

Abstract
Interactions of amyloid-β (Aβ) peptides and cellular membranes are proposed to be closely related with Aβ neurotoxicity in Alzheimer’s disease. In this study, we systematically investigated the effect of the N-terminal hydrophilic region of Aβ40 on its amyloidogenesis and interaction with supported phospholipid bilayer. Our results show that modulation of the charge properties of the dynamic N-terminal region dramatically influences the aggregation properties of Aβ. Furthermore, our results demonstrate that the N-terminal charged residues play a crucial role in driving the early adsorption and latter remobilization of the peptide on membrane bilayer, and mediating the rigidity and viscoelasticity properties of the bound Aβ40 at the membrane interface. The results provide new mechanistic insight into the early Aβ-membrane interactions and binding, which may be critical for elucidating membrane-mediated Aβ amyloidogenesis in a physiological environment and unravelling the origin of Aβ neurotoxicity.
Keywords: adsorption kinetics, aggregation, electrostatic interactions, lipids, peptides
Role of the N-terminal charged region
Interactions between amyloid-β (Aβ) peptides and cellular membranes are closely related with Aβ neurotoxicity in Alzheimer’s disease. We systematically investigated the effect of the dynamic N-terminal region of Aβ40 on its amyloidogenesis and interactions with supported phospholipid bilayer (see figure).
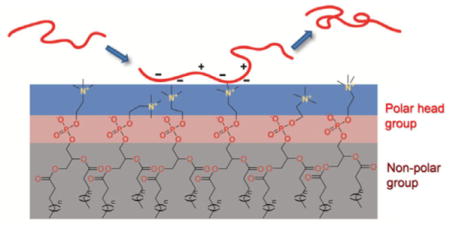
Accumulation and amyloid fibril formation of amyloid-β (Aβ) peptides is associated with the pathogenesis of Alzheimer’s disease (AD).[1] Although the fundamental mechanism by which the assembly of Aβ leads to neuronal death is still unclear, a prominent focus of mechanistic studies has centered on the interaction of Aβ with lipid bilayer of neuronal cells, and the subsequent damage of membrane.[2] The cell membrane bilayer provides an extensive surface with which Aβ can interact, and may play a critical role in mediating Aβ self-assembly and toxicity.[3] However, the critical interactions that drive and regulate the adsorption and binding of Aβ at the surface of membrane are not fully characterized. Although electrostatic interactions have been recognized to play a pivotal role in mediating Aβ-membrane interactions and binding,[4] the detailed mechanistic view of such interactions in directing early Aβ-membrane interaction and adsorption, as well as the crucial residues that participate in these interactions, have yet to be fully explored experimentally.
The N-terminal region (D1–K16) of Aβ is mainly composed of hydrophilic amino acids, amounting to approximately 60 % of the total residues. Six residues in this region contain charged side chains under physiological conditions. A variety of NMR structural studies show that the N-terminal region of Aβ remains disordered and flexible in the final fibrillar state.[5] However, recent emerging studies have suggested that this conformationally dynamic region has substantial influence on Aβ aggregation properties.[6] While these pioneering studies underscore the importance of this dynamic region in Aβ self-association, there are little systematic studies on the mechanistic roles of the properties of the N-terminal residues, for example, charge properties and hydrophobicity, in determining Aβ aggregation and guiding Aβ-membrane interactions and binding. There is in vitro and in silico evidence suggesting that electrostatic interactions play important roles in Aβ fibril formation and stabilization.[7] In the present work, we perform a systematic mutation study on the N-terminal charged amino acid residues of Aβ40, and investigate the role of these residues in regulating Aβ40 amyloidogenesis, as well as in mediating adsorption kinetics, binding properties, and remobilization of Aβ on a supported membrane bilayer.
To modulate the N-terminal charge properties of Aβ40, we made eight mutants by substituting certain charged residues with amino acids of different charge properties, as shown in Figure 1 (physical properties of the mutants summarized in Table S1). The unnatural amino acids ornithine (Orn), 2,4-diaminobutyric acid (Dab), and norleucine (Nle) were used to minimize mutation-induced steric interference because of the similar size of their side chains to the corresponding natural amino acids.
Figure 1.
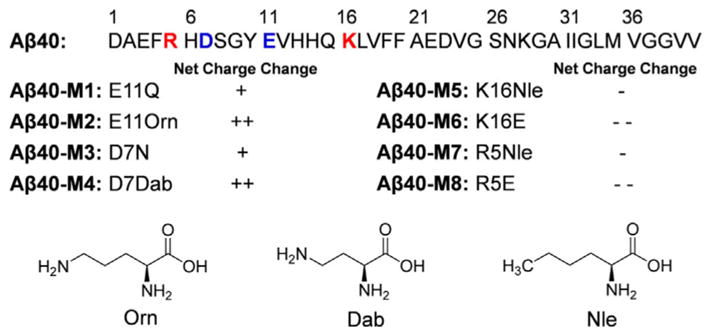
Primary sequence of Aβ40 and the mutations of the N-terminal charged residues. “ +”, “ + +”, “−”, and “− −” denote the mutation-induced change in net charge at pH 7.4. The molecular structures of the unnatural amino acids used in the study are shown.
The morphology of the peptide aggregates was first examined using atomic force microscopy (AFM) imaging. Seven of the mutants, except Aβ40-M5, aggregated into amyloid fibrils after incubation (Figure 2). Aβ40-M1, M3, M4, M6-M8, all formed long and curly fibrils, similar to the wild type Aβ40. Aβ40-M2, on the contrary, mainly formed short and straight fibrillar structures. The aggregation kinetics of the Aβ40 mutants were monitored using thioflavin T (ThT) fluorescence (Figure S1). As summarized in Figure 3A, while the ThT fluorescence intensity at the stationary phase of most peptides is not dramatically different, the final ThT fluorescence intensity of Aβ40-M2 is only approximately 42 % of that of Aβ40, significantly lower than other homologues (except Aβ40-M5). This is in accord with what was observed in AFM imaging. Furthermore, Aβ40-M2 shows the shortest aggregation half time (t50) of approximately 2.6 h (Figure 3B). The results of Aβ40-M2 underscore the importance of charged amino acids at the N-terminal region in affecting amyloid formation. Substituting the Glu11 residue to a positively charged Orn dramatically facilitates peptide amyloidogenesis. The shorter length of the fibrils formed by Aβ40-M2 indicates the sensitivity of morphology and stability of amyloid structures to mutation-induced primary sequence change. Interestingly, a similar mutation of replacing Asp7 residue with Dab (Aβ40-M4) leads to a noticeable while less dramatic aggregation rate change (t50 of Aβ40 vs. Aβ40-M4), revealing the sensitivity of such influence to the specific position of the residue in the primary sequence.
Figure 2.

Tapping mode AFM images of Aβ40 and the mutants. The AFM images were acquired after incubating the samples (30 μM) for 6 d at 37 °C in Tris buffer (50 mM Tris, 150 mM NaCl) of pH 7.4.
Figure 3.
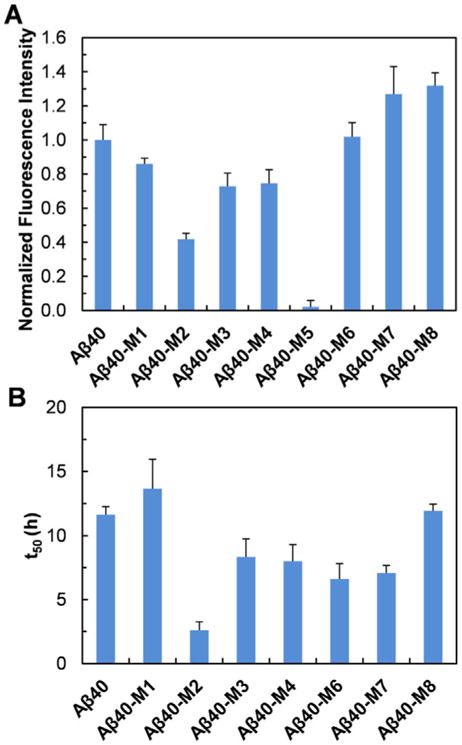
(A) Normalized ThT fluorescence intensity at the stationary phase of the aggregation of Aβ40 and the mutants (10 μM, pH 7.4) at 37 °C. (B) The aggregation half time (t50) of Aβ40 and the mutants (10 μM, pH 7.4) followed by ThT fluorescence at 37 °C.
Mutation of the Lys16 residue to a neutral Nle in Aβ40-M5, dramatically disrupts fibrillation of the peptide in Tris buffer. No fibrils were observed in AFM imaging of Aβ40-M5 (Figure 2); instead, appreciable amount of small oligomeric aggregates formed. There is negligible ThT fluorescence intensity after 30 h of incubation (Figure S1). These are consistent with our previous report that the aggregation of this peptide produces stable oligomers instead of proceeding to form fibrils in phosphate buffer.[8] The Lys16 residue is in close proximity to the hydrophobic core “L17VFFA21” region, which is essential for Aβ oligomer and fibril formation.[9] K16Nle mutagenesis dramatically increases the hydrophobicity of the peptide (Table S1). It is conceivable that in the initial collapse process of Aβ self-association,[10] the K16Nle mutation may strongly favor the intermolecular hydrophobic interactions, thus enhancing the thermostability of oligomers. This would further increase the energy cost of conformational conversion of the spherical oligomers to form nucleus seeds, which is considered to be the rate-limiting step towards fibril formation.[11] Kaden et al. recently reported that an Aβ40 derivative with K16N mutation mainly forms low-n oligomers,[12] in agreement with our results. The Aβ40-M6 peptide, in which the Lys16 residue is mutated to a negatively charged Glu, steadily aggregates to form fibrillar structures, similar to the wild type Aβ40. This further validates the crucial role of the 16th amino acid in mediating the local polarity property next to the central hydrophobic core, which is critical in directing the oligomer formation and subsequent conformational conversion to fibrils. Together, our results show that the charged residues in the N-terminal region are actively involved in mediating the self-association pathway and the morphology of the aggregated state. While the detailed interactions that these residues are involved in along the aggregation pathway cannot be specified from our results, they may form non-specific inter- and intramolecular electrostatic interactions and hydrogen bonding that can alter the aggregation characteristics of peptides.[7c, 13]
To gain insight into the role of the charge-rich N-terminal region of Aβ40 in mediating Aβ-membrane interaction and binding, we studied the deposition kinetics of Aβ40 and its mutants on a model 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) membrane using a quartz crystal microbalance with dissipation monitoring (QCM-D). Phosphatidylcholine, the most abundant phospholipid of mammalian membranes,[14] is one of the main compositions in brain cell membranes.[15] POPC is used in this study to prepare a supported lipid bilayer on silica surface. The adsorbed mass of peptides during adsorption process was calculated by fitting the frequency and dissipation shifts obtained in QCM-D (Figure S2). As shown in Figure 4A, the deposition rate of Aβ40 on POPC bilayer is approximately 24 ng cm−2 min−1. The deposition rate of Aβ40-M1 is approximately 3.8 ng cm−2 min−1, accounting for only approximately 16 % of that of Aβ40. The deposition rate of Aβ40-M2 is approximately 1.3 ng cm−2 min−1, showing a more significant decrease. These results suggest that the two mutants have significantly lower affinity on POPC membrane surface compared to the wild type analogue. POPC contains a zwitterionic head group composed of a positively charged quaternary amine group and a negatively charged phosphate (Figure 4B). When POPC forms bilayers, the amine groups are protruding into solvent while the phosphate groups are relatively more buried within the bilayer. The positively charged amine groups are likely more accessible for first interacting with the peptides in close proximity to the bilayer surface via electrostatic interactions. Substitution of the Glu11 residue to Gln, in the case of Aβ40-M1, likely eliminates the favorable attractive electrostatic interactions between Glu11 and the amine groups on the surface of the bilayer, leading to drastic decrease of the deposition rate. The substitution of Glu11 with an ornithine residue, which contains a positively charged side chain at pH 7.4, may introduce unfavorable repulsive electrostatic interactions between the residue and the choline head group at the bilayer surface,[16] accounting for the lowest deposition rate of Aβ40-M2.
Figure 4.
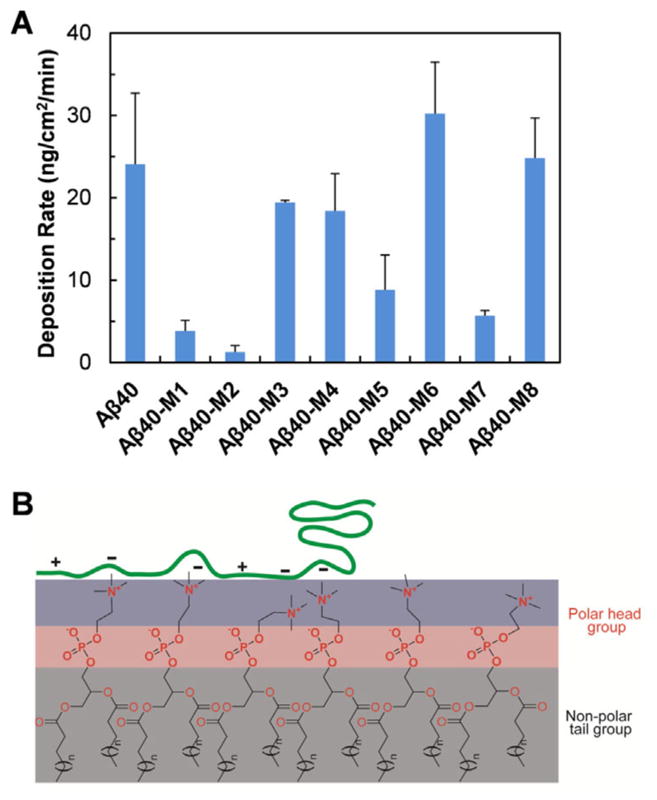
(A) Deposition rate of Aβ40 and the mutants on supported POPC lipid bilayer measured using QCM-D. (B) Schematic representation of electrostatic interactions between the flexible and charge-rich N-terminal region of Aβ40 and the POPC lipid bilayer.
Similar studies were also carried out on Aβ40-M3 and Aβ40-M4, in which Asp7 is substituted to Asn and Dab, respectively. Interestingly, although a decrease of the deposition rate in comparison to Aβ40 is also observed (Figure 4A), the amplitude of the decrease is significantly smaller compared to that of Aβ40-M1 and Aβ40-M2. This result indicates the sensitivity of specific position of the N-terminal charged residues in mediating binding with phospholipid membrane. Although Aβ is generally considered to be intrinsically disordered, previous studies suggest that the peptide can adopt fluctuating residual structures and be partially folded in aqueous solution.[17] The formation of local residual structures may put Glu11 at a conformation favorable for initial interaction with the surface of the membrane bilayer. In addition, as Asp7 is closer to the N-terminus, the highly dynamic characteristics of the residue may also in part be responsible for its lesser effect on regulating binding of the peptide with membrane. Together, these results indicate the important influence of the charge properties of the residues at the N-terminal region, as well as their specific positions, on peptide deposition onto phospholipid membranes. In addition, interactions of lipid membrane and amyloidogenic proteins are significantly influenced by the properties of lipids, such as the type of lipids, hydrophobicity, and length of acyl chain.[18] Our results suggest that the amine moiety in the head group of POPC, instead of the phosphate group, is more responsible for early interacting with Aβ peptide, reinforcing the importance of chemical properties of lipids in affecting interactions of membrane and amyloidogenic proteins.[19]
The deposition rate of Aβ40-M6, in which the positively charged Lys16 is mutated to Glu, is approximately 30 ng cm−2 min−1 (Figure 4), slightly faster than that of Aβ40. This could be reasonably attributed to a more favorable electrostatic interaction between Glu and the positively charged choline group on POPC bilayer surface. Lys16 is one of the crucial amino acids responsible for anchoring the peptide onto phospholipid membrane.[12, 20] Interestingly, when Lys16 is mutated to a neutral Nle residue, the deposition rate is dramatically decreased compared to the wild type analogue (Aβ40-M5 vs. Aβ40 in Figure 4A), likely due to the increased hydrophobicity of the region caused by mutation (Table S1), thus weakening interactions with the hydrophilic membrane surface. Mutation of Arg5 to an oppositely charged Glu (Aβ40-M8) does not change the deposition rate of the peptide dramatically (Figure 4A), possibly because Arg5 is close to the N-terminal of the sequence and does not contribute much to the electrostatic interactions between the peptide and membrane. Mutation of the Arg5 residue to Nle leads to a decrease of the deposition rate of Aβ40-M7. This is in agreement with the results of Aβ40-M5, indicating the importance of the hydrophobic properties of the residues in influencing peptide adsorption on membranes.
The viscoelasticity properties of the adsorbed peptide on membrane surface were studied by analyzing the shear moduli and viscosities of peptide layers at the end of the deposition period via fitting frequency and dissipation shifts using Voigt model (results of Aβ40 shown in Figure S3).[21] As depicted in Figures 5A and 5B, elimination of negative charge and/or introduction of additional positive charge at position 7 or 11 results in lower shear moduli and viscosities than that of Aβ40. These mutations likely interfere with the original electrostatic interaction of the peptide with the membrane surface, resulting in less firm contact with the surface of POPC membrane. No reliable values of shear modulus and viscosity of Aβ40-M2 could be obtained, possibly because of the fast fibrillation rate of this mutant (Figure 3B and S1) which leads to the formation of more complicated assembled structures on the surface of POPC membrane. K15E and R5E mutations in Aβ40-M6 and Aβ40-M8, do not change the shear modulus and viscosity values dramatically (Figures 5C and 5D). This suggests that these mutations do not dramatically influence the rigidity of the peptide on the surface of the model membrane. The shear moduli and viscosities of Aβ40-M5 (K16Nle) and Aβ40-M7 (R5Nle), however, are smaller compared to Aβ40, likely due to the combinational effects of elimination of unfavorable electrostatic interaction which favors firm contact and introduction of hydrophobic side chains which weakens the contact of peptides with the hydrophilic surface of membrane.
Figure 5.
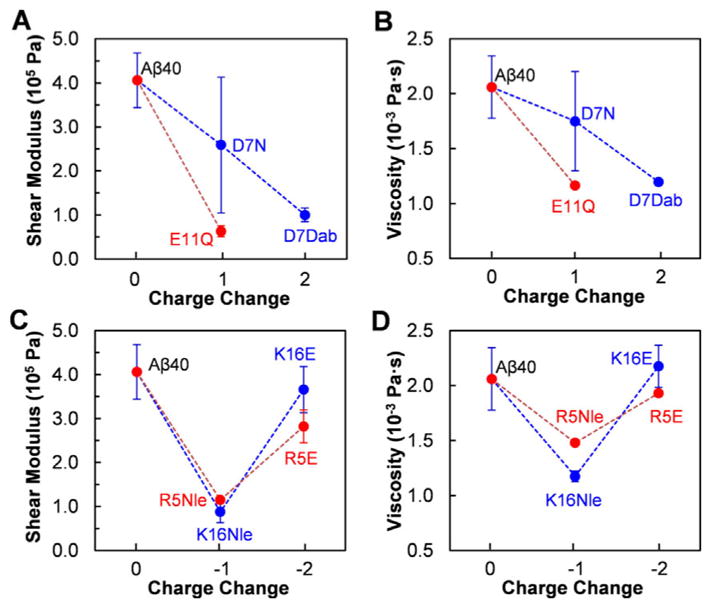
Shear modulus (A and C) and viscosity (B and D) of the deposited peptide layers on POPC model membrane at the end of deposition period.
The reversibility of peptide adsorption on the POPC membrane was also investigated by QCM-D. In general, the deposition reversibility of the peptides correlates inversely with the initial deposition rate of the peptides on POPC membrane (Figure 4A vs. Figure 6A). The mutants with low deposition rates, i.e., Aβ40-M1, Aβ40-M2, Aβ40-M5, Aβ40-M7, are easier to be released from membrane surface. For Aβ40-M1 and Aβ40-M2, the reversibility is higher than 100 %. This could be a result of the lipid of the supported bilayer washed off the silica surface. In particular, Aβ40-M2 shows the highest reversibility of around 347 %. The fast aggregation rate of Aβ40-M2, and the likely firm contact of Aβ40-M2 with membranes, may result in significant amount of lipids carried away by the released peptides via a “detergent-like” mechanism.[22] The peptides that exhibit high deposition rates (i.e., Aβ40, Aβ40-M3, Aβ40-M4, Aβ40-M6, Aβ40-M8) tend to remain on the membrane surface and have low reversibility due to their higher binding affinities on membrane.
Figure 6.
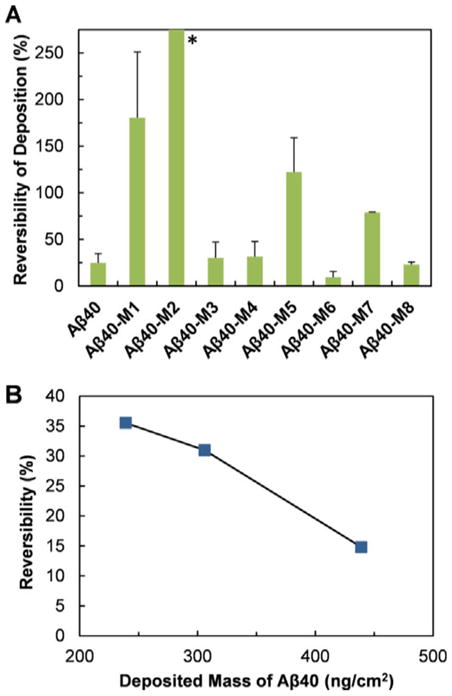
(A) Reversibility of Aβ40 and the mutants after deposition on POPC lipid bilayer. (B) Reversibility of Aβ40 deposition on POPC lipid bilayer as a function of the initial deposited mass. *The average reversibility of Aβ40-M2 is 347 %, which is not shown for better presentation of the reversibility data of other peptides.
The influence of the surface concentration of the deposited Aβ40 on peptide reversibility from membrane surface was further studied. As shown in Figure 6B, the reversibility decreases from 36 to 15 % as the surface concentration of the peptide increases from 239 to 439 ng cm−2. The decrease of reversibility with increasing surface density of deposited peptide is reasonably due to stacking of the deposited peptide at higher surface densities, and the resultant difficulty of the peptide in diffusing away from the membrane surface.[23] This finding has significant implication, as it will be increasingly difficult to reverse the binding of Aβ peptides on cell membranes if the accumulation of peptides on membranes is not alleviated at the early stage. It has been reported that membrane disruption by Aβ may occur by a two-step mechanism, with the initial formation of pores followed by nonspecific fragmentation of the lipid membrane during amyloid fiber formation.[24] Preventing the initial adsorption of the peptide on membrane by targeting on the crucial charged residues at the N-terminal region may be an alternative strategy for ameliorating Aβ-induced cellular membrane damage.
In summary, we have systematically investigated the roles of the N-terminal charged residues of Aβ40 in regulating the intrinsic aggregation properties of Aβ and interactions with lipid membrane. Mutations on critical charged residues, e.g., Glu11 and Lys16, dramatically interfere with Aβ amyloid fibril formation. Furthermore, our results show that the electrostatic interactions between the N-terminal crucial charged residues and membrane surface are crucial in determining the early adsorption kinetics and later detachment property of Aβ on membrane bilayer, and mediating the rigidity and viscoelasticity of the membrane-bound Aβ. The results provide novel insight into the mechanistic functions of the dynamic N-terminal region in the early Aβ-membrane interactions and binding. These findings may inspire development of novel strategies for blocking formation of toxic Aβ structures responsible for membrane damage, by modulating crucial interactions between the N-terminal charged residues and lipid membranes.
Supplementary Material
Acknowledgments
D.D. gratefully acknowledges the financial support from the National Institutes of Health (R15GM116006) and the Alzheimer’s Association (AARG-17–531423).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/chem.201801805.
References
- 1.a) Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]; b) Selkoe DJ. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]; c) Tanzi RE, Bertram L. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.a) Arispe N, Pollard HB, Rojas E. Proc Natl Acad Sci USA. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kotler SA, Walsh P, Brender JR, Ramamoorthy A. Chem Soc Rev. 2014;43:6692–6700. doi: 10.1039/c3cs60431d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Serra-Batiste M, Ninot-Pedrosa M, Bayoumi M, Gairí M, Maglia G, Carulla N. Proc Natl Acad Sci USA. 2016;113:10866–10871. doi: 10.1073/pnas.1605104113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Waschuk SA, Elton EA, Darabie AA, Fraser PE, McLaurin J. J Biol Chem. 2001;276:33561–33568. doi: 10.1074/jbc.M103598200. [DOI] [PubMed] [Google Scholar]; b) Lin H, Bhatia R, Lal R. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]; c) Diaz JC, Simakova O, Jacobson KA, Arispe N, Pollard HB. Proc Natl Acad Sci USA. 2009;106:3348–3353. doi: 10.1073/pnas.0813355106. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Proc Natl Acad Sci USA. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) MacManus A, Ramsden M, Murray M, Henderson Z, Pearson HA, Campbell VA. J Biol Chem. 2000;275:4713–4718. doi: 10.1074/jbc.275.7.4713. [DOI] [PubMed] [Google Scholar]
- 4.a) McLaurin J, Chakrabartty A. FASEB J. 1997;245:355–363. doi: 10.1111/j.1432-1033.1997.t01-2-00355.x. [DOI] [PubMed] [Google Scholar]; b) Tofoleanu F, Buchete NV. J Mol Biol. 2012;421:572–586. doi: 10.1016/j.jmb.2011.12.063. [DOI] [PubMed] [Google Scholar]; c) Pannuzzo M, Milardi D, Raudino A, Karttunen M, La Rosa C. Phys Chem Chem Phys. 2013;15:8940–8951. doi: 10.1039/c3cp44539a. [DOI] [PubMed] [Google Scholar]
- 5.a) Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Maji SK, Ogorzalek Loo RR, Inayathullah M, Spring SM, Vollers SS, Condron MM, Bitan G, Loo JA, Teplow DB. J Biol Chem. 2009;284:23580–23591. doi: 10.1074/jbc.M109.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qahwash I, Weiland KL, Lu Y, Sarver RW, Kletzien RF, Yan R. J Biol Chem. 2003;278:23187–23195. doi: 10.1074/jbc.M213298200. [DOI] [PubMed] [Google Scholar]; c) Brännström K, Ohman A, Nilsson L, Pihl M, Sandblad L, Olofsson A. J Am Chem Soc. 2014;136:10956–10964. doi: 10.1021/ja503535m. [DOI] [PubMed] [Google Scholar]
- 7.a) Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. Biochemistry. 2005;44:6003–6014. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]; b) Petkova AT, Yau WM, Tycko R. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yun S, Urbanc B, Cruz L, Bitan G, Teplow DB, Stanley HE. Biophys J. 2007;92:4064–4077. doi: 10.1529/biophysj.106.097766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elbassal EA, Morris C, Kent TW, Lantz R, Ojha B, Wojcikiewicz EP, Du D. J Phys Chem C. 2017;121:20007–20015. doi: 10.1021/acs.jpcc.7b05169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Williams AD, Shivaprasad S, Wetzel R. J Mol Biol. 2006;357:1283–1294. doi: 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]; b) Cheon M, Chang I, Mohanty S, Luheshi LM, Dobson CM, Vendruscolo M, Favrin G. PLoS Comput Biol. 2007;3:e173. doi: 10.1371/journal.pcbi.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Iwata K, Lachenmann M, Peng J, Li S, Stimson E, Lu Y-A, Felix A, Maggio J, Lee J. J Struct Biol. 2000;130:130–141. doi: 10.1006/jsbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- 11.Lee J, Culyba EK, Powers ET, Kelly JW. Nat Chem Biol. 2011;7:602–609. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaden D, Harmeier A, Weise C, Munter LM, Althoff V, Rost BR, Hildebrand PW, Schmitz D, Schaefer M, Lurz R. EMBO Mol Med. 2012;4:647–659. doi: 10.1002/emmm.201200239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meli M, Morra G, Colombo G. Biophys J. 2008;94:4414–4426. doi: 10.1529/biophysj.107.121061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadowaki H, Grant MA. J Lipid Res. 1995;36:1274–1282. [PubMed] [Google Scholar]
- 15.Kosicek M, Hecimovic S. Int J Mol Sci. 2013;14:1310–1322. doi: 10.3390/ijms14011310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seelig J, MacDonald PM, Scherer PG. Biochemistry. 1987;26:7535–7541. doi: 10.1021/bi00398a001. [DOI] [PubMed] [Google Scholar]
- 17.a) Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJL, Ray DG, Vitek MP. J Am Chem Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]; b) Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. Biochem Biophys Res Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) La Rosa C, Scalisi S, Lolicato F, Pannuzzo M, Raudino A. J Chem Phys. 2016;144:184901–184911. doi: 10.1063/1.4948323. [DOI] [PubMed] [Google Scholar]; b) Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee YH, La Rosa C, Lim MH. J Biol Chem. 2017;292:4638–4650. doi: 10.1074/jbc.M116.764092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galvagnion C, Brown JW, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, Sparr E, Dobson CM. Proc Natl Acad Sci USA. 2016;113:7065–7070. doi: 10.1073/pnas.1601899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poojari C, Strodel B. PLoS One. 2013;8:e78399. doi: 10.1371/journal.pone.0078399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voinova MV, Rodahl M, Jonson M, Kasemo B. Phys Scr. 1999;59:391–396. [Google Scholar]
- 22.Martel A, Antony L, Gerelli Y, Porcar L, Fluitt A, Hoffmann K, Kiesel I, Vivaudou M, Fragneto G, De Pablo JJ. J Am Chem Soc. 2017;139:137–148. doi: 10.1021/jacs.6b06985. [DOI] [PubMed] [Google Scholar]
- 23.Yi P, Chen KL. Environ Sci Technol. 2013;47:12211–12218. doi: 10.1021/es403133r. [DOI] [PubMed] [Google Scholar]
- 24.Sciacca MF, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. Biophys J. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.