

Abstract
Background & Aims
Little is known about how the immune system affects stem cell features of pancreatic cancer cells. Immune cells that produce interleukin 17 A (IL17A) in the chronically inflamed pancreas (chronic pancreatitis) contribute to PanIN initiation and progression. We investigated the effects of IL17A signaling exerts on pancreatic cancer progenitor cells and the clinical relevance of this phenomena.
Methods
We performed studies with Mist1Cre;LSLKras;Rosa26mTmG (KCiMist;G) and Kras(G12D); Trp53(R172H); Pdx1-Cre (KPC) mice (which upon tamoxifen induction spontaneously develop pancreatic intraepithelial neoplasias, PanINs) and control littermates. Some mice were injected with neutralizing antibodies against IL17A or control antibody. Pancreata were collected, PanIN epithelial cells were isolated by flow cytometry based on lineage tracing, and gene expression profiles were compared. We collected cells from pancreatic tumors of KPC mice, incubated them with IL17 or control media, measured expression of genes regulated by IL17 signaling, injected the cancer cells into immune competent mice, and measured tumor growth. IL17A was overexpressed in pancreata of KCiMist mice from an adenoviral vector. Pancreata were collected from all mice and analyzed by histology and immunohistochemistry. Levels of doublecortin like kinase 1 (DCLK1) and other proteins were knocked down in KPC pancreatic cancer cells using small interfering or small hairpin RNAs; cells were analyzed by immunoblotting. We obtained 65 pancreatic tumor specimens from patients, analyzed protein levels by immunohistochemistry, and compared results with patient survival times. We also analyzed gene expression levels and patient outcome using the Cancer Genome Atlas database.
Results
PanIN cells from KCiMist;G mice had a gene expression pattern associated with embryonic stem cells. Mice given injections of IL17 neutralizing antibodies, or with immune cells that did not secrete IL17, lost this expression pattern, and significantly decreased expression of DCLK1 and POU class 2 homeobox 3 (POU2F3), which regulate tuft cell development. KCiMist mice that overexpressed IL17 formed more PanINs, with more DCLK1-positive cells, than control mice. Pancreatic tumor cells from KPC mice and human Capan-2 cells exposed to IL17A had increased activation of NF-κB and MAPK signaling, and increased expression of DCLK1 and ALDH1A1 (a marker of embryonic stem cells), compared to cells in control media. These cells also formed tumors faster that cells not exposed to IL17 when they were injected into immunocompetent mice. KPC cells with knockdown of DCLK1 expressed lower levels of ALDH1A1 following incubation with IL17 than cells without knockdown. Expression of the IL17 receptor C (IL17RC) was higher in DCLK1-positive PanIN cells from mice compared to DCLK1-negative PanIN cells. In human pancreatic tumor tissues, high levels of DCLK1 associated with a shorter median survival time of patients (17.7 months, compared with 26.6 months of patients whose tumors had low levels of DCLK1). Tumor levels of POU2F3 and LAMC2 also associated with patient survival time.
Conclusions
In studies of mouse and human pancreatic tumors and precursors, we found immune cell-derived IL17 to regulate development of tuft cells and stem cell features of pancreatic cancer cells via increased expression of DCLK1, POU2F3, ALDH1A1, and IL17RC. Strategies to disrupt this pathway might be developed to prevent pancreatic tumor growth and progression.
Keywords: Kras, PDAC, tumorigenesis, immune response
Graphical abstract
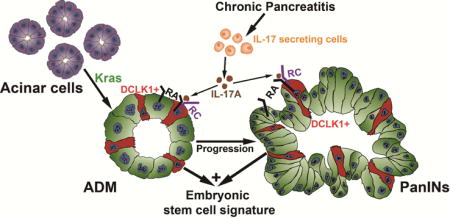
INTRODUCTION
The aggressive nature and poor prognosis of pancreatic cancer are well known1. Recently, genomic analysis of hundreds of pancreatic cancers revealed molecular subtypes which include an Immunogenic type characterized by upregulation of immune networks2. Further understanding of the early immunological events surrounding pancreatic tumor initiation and progression could result in effective novel immunopreventive strategies, potentially resulting in a stronger impact on this disease.
The interaction between immune cells and tumor cells can result in either tumor development and progression or effective immunosurveillance. In particular, IL17-secreting immune cells have been described as having mostly tumor promoting activity3, 4 although there are reports of anti-tumoral activity in specific context and model systems5. In pancreatic cancer, using a genetically engineered mouse model (GEMM) which mimics the human disease6, we previously reported that IL17-producing CD4+ (T Helper-17 or TH17 cells) and gamma-delta-T cells (γδ/IL17 cells) induced pancreatic premalignant lesions development and progression7. These phenomena is futher augmented in the presence of concomitant chronic inflammation, a well recognized risk factor for pancreatic cancer. However, the mechanisms by which IL17-secreting immune cells ultimately accelerate pancreatic intraepithelial neoplasia (PanIN) initiation and progression are not well defined.
IL17-secreting immune cells play an essential role in host defense at mucosal surfaces by inducing acute phase proteins (IL6, LCN2), neutrophils-recruiting chemokines and antimicrobial peptides (defensins, mucins)8, 9. Recent evidence suggests that the immune component of the microenvironment can affect the stem cell niche10–12. As examples of this, Th22, CD4+T cells capable of secreting IL22, have been recently found to promote colorectal cancer stemness through activation of STAT3 and expression of the metyltransferase DOT1L, which induces core stem cell genes NANOG, SOX2 and POU5F110. In a similar manner, IL17B secreted by non-tumor cells has been reported to promote gastric cancer stemness by signaling with IL17RB through activation of AKT/β-catenin pathway12.
The presence and role of stem cells in pancreatic cancer is well established13, 14, contributing to tumor relapse15 and resistance to chemotherapy16. Tuft cells have been described originally as unique solitary chemosensory cells that are analogous to taste cells17 with tumor initiating capacity in the colon18 and their presence in both normal and preneoplastic pancreatic lesions has been recently rigurously described19, 20, 17. Delgiorno et al have shown that KrasG12D activation is associated with aberrant genesis of pancreatic tuft cells which are identified by a specific array of markers17. DCLK1 has been proposed as a marker of tuft cells in the stomach, intestine and pancreas18, 21 Furthermore, the role of DCLK1-expressing cells as long-lived progenitor cells as well as potent tumor-initiating cells following injury has been recently elegantly demonstrated22. However, the upstream regulation of DCLK1 and pancreatic cancer stem cells is not well understood.
In this paper, we found that IL17 induces an embryonic stem cell signature in the PanINs. Using in vivo and in vitro models, we confirmed that IL17/IL17R promotes stemness functionally and regulates DCLK1 through activation of NFkB via the canonical pathway. We also determined that the inducible IL17RC is differentially expressed in PanIN DCLK1+ cells which may contribute to the expansion of these cells upon IL17 signaling. Finally, we explored prognostic relevance of IL17-induced ESC signature genes for patients with pancreatic cancer.
Materials and Methods
Genetically engineered mice
All animal experiments were conducted in compliance with the National Institute of Health guidelines for animal research, and approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center (MDACC). The tamoxifen-inducible Mist1Cre;LSLKras (KCiMist) and Mist1Cre;LSLKras;Rosa26mTmG (KCiMist;G) mice were used as previously described19, 56. KCiMist;G enabled FACS-based isolation of KrasG12D-expressing cells by virtue of simultaneous GFP activation. IL17 knockout (IL17KO) mice were kindly obtained from Prof. Yoichiro Iwakura (Center for Experimental Medicine and Systems Biology, The Institute of Medical Science, The University of Tokyo, Tokyo, Japan). C57BL/6 mice purchased from Taconic Biosciences (Hudson, New York) were used for subcutaneous mouse models.
Cell Lines
Murine pancreatic adenocarcinoma cells (KPC cells) derived from a spontaneous tumor in a KRASG12D; Trp53R172H; Pdx1-Cre (KPC) mouse were used. The originating mice was backcrossed to B6 mice for several generations. KPC-Tak1f/f and its KPC control cells were developed from Pdx-D-Cre;KrasG12 D/+; Trp53 R172H/+ ;Tak1F/F and Pdx-D-Cre; Kras G12 D/+; Trp53 R172H/+ ;Tak1+/+ mice. Human pancreatic cancer Capan-2 cells were purchased from ATCC (ATCC HTB-80). Cells were cultivated in Dulbecco’s Modification of Eagle’s Medium (DMEM) with 4.5 g/L glucose (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Sigma, St. Louis, Mo) and 1% Penicillin/Streptomycin (Pen/Strep, HyClone, South Logan, UT) at 37 °C and 5% CO2 in a humidified atmosphere.
Adenoviral infection
Adenovirus (5×109 pfu suspended in 50 μl) encoding either GFP (Ad-EGFP), Luciferase (Ad-Luc) or IL-17A (Ad-IL-17A) (Schwarzenberger et al., 1998) were injected directly into the pancreatic parenchyma as as previously described7.
Neutralizing antibody administration
Monoclonal neutralizing antibodies against IL-17 signaling (IL-17RA, IL-17A and IL-17F) generously provided by Amgen were used as previously described for short term (1 week) or long term (7 weeks) treatments7.
Bone marrow preparation and transplantation
Femurs and tibias of donor mice were harvested and processed. Obtained bone marrow cells were transplanted into recipient KCiMist1 mice as previously described7.
Immunofluorescence
Paraformaldehyde-fixed and paraffin-embedded sections were subjected to antigen retrieval and nonspecific epitopes blocking and incubated with antibodies against E-cadherin (Santa Cruz Biotechnology, Santa Cruz, CA) and Dclk1 (Abcam, Cambridge, MA) as previously described19.
In vitro tumorsphere formation assay
Cells were pre-treated with IL-17A cytokine 0 or 10 ng/ml for 7 days, then plated in ultra-low attachment 6-well plates (Corning, Corning, N.Y) in 2ml serum-free DMEM/F12 media (Invitrogen, Frederick, MD) supplemented with 20ng/ml EGF (Sigma, St Louis, MO), 10ng/ml basic fibroblast growth factor (bFGF; Sigma), 5 μg/ml insulin (Sigma), 1×B27 supplement (Invitrogen) and 0.4% bovine serum albumin (BSA; Sigma). Cells were cultured under 5% CO2 at 37°C for 7 days. The tumorspheres were visualized and counted under a light microscope. Assays were performed in triplicate and repeated three times.
Tumor initiation assay
1, 5, or 10 ×103 KPC cells or KPC cells treated with IL17 cytokine for 7 days suspended in 100 μl of PBS were injected subcutaneously into the flank of C57BL/6 mice. See more details in Supplementary Materials.
RNA Sequencing
Total RNA from KPC cells exposed to IL17 or media was extracted. Each sample was assessed using Qubit 2.0 fluorometer and Agilent TapeStation 2200 for RNA quantity and quality. The cDNA libraries were pooled at a final concentration 1.8pM. Cluster generation and 75 bp single-read dual-indexed sequencing was performed on Illumina NextSeq 500’s. See more details in Supplementary Materials.
Human pancreatic cancer tissue
Pancreatic adenocarcinoma samples were obtained from MDACC tissue Biobank and Human pancreas intraepithelial neoplasia tissue microarrays were constructed. Human studies were approved by The MDACC institutional review board (IRB protocol number: LAB05-0854) and informed consent was obtained from all subjects.
TCGA Survival analysis
RNAseq V2 mRNA expression Z-scores and clinical information of primary PDAC tumors were downloaded from CbioPortal57. Tumor samples were designated as “upregulated” if z-score > 1; else samples were designated as “other”. For each gene of interests, we compared the survival curves between “upregulated” and “other” samples using a cox proportional hazards regression model implemented in TCGAbiolinks_2.2.658. Resulting Kaplan-Meier Survival Curves were plotted in R version 3.3.1. Log-rank p-value < 0.05 were indicated as significant results.
Statistical Analysis
Data were expressed as the mean ± standard deviation (SD). Data were analyzed using GraphPad Prism (GraphPad Software, Inc., San Diego, CA). Statistical analysis was performed using the Student’s t-test when only two value sets were compared, and one-way ANOVA followed by Dunnett’s test when the data involved three or more groups. P < .05, P < .01 or P < .001 were considered statistically significant.
RESULTS
IL17 induces pancreatic cancer stemness
Following in vivo IL17 signaling blockade, transcriptional analysis compared mPanIN cells isolated from KCiMist;G mice treated with anti-IL17 antibodies versus cells from mice who received IgG isotype (Fig. 1A). Differentially expressed genes were assessed by Ingenuity Pathway Analysis (IPA) which revealed significant down-regulation of genes belonging to the following biological functional categories: cellular movement, cellular development, cellular growth and proliferation, cell-to-cell signaling and cellular assembly and organization (Fig. 1B, Table S1).
Figure 1. IL17 induces pancreatic cancer stemness and an Embryonic Stem Cell (ESC) Genes Signature.
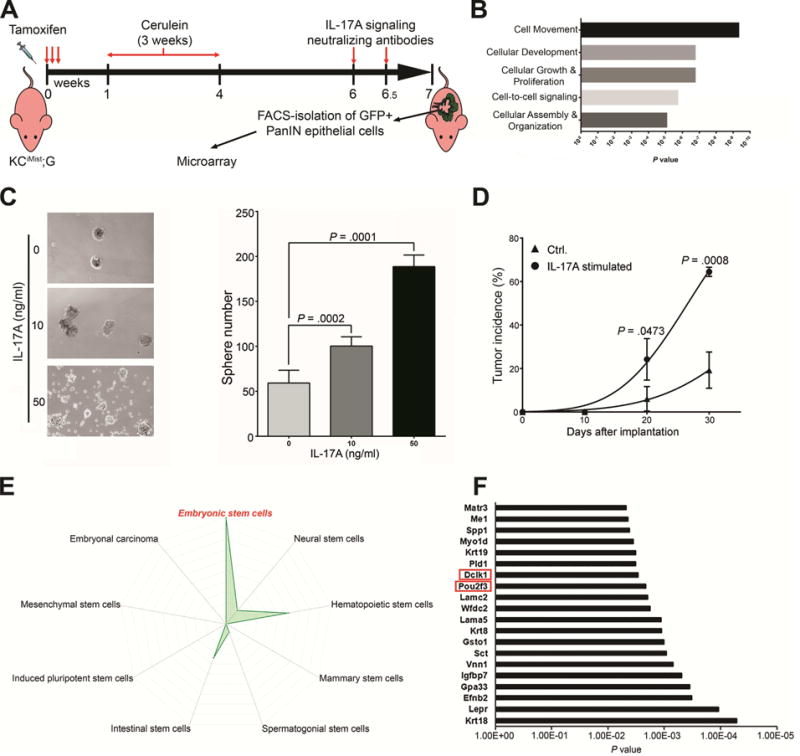
(A) Protocol for KCiMist1G mice treated with antibodies to block IL17-signaling pathway.
(B) Ingenuity Pathway Analysis showing top four molecular and cellular functions enriched among genes significantly down-regulated in GFP+ cells sorted from KCiMist1G mice treated with IL17 signaling neutralizing antibodies. As indicated on Y-axis, biological functions are sorted based on P value.
(C) Representative pictures (left) and quantification of tumorspheres (right) developed from KPC cells in vitro stimulated with IL17 (0, 10 and 50 ng/ml). The tumorsphere assay has been repeated 3 times. Results of one representative experiment are shown.
(D) Tumor incidence with subcutaneously injected 5,000 IL17-stimulated KPC cells vs. control into syngeneic immune-competent mice. The tumor Initiation experiment has been repeated 3 times. A total of 7 mice per group were injected in each experiment. Results of one representative experiment are shown.
(E) Radar chart displaying the overlap between the IL17-regulated genes and the stem cell types. The values plotted represent the significance of the overlap (with each radar representing −log10 P-value).
(F) List of the top 20 genes belonging to the ESC signature (total: 94 genes) that were downregulated (threshold: <−1.5 fold changes) in PanINs from mice exposed to IL17 blockade in vivo. The list with 20 genes was ordered based on their p values. List of genes was selected based on P value <0.005.
We then tested if IL17 is able to directly induce pancreatic epithelial migration, proliferation and stemness by exposing KPC cells to IL17-containing or control media. To validate the treatment response, we first confirmed upregulation of two known target genes of IL17, LCN2 and TNFα23,24 (Fig. S1A). Cells stimulated with IL17 exhibited a significant, dose-dependent, induction in tumorspheres formation (Fig. 1C), albeit proliferation and migration were unaffected (Fig. S1B, C). Furthermore, KPC cells incubated with IL17 for 7 days exhibited significantly accelerated capacity for in vivo- tumor initiation when subcutaneously injected into immune-competent mice (Fig. 1D), suggesting that IL-17 promotes stemness. To further confirm the tumor initiating potentiation, we implanted limiting dilutions of IL-17-treated vs untreated KPC cells into syngeneic WT mice and monitored tumor initiation over a period of 21 days. In these experiments, IL-17-treated KPC cells formed tumors with a calculated tumor-initiating cell frequency of 1/29614 (95% confidence limit, 1/3523–1/600) compared with 1/94701 (95% confidence limit, 1/1248–1/234) for untreated KPC cells (Fig. S1D). Furthermore, we have deleted the IL-17RA from KPC cells by CRISPR/Cas9 and used scramble-treated KPC cells as control (Fig. S1E). A limiting dilution tumor initiation in vivo assay was performed by implanting these cells into syngeneic mice and we found that IL-17RA-deficient KPC cells presented significant decrease in tumor incidence (Fig. S1F). Together, these findings strongly suggest that IL-17 signaling in the pancreatic oncogenic epithelium is capable of inducing stemness. When subcutaneous tumors developed from IL17-stimulated KPC cells were compared with controls, no differences were detected in the oncogenic epithelial proliferation and apoptosis as measured by Ki67 and cleaved caspase-3 expression (Fig. S1G). Human pancreatic cancer Capan-2 cells were also tested and IL17-mediated significant induction of tumorspheres formation was observed while no effect was seen in proliferation (Fig. S7A,B).
IL17 induces an Embryonic Stem cells (ESC) gene signature that includes two genes key for tuft cells specification during pancreatic tumorigenesis
We then used a novel stemness bioinformatics tool for detection of IL17-induced stemness signatures25–28 by analyzing 424 down-regulated genes detected on PanINs following in vivo IL17 blockade (Fig. 1A). Remarkably, we found that 94 genes were included in the murine Embryonic Stem Cells (ESC) signature, representing a significant enrichment (adjusted p-value: 9.84×10−10) of ESC associated up-regulated genes (Fig. 1E, S1H). We also found a significant enrichment for hematopoietic (Adjusted P= 1.14×10−5) and intestinal (Adjusted P= .004) stem cells-associated genes (Fig. 1E, S1H). Using the same tool and genes set, we then looked for transcriptional factors that regulate target genes with ESC identity and pluripotency. We found a significant enrichment (Adjusted P < .005) for the target genes of the following stemness transcriptional regulators: EZH2, SUZ12, NANOG, OCT4 and FLI1 (Fig. S1I, S1J).
We then validated the top-20 individual ESC signature genes significantly regulated by IL17 in the pancreatic oncogenic epithelium (Fig. 1F) in two independent in vivo experiments: IL-17-overexpression mediated by adenovirus (Fig S2A) and long-term IL-17-blockade with monoclonal antibodies (Fig S2B) performed also on KCiMist1 mice. In these experiments, we found that 25-30% of the signature genes were significantly regulated by IL-17. Of note, three genes were consistently modulated by IL-17 in all tested models: DCLK1, POU2F3 and EFNB2.
POU2F3 and DCLK1 have been described as essential for tuft cells development and function. POU2F3 has been recently found to be the transcription factor required for tuft cells differentiation and expansion in response to infection29. DCLK1 has been recognized as a useful marker of tuft cells17, 19 and a unique kinase responsible for pancreatic tumor initiation and development22, 30.
While DCLK1 was recently shown to be a downstream effector of Kras22, many of its upstream regulators have yet to be identified. Since DCLK1 was one of the PanINs genes regulated by IL-17 in vivo, we focused on further dissecting IL17-mediated regulation of DCLK1+ cells.
IL17 regulates DCLK1 and ALDH1A1 in pancreatic cancer cells
To understand if upregulation of DCLK1 is directly induced by IL17, we stimulated KPC cells with IL17 and observed time-dependent (Fig. 2A, S3A, S3B) and dose-dependent increases in DCLK1 protein expression (Fig. 2B upper panel). The DCLK1 induction was also observed in human Capan-2 cells exposed to IL17 (Fig. S8C). Interestingly, we also found a consistent time-dependent and dose-dependent upregulation of ALDH1A1 (Fig. 2A, 2B lower panel and S3A), a known marker and functional mediator of pancreatic cancer stem cells31, 32, suggesting that IL17 may regulate both DCLK1 and ALDH1A1.
Figure 2. IL17 regulates DCLK1 and ALDH1A1 in pancreatic cancer cells.
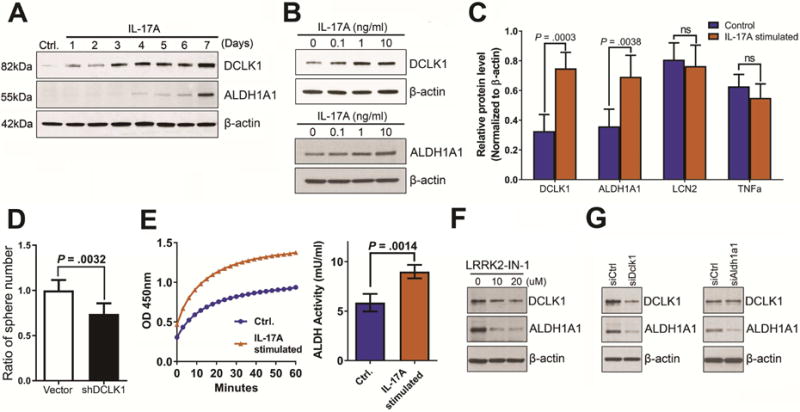
(A) Immunoblotting for DCLK1 and ALDH1A1 on KPC cells stimulated with IL17 in vitro for up to 7 days.
(B) Immunoblotting for DCLK1(upper) and ALDH1A1(lower) on KPC cells stimulated with IL17 at increasing concentrations.
(C) Quantification of immunoblotting for DCLK1, ALDH1A1, LCN2, and TNFα on tumors formed with subcutaneously injected KPC cells pre-incubated with IL17 vs control. Tumors were removed 10 days after KPC cells injections.
(D) Quantification of tumorspheres developed from KPC cells treated with DCLK1 shRNA vs control vector. The tumorsphere assay has been repeated 3 times. Results of one representative experiment are shown.
(E) ALDH Activity Assay on lysates obtained from KPC cells treated with IL-17 vs control. ALDH Kinetic curves measured during 60 minutes (left panel) and Quantification of last kinetic point (60 minutes) in both groups (right panel).
(F) Immunoblotting for DCLK1 and ALDH1A1 with KPC cells treated with increasing doses of LRRK2-IN-1 (0,10 and 20 uM).
(G) Immunoblotting for DCLK1 and ALDH1A1 with KPC cells treated with siDCLK1 and siControl (left panel). Immunoblotting for DCLK1 and ALDH1A1 with KPC cells treated with siALDH1A1 and siControl (right panel). β-actin was used as loading control for F,G.
Since we already found that KPC cells in vitro exposed to IL17 had accelerated capacity for in vivo- tumor initiation than untreated cells, we then aimed to discover if DCLK1 and ALDH1A1 were involved in this effect.
We performed subcutaneous injections of IL17-stimulated vs control KPC cells into syngeneic mice and removed the tumors at 10 days. We analyzed DCLK1 and ALDH1A1 protein expression in the tissue and found that both were significantly increased in the tumors formed with IL17-stimulated KPC cells as shown by immunohistochemistry (Fig S3C) and quantified by immunoblotting of the tissue for the respective proteins (Fig 2C,S3D). Of note, the immunohistochemical pattern of tumoral epithelial DCLK1 expression was diffuse and did not colocalize with known tuft cells markers (data not shown). In contrast, the two pro-inflammatory IL17 target genes, LCN2 and TNFα, previously validated as differentially increased in IL17 treated cells- in vitro (Fig S1A) were found decreased to similar levels that control tumors formed with non-treated KPC cells (Fig.2D, S3D), suggesting that the IL17-mediated effect on stemness might be longer-lasting that the induction in proinflammatory genes.
We then confirmed DCLK1 functional relevance in stemness by performing tumorsphere assays with KPC cells treated with DCLK1 shRNA which resulted in significant decrease in spheres number versus control shRNA treated cells (Fig. 2D). Also, we further confirmed the presence of significantly increased ALDH activity in IL-17-treated cells vs control (Fig 2E).
We observed a temporal difference in the DCLK1 and ALDH1A1 induction by IL17 stimulation, with DCLK1 expression increasing 24 hrs after IL17 stimulation while ALDH1A1 expression increases by day 6 (Fig 2A, S3A). Therefore, we hypothesized that IL17 directly induces DCLK1 which in turn regulates ALDH1A1 rather than IL17 directly regulating both. As a first approach to test this hypothesis we used the small molecule inhibitor of Leucin-rick repeat kinase 2 (LRRK2-IN-1) which has significant affinity for DCLK1, besides inhibiting LRKK233, 34. We first confirmed the dose-dependent inhibition of DCLK1 and then assessed for ALDH1A1 expression upon LRRK2 treatment and observed a significant decrease in ALDH1A1 expression (Fig. 2F). To confirm these results in a more specific system, we then treated KPC cells with DCLK1 siRNA. We validated the assay and proved DCLK1 loss with siRNA treatment and then measured ALDH1A1 by immunoblotting and observed inhibition of its expression upon DCLK1 loss (Fig.2G left panel). As a control, we treated KPC cells with siRNA for ALDH1A1, which selectively inhibited ALDH1A1, and did not observe changes in DCLK1 expression when compared with control siRNA treated KPC cells (Fig.2G right panel). Jointly, the data suggests that IL17 directly regulates DCLK1 which, in turn, may regulate ALDH1A1.
IL17 regulates expansion of DCLK1+ pancreatic progenitors in the presence of oncogenic Kras
To investigate the in vivo role of IL17 in inducing the development of DCLK1+ cells in the pancreatic oncogenic epithelium, we achieved IL17 over-expression by adenovirus transfer into the pancreas of KCiMist1 mice (Fig. S6A). Mice with pancreatic IL-17-overexpression had more extensive PanINs, as previously reported7, but they also had a significant increase in the relative abundance of DCLK1+ cells in ADMs and PanINs when compared with mice injected with control virus (Fig. 3A, B, S4). To determine if DCLK1+ cells over-represented in PanINs formed in the presence of higher IL17 levels were actually tuft cells we performed co-staining with known tuft cells markers: Acetylated-α-tubulin (Ac-α-tubulin), Transient receptor potential cation channel subfamily M member 5 (TRPM5) and cyclooxygenase-2 (COX2). We found that ~98% of DCLK1+ cells in PanINs co-express the tuft cell structural component Ac-α-tubulin while 92% and 94% of them co-express TRPM5 and COX2, respectively, confirming that IL17 induces expansion of pancreatic DCLK1+ tuft cells (Fig. S5A,B). Of note, no significant differences were noted in the PanIN DCLK1 co-labeling with tuft cells markers between KCiMist1 Ad Luc and KCiMist1 AdIL17, suggesting that the number of DCLK1+ pancreatic progenitor cells is increased by IL17 but not their phenotype.
Figure 3. IL17 regulates expansion of DCLK1+ pancreatic progenitors in presence of oncogenic Kras.
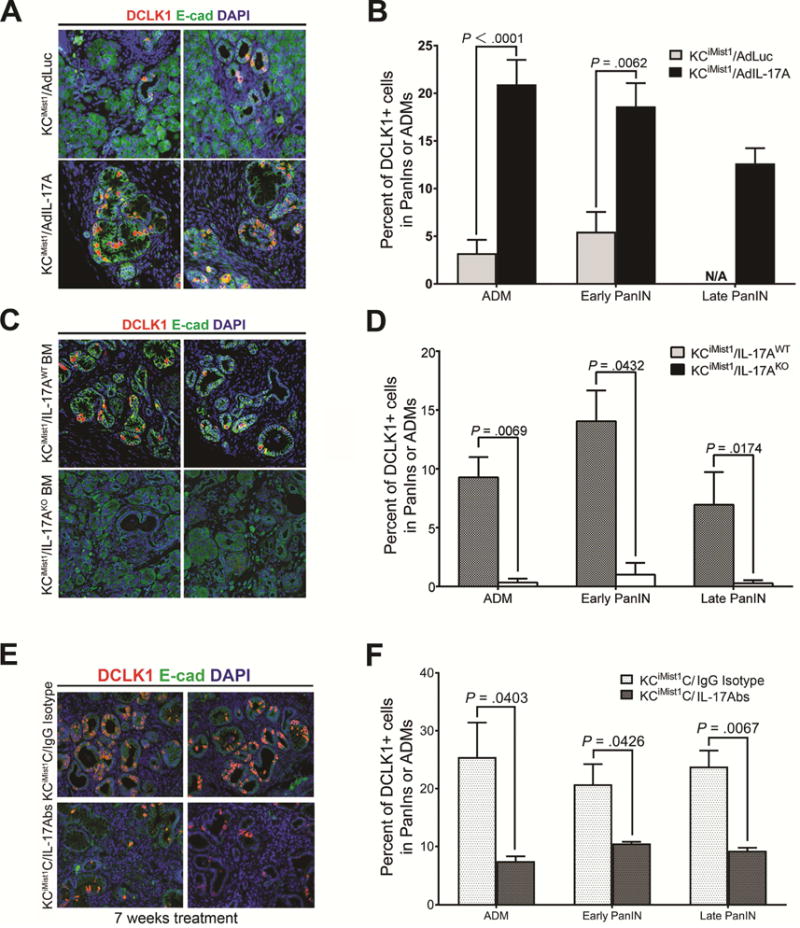
(A,C,E) Representative pictures of DCLK1 and e-cadherin immunofluorescence staining in ADMs or PanINs from AdLuc vs AdIL17 injected KCiMist1 mice (A), KCiMist mice transplanted with IL17-deficient vs WT bone marrow (C) and KCiMist mice that after cerulean inoculations were treated with IL17 neutralizing antibodies vs IgG isotype (E).
(B, D, F) Quantification of the number of cells expressing Dclk-1 in lesions from AdLuc vs AdIL17 injected KCiMist1 mice (B), KCiMist mice transplanted with IL17-deficient vs WT bone marrow (D) and KCiMist + CP mice treated for 7 weeks with IL17 neutralizing antibodies vs IgG isotype (F). Results expressed as percent of Dclk-1 positive cells/ADM or PanIN.
To test if absence of IL17 signaling would decrease DCLK1+ cells, we then performed genetic ablation of IL17 from the hematopoietic compartment of KCiMist1 mice (Fig. S6B). This model resulted in ADMs and PanINs with significantly lower numbers of DCLK1+ cells when compared with lesions from mice transplanted with wild type (WT) bone marrow (Fig. 3C,D). Secondly, we analyzed KCiMist1 mice that had undergone pharmacological inhibition of the IL17 pathway (Fig S6C) and found that DCLK1+ cells were significantly decreased in ADMs and PanINs from mice that received IL17 neutralizing antibodies for seven weeks (Fig.3E,F). Of note, when we quantified DCLK1+ cells in the short term neutralization model (Fig 1A), no significant differences in DCLK1 expression were found between the groups (Fig S6D), suggesting that neutralization of the IL17 pathway for sufficient time may be able to revert the embryonic stem cell signature that aids tumor initiation and progression.
Taken together, we conclude that IL17 is able to regulate the expansion of DCLK1+ pancreatic progenitors in the oncogenic epithelium.
Regulation of DCLK1 by IL17 is mediated by NFkB canonical pathway activation
To gain further understanding about early signaling events involved in the IL17-mediated induction of DCLK1 and other stemness-related genes, we performed RNA Sequencing (RNAseq) on KPC cells stimulated for only 24 hrs with IL17-enriched or control media. Unsupervised principal component analysis (PCA) was performed on gene expression profiles from the groups, and distinctive clusters were obtained, indicative of a strong intragroup gene expression correlation but differences in intergroup gene expression signatures (Fig. S7A). We performed a statistical comparison to identify genes that were differentially expressed in the IL17-treated-KPC cells versus untreated ones. Based on a false discovery rate (FDR) corrected p-value of less than 0.05 and fold-changes of greater or equal to 1.5, a total of 117 genes were found differentially regulated, from which 99 were upregulated and 18 downregulated (Table S2). We then searched for common genes upregulated early in KPC cells treated in vitro with IL17 vs genes downregulated in PanIN cells sorted from mice treated with monoclonal antibodies against IL17, and found that 16 genes were commonly regulated by IL17 (Fig. 4A). To gain further understanding into the regulatory mechanisms, we utilized IPA to identify gene networks. The main network identified by analysis of commonly regulated genes had central nodes of interconnected regulatory system integrated by NFkB, ERK1/2, AKT, JNK and p38 MAPK (Fig 4B), as previously described in other cell types35–37. To validate these findings, immunoblotting was used to detect expression of these pathways in IL17-treated KPC cells. NFkB p65 nuclear translocation and p-ERK expression were increased as early as 15 min after IL17 administration (Fig. 4C, S7B, S7C). In contrast, p38, JNK and AKT were not activated by IL17 treatment of KPC cells (Fig. S7D). To confirm IL17 mediated transcriptional activation of NFkB we measured NFkB DNA binding activity in KPC and Capan-2 cells exposed to IL17 stimulation in vitro and found significant increase in binding activity with peak at 30 minutes for KPC and 15 minutes for Capan-2 cells (Fig. 4D, S8D).
Figure 4. Regulation of DCLK1 by IL17 is mediated by NFκB canonical pathway activation.
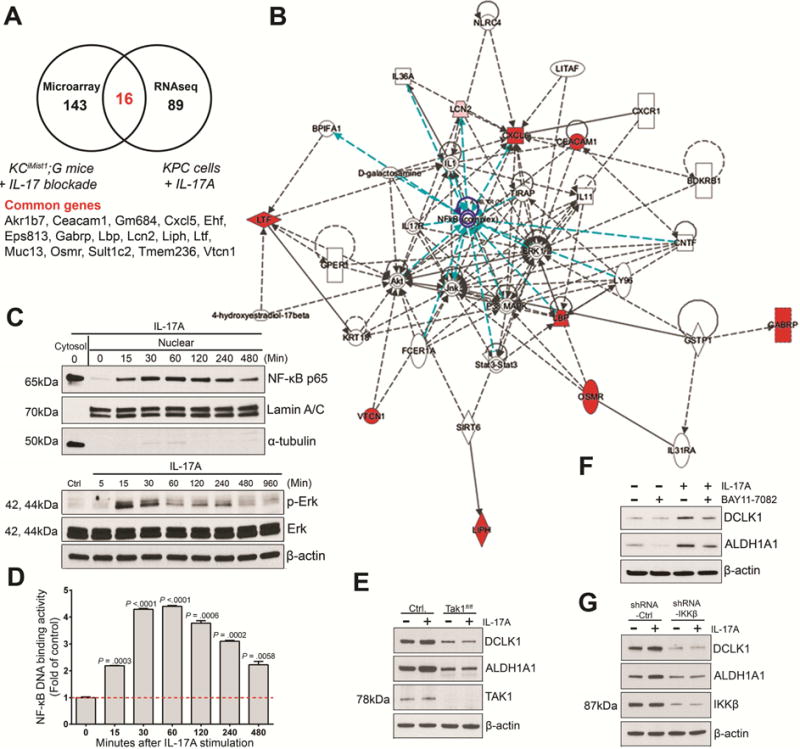
(A) Venn’s diagram of the cross comparison of significantly changed genes based on data from microarray analysis (genes downregulated in PanINs upon in vivo IL17 blockade) and RNA-seq (genes upregulated by IL17-mediated stimulation of KPC cells) and Compendium of genes commonly regulated by IL17 in the RNAseq vs microarray data.
(B) Network analysis on commonly changed genes using Ingenuity Pathway Analysis. NFκB node and connections are highlighted in blue.
(C) Immunoblotting for NFkBp65 (top) in nucleus and cytoplasm of KPC cells stimulated with IL17. The purity of nuclear extracts was confirmed by immunoblotting with anti-lamin A/C and α-tubulin. Immunoblotting for p-Erk (bottom) activation and total Erk on KPC cells stimulated with IL17.
(D) Time-dependent NFκB DNA binding activity measured in KPC cells stimulated with IL17. Results are expressed as fold changes over control KPC cells untreated. This experiment was repeated 2 times and a representative experiment is shown.
(E) Immunoblotting for DCLK1 on Tak1-competent (Control) and Tak1-deficient (Takf/f) KPC cells in the presence and absence of IL17 (10 ng/ml) for 7 days.
(F) Immunoblotting for DCLK1 on KPC cells stimulated with IL17 upon NFkB inhibitor (Bay11-7082) for 5 days. β-actin was used as loading control in C,E,F.
(G) Immunoblotting for DCLK1 and IKKβ on KPC cells treated with IKKβ shRNA vs control shRNA, in the presence and absence of IL17 (10 ng/ml) for 7 days.
Based on these experimental results, we hypothezised that the activation of the NFkB and MAPK/ERK pathways may potentially be involved in IL17-mediated induction of DCLK1 and stemness in neoplastic pancreatic epithelial cells. Since TGF-β activated kinase (TAK1) has been described as a critical regulator of both NFκB and MAPK/ERK pathways38, we utilized KPC-Tak1f/f cells treated with IL17 to confirm if the IL17-dependent induction of DCLK1 was mediated by NFκB and/or MAPK/ERK pathways. We found that Tak1-deficient KPC cells were unable to upregulate DCLK1 in response to IL17 stimulation (Fig. 4E, S7E), consistent with a role for NFκB and/or MAPK/ERK pathways in IL17-stimulation responses. To determine if either or both NFκB and/or ERK activation were required for IL17-mediated induction of DCLK1, we used inhibitors of both pathways. Only the NFκB activation inhibitor (BAY11-7082), but not the MAPK/ERK inhibitor (U0126), was able to supress IL17-dependent increase in DCLK1 expression in KPC cells (Fig. 4F, S7F, S7G). To assess for the pathway of activation of NFκB, we used IKKβ shRNA knockdown in KPC cells, since phosphorylation of IKKβ is required for activation of the NFkB via the canonical pathway39, and found that DCLK1 upregulation was repressed upon IL17 stimulation (Fig. 4G, S7H). Therefore, we concluded that IL17 induces DCLK1 transcription through activation of NFκB via the canonical pathway.
We then hypothesized that IL17-induced MAPK/ERK activation is a consequence of elevated DCLK1 expression induced by IL17, based on a recent publication which showed that DCLK1 loss was associated with significant reduction in ERK phosphorylation22. To test this, we performed an experiment using DCLK1 siRNA and after validating the functional inhibition of DCLK1 (Fig 2G left panel), we tested for ERK phosphorylation upon IL17 induction and found that Erk activation is unchanged upon DCLK1 inhibition, concluding that DCLK1 induction is only mediated by NFκB activation and ERK is directly activated by IL17 but does not mediate DCLK1 induction (Fig. S7I).
IL17RC is differentially overexpressed in murine pancreatic DCLK1+ tuft cells and human pancreatic DCLK1+ cancer cells
We have previously shown that IL17RA is overexpressed in pancreatic premalignant lesions7. We now focused on assessing for the differential expression of IL17 receptors within mPanINs DCLK1+ cells induced by Kras activation (Fig 5A). To this end, PCR was conducted on DCLK1+ vs DCLK1− sorted GFP+ cells (Fig S9A) and IL17RA expression level was found comparable in both fractions (data not shown). Since IL17 signals through IL17RA which dimerizes with the inducible IL17RC40, 41, we then assessed for IL17RC relative abundance in the same cellular populations and found significantly higher expression of IL17RC in the mPanIN DCLK1+ cells compared with DCLK1− cells (P= .0008) (Fig. 5B). To confirm if the IL17RC is specifically induced in PanIN tuft cells, we assessed for IL17RC levels in GFP+ cells sorted based on Ac-α-tubulin and DCLK1 staining (Fig. S9B) and found significantly higher expression of IL17RC in Ac-α-tubulin+ cells that in Ac-α-tubulin− sorted GFP+ cells (Fig S9C). Importantly from mPanINs Ac-α-tubulin+ cells, only those expressing DCLK1 (GFP+/Ac-α-tubulin+/DCLK1+) had higher expression of IL17RC (Fig. S9D).
Figure 5. IL17RC is differentially overexpressed in DCLK1+ mPanIN cells as well as in human pancreatic cancer DCLK1+ cells.
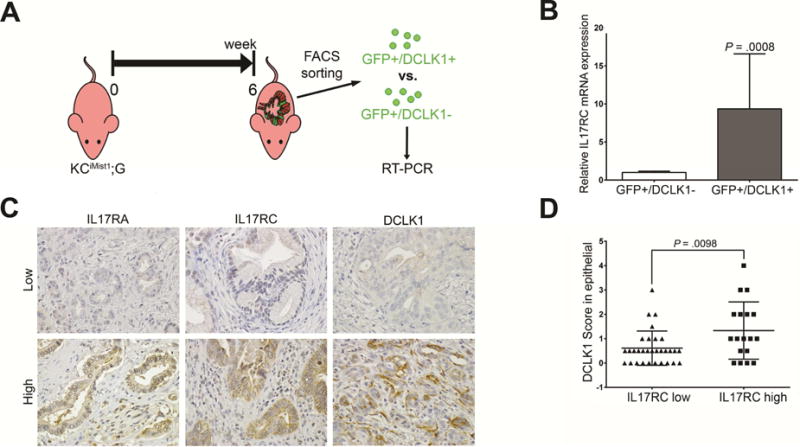
(A) Protocol for sorting GFP+/Dclk+ vs GFP+/DCLK1− cells
(B) Relative expression of IL17RC mRNA expression quantified by RT-PCR from GFP+/DCLK1+ vs GFP+/DCLK1− cells based on DCLK1 antibody staining. Results show the mean ± SD (n=2) of fold changes from GFP+/DCLK1+ cells over GFP+/DCLK1−, (*P < .05).
(C) Representative photos of Low and High expression of IL17RA, IL17RC, and DCLK1 in human PDAC.
(D) Comparison of DCLK1 expression between PDAC patient tissues with high IL17RC expression vs. low IL17RC expression, based on immunohistochemistry staining score (n=65).
Furthermore, in an attempt to test if the differential expression of IL17-receptors in murine DCLK1+ cells can also be detected in human pancreatic tumoral epithelium, we classified PDAC tissue based on levels of expression of IL17RA, IL17RC and DCLK1 (Fig. 5C). Importantly, the pattern of DCLK1 staining on human PDAC was very different from the one observed in PanINs, in which it was primarily staining cells with the typical shape of tuft cells.
We then compared DCLK1 scores in IL17RC low-expressing vs high expressing tissue and found significantly higher DCLK1 scores in the IL17RC high-expressing tissue (Fig. 5D). Of note, no differences in DCLK1 scores were noted between tissues with IL17RA high and low expression (Fig. S9E).
High expression of three IL17-regulated ESC signature genes in PDAC patients predict poor survival
We performed immmunohistochemical staining for DCLK1 in tissue from 65 resected pancreatic cancer cases and found the expression of this marker to have predictive value for poor survival (Fig. 6A). In our patients cohort, the median survival of DCLK1-low patients was 26.6 months vs 17.7 months in the DCLK1-high patients (P= .0136).
Figure 6. High expression of three IL17-regulated ESC signature genes in PDAC patients predict poor survival.
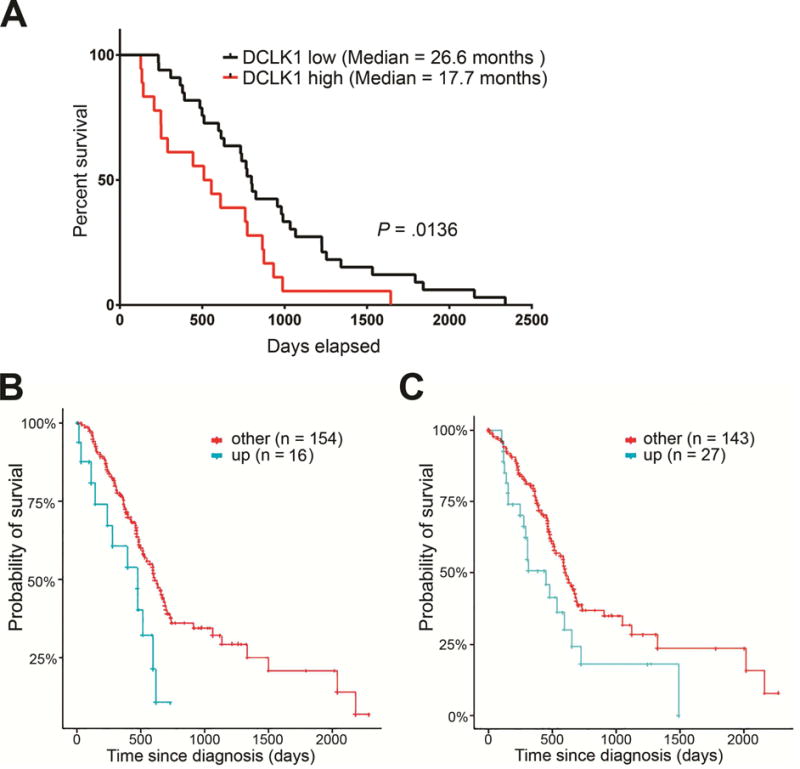
(A) Kaplan-Meier survival curve of PDAC patients with DCLK1 high vs. low expression based on stainings score (n=65).
(B,C) Kaplan-Meier survival curve based on expression of POU2F3 (B) and LAMC2 (C) on 170 patients with PDAC from TCGA (upregulated vs others).
Finally, we sought to determine the potential clinical relevance of the top 20 individual ESC signature genes regulated by IL17 in pancreatic oncogenic epithelium by performing survival analysis using data from PDAC patients (n=170) from the TCGA database. We compared patients with upregulated gene expression versus either downregulation or no change. We found that two genes had significant prognostic value in patients with PDAC: POU2F3 (P= .025) and LAMC2 (P= .03) (Fig. 6B, C). Importantly, a total of 9% of analyzed PDAC patients had upregulation of POU2F3 whereas ~16% of patients had LAMC2 upregulated.
The data strongly suggest that IL17 signaling promotes stemness in pancreatic oncogenic epithelium through induction of genes which expression is predictive of survival. LAMC2 and POU2F3 are novel oncogenic predictors of poor survival in pancreatic cancer patients.
DISCUSSION
The link between chronic inflammation and cancer has been extensively studied and has strongly implicated immune cells. In particular, IL17 secreting cells have been associated with chronic inflammation42 and cancer development through diverse mechanisms that include recruitment of myeloid cells43, angiogenesis and epithelial proliferation promotion8, 44,45. In this manuscript we describe a novel mechanism for IL17-mediated induction in pancreatic tumorigenesis. We found that several global functions were in vivo induced by IL17 but when we functionally in vitro interrogated cancer cells for each of those functions, we found that only stemness was consistently induced by IL17 stimulation. The lack of in vitro effect in some of those functions, like proliferation or cellular movement, might be explained by the requirement of other factors, which may only be provided in the in vivo model, as previously described for other IL17-related effects8, 46. Another explanation could lay on the requirement of further epigenetic reorganization for stem cells to acquire an EMT phenotype as reported by Gaeta et al47. We have shown that IL17 can induce direct regulation of DCLK1, a process dependent on the activation of NFκB via canonical pathway. Using several in vivo approaches, we showed that Il17 mediates expansion of DCLK1 cells in pancreatic progenitors. The IL17RA is an ubiquitous receptor that we have already described as overexpressed in premalignant epithelial lesions. We now determined that PanINs DCLK1+ cells have differential upregulation in the IL17RC which has been previously described as an inducible receptor. The over-expression of IL17RC in PanIN DCLK1+ cells might be induced by Kras-dependent inflammation.
Chronic pancreatitis is a risk factor for pancreatic cancer but the underlying mechanisms remain unknown. We propose that pancreatic chronic inflammation induces recruitment of immune cells capable of IL17 secretion which induces embryonic stem cells reprogramming with the primary goal of promoting epithelia regeneration but in the context of oncogenic Kras activation the stemness activation further promotes cancer initiation and progression. A recent study describes a similar phenomena in which IL17 is involved in colon tissue repair and tumorigenesis as IL17-signaling-deficient mice had abnormal epithelial regeneration and reduced tumor burden upon colorectal chemical carcinogenesis induction48. Furthermore, that study describes a novel IL17-target gene, PLET1, a progenitor cell marker involved with wound healing, which is induced in the colon upon injury in an IL17RC-dependent manner48.
Previous studies have shown that human PDAC is characterized by expression of stemness signature. Using the same stemness analytical tool that we used, Haider et al have recently analyzed genetic expression profiles from 466 patients with PDAC and have found that the genes that inversely correlated with survival were enriched for ESC signature49, 50. We have now found that upregulation of three genes included in our IL17-induced stemness pattern, DCLK1, POU2F3 and LAMC2, are predictors of worse survival in human PDAC. Recently, a monoclonal antibody against DCLK1 (CBT-15G) has been used for pancreatic cancer treatment in preclinical setting with very promising results51. POU2F3 has been postulated as a candidate tumor suppressor gene in cervical cancer52 but it may play a different role in pancreatic cancer as it is upregulated in almost 10% of the patients. LAMC2 encodes for laminin gamma-2, a glycoprotein that forms the basal lamina recently proposed as a PDAC early detection and poor prognosis prognostic biomarker in colon cancer patients53,54. Furthermore, since LAMC2 can interact with cell-surface receptors to direct tumor migration or invasion, it could be a potential effective cancer target55.
In conclusion, the regulation that IL17-secreting immune cells impose on the oncogenic epithelium by activating an embryonic stem cell program has wide implications. Further understanding of the effect of TH17 cells on pancreatic cancer stemness and invasiveness induction would be highly significant, not only because it illustrates novel mechanisms underlying the highly aggressive characteristics of pancreatic cancer but also, given the immediate availability of commercially available IL17 monoclonal antibodies, it will provide a practical approach to preventing or treating this lethal disease.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Table 1. Provided as an Excel file.
Supplementary Table 2. Provided as an Excel file.
Acknowledgments
The authors wish to thank Mien-Chie Hung, PhD, for helpful discussions on the manuscript and Amgen for the generous provision of neutralizing antibodies.
Grant support
Dr McAllister received support from the PanCAN/AACR Career Development Award (14-20-25 MCAL), National Pancreas Foundation and V Foundation (V Scholar). Dr McAllister is also a Paul Calabresi K12 clinical scholar (NCI grant awarded to MDACC K12CA088084-16A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors Contribution: Yu Zhang was involved in the study design, acquisition, analysis and interpretation of data as well as writing and revision of the manuscript. Michelle Zoltan, Erick Riquelme, Ismet Sahin, Hanwen Xu, Maria Fernanda Montiel, Susana Castro-Pando, Kyle Chang, Melissa Pruski, Zhengyu Jiang, Jianhua Ling, Sonal Gupta and William Horne have been involved in data acquisition, analysis and interpretation. Huamin Wang has provided material support. Shao-Cong Sun, Guillermina Lozano, Anirban Maitra, Paul Chiao, Steven D. Leach, Jay K. Kolls, Eduardo Vilar Sanchez, Timothy Wang and Jennifer Bailey have been involved in analysis and interpretation of data as well as critical revision of manuscript. Florencia McAllister has been involved in study conception and design, data analysis and interpretation, statistical analysis and manuscript writing.
Disclosures: The authors have nothing to disclose.
References
- 1.Kamisawa T, Wood LD, Itoi T, et al. Pancreatic cancer. Lancet. 2016;388:73–85. doi: 10.1016/S0140-6736(16)00141-0. [DOI] [PubMed] [Google Scholar]
- 2.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 3.Numasaki M, Watanabe M, Suzuki T, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol. 2005;175:6177–89. doi: 10.4049/jimmunol.175.9.6177. [DOI] [PubMed] [Google Scholar]
- 4.Benevides L, da Fonseca DM, Donate PB, et al. IL17 Promotes Mammary Tumor Progression by Changing the Behavior of Tumor Cells and Eliciting Tumorigenic Neutrophils Recruitment. Cancer Res. 2015;75:3788–99. doi: 10.1158/0008-5472.CAN-15-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kryczek I, Wei S, Szeliga W, et al. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114:357–9. doi: 10.1182/blood-2008-09-177360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Habbe N, Shi G, Meguid RA, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–8. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAllister F, Bailey JM, Alsina J, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell. 2014;25:621–37. doi: 10.1016/j.ccr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAllister F, Henry A, Kreindler JL, et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol. 2005;175:404–12. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kryczek I, Lin Y, Nagarsheth N, et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40:772–84. doi: 10.1016/j.immuni.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang M, Li Y, Huang K, et al. IL-33 promotes colon cancer cell stemness via JNK activation and macrophage recruitment. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-16-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bie Q, Sun C, Gong A, et al. Non-tumor tissue derived interleukin-17B activates IL-17RB/AKT/beta-catenin pathway to enhance the stemness of gastric cancer. Sci Rep. 2016;6:25447. doi: 10.1038/srep25447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Lee CJ, Simeone DM. Identification of human pancreatic cancer stem cells. Methods Mol Biol. 2009;568:161–73. doi: 10.1007/978-1-59745-280-9_10. [DOI] [PubMed] [Google Scholar]
- 14.Rasheed ZA, Yang J, Wang Q, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102:340–51. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Rogoff HA, Keates S, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A. 2015;112:1839–44. doi: 10.1073/pnas.1424171112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duong HQ, Hwang JS, Kim HJ, et al. Aldehyde dehydrogenase 1A1 confers intrinsic and acquired resistance to gemcitabine in human pancreatic adenocarcinoma MIA PaCa-2 cells. Int J Oncol. 2012;41:855–61. doi: 10.3892/ijo.2012.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delgiorno KE, Hall JC, Takeuchi KK, et al. Identification and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology. 2014;146:233–44 e5. doi: 10.1053/j.gastro.2013.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westphalen CB, Asfaha S, Hayakawa Y, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest. 2014;124:1283–95. doi: 10.1172/JCI73434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey JM, Alsina J, Rasheed ZA, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology. 2014;146:245–56. doi: 10.1053/j.gastro.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May R, Sureban SM, Lightfoot SA, et al. Identification of a novel putative pancreatic stem/progenitor cell marker DCAMKL-1 in normal mouse pancreas. Am J Physiol Gastrointest Liver Physiol. 2010;299:G303–10. doi: 10.1152/ajpgi.00146.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saqui-Salces M, Keeley TM, Grosse AS, et al. Gastric tuft cells express DCLK1 and are expanded in hyperplasia. Histochem Cell Biol. 2011;136:191–204. doi: 10.1007/s00418-011-0831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westphalen CB, Takemoto Y, Tanaka T, et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell. 2016;18:441–55. doi: 10.1016/j.stem.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen F, Ruddy MJ, Plamondon P, et al. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J Leukoc Biol. 2005;77:388–99. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Liao MY, Gao XL, et al. IL-17A induces pro-inflammatory cytokines production in macrophages via MAPKinases, NF-kappaB and AP-1. Cell Physiol Biochem. 2013;32:1265–74. doi: 10.1159/000354525. [DOI] [PubMed] [Google Scholar]
- 25.Fortunel NO, Otu HH, Ng HH, et al. Comment on “‘Stemness’: transcriptional profiling of embryonic and adult stem cells” and “a stem cell molecular signature”. Science. 2003;302:393. doi: 10.1126/science.1086384. author reply 393. [DOI] [PubMed] [Google Scholar]
- 26.Ramalho-Santos M, Yoon S, Matsuzaki Y, et al. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 27.Ivanova NB, Dimos JT, Schaniel C, et al. A stem cell molecular signature. Science. 2002;298:601–4. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- 28.Munoz J, Stange DE, Schepers AG, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012;31:3079–91. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerbe F, Sidot E, Smyth DJ, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature. 2016;529:226–30. doi: 10.1038/nature16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westphalen CB, Quante M, Wang TC. Functional implication of Dclk1 and Dclk1-expressing cells in cancer. Small GTPases. 2016:1–8. doi: 10.1080/21541248.2016.1208792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rovira M, Scott SG, Liss AS, et al. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A. 2010;107:75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomita H, Tanaka K, Tanaka T, et al. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget. 2016;7:11018–32. doi: 10.18632/oncotarget.6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weygant N, Qu D, Berry WL, et al. Small molecule kinase inhibitor LRRK2-IN-1 demonstrates potent activity against colorectal and pancreatic cancer through inhibition of doublecortin-like kinase 1. Mol Cancer. 2014;13:103. doi: 10.1186/1476-4598-13-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng X, Dzamko N, Prescott A, et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol. 2011;7:203–5. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie S, Li J, Wang JH, et al. IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J Immunol. 2010;184:2289–96. doi: 10.4049/jimmunol.0903133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu FM, Li QL, Gao Q, et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer. 2011;10:150. doi: 10.1186/1476-4598-10-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li JK, Nie L, Zhao YP, et al. IL-17 mediates inflammatory reactions via p38/c-Fos and JNK/c-Jun activation in an AP-1-dependent manner in human nucleus pulposus cells. J Transl Med. 2016;14:77. doi: 10.1186/s12967-016-0833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ajibade AA, Wang Q, Cui J, et al. TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity. 2012;36:43–54. doi: 10.1016/j.immuni.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho AW, Shen F, Conti HR, et al. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J Immunol. 2010;185:1063–70. doi: 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Y, Ota N, Peng I, et al. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4307–16. doi: 10.4049/jimmunol.0903614. [DOI] [PubMed] [Google Scholar]
- 42.Beringer A, Noack M, Miossec P. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol Med. 2016;22:230–41. doi: 10.1016/j.molmed.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 43.He D, Li H, Yusuf N, et al. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol. 2010;184:2281–8. doi: 10.4049/jimmunol.0902574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Numasaki M, Fukushi J, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–7. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 45.Wang K, Kim MK, Di Caro G, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity. 2014;41:1052–63. doi: 10.1016/j.immuni.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang CQF, Akalu YT, Suarez-Farinas M, et al. IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: potential relevance to psoriasis. J Invest Dermatol. 2013;133:2741–2752. doi: 10.1038/jid.2013.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaeta X, Xie Y, Lowry WE. Sequential addition of reprogramming factors improves efficiency. Nat Cell Biol. 2013;15:725–7. doi: 10.1038/ncb2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zepp JA, Zhao J, Liu C, et al. IL-17A-Induced PLET1 Expression Contributes to Tissue Repair and Colon Tumorigenesis. J Immunol. 2017;199:3849–3857. doi: 10.4049/jimmunol.1601540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haider S, Wang J, Nagano A, et al. A multi-gene signature predicts outcome in patients with pancreatic ductal adenocarcinoma. Genome Med. 2014;6:105. doi: 10.1186/s13073-014-0105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinto JP, Kalathur RK, Oliveira DV, et al. StemChecker: a web-based tool to discover and explore stemness signatures in gene sets. Nucleic Acids Res. 2015;43:W72–7. doi: 10.1093/nar/gkv529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nathaniel Weygant DQ, May Randal, Chandrakesan Parthasarathy, Ge Yang, Ryan Chelsea D, An Guangyu, Schlosser Michael J, Bannerman-Menson Edwin, Houchen Courtney W. Systemic delivery of CBT-15G DCLK1-targeted monoclonal antibody dramatically decreases tumorigenesis in a xenograft model of pancreatic cancer (Abstract 577). Cancer Research- Proceedings: AACR 107th Annual Meeting 2016; New Orleans, LA. 2016. [Google Scholar]
- 52.Zhang Z, Huettner PC, Nguyen L, et al. Aberrant promoter methylation and silencing of the POU2F3 gene in cervical cancer. Oncogene. 2006;25:5436–45. doi: 10.1038/sj.onc.1209530. [DOI] [PubMed] [Google Scholar]
- 53.Kosanam H, Prassas I, Chrystoja CC, et al. Laminin, gamma 2 (LAMC2): a promising new putative pancreatic cancer biomarker identified by proteomic analysis of pancreatic adenocarcinoma tissues. Mol Cell Proteomics. 2013;12:2820–32. doi: 10.1074/mcp.M112.023507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang D, Du C, Ji D, et al. Overexpression of LAMC2 predicts poor prognosis in colorectal cancer patients and promotes cancer cell proliferation, migration, and invasion. Tumour Biol. 2017;39:1010428317705849. doi: 10.1177/1010428317705849. [DOI] [PubMed] [Google Scholar]
- 55.Garg M, Kanojia D, Okamoto R, et al. Laminin-5gamma-2 (LAMC2) is highly expressed in anaplastic thyroid carcinoma and is associated with tumor progression, migration, and invasion by modulating signaling of EGFR. J Clin Endocrinol Metab. 2014;99:E62–72. doi: 10.1210/jc.2013-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zubarik R, Gordon SR, Lidofsky SD, et al. Screening for pancreatic cancer in a high-risk population with serum CA 19-9 and targeted EUS: a feasibility study. Gastrointest Endosc. 2011;74:87–95. doi: 10.1016/j.gie.2011.03.1235. [DOI] [PubMed] [Google Scholar]
- 57.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Table 1. Provided as an Excel file.
Supplementary Table 2. Provided as an Excel file.