

Abstract
Biosynthesis of the gibberellin A (GA) plant hormones evolved independently in plant-associated fungi and bacteria. While the relevant enzymes have distinct evolutionary origins, the pathways proceed via highly similar reactions. One particularly complex transformation involves combined demethylation and γ-lactone ring formation, catalyzed in bacteria by the cytochrome P450 CYP112 in three individual steps, which involves large structural changes in the transition from substrate to product, with further divergence in the recently demonstrated use of two separate mechanistic routes. Here the substrate specificity of the isozyme from Erwinia tracheiphila, EtCYP112, was probed via UV-Vis spectral binding studies and activity assays with alternate substrates from the GA biosynthetic pathway. EtCYP112 tightly binds its native substrate GA12 and the intermediates GA15 and GA24, as well as the methylated derivatives of GA12 and GA15. It however only poorly binds methylated GA24, its GA9 final product and the C-20 carboxylate side-product GA25. These distinct affinities are consistent with the known reactivity of EtCYP112. However, while it binds to the immediately preceding pathway metabolite GA12-aldehyde and even earlier oxygenated ent-kaurene precursors, EtCYP112 only reacts with GA12-aldehyde, and not the earlier ent-kaurene derived metabolites. But even with GA12-aldehyde conversion is limited to the first two steps and the final combined demethylation and γ-lactone ring forming reaction is not catalyzed. Thus, CYP112 has evolved specificity at the catalytic rather than substrate binding level to enable its role in GA biosynthesis.
Introduction
Gibberellins (GAs) are essential hormones in plants, and certain plant-associated fungi and bacteria also have acquired the ability to produce GAs in order to manipulate their host plants (1). The bacterial gibberellin biosynthetic pathway was elucidated very recently (Figure 1)(2–4). The steps of the individual pathways are quite similar between plants, fungi and bacteria. Nevertheless, at the enzyme level it is evident from the low sequence identity that GA biosynthesis evolved separately in plants, fungi and bacteria (1–4). One hallmark transformation in GA biosynthesis is the conversion of 20-carbon GAs to the 19-carbon GAs, involving the combined loss of a methyl group and the formation of an intramolecular γ-lactone bridge. Plants mediate this complex multi-step reaction with 2-oxoglutarate dependent dioxygenases (2ODDs) termed GA 20-oxidase (GA20ox) and, although this transformation is mediated by cytochromes P450 (CYPs) in both fungi (CYP68B) and bacteria (CYP112), these two CYP sub-families share less than 15% sequence identity (1, 4). Moreover, while the relevant fungal CYP68B is membrane-bound, the bacterial CYP112 is soluble. Nonetheless, the chemical transformations catalyzed by the three individual enzymes appear to be identical, involving stepwise oxidation of C-20. Specifically hydroxylation of GA12 at this position to form GA15, followed by further oxidation to the aldehyde equivalent GA24 and, finally, loss of C-20 as CO2 with formation of the intramolecular γ-lactone bridge that yields GA9 (5–9)(Figure 1). While the fungal enzyme does not react with the intermediates GA15 or GA24 and will only release them under sub-optimal conditions, the bacterial and plant enzymes release their intermediates more readily, and can use GA15 and GA24 as substrates to produce GA9 (1, 4, 7, 9). Although all of these oxidases produce the C-20 carboxylate GA25, this seems to be a side-product rather than intermediate, as it cannot be transformed to GA9 by any of them.
Figure 1.

Bacterial gibberellin operon (A) Schematic representation of the GA biosynthetic operon. Arrows indicate the direction of translation, abbreviations for the genes are CYP, cytochrome P450; Fd, ferredoxin; SDR, short-chain alcohol dehydrogenase/reductase; IDS, isoprenyl diphosphate synthase; CPS, copalyl diphosphate synthase; KS, ent-kaurene synthase; and IDI, isopentenyl diphosphate isomerase. (B) Catalyzed reactions, with emphasize on the sequential oxidation and elimination of C-20 from GA12, to produce GA9, by CYP112.
The recently discovered bacterial CYP112 offers distinct advantages for study as, in contrast to the 2ODDs utilized by plants, CYPs change their spectral properties upon substrate binding. Unbound CYPs have a characteristic absorbance maximum around 420 nm, called the Soret-band, which shifts to around 390 nm upon binding of their substrates, and this spectral shift can be used to measure affinity (10). While the fungal CYP68B presumably undergoes a similar spectral shift, it is membrane bound, which significantly complicates expression and purification. Moreover, the fungal CYP68B does not release its reaction intermediates or exhibit reactivity with these, at least in the context of incubation with cultures of Gibberella fujikuroi to which these studies have been limited (11, 12). In contrast, the CYP112 from Erwinia tracheiphila (EtCYP112) can be readily recombinantly expressed in Escherichia coli, purified and assayed in vitro, with characterization of its reaction mechanism via 18O labeling experiments recently reported (9). Specifically, EtCYP112 variably employs two diverging reaction mechanisms, with some significant difference in intermediates – i.e., either the open or closed (δ-lactone and lactol, respectively) forms of the intermediates GA15 and GA24 – that reconverge at a transient cyclic anhydride C-20 geminal-diol intermediate (Figure 1B). Thus, EtCYP112 is able to react with all potential intermediates, including those not utilized by either of the functionally equivalent enzymes from fungi or plants. In particular, as the fungal CYP68B has not been shown to react with any intermediate (11), while the plant GA20ox does not react with the closed lactone of GA15 (7). This sparked interest as to the substrate specificity of EtCYP112, which was investigated here by UV-Vis monitored binding studies and in vitro activity assays with alternate substrates. The results demonstrate that, although EtCYP112 is able to bind multiple upstream metabolites, it only reacts with GA12-aldehyde. However, the resulting transformation is limited to production of GA15-aldehyde and GA24-aldehyde.
Experimental
Cloning, Expression and Purification of EtCYP112 and EtFdR
Cloning of EtCYP112 and EtFdR was described previously (9). Plasmids containing either EtCYP112 or EtFdR were transformed into E. coli BL21-Star (Invitrogen) for expression. Starter cultures of 25 ml liquid NZY medium (10 g/L NaCl, 10 g /L casein, 5 g/L yeast extract, 1 g/L MgSO4 (anhydrous), pH 7.0), including 50 μg/mL carbenicillin, were inoculated with 3 individual colonies and incubated at 18 °C for 2 days with constant shaking at 200 rpm. Theses starter culture were used to inoculate 1 L of NZY with 50 μg/mL carbenicillin, which was kept at 18 °C with constant shaking at 200 rpm until an OD600 of 0.8 was reached. At this point the culture was induced with 1 mM IPTG and, for cultures expressing EtCYP112, 1 mM aminolevulinic acid, 1 mM riboflavin and 0.1 mM FeCl3 were added. After 36 hours, the cells were harvested by centrifugation at 5000 x g for 20 min. The EtCYP112 cell pellet was re-suspended in 10 mL 25 mM 3-(N-morpholino)-2-hydroxypropanesulfonic acid (MOPSO), pH 7.2, 10% (v/v) glycerol), while the EtFdR cell pellet was re-suspended in 10 mL 50 mM phosphate, pH 7.5, 10% glycerol. Both cell suspensions were homogenized with an EmulsiFlex C-3 (Avestin, Canada). The lysate was centrifuged at 17,000 x g for 30 min. The cleared lysate was then added to 1 mL of Ni-NTA Agarose (Qiagen), with the corresponding buffer containing 1 M imidazole added to reach a final concentration of 20 mM imidazole. Columns were incubated with gentle shaking at 4 °C for 1 h. The column was washed with 5 mL buffer containing 20 mM imidazole and then 5 mL with 60 mM imidazole, before being eluted with the corresponding buffer with 250 mM imidazole. The eluted protein was then transferred to dialysis tubing with a molecular weight cutoff of 15 kDa and dialyzed 3 times against 1L of 10 mM MOPSO, pH 7.2, 5% (v/v) glycerol in the case of EtCYP112 when used for spectral binding studies or 25 mM phosphate, pH 7.5, 10% glycerol in the case of EtFdR, as well as EtCYP112 when used for enzymatic activity assays. The resulting enzymes were either used fresh or stored at −80 °C after flash freezing by brief immersion in liquid nitrogen of 0.5 mL aliquots in 1.5 mL Eppendorf tubes. Only fractions with ≥95% purity, as assessed by SDS-PAGE analysis, were used. The concentration of EtCYP112 was measured by optical absorbance at 280 nm, using the calculated extinction coefficient of 43,555 M−1 cm−1.
Enzyme assays
Enzyme assays under regular atmosphere or 18O2 were performed as described previously (9). Briefly, 0.01 mL each of purified EtCYP112, spinach ferredoxin (Sigma-Aldrich), and EtFdR (all at 1 mg/mL in 25 mM phosphate, pH 7.5, 10% glycerol) were diluted with 1.5 mL of 25 mM phosphate, pH 7.5, 10% glycerol in 4 mL clear glass vials, and 20 μM of the specified substrate and 60 μM NADPH were added. The vial was sealed with a Teflon-septum cap and incubated for 12 h at 25 °C. For 18O2 labeling experiments these vials were first flushed with nitrogen for 5 min, then 0.05 mL each of purified EtCYP112, spinach ferredoxin (Sigma-Aldrich), and EtFdR (all at 1 mg/mL in 25 mM phosphate, pH 7.5, 10% glycerol), and 1.7 mL of 25 mM phosphate, pH 7.5, 10% glycerol were added using a syringe. 18O2 (Sigma-Aldrich, 97% labeled; max loaded pressure less than 2.4 bar) was added from the gas cylinder via a needle through the septum until the pressure equilibrated between the tank and the vial. The reaction was started by the addition of substrate to a final concentration of 40 μM. Assays from 18O2 labeling experiments were analyzed with an Agilent 6540 GC coupled to a Waters GCT Premier mass spectrometer and a 3900 Saturn GC coupled to a 2100T ion trap mass spectrometer (Varian, Palo Alto, CA, USA), enzyme assays under regular atmosphere were only analyzed with the 3900 Saturn GC coupled to a 2100T ion trap mass spectrometer. Separation was achieved over a DB-5MS column (30 m, 250 μm, 0.25 μm) with a previously described temperature gradient (3). For further conversion of the GA12-aldehyde products, two-thirds of the organic solvent extract from the primary in vitro enzyme assay were split off and, after evaporation, dissolved in 300 μL of methanol:DMSO (1:1, v/v) without methylation. This material fed to the GA operon SDR from E. tracheiphila (EtSDR), much as previously described (2). Briefly, this was done via whole-cell feeding for three days to a recombinant culture of E. coli expressing EtSDR, with subsequent extraction and analysis by GC-MS.
UV-Vis spectroscopy
Purified EtCYP112 in 10 mM MOPSO, pH 7.2, 5% (v/v) glycerol was diluted in the same buffer so that absorbance at 419 nm was 0.6 – 0.7 and spectra from 300 to 700 nm were collected on a Cary 50 UV-VIS spectrometer (Varian) at a scan rate of 60 nm per minute. Substrates were added stepwise from a stock solution of 0.1 or 1 mg/mL dissolved in methanol. Methanol was added to EtCYP112 as a control for spectral shifts originating from the solvent alone, although no changes in the UV-Vis spectrum were observed with up to 8% methanol (v/v). Kd for individual binding curves were calculated using plots of the added substrate concentration versus the induced spectral shifts, the curve was then fitted using the model for tight binding ligands with the following formula.
The mean and standard error were calculated from three individual experiments and reported in Table 1.
Table 1.
Et CYP112 affinity for GA metabolites and methylated derivatives.
| Substrate | Kd [μM] | High Spin State | α-band Absorbtion |
|---|---|---|---|
| GA12 | 2.6 ± 0.3 | > 90% | decreased |
| GA15 (open lactone) | 0.11 ± 0.02 | > 90% | decreased |
| GA15 (closed lactone) | 0.6 ± 0.1 | > 90% | decreased |
| GA24 | 0.32 ± 0.07 | 80 – 90% | decreased |
| GA9 | n.a. | < 5% | unchanged |
| GA25 | n.a. | 10 – 20% | unchanged |
| GA12-aldehyde | 2.0 ± 0.3 | > 90% | decreased |
| MeGA12 | 14 ± 3 | 50 – 60% | decreased |
| MeGA15 | 0.11 ± 0.03 | 80 – 90% | decreased |
| MeGA24 | n.a. | 5 – 10% | unchanged |
| ent-kaurenoic acid | 2.7 ± 0.6 | 50 – 60% | decreased |
| methyl-ent-kaurenoate | 0.5 ± 0.1 | 10 – 15% | decreased |
| ent-7α-hydroxy kaurenoic acid | 3.4 ± 0.6 | 50 – 60% | decreased |
| ent-kaurenal | 18 ± 2 | 5 – 10% | decreased |
| ent-kaurenol | 27 ± 7 | 20 – 30% | decreased |
| ent-kaurene | n.a. | < 5% | unchanged |
The Kd and the standard error were calculated from 3 independently performed titration experiments.
Results
The previously described N-terminal His-tagged EtCYP112 construct was found to be expressed to about 15 mg/ml in E. coli and easily purified by nickel affinity chromatography. UV-Vis spectral analysis found that the eluted preparation exhibited a Soret-band peak at 424 nm. Following dialysis, the substrate-free enzyme showed a more characteristic Soret-band maximum at 419 nm. Upon reduction and binding of carbon monoxide, purified EtCYP112 also exhibited the eponymous absorption peak at 450 nm (Supplemental Figure S1).
Upon addition of substrate or intermediates to purified EtCYP112, typical Type I binding spectra were observed. These are characterized by an almost complete shift of the Soret-band peak to 391 nm, as well as decreased α-band absorption at 570 nm. Given the evident high-affinity of EtCYP112 for these, their Kd values were calculated from Soret-band shift data using the equation that applies to such tight-binding ligands (13). Kd values for GA12, GA15 (open lactone, added from a stock with 1 M KOH), GA15 (closed δ-lactone) and GA24 were calculated to be 2.6, 0.11, 0.6 and 0.3 μM respectively (Figures 2, S2 and Table 1). By contrast, the GA25 side-product and the GA9 final product only shift the Soret-band from 419 to 391 nm about 10–20% or 5% respectively, without any decrease of the α-band.
Figure 2.
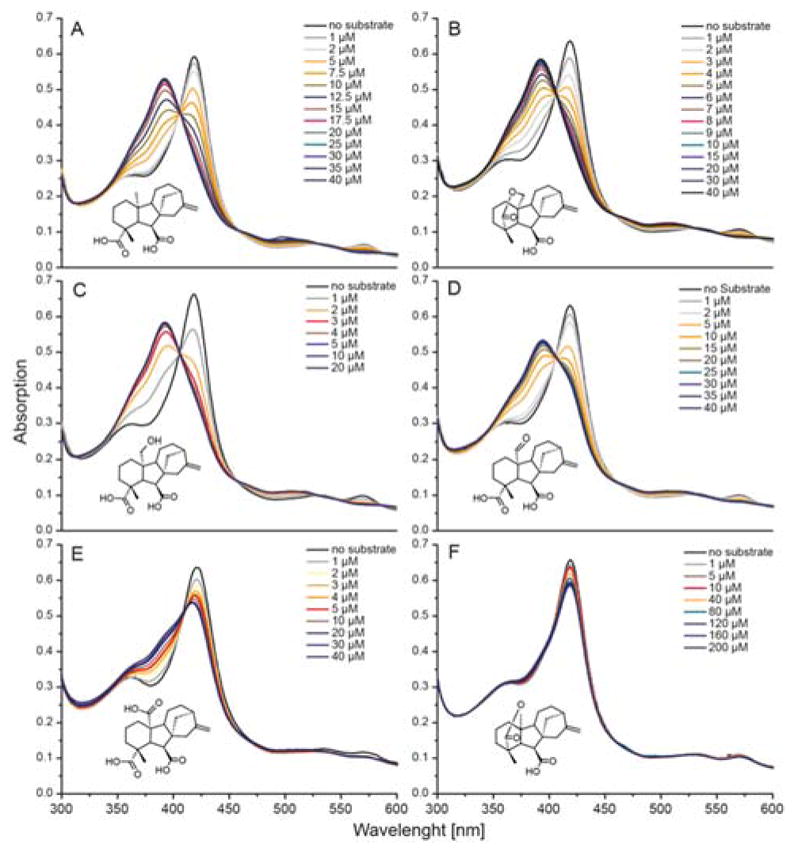
Spectral titration of EtCYP112 with substrates and products. UV-Vis spectra of EtCYP112 in the absence and with increasing concentrations of (A) GA12, (B) GA15 (closed lactone), (C) GA15 (open lactone), (D) GA24, (E) GA25 and (F) GA9.
Previously, to elucidate the mechanism of CYP112 the carboxylates at C-7 and C-19 were methylated and the resulting compounds tested as substrates. While MeGA12 and MeGA15 (δ-lactone form) are converted by EtCYP112 to GA9, MeGA24 was not (9). Binding of these methylated GAs also was investigated here by UV-Vis spectral analysis (Table 1, Figures 3 and S3). Methylation of GA12 (MeGA12) reduced its affinity for EtCYP112, with a 5-fold higher Kd observed than that measured for free GA12. By contrast, MeGA15 (closed δ-lactone) is bound about 6-fold more tightly by EtCYP112 than the analogous form of free GA15, and its Kd is more similar to the open form of GA15. MeGA24 on the other hand only induces a partial shift of the Soret-band, the binding curve could not be fit to the model for tight binding ligands, and there was no decrease in the absorption of the α-band.
Figure 3.
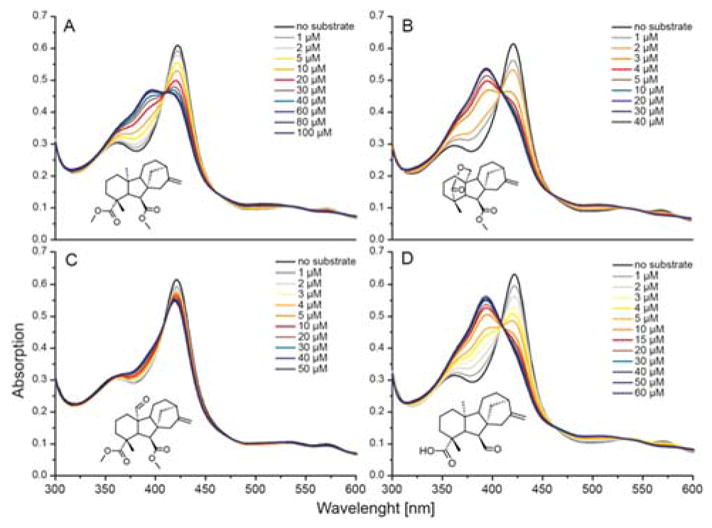
Spectral titration of EtCYP112 with methylated substrates, as well as immediate precursor GA12-aldehyde. UV-Vis spectra of EtCYP112 in the absence and with increasing concentrations of (A) MeGA12, (B) MeGA15 (closed lactone), (C) MeGA24 and (D) GA12-aldehyde.
Given the ability of EtCYP112 to act upon C-7 methyl ester derivatives, it seemed possible that it further might exhibit activity with the immediate precursor to GA12 – GA12-aldehyde – in which C-7 is an aldehyde rather than a carboxylate. Indeed, GA12-aldehyde was found to be tightly bound by EtCYP112 in UV-Vis spectral studies, with a Kd of 2.0 μM (Table 1 and Figures 3 and S3). When fed to the previously reported reconstituted enzymatic system, GA12-aldehyde is transformed into two products, although conversion was not complete even in the presence of a 10-fold excess of NADPH (Figure 4). The two products exhibited molecular ions with 30 mass units less than those of one of the corresponding C-7 carboxylate containing GAs, consistent with the expected mass difference between an aldehyde and a (methylated) acid function (note that all of the compounds examined here were methylated for analysis by GC-MS)(Figure 5). Based on the ability of the short-chain alcohol dehydrogenase/reductase (SDR) found in the bacterial GA biosynthetic operon to convert GA12-aldehyde to GA12 (2–4), the observed products were incubated with E. coli expressing this SDR from E. tracheiphila (EtSDR). This led to conversion of one product to GA24, indicating that this was GA24-aldehyde. These had very similar retention times (Figure 4). Another product exhibited a similarly close retention time to GA15, and has a very similar mass fragmentation pattern to GA15 as well, suggesting that it is GA15-aldehyde. This tentative assignment was further supported by 18O labeling experiments, as the mass of the putative GA15-aldehyde was increased by 2 mass units, while the mass of GA24-aldehyde was increased by 2 mass units (about 30%) or 4 mass units (about 66%)(Figure 5). This labeling pattern is similar to that previously reported for the production of GA24 from GA12 (9, 13). The inability of EtSDR to convert the putative GA15-aldehyde to the corresponding C-7 carboxylate (i.e., GA15) is presumably due to the presence of the δ-lactone ring. However, no peak exhibiting the expected molecular mass and/or retention time for GA9-aldehyde was observed.
Figure 4.
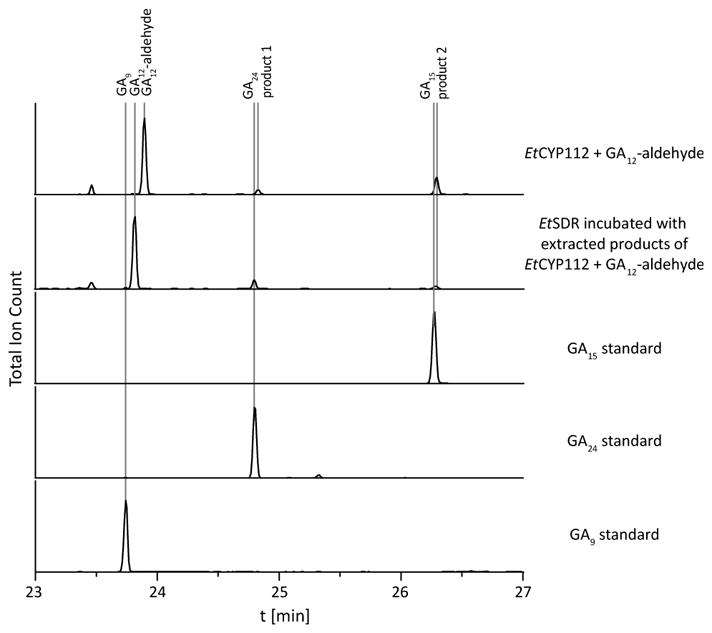
Conversion of GA12-aldehyde by EtCYP112. GC-MS-chromatograms of in vitro assays with EtCYP112 and GA12-aldehyde, incubation of the products with EtSDR and GA15, GA24 and GA9 standards.
Figure 5.

Mass spectra of GA15- and GA24-aldehyde. MS spectra of standard compounds and the unknown products of enzyme assays with EtCYP112 and GA12-aldehyde, either under regular atmosphere or 18O2. Presented mass spectra were acquired with a Varian 2100T ion trap mass spectrometer.
GA biosynthetic intermediates prior to GA12-aldehyde do not have the characteristic gibberellane 6-5-6-5 ring structure, but instead a 6-6-6-5 ent-kaurane ring structure. While addition of the olefin precursor ent-kaurene to EtCYP112 only induces a small shift of the Soret-band, with no decrease in α-band absorption, surprisingly, the oxygenated derivatives induced typical type I binding spectra. In particular, ent-kaurenoic acid, its methylated derivative (methyl ent-kaurenoate), ent-7α-hydroxy kaurenoic acid, ent-kaurenal and ent-kaurenol, for which the Kd values were calculated to be 2.7, 0.5, 3.4, 18 and 27 μM, respectively (Table 1 and Figures 6 and S4). However, no products were observed from assays with these compounds (data not shown).
Figure 6.
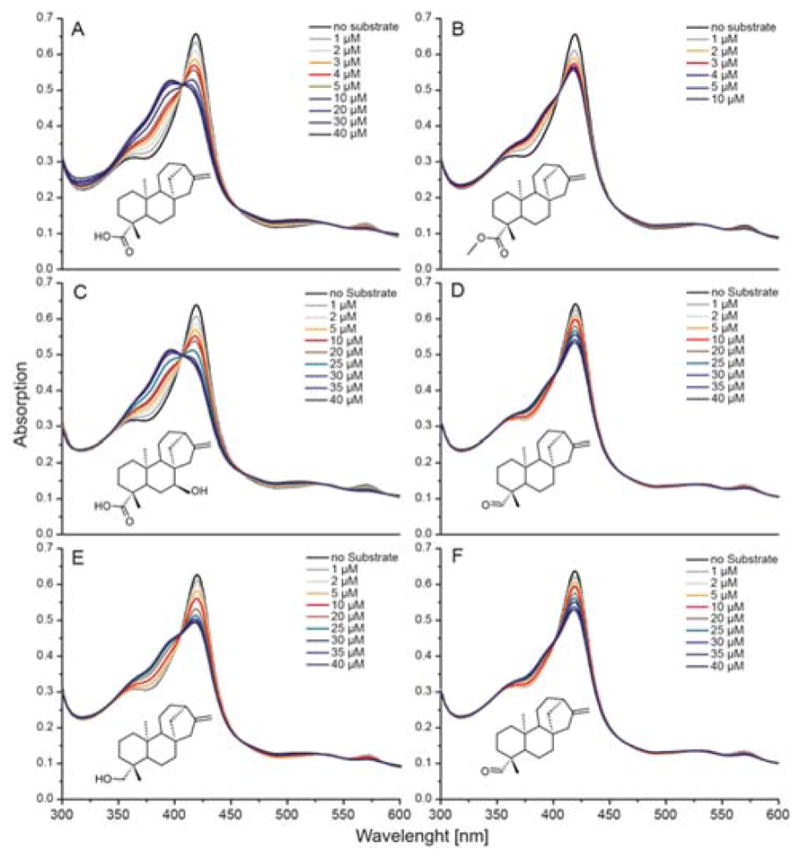
Spectral titration of EtCYP112 with kaurane precursors. UV-Vis spectra of EtCYP112 in the absence and with increasing concentrations of (A) ent-kaurenoic acid, (B) methyl ent-kaurenoate, (C) ent-7α-hydroxy-kaurenoic acid, (D) ent-kaurenal, (E) ent-kaurenol, and (F) ent-kaurene.
Discussion
The substrate specificity of the E. tracheiphila CYP112 isozyme was tested here. This was enabled by its ready over-expression in E. coli, perhaps reflecting their relatively close phylogenetic relationship (i.e., both species fall within the Enterobacteriaceae family). However, unlike EtCYP117, which catalyzes the first oxidative transformation in the GA biosynthetic pathway (Figure 1) and also is readily over-expressed in E. coli (14), EtCYP112 can be purified with retention of catalytic activity and exhibits the characteristic Soret-band at about 420 nm, as well as the characteristic Soret peak at 450 nm after reduction and CO binding.
Based on the ability to measure binding with straightforward UV-Vis spectral studies it was possible to examine the affinity of EtCYP112 for a number of GA biosynthetic intermediates, as well as selected methylated derivatives (Table 1). In contrast to at least a GA20ox from wheat (Triticum aestivum) that exhibited increased KM values for the catalyzed reaction intermediates GA15 (open) and GA24 relative to GA12 (15), EtCYP12 exhibits decreased Kd (i.e., higher affinity) for these. This is consistent with their observed retention in the active site during the course of the catalyzed series of reactions (9). While the KM values for GA20ox and Kd values for EtCYP112 cannot be directly compared, they are both in the upper nM to lower μM range (no binding affinities have been reported for the fungal equivalent CYP68B). Given the highly bioactive nature of GAs low production levels are sufficient to induce an effect, consistent with the tight binding observed here. In addition to GA12, GA15 and GA24, EtCYP112 was found to bind, at least to some degree, essentially all precursors in the bacterial GA biosynthetic pathway, as well as the methylated derivatives of its substrates and reaction intermediates. Surprisingly, this included precursors with the 6-6-6-5 ent-kaurane rather than 6-5-6-5 gibberellane ring structure. Although ent-kaurene and its derivatives did not serve as substrates, some spectral change was observed in Soret-band absorption upon their addition to EtCYP112. Accordingly, these precursors can bind and displace the water molecule that coordinates the heme-Fe(III) as its sixth ligand, leading to some conversion to the spectrally observed high-spin state. However, unlike the native reactants, these precursors do not fully convert EtCYP112 to the high-spin state (i.e., this is generally below <60%). This promiscuous binding may be linked to the complex reactions catalyzed by EtCYP112, as these involve relatively large changes in the structure of the reactant. Nonetheless, EtCYP112 exhibits higher fidelity in terms of activity, since only the natural substrate and intermediates are efficiently reacted upon. Indeed, even the immediate precursor GA12-aldehyde, which particularly in its geminal-diol form is a close structural analog of GA12, is not efficiently acted upon by EtCYP112. Accordingly, the different ring structure of ent-kaurene and its derivatives must alter their positioning in the EtCYP112 active site such that they are not correctly oriented for catalysis (i.e., relative to the thiolate heme-iron), despite their ability to (partially) displace the water molecule otherwise serving as the 6th ligand.
It was previously shown that EtCYP112 does not react with either the C-20 carboxylate GA25 or the 7,19-dimethyl ester derivative of MeGA24 (9). Here this was found to be at least partially due to the fact that neither compound binds to EtCYP112 with a significant shift of the Soret-band and decrease of the α-band. Together these findings support the previously indicated use of a cyclic anhydride C-20 gem-diol intermediate, as methylation of the C-19 carboxylate in MeGA24 prevents cyclization and GA25, while theoretically able to cyclize to the anhydride, is energetically unlikely do so in an aqueous environment.
In conclusion, the studies reported here provide insight into the impact of the complex transformation of GA12 to GA9 on the substrate structure-function relationship of CYP112, which must accommodate significant changes in its substrate/reactants (see Figure 1B). This is shown to be reflected, in promiscuous binding of upstream precursors. Nevertheless, although these preceding metabolites can bind to CYP112, they only do so with a higher Kd or a reduced ability to convert the thiolate heme-iron to its high-spin state. Accordingly, CYP112 reactivity seems to be quite specific. Of the various biosynthetic precursors bound by CYP112, only GA12-aldehyde is reacted upon, and even this is only slowly and partially transformed, with no catalysis of the final combined demethylation and γ-lactone ring forming reaction. This presumably is a function of the complexity of this final transformation (i.e., from GA24 to GA9), and corresponding requirement for precise positioning of the substrate/reactant within the active site. Regardless, these results indicate that, despite the need to accommodate significant alteration of its substrate/reactants during transformation of GA12 to GA9, leading to promiscuous binding of upstream metabolites, CYP112 only readily reacts with its natural substrate and reaction intermediates. Accordingly, the role of CYP112 in bacterial GA biosynthesis is specified at the catalytic rather than substrate binding level.
Supplementary Material
Acknowledgments
We wish to thank ISU Chemical Instrumentation Facility staff member Steve Veysey for assistance with the Waters GCT GC-MS analyses, and Prof. Peter Hedden (Rothamsted Research) for GA12 and GA25 standards.
Funding
This work was supported by grants from the NIH (GM109773) and NSF (CHE-1609917) to R.J.P., along with a postdoctoral fellowship to R.N. from the Deutsche Forschungsgemeinschaft (DFG) NA 1261/1-2.
Abbreviations List
- GA
Gibberellin
- CYP
cytochrome P450
- 2ODD
2-oxoglutarate dependent dioxygenases
- GA20ox
GA 20-oxidase
- EtCYP112
Erwinia tracheiphila CYP112
- EtFdR
E. tracheiphila ferredoxin reductase
- EtSDR
E. tracheiphila short-chain alcohol dehydrogenase/reductase
- GC-MS
gas chromatography mass spectrometry
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions
R.N. performed experiments and analyzed data; R.N. and R.J.P. conceived the experiments and wrote the manuscript.
References
- 1.Hedden P, Sponsel V. A Century of Gibberellin Research. J Plant Growth Regul. 2015;34(4):740–60. doi: 10.1007/s00344-015-9546-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagel R, Peters RJ. Investigating the Phylogenetic Range of Gibberellin Biosynthesis in Bacteria. Molecular Plant-Microbe Interactions. 2017;30(4):343–9. doi: 10.1094/MPMI-01-17-0001-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagel R, Turrini PC, Nett RS, Leach JE, Verdier V, Van Sluys MA, et al. An operon for production of bioactive gibberellin A4 phytohormone with wide distribution in the bacterial rice leaf streak pathogen Xanthomonas oryzae pv. oryzicola. New Phytologist. 2017;214(3):1260–6. doi: 10.1111/nph.14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nett RS, Montanares M, Marcassa A, Lu X, Nagel R, Charles TC, et al. Elucidation of gibberellin biosynthesis in bacteria reveals convergent evolution. Nature Chemical Biology. 2017;13(1):69–74. doi: 10.1038/nchembio.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamiya Y, Takahashi N, Graebe JE. The loss of carbon-20 in C19-gibberellin biosynthesis in a cell-free system from Pisum sativum L. Planta. 1986;169(4):524–8. doi: 10.1007/BF00392102. [DOI] [PubMed] [Google Scholar]
- 6.Ward JL, Gaskin P, Brown RGS, Jackson GS, Hedden P, Phillips AL, et al. Probing the mechanism of loss of carbon-20 in gibberellin biosynthesis. Synthesis of gibberellin 3α,20-hemiacetal and 19,20-lactol analogues and their metabolism by a recombinant GA 20-oxidase. Journal of the Chemical Society, Perkin Transactions 1. 2002;(2):232–41. [Google Scholar]
- 7.Ward JL, Jackson GJ, Beale MH, Gaskin P, Hedden P, Mander LN, et al. Stereochemistry of the oxidation of gibberellin 20-alcohols, GA15 and GA44, to 20-aldehydes by gibberellin 20-oxidases. Chemical Communications. 1997;(1):13–4. [Google Scholar]
- 8.Dockerill B, Hanson JR. The fate of C-20 in C19 gibberellin biosynthesis. Phytochemistry. 1978;17(4):701–4. [Google Scholar]
- 9.Nagel R, Peters RJ. Diverging Mechanisms: Cytochrome-P450-Catalyzed Demethylation and gamma-Lactone Formation in Bacterial Gibberellin Biosynthesis. Angew Chem Int Ed Engl. 2018;57(21):6082–5. doi: 10.1002/anie.201713403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLean KJ, Warman AJ, Seward HE, Marshall KR, Girvan HM, Cheesman MR, et al. Biophysical characterization of the sterol demethylase P450 from Mycobacterium tuberculosis, its cognate ferredoxin, and their interactions. Biochemistry. 2006;45(27):8427–43. doi: 10.1021/bi0601609. [DOI] [PubMed] [Google Scholar]
- 11.Tudzynski B, Rojas MC, Gaskin P, Hedden P. The gibberellin 20-oxidase of Gibberella fujikuroi is a multifunctional monooxygenase. J Biol Chem. 2002;277(24):21246–53. doi: 10.1074/jbc.M201651200. [DOI] [PubMed] [Google Scholar]
- 12.Bearder JR, MacMillan J, Phinney BO. Fungal products. Part XIV. Metabolic pathways from ent-kaurenoic acid to the fungal gibberellins in mutant B1-41a of Gibberella fujikuroi. Journal of the Chemical Society, Perkin Transactions 1. 1975;(8):721–6. [Google Scholar]
- 13.Gaskin P, MacMillan J. GC-MS of the gibberellins and related compounds : methodology and a library of spectra. Bristol: Univ. of Bristol (Cantocks Enterprises Ltd); 1991. [Google Scholar]
- 14.Nagel R, Peters RJ. 18O2 labeling experiments illuminate the oxidation of ent-kaurene in bacterial gibberellin biosynthesis. Organic & Biomolecular Chemistry. 2017;15(36):7566–71. doi: 10.1039/c7ob01819c. [DOI] [PubMed] [Google Scholar]
- 15.Appleford NEJ, Evans DJ, Lenton JR, Gaskin P, Croker SJ, Devos KM, et al. Function and transcript analysis of gibberellin-biosynthetic enzymes in wheat. Planta. 2006;223(3):568–82. doi: 10.1007/s00425-005-0104-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.