) and translocation of nucleosomes to expose DNA to initiate transcription, functions attributed to KDM6A, P300, CHD7 and PUF60 respectively. A shared molecular biology is also supported by evidence of direct interaction between CHD7 and P300-incorporating complexes 34.
Figure 1.
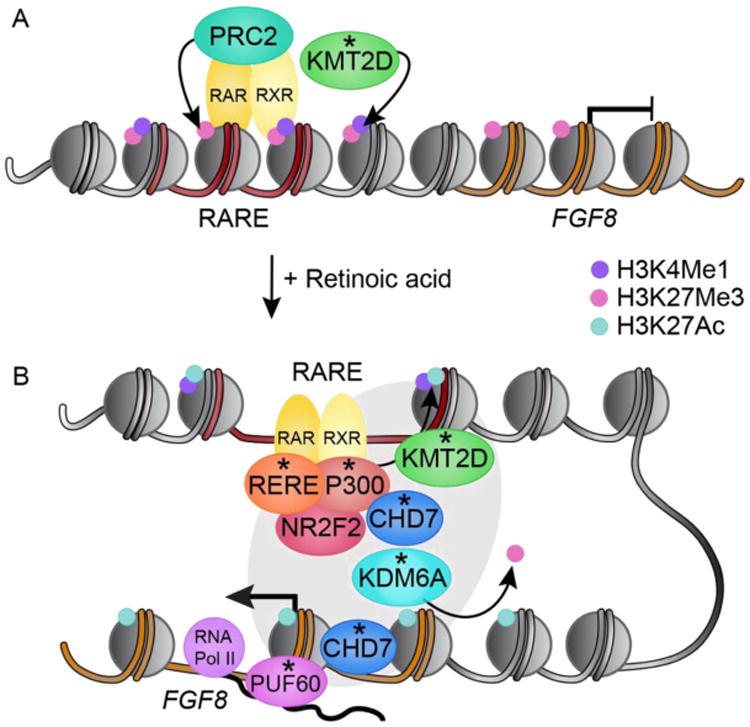
Model of molecular synergy implicated by genetic determinants (asterisks) that share clinical features of CHARGE syndrome. Cis-regulatory retinoic acid response elements (red DNA; RAREs) control cell-specific transcription of retinoic acid (RA) responsive genes, like FGF8 (orange DNA). (A) In the absence of RA, Polycomb repressive complex 2 (PRC2) and KMT2D are recruited to RAREs, where they catalyze H3K27Me3 and H3K4Me1 respectively, promoting FGF8 transcriptional repression. (B) In response to RA, the RERE/NR2F2/P300 complex forms, binding nuclear heterodimers of retinoic acid receptors (RAR/RXR) at RAREs. Histone acetyltransferase P300 and KMT2D cooperate to activate enhancer and enhancer-promoter looping that requires nucleosome translocation along the DNA by CHD7. KDM6A demethylates the repressive H3K27Me3 histone modifications. Enhancer-promoter looping is stabilized by the mediator complex (grey oval). CHD7 translocates nucleosomes to permit transcription of FGF8 by RNA pol II. PUF60 helps form the transcription bubble required for RNA Pol II transcription and pre-mRNA splicing.
This functional synergy may be pertinent to understand the epistatic relationship between CHD7 and FGF8 discovered in the Chd7 knockout mouse35. While mice lacking one working copy of Chd7 or Fgf8 exhibit normal cerebellar vermis development, those lacking one functional copy each of Chd7 and Fgf8 exhibit cerebellar vermis hypoplasia. Importantly, CHD7 and FGF8 do not directly interact and the molecular underpinning of this epistatic effect have not been described. Fgf8 is a retinoic acid (RA) responsive gene. In the absence of RA, RAR–RXR heterodimers bind to RARE sequences, accompanied by histone deacetylases, H3K27 methytransferase polycomb repressive complex 2 (PRC2) and histone H3K4 mono- and di- methyltransferase KMT2D to repress transcription of RA responsive genes31. In response to RA the RERE/NR2F2/P300 complex binds retinoic acid receptors at RAREs32. Activation of the RAREs promotes nucleosome translocation, an important function of CHD7 that allows for KDM6A occupancy concomitant with H3K27 demethylation and activating H3K27 acetylation by P300, setting the stage for coordinated transcriptional regulation of Fgf830; 36.These findings also provide insights into the ability of RA to rescue structural inner ear defects in the conditional Chd7 knockout mouse models 37. Future studies will be required to determine if chromatin based co-regulators of RA responsive transcription may cooperate to mediate the Chd7-Fgf8 epistatic effect, and underlie a shared molecular pathology between these developmental disorders.
Nuclear receptor NR2F2 is a ligand-dependent transcription factor that regulates expression of genes critical for a variety of biological processes, including development, growth, and differentiation 33. The observation that interaction between RERE, P300 and NR2F2 is required for body patterning and somite formation early in embryogenesis implicates NR2F2 in the etiology of developmental disorders (Fig 1). While NR2F2 variants were not detected in our study, they have been identified in individuals with congenital heart defects (MIM 615779) which are also prominent in CHARGE syndrome38.
Classifying variants is an inherent challenge of WES interpretation, as demonstrated by the FLNA variant identified in proband 522. Such an example illustrates the value of testing for segregation of the allele with the disorder among family members, especially for genes like FLNA that are associated with diverse malformations and exhibit complicated patterns of variation and X-inactivation 23. In total, 8/28 (28.6%) probands in our cohort had no pathogenic variant detected by WES. Further genome-wide analysis may be instrumental in identifying an underlying genetic etiology for these individuals. It is possible that pathogenic variants in non-coding regions may be identified to cause CHARGE syndrome. This potential etiology is particularly relevant for CHARGE, since human CHD7 is a large gene (188 kb) and CHD7 is known to bind preferentially to tens of thousands of H3K4me-enriched regions of the genome, including promoters, enhancers, and super-enhancers39; 40. Other potential causes of CHARGE syndrome that could escape detection by standard chromosomal microarray and single gene, gene panel or whole exome sequencing include (a) differences in DNA methylation, (b) cryptic chromosomal rearrangements, and (c) somatic mosaicism. A recent study showed, for example, that DNA methylation signatures differ greatly between individuals with CHARGE and Kabuki syndromes28, despite extensive overlap in clinical presentation. Potential environmental causes of features of CHARGE syndrome may include retinoic acid exposure or vitamin D deficiency, which also present with craniofacial malformations, ocular and auricular anomalies, and heart defects. Further studies using larger cohorts of individuals with CHARGE and those with related clinical features, accompanied by methylation and genome sequencing studies will help explore and distinguish among these possibilities.
Supplementary Material
Acknowledgments
We thank the CHARGE Syndrome Foundation for allowing research and study recruitment at their conference. We also appreciate Dr. Marwan Tayeh, Rebecca Lank and Noor Ghali for their advice and assistance throughout this process. This work was supported by the NIH (R00HD069624 to S.L.B), (R01 DC009410, R01 DC014456 to D.M.M.), Donita B. Sullivan, MD Research Professorship in Pediatrics and Communicable Disease (D.M.M), and the NIH training grant Michigan Predoctoral Training in Genetics (T32GM007544 to A.M.). Whole exome sequencing was performed by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant HG006493 to D.N., M.B., and S.L.
References
- 1.Pagon RA, Graham JM, Jr, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. Journal of Pediatrics. 1981;99:223–227. doi: 10.1016/s0022-3476(81)80454-4. [DOI] [PubMed] [Google Scholar]
- 2.Issekutz KA, Graham JM, Jr, Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A. 2005;133:309–317. doi: 10.1002/ajmg.a.30560. [DOI] [PubMed] [Google Scholar]
- 3.Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 4.Hughes SS, Welsh HI, Safina NP, Bejaoui K, Ardinger HH. Family history and clefting as major criteria for CHARGE syndrome. Am J Med Genet A. 2014;164A:48–53. doi: 10.1002/ajmg.a.36192. [DOI] [PubMed] [Google Scholar]
- 5.Snijders Blok C, Corsten-Janssen N, FitzPatrick DR, Romano C, Fichera M, Vitello GA, Willemsen MH, Schoots J, Pfundt R, van Ravenswaaij-Arts CM, et al. Definition of 5q11.2 microdeletion syndrome reveals overlap with CHARGE syndrome and 22q11 deletion syndrome phenotypes. Am J Med Genet A. 2014;164A:2843–2848. doi: 10.1002/ajmg.a.36680. [DOI] [PubMed] [Google Scholar]
- 6.Sanyanusin P, McNoe LA, Sullivan MJ, Weaver RG, Eccles MR. Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum Mol Genet. 1995;4:2183–2184. doi: 10.1093/hmg/4.11.2183. [DOI] [PubMed] [Google Scholar]
- 7.Gordon CT, Petit F, Oufadem M, Decaestecker C, Jourdain AS, Andrieux J, Malan V, Alessandri JL, Baujat G, Baumann C, et al. EFTUD2 haploinsufficiency leads to syndromic oesophageal atresia. J Med Genet. 2012;49:737–746. doi: 10.1136/jmedgenet-2012-101173. [DOI] [PubMed] [Google Scholar]
- 8.Schulz Y, Freese L, Manz J, Zoll B, Volter C, Brockmann K, Bogershausen N, Becker J, Wollnik B, Pauli S. CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum Mol Genet. 2014;23:4396–4405. doi: 10.1093/hmg/ddu156. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krumm N, Sudmant PH, Ko A, O'Roak BJ, Malig M, Coe BP, Quinlan AR, Nickerson DA, Eichler EE, Project NES. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9:e1003153. doi: 10.1371/journal.pcbi.1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Directors ABo. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med. 2015;17:68–69. doi: 10.1038/gim.2014.151. [DOI] [PubMed] [Google Scholar]
- 16.Hale CL, Niederriter AN, Green GE, Martin DM. Response to correspondence to Hale et al. atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016 doi: 10.1002/ajmg.a.37435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hale CL, Niederriter AN, Green GE, Martin DM. Atypical Phenotypes Associated With Pathogenic CHD7 Variants and a Proposal for Broadening CHARGE Syndrome Clinical Diagnostic Criteria. American Journal of Medical Genetics. 2015 doi: 10.1002/ajmg.a.37435. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fregeau B, Kim BJ, Hernández-García A, Jordan VK, Cho MT, Schnur RE, Monaghan KG, Juusola J, Rosenfeld JA, Bhoj E, et al. De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am J Hum Genet. 2016;98:963–970. doi: 10.1016/j.ajhg.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bögershausen N, Gatinois V, Riehmer V, Kayserili H, Becker J, Thoenes M, Simsek-Kiper P, Barat-Houari M, Elcioglu NH, Wieczorek D, et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum Mutat. 2016;37:847–864. doi: 10.1002/humu.23026. [DOI] [PubMed] [Google Scholar]
- 20.Negri G, Magini P, Milani D, Colapietro P, Rusconi D, Scarano E, Bonati MT, Priolo M, Crippa M, Mazzanti L, et al. From Whole Gene Deletion to Point Mutations of EP300-Positive Rubinstein-Taybi Patients: New Insights into the Mutational Spectrum and Peculiar Clinical Hallmarks. Hum Mutat. 2016;37:175–183. doi: 10.1002/humu.22922. [DOI] [PubMed] [Google Scholar]
- 21.Hastings ML, Allemand E, Duelli DM, Myers MP, Krainer AR. Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF(65) PLoS One. 2007;2:e538. doi: 10.1371/journal.pone.0000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Chehadeh S, Kerstjens-Frederikse WS, Thevenon J, Kuentz P, Bruel AL, Thauvin-Robinet C, Bensignor C, Dollfus H, Laugel V, Rivière JB, et al. Dominant variants in the splicing factor PUF60 cause a recognizable syndrome with intellectual disability, heart defects and short stature. Eur J Hum Genet. 2016;25:43–51. doi: 10.1038/ejhg.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet. 2003;33:487–491. doi: 10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- 24.Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child. 1963;105:588–608. doi: 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- 25.Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, Yau S, Lillis S, Hurst JA, Clement E, Reardon W, Joss S, Hobson E, et al. Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol. 2016;25:135–145. doi: 10.1097/MCD.0000000000000143. [DOI] [PubMed] [Google Scholar]
- 26.Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott D, et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 2016;12:e1005963. doi: 10.1371/journal.pgen.1005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, Beenken A, Clarke J, Pers TH, Dworzynski P, et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet. 2013;92:725–743. doi: 10.1016/j.ajhg.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, Mendoza-Londono R, Chitayat D, Walker S, Machado J, Caluseriu O, et al. CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am J Hum Genet. 2017;100:773–788. doi: 10.1016/j.ajhg.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verhagen JM, Oostdijk W, Terwisscha van Scheltinga CE, Schalij-Delfos NE, van Bever Y. An unusual presentation of Kabuki syndrome: clinical overlap with CHARGE syndrome. Eur J Med Genet. 2014;57:510–512. doi: 10.1016/j.ejmg.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, Duester G. Retinoic acid controls body axis extension by directly repressing Fgf8 transcription. Development. 2014;141:2972–2977. doi: 10.1242/dev.112367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocha-Viegas L, Villa R, Gutierrez A, Iriondo O, Shiekhattar R, Di Croce L. Role of UTX in retinoic acid receptor-mediated gene regulation in leukemia. Mol Cell Biol. 2014;34:3765–3775. doi: 10.1128/MCB.00839-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vilhais-Neto GC, Maruhashi M, Smith KT, Vasseur-Cognet M, Peterson AS, Workman JL, Pourquié O. Rere controls retinoic acid signalling and somite bilateral symmetry. Nature. 2010;463:953–957. doi: 10.1038/nature08763. [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Feng S, Tang K. COUP-TF Genes, Human Diseases, and the Development of the Central Nervous System in Murine Models. Curr Top Dev Biol. 2017;125:275–301. doi: 10.1016/bs.ctdb.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Zentner GE, Hurd EA, Schnetz MP, Handoko L, Wang C, Wang Z, Wei C, Tesar PJ, Hatzoglou M, Martin DM, et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum Mol Genet. 2010;19:3491–3501. doi: 10.1093/hmg/ddq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basson MA. Epistatic interactions between Chd7 and Fgf8 during cerebellar development: Implications for CHARGE syndrome. Rare diseases. 2014;2:e28688. doi: 10.4161/rdis.28688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol. 2015;16:110–123. doi: 10.1038/nrm3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Micucci JA, Layman WS, Hurd EA, Sperry ED, Frank SF, Durham MA, Swiderski DL, Skidmore JM, Scacheri PC, Raphael Y, et al. CHD7 and retinoic acid signaling cooperate to regulate neural stem cell and inner ear development in mouse models of CHARGE syndrome. Hum Mol Genet. 2014;23:434–448. doi: 10.1093/hmg/ddt435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, D'Alessandro LC, Swaminathan GJ, Bentham J, Arndt AK, et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. 2014;94:574–585. doi: 10.1016/j.ajhg.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niederriter AR, Varshney A, Parker SC, Martin DM. Super Enhancers in Cancers, Complex Disease, and Developmental Disorders. Genes (Basel) 2015;6:1183–1200. doi: 10.3390/genes6041183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schnetz MP, Handoko L, Akhtar-Zaidi B, Bartels CF, Pereira CF, Fisher AG, Adams DJ, Flicek P, Crawford GE, Laframboise T, et al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 2010;6:e1001023. doi: 10.1371/journal.pgen.1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.