

Abstract
A stereoselective synthesis of the AB ring of the complex sesterterpenoid variecolin is presented. Our strategy features the development of a tandem Wolff/Cope rearrangement of α-diazo cyclobutyl ketones for the construction of fused, eight-membered carbocycles. Preliminary studies revealed a facile Wolff rearrangement but a difficult vinyl ketene cyclobutane Cope rearrangement. We have leveraged an efficient microwave-promoted tandem rearrangement to prepare the desired functionalized cyclooctadienones that we envision as potential key intermediates in the convergent synthesis of variecolin.
Graphical Abstract
Introduction
The complex sesterterpenoid variecolin (1) was isolated in 1991 by Hensens and co-workers from the fungal fermentation extracts of Aspergillus variecolor.1 The fascinating polycyclic structure of 1 was elucidated by NMR methods and later confirmed by X-ray crystallography,2 with the natural antipode3 represented in Figure 1. Preliminary biological evaluation has established an array of activities for variecolin. The original report identified 1 as a modest angiotensin II receptor binding inhibitor with potential application in the treatment of hypertension.1 Subsequent studies demonstrated 1 to suppress the proliferation of induced mouse splenic lymphocytes.3 Variecolin has also been shown to compete with macrophage inflammatory protein (MIP)-1α for binding to CCR5,2 a key chemokine receptor involved in the uptake of HIV-1 into target cells. Other reports have identified general antifungal properties.4 Several variecolin congeners have also been identified from other fungal species, with many exhibiting a similar biological profile (Figure 1).2,3,4,5,6 The varied activity and intriguing molecular architecture of this family of sesterterpenes serves to stimulate new strategies targeting their chemical synthesis. Our interest in the pursuit of an effective synthetic strategy toward these bioactive natural products focused on variecolin given that it is the most widely studied and biologically relevant member.
Figure 1.

Variecolin family of sesterterpenes.
Synthesis Design
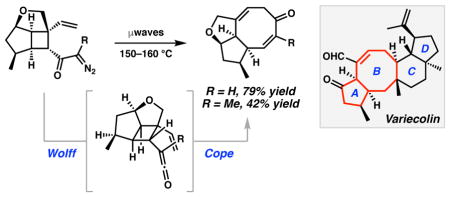
The 25-carbon tetracyclic core of variecolin (1) consists of a central eight-membered ring and possesses eight stereocenters, including two all-carbon quaternary stereocenters at C(11) and C(14) of the BC and CD ring fusions, respectively. Synthetic control of the relative and absolute stereochemistry of these trans-fused rings that form a stereopentad poses a notable design challenge for critical bond-forming reactions. We therefore envisioned that the assembly of these key features of variecolin in a convergent manner would provide opportunities to explore new reactivity. When considering key elements toward our synthetic design, we were drawn to the eight-membered B ring. Eight-membered rings are common structural motifs that occur in widely diverse terrestrial plants, insects, marine organisms, and fungi. The theoretical and synthetic intrigue of these medium-sized carbocyclic structures has stimulated the development of various strategies for their preparation, many of which have been applied toward the synthesis of complex molecular targets.7 Inasmuch as we are restricted by the limitations of reaction scope in the state of the art, the selective preparation of eight-membered carbocycles remains a noteworthy and continuing challenge to modern chemical methods.8 No total synthesis of variecolin or any family members has been reported to date, however, significant progress toward this end has been achieved by the Piers9 and Molander10 labs. Related general strategic advancements have also been reported.11,12,13
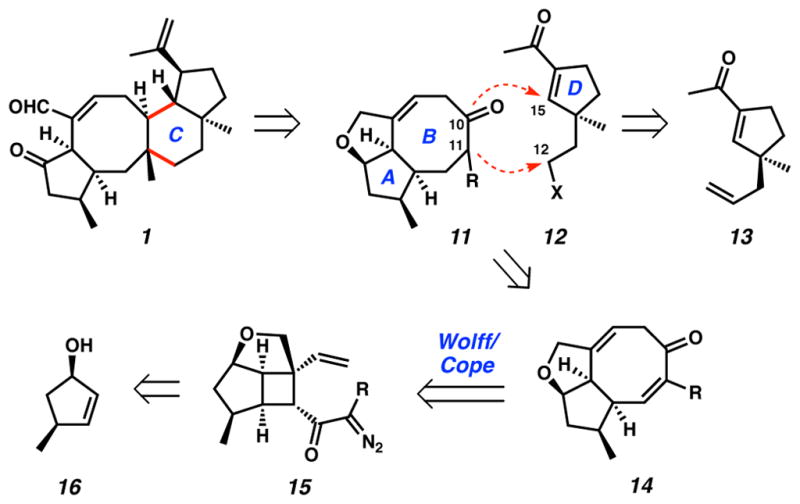
The formulation of our synthetic approach toward variecolin (1) focused on the construction of the central eight-membered ring, revealing a critical C ring disconnection that could simplify the target into two functional AB (11) and D ring (12) fragments (Scheme 1). The scalable synthesis of D ring fragment 13 has been advanced by our lab using an asymmetric alkylation/ring contraction technology.14 Notably, this acylcyclopentene motif is amenable to selective functionalization. A suitable AB ring fragment could comprise functionalized cyclooctadienone 14 in order to facilitate selective bond forming reactions at the C(10) and C(11) positions. This structural unit is comparable to fused bicyclic cycloheptadienones that have been constructed using a powerful tandem Wolff/Cope rearrangement developed in our lab.15 Extending this technology to eight-membered carbocycles, we anticipated that AB ring synthon 14 could be accessed through a Wolff/Cope rearrangement of highly functionalized α-diazo cyclobutyl ketone 15, which may in turn be assembled via a cycloaddition of allylic alcohol 16 and an appropriate functionalized olefin component. The design of tandem reaction sequences for the rapid generation of molecular complexity is an area of constant investigation in our laboratory,16 and therefore a primary objective of our synthetic endeavor toward variecolin (1) is the development of a tandem Wolff/Cope rearrangement to forge fused eight-membered carbocyclic systems. Application of this key transformation for the construction of the central B ring of 1 would expand the reaction scope, and furthermore, would create a new tool for the general preparation of natural and non-natural substances containing this eight-membered ring motif. Herein we report the asymmetric synthesis of the AB ring of variecolin employing a tandem Wolff/Cope rearrangement to forge the eight-membered carbocycle.
Scheme 1.
Retrosynthetic analysis of variecolin (1)
Results and Discussion
A. Synthesis of a Wolff/Cope Model Substrate
The development of a tandem Wolff/Cope rearrangement toward the fused bicyclic eight-membered AB ring fragment (14) required an expedient, regio and stereoselective synthesis of a highly substituted cyclobutane substrate (e.g., 15). During the course of our efforts, we elected to employ an efficacious intramolecular cyclobutadiene–olefin cycloaddition for the rapid construction of this moiety.17,18,19 Model studies pursued toward this goal provided insight into the identification and physical properties of reaction intermediates using a readily accessible cyclopentenol analogue.
Our investigations toward an AB ring model system began with the preparation of a suitable cyclopentenol that would engage cyclobutadiene via a tethered20 cycloaddition. Convenient access to stereodefined 4-substituted cyclopentenols is possible using copper-catalyzed allylic substitution chemistry. Exposure of allylic monoacetate (±)-1721 to a mixture of CuCN and p-tolylmagnesium bromide generated anti-cyclopentenol 1822 in excellent yield (Scheme 2). We then sought the alkylation of this alcohol with (cyclobutadiene)tricarbonyliron complexes that possess sufficient electrophilicity to react under mild conditions.23 This was achieved by the zinc(II)-catalyzed alkylation24 of cyclopentenol 18 with trichloroacetimidate 19 to prepare the requisite intramolecular cycloaddition substrate 20 in 76% yield. Oxidative liberation of cyclobutadiene promoted by ceric ammonium nitrate (CAN) and subsequent [4+2] cycloaddition rapidly assembled cyclobutene 21 as a single diastereomer in 76% yield.
Scheme 2.
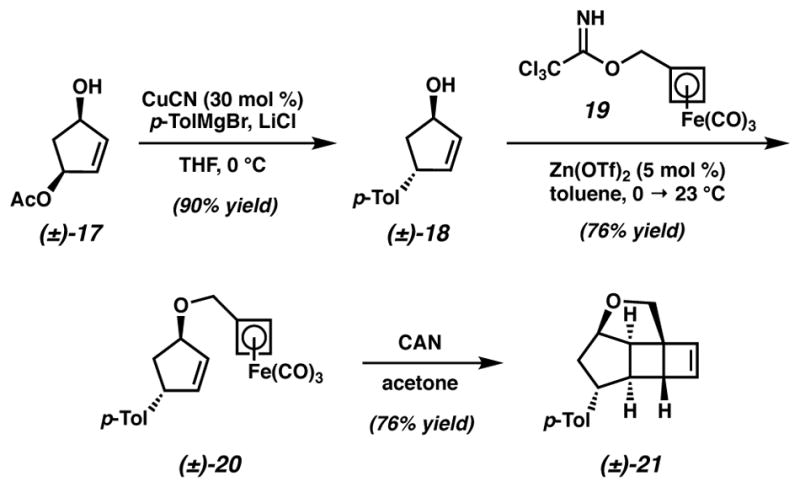
Tethered olefin-cyclobutadiene cycloaddition of alcohol (±)-18
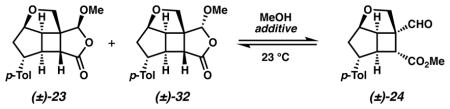
Advancement of cycloadduct 21 toward a Wolff/Cope substrate necessitated a regioselective olefin functionalization with our aim of generating a C(8) aldehyde and C(10) ester (i.e., 24).25 We were intrigued by Schreiber’s method26,27 for the ozonolytic cleavage of olefins to terminally differentiated products as it has been demonstrated as a powerful tool for the regioselective cleavage of advanced synthetic intermediates in complex molecule synthesis.28 In the event, ozonolysis of cycloadduct 21 in CH2Cl2/MeOH followed by an acylation and base induced elimination provided a mixture of several of compounds, including undesired acetal 22 in 13% yield and an inseparable 9:1 mixture of acetal 23 and desired aldehyde 24 in 68% yield (Scheme 3). Direct Wittig methylenation of this mixture afforded pure acetal 2329 in addition to minor quantities of desired olefin 25.
Scheme 3.

Termini-differentiating ozonolysis of cyclobutene 21
The production of acetals 22 and 23 from this unsymmetrical ozonolysis indicates diverging reaction pathways. Cycloaddition of ozone and cyclobutene 21 likely generates primary ozonide 26, which could fragment in two possible ways (Scheme 4, paths a and b). Cleavage of the primary ozonide to 27 (path a) positions the carbonyl oxide on the fully substituted carbon. Subsequent addition of methanol to this intermediate and dehydration of 28 with Ac2O/Et3N generates acetal 22. Conversely, primary ozonide cleavage in the opposite manner produces 29 (path b) with the carbonyl oxide positioned on the less-substituted carbon. This intermediate further reacts by another of two possible pathways: (1) addition of methanol from the reaction medium (path c) and dehydration of 30 to generate acetal 23, or (2) methanol addition to the carbonyl oxide (path d) and dehydration of 31 to furnish aldehyde 24. Although desired aldehyde 24 is a minor component of this reaction, the selectivity for the cleavage of primary ozonide 26 via the desired path b is favored in an approximate 5:1 ratio and is presumably the result of steric influences on the primary ozonide moiety.30
Scheme 4.

Diverging pathways for the termini-differentiating ozonolysis of cycloadduct 21
The identification of aldehyde 24 as a minor product from the unsymmetrical ozonolysis of cyclobutene 21 hindered progress toward a model Wolff/Cope substrate. Fortuitously, we recognized that acetal 23 and aldehyde 24 arise from the same fragmentation pathway (path b) and thus possess the same C(8) aldehyde oxidation state. Encouraged by this result, we explored potential equilibration conditions to determine the propensity for formation of aldehyde 24 from the isomeric aldehyde/acetal mixture (Table 1). We were gratified to find that solvation of pure acetal 23 in methanol effected the equilibration in favor of aldehyde 24 and produced minor quantities of acetal diastereomer 3229 (entries 1 and 2). A survey of various Lewis acids identified the proficiency of divalent triflate salts in shifting the equilibrium to further favor 24 (entries 3–6).31 Similarly, molecular sieves and combinations thereof with Lewis acids proved to be efficient for the conversion to 24 (entries 7–10) to reach the apparent equilibrium of ca. 3:1 aldehyde 24 to acetals 23 and 32. We elected to proceed in our synthesis using 4 Å MS due to operational simplicity.
Table 1.
Equilibration of acetal 23 to aldehyde 24
| ||
|---|---|---|
| entrya | additiveb | 23/32: 24c |
| 1 | — | 66 : 34 |
| 2 | —d | 39 : 61 |
| 3 | CuCl2 | 75 : 25 |
| 4 | ZnCl2 | 41 : 59 |
| 5 | Cu(OTf)2 | 36 : 64 |
| 6 | Zn(OTf)2 | 30 : 70 |
| 7 | 3 Å MS | 32 : 68 |
| 8 | 4 Å MS | 29 : 71 |
| 9 | 4 Å MS/Zn(OTf)2 | 27 : 73 |
| 10 | 4 Å MS/Cu(OTf)2 | 28 : 72 |
Each entry started from pure acetal 23.
Lewis acids were used in 20 mol %; molecular sieves were used in 0.5 g/mmol.
Ratio determined by 1H NMR analysis of crude reaction filtrate after 20–24 h.
At 50 °C.
The equilibration of acetal 23 to desired aldehyde 24 considerably improved the overall reaction sequence for the preparation of a Wolff/Cope α-diazo cyclobutyl ketone substrate. An optimized sequence involves the unsymmetrical ozonolysis of cyclobutene 21 followed by equilibration with 4 Å MS in methanol to afford ca. 1:3 ratio of acetals 23/32 and aldehyde 24 (Scheme 5a). Wittig methylenation of this crude reaction mixture produced the desired olefin (25) in 40% yield over three steps and the recovery of acetal diastereomers 23 and 32 in 14% yield. The recovered acetal mixture enabled recycling of material to augment our quantity of olefin 25 (Scheme 5b). The model Wolff/Cope substrate synthesis was completed by the high-yielding hydrolysis of ester 25 with KOTMS and conversion to α-diazoketone 34 by way of an acid chloride and diazomethane.
Scheme 5.

a) Optimized synthesis of model α-diazo cyclobutyl ketone 34; b) Recycling of acetal 23/32 to increase the production of olefin 25
B. Wolff-Cope Examination
The synthesis of α-diazo cyclobutyl ketone 34 enabled the examination of our key Wolff/Cope rearrangement toward the eight-membered B ring of variecolin. A critical component to the success of our previously reported tandem Wolff/Cope rearrangement was the identification of photochemical or silver-catalyzed sonochemical conditions to allow direct access to a variety of [n–7] fused bicyclic systems in excellent yields.15 Thorough investigations of this transformation using substrate 34 under various photochemical or silver(I)-catalyzed sonochemical conditions afforded only intractable mixtures and no evidence of the desired cyclooctadienone 35. The lack of useful information acquired from these initial experiments required us to examine the tandem process in a stepwise manner. Accordingly, irradiation of α-diazo cyclobutyl ketone 34 in methanol at 350 nm induced the photochemical Wolff rearrangement to form homologated ester 37 as the sole product, confirming the intermediacy of ketene 36 (Scheme 6). This critical result indicated that we were likely accessing a facile Wolff rearrangement, however, the ketene vinyl cyclobutane rearrangement does not readily occur under the conditions surveyed.
Scheme 6.
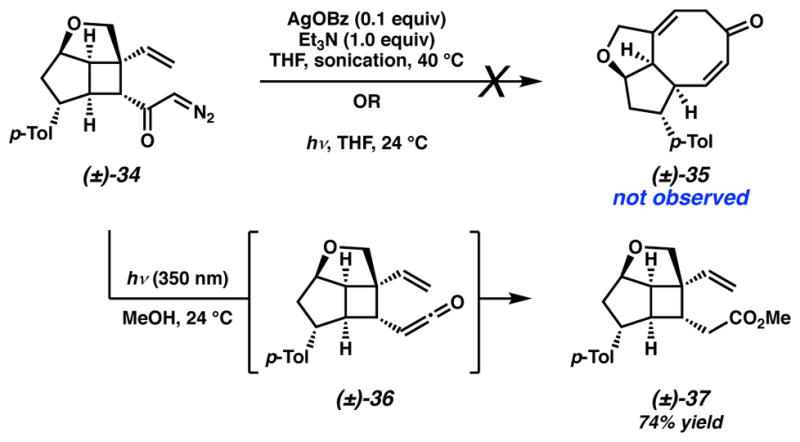
Preliminary Wolff/Cope rearrangement studies of α-diazo cyclobutyl ketone 34
In order to rationalize the difficulty of the cis-ketene vinyl cyclobutane rearrangement of 36 we considered the analogous cis-divinyl cyclobutane rearrangement.32 A comparison of the experimental activation energy for a strain-releasing Cope rearrangement of cis-divinyl cyclobutane is approximately 4–5 kcal/mol higher than the barrier of the cyclopropane analogue,33 and is consistent with the modestly elevated reaction temperatures known to be required for cis-divinyl cyclobutane rearrangements. This reactivity difference, when coupled with our observations that the ketene vinyl cyclopropane rearrangement to afford substituted cycloheptadienones occurs under mild conditions,15 suggested that thermolysis of the intermediate ketene should facilitate the rearrangement. In the event, a photochemical Wolff rearrangement with subsequent thermolysis at 80 °C provided the desired cyclooctadienone 35 in 59% yield (Scheme 7). With the success of this tandem reaction, we recognized that the general instability of a ketene intermediate and the time between photolysis/thermolysis could account for the moderate yield of 35 and furthermore might hinder material throughput. Our search for alternative conditions was bolstered by reports harnessing microwave irradiation to promote Wolff rearrangements,34 where we anticipated the surplus energy could facilitate a Cope rearrangement. Indeed, microwave irradiation of α-diazo cyclobutyl ketone 34 for 20 minutes at 140 °C in toluene afforded cyclooctadienone 35 in a remarkable 95% yield. The results of our model system explorations thereby confirm the Wolff/Cope strategy for the synthesis of the AB fragment of variecolin (1), and provide new tools for the construction of substituted eight-membered rings. Having established a viable route toward the AB ring system of variecolin through model system studies, we then pursued an asymmetric synthesis of this desirable fragment.
Scheme 7.
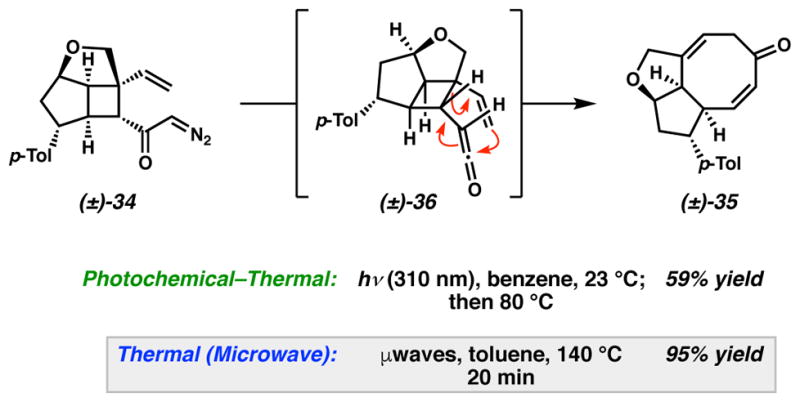
Optimal tandem Wolff/Cope rearrangement of α-diazo cyclobutyl ketone 34
C. Asymmetric Synthesis of the AB ring of Variecolin
The application of our proven intramolecular cycloaddition strategy for an asymmetric synthesis of the AB ring fragment (i.e., 14) originated from a chiral cyclopentenol possessing syn stereochemistry. Allylic substitution of monoacetate (+)-1735 using CuCN and methylmagnesium chloride afforded a 95:5 mixture of alcohols 38 and 39 in 91% yield (Scheme 8).22 Mitsunobu inversion of this alcohol mixture36 using benzoic acid and subsequent methanolysis of the resulting ester produced allylic alcohol 16 in 81% yield over two steps with the desired syn stereochemistry between C(3) and C(5). Lewis acid-catalyzed coupling24 with trichloroacetimidate 19 gave the requisite intramolecular cycloaddition substrate 40 in good yield.
Scheme 8.
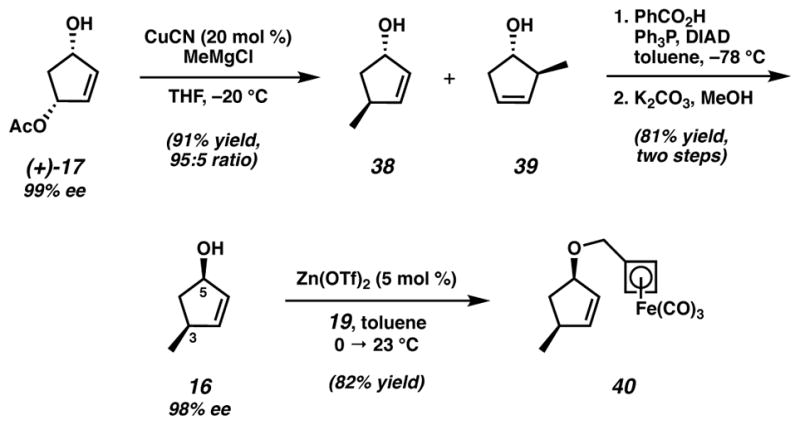
Asymmetric synthesis of intramolecular cycloaddition substrate 40
Initial efforts to implement the CAN-promoted intramolecular cyclobutadiene–olefin cycloaddition of 40 resulted in low yields and complex mixtures of products, presumably the result of competing intermolecular dimerization reactions. Snapper has demonstrated that the rapid oxidative decomplexation of (cyclobutadiene)tricarbonyliron complexes using CAN provides sufficient access to cycloadducts of substrates predisposed for the intramolecular cycloaddition.37 This works well for anti-substrate 20, however, syn-substrate 40 possesses a C(3) methyl group that sterically impedes cyclobutadiene access to the olefin. The alternative use of mild oxidants such as trimethylamine-N-oxide enact a slow release of highly reactive cyclobutadiene to facilitate access to cycloadducts of substrates with a lower propensity for the cycloaddition (e.g., 40). Application of trimethylamine-N-oxide to facilitate the oxidative decomplexation of 40 in refluxing acetone smoothly generated cycloadduct 41 (Scheme 9).29,38 Subsequent unsymmetrical ozonolysis of 41, acetal equilibration promoted by 4 Å MS in refluxing methanol and Wittig methylenation afforded a 2.7:1 ratio of olefins 44 and 45, in addition to acetals 42 and 43.29 This reaction sequence provided desired olefin 44 in 16% yield over four steps, together with 17% yield of recyclable acetal 42.39 Acetal 43 was sufficiently crystalline to enable X-ray analysis, providing further confirmation of the desired relative stereochemistry of this polycyclic fragment (Figure 2).40 It is notable that the termini-differentiating ozonolysis of 41 provided ~ 3:1 ratio of products 44:45, whereas this differentiation strategy with cyclobutene substrate 21 yielded ~ 5:1 ratio of products. The C(3) stereochemistry is the major structural difference between the two fused polycyclic substrates, and it is plausible that subtle changes of the fused ring conformation are impacting the primary ozonide fragmentation pathway (paths a and b, Scheme 4) in an unknown way.
Scheme 9.

Cycloaddition, oxidative cleavage and olefination sequence toward an AB ring Wolff/Cope substrate
Figure 2.
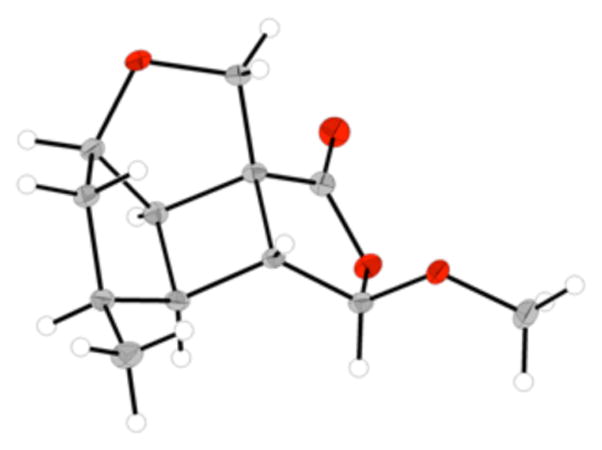
X-ray crystal structure of acetal 43. The molecular structure is shown with 50% probability ellipsoids.
We desired cyclooctadienone products in our synthesis of asymmetric AB ring fragment 14 where R = H, Me or alkyl. Model studies have demonstrated the tandem rearrangement where R = H (i.e., 34 → 35). However, alkyl substitution had not yet been explored for the Wolff/Cope rearrangement and thus represented an unprecedented extension of this method. Advancing to the target α-diazoketones, hydrolysis of ester 44 to acid 46, conversion to the acid chloride and treatment with either diazomethane (RCH2N2 where R = H) or diazoethane (R = Me) produced α-diazoketones 47 and 48, respectively (Scheme 10). The efficiency of diazomethane in the acylation of the acid chloride of 46 was notable (91% yield), however, we were surprised by the capricious and lower-yielding results obtained using diazoethane.41 Unfortunately, improvements in yield could not be realized despite efforts toward further optimization.42 We were nonetheless delighted to find that the microwave-promoted tandem Wolff/Cope rearrangement of both substrates resulted in the successful construction of their respective cyclooctadienones (49 and 50).
Scheme 10.

AB ring α-diazo cyclobutyl ketone synthesis and Wolff/Cope rearrangement
The low yield of α-substituted cyclooctadienone 50 (R = Me) using microwave irradiation is a result of the formation of numerous byproducts.43 Inspection of these various compounds revealed that cyclopropane 5129 is a major side product (ca. 1:1 ratio of 50:51) of this reaction, ostensibly formed through a carbene intermediate. Two mechanisms have been advanced for the Wolff rearrangement (Scheme 11), comprising a concerted group migration with nitrogen expulsion to a ketene (53 → 56) or the stepwise loss of nitrogen to generate an α carbonyl carbene intermediate (54 → 57).42,44 The resulting carbene intermediate can then undergo the desired Wolff rearrangement to ketene 56, or it can participate in other intra- or intermolecular reactions characteristic of carbenes, such as cyclopropanations, hydride shifts and C–H insertions. Substrate conformation has been demonstrated to be pivotal in the mechanistic outcome, with the s-Z conformation typically resulting in products consistent with the concerted pathway.44,45 Moreover, the large barrier of rotation around the C–C bond imposes a balance between electrostatic attractions and steric repulsions that influence conformation. For example, substrates where R2 = H exist mostly in the s-Z (53) conformation, whereas substrates where R2 = Me exist predominately in the s-E conformation (54).44,45
Scheme 11.
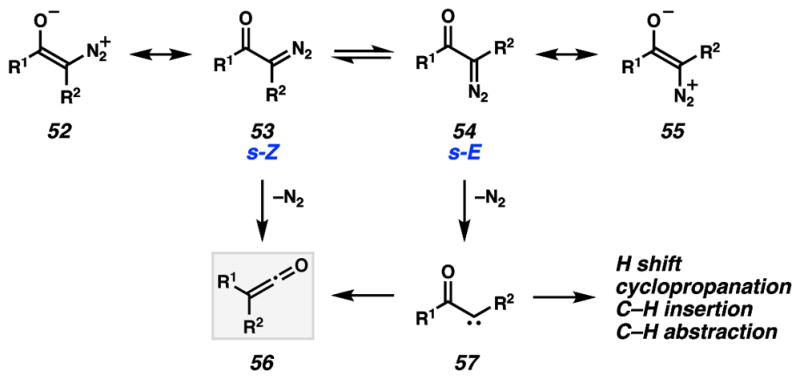
Mechanistic pathways for a Wolff rearrangement
A complicating factor in our analysis of the rearrangement of 48 is that the two mechanisms often operate competitively with high substrate dependence. Given this insight and the interactions that affect conformation, we reasoned that solvent polarity was a plausible parameter to explore. We posited that non-polar solvents might enhance the electrostatic attraction in 52, favoring the s-Z conformation and promoting the concerted rearrangement pathway to increase the production of our targeted cyclooctadienone 50 (Table 2).46 We observed that the tandem rearrangement in higher dielectric solvents such as acetonitrile or 1,2-dichloroethane favored cyclopropane formation decidedly over the Wolff rearrangement (entries 1 and 2). The data suggests that polar solvents could stabilize the charge-separated conformation (55), leading to greater population of s-E and therefore increasing amounts of carbene-related products. Lower dielectric solvents, such as THF, ethyl acetate, and toluene, modulated the selectivity and improved the formation of desired product 50 (entries 3–6). Furthermore, the nonpolar solvents methylcyclohexane and heptane reversed the reaction selectivity to favor the desired 50 as a major product in a 3:1 ratio (entries 7 and 8). Although a trend in solvent dielectric roughly reflects product selectivity in which lower dielectric solvents seemingly favor the concerted Wolff rearrangement, the complex reaction profile and other unidentified reaction products make it difficult to conclusively correlate solvent polarity to the mechanistic pathway. Moreover, we do not know if the conformational interconversion is fast relative to decomposition of 48. This is nonetheless an intriguing result that has broader implications for future applications of the Wolff/Cope method.
Table 2.
Wolff/Cope solvent studies of substrate 48
| |||
|---|---|---|---|
| entrya | solvent | dielectric constant (ε) | 50 : 51b |
| 1 | MeCN | 37.5 | 1 : 4 |
| 2 | DCE | 10.4 | 1 : 2.8 |
| 3 | THF | 7.58 | 1 : 1.1 |
| 4 | EtOAc | 6.02 | 1 : 1.3 |
| 5 | toluene | 2.38 | 1.1 : 1 |
| 6 | 1,4-dioxane | 2.21 | 1 : 1.7 |
| 7 | methylcyclohexane | 2.02 | 3 : 1 |
| 8 | heptane | 1.92 | 3 : 1 |
α-Diazoketone 48 was consumed in all reactions.
Ratios determined by 1H NMR analysis of crude reaction mixtures.
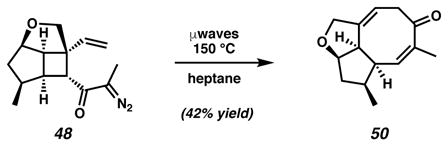
The application of solvent-optimized reaction conditions with heptane enabled the microwave-promoted rearrangement of 48 to produce α-methyl cyclooctadienone 50 in 42% isolated yield (Equation 1). The success of this rearrangement is significant as it represents the first example of the tandem Wolff/Cope rearrangement of a substrate possessing α-alkyl functionality. Moreover, the combined results from all α-diazo cyclobutyl ketone substrates in this study (i.e., 34, 47 and 48) highlight the utility of microwave irradiation to facilitate tandem rearrangements and expand the collection of eight-membered rings available by this method.
 |
(1) |
Conclusion
We have described our efforts to develop a Wolff/Cope strategy for the asymmetric synthesis of the AB ring of the complex sesterterpenoid variecolin. Our approach has employed an intramolecular cyclobutadiene–olefin cycloaddition to control product regio and stereoselectivity, as well as an advanced application of a termi-differentiating ozonolytic cleavage of a cyclobutene intermediate toward α-diazo cyclobutyl ketone substrates. These investigations have demonstrated an efficient microwave-promoted tandem Wolff/Cope rearrangement to forge highly-substituted, fused cyclooctadienone structures. Efforts are underway in our laboratory to leverage Wolff/Cope products 49 and 50 in coupling reactions with suitable D ring fragments toward completion of variecolin and will be reported in due course.
Experimental Section
General Methods
Reactions, reagents and solvents
Unless otherwise stated, reactions were performed in flame-dried glassware under an argon or nitrogen atmosphere using dry, deoxygenated solvents. Solvents were dried by passage through an activated alumina column under argon. Water (18 MΩ) used as reaction medium was obtained from a Millipore MiliQ water purification system. All starting materials were purchased from commercial sources and used as received, unless otherwise stated. Liquids and solutions were transferred via syringe or positive-pressure cannulation. Triethylamine was distilled from CaH2 prior to use. Oxalyl chloride was purified by distillation and stored in a Schlenk tube under nitrogen. Acetic acid was distilled from CrO3. Solutions of p-TolMgBr, and MeMgCl were titrated47 prior to use. Molecular sieves were dried and stored in a 115 °C oven. Diazomethane was freshly prepared from N-methyl-N-nitroso-p-toluenesulfonamide (Diazald) as a solution in Et2O using a Diazald kit. Diazoethane was freshly prepared from N-ethyl-N-nitrosourea48 as a solution in Et2O using a Diazald kit. Diazoalkane solutions were dried over KOH pellets for ca. 30 min at or below 0 °C and cannula (Teflon) transferred under nitrogen to a dry Erlenmeyer flask prior to use.
Instruments, purification and analysis
Reaction temperatures were controlled by an IKAmag temperature modulator. Ozonolysis reactions were performed with an OzoneLab OL80 Desktop ozone generator. Photochemical irradiation was performed in septum sealed quartz tubes with a Luzchem Photochemical reactor or with a water-cooled Hanovia 450 W medium pressure mercury-vapor immersion lamp. Microwave reactions were performed with a Biotage Initiator Eight 400 W apparatus at 2.45 GHz and reaction temperatures were measured with an external surface sensor. Thin-layer chromatography was performed using E. Merck silica gel 60 F254 precoated plates (0.25 mm) and visualized by UV fluorescence quenching, p-anisaldehyde, potassium permanganate, or ceric ammonium molybdate staining. SiliCycle SiliaFlash P60 Academic Silica Gel (particle size 40–63 μm; pore diameter 60 Å) was used for flash chromatography. Analytical chiral HPLC was performed with an Agilent 1100 Series HPLC utilizing a Chiralpak OD-H column (4.6 mm × 25 cm) obtained from Daicel Chemical Industries, Ltd. with 1 mL/min mobile phase and visualization at 254 nm. Analytical achiral GC was performed on an Agilent 6850 GC with FID detector using an Agilent DB-WAX (30.0 m × 0.25 mm) column at 1.0 mL/min He carrier gas flow. Chiral GC was performed on an Agilent 6850 GC with FID detector using a Chiraldex GTA column (30.0 m × 0.25 mm, purchased from Bodman Industries) at 1.0 mL/min He carrier gas flow. Optical rotations were measured with a Jasco P-1010 polarimeter at 589 nm using spectrophotometric grade solvents. 1H and 13C NMR spectra were recorded on a Varian Mercury 300 (at 300 MHz and 75 MHz respectively), Varian Inova 500 (at 500 MHz and 126 MHz, respectively) or Varian Inova 600 (at 600 MHz), and are reported relative to residual protio-solvent for 1H (C6D5H at δ 7.16 ppm, CHCl3 at δ 7.26 ppm) and solvent for 13C (C6D6 at δ 128.06 ppm, CDCl3 at δ 77.16 ppm). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). IR spectra were recorded on a Perkin Elmer Paragon 1000 spectrometer and are reported in wavenumbers (cm−1). Melting points were acquired using a Buchi Melting Point B-545 instrument and the values are uncorrected. High-resolution mass spectra were acquired using an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in ESI, APCI, or MM (ESI/APCI) ionization mode, in addition to the Caltech Mass Spectral Facility. Crystallographic data have been deposited at the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, and copies can be obtained on request, free of charge, by quoting the publication citation and the deposition number.
Trichloroacetimidate 19
To a round-bottom flask charged with hexanes-washed KH (41 mg, 1.0 mmol, 0.057 equiv) and Et2O (43 mL) at 0 °C was added a solution of (hydroxymethyl cyclobutadiene)tricarbonyliron (SI3)49 (3.86 g, 17.4 mmol, 1.0 equiv) in Et2O (43 mL, 0.2 M total) by cannula transfer. After 10 min, trichloroacetonitrile (8.7 mL, 87 mmol, 5.0 equiv) was added dropwise by syringe to the light orange solution. The reaction turned dark brow over the course of the addition. After 15 min, the ice bath was removed and upon reaching ambient temperature the volatiles were removed in vacuo. The remaining dark brown oil was taken up in hexane (20 mL, anhydrous) with vigorous shaking, filtered through a pad of Celite, and the reaction flask was washed with an additional portion of hexane (20 mL) and filtered. The combined filtrate was concentrated in vacuo to afford 19 (6.38 g, 17.4 mmol, 100% yield) as a clear, pale red oil. This oil was immediately used in the next step without further purification and it is not stable to prolonged storage. Rf = unstable to SiO2; 1H NMR (500 MHz, C6D6) δ 8.22 (br s, 1H), 4.25 (s, 2H), 3.49 (s, 2H), 3.30 (s, 1H); 13C{1H} NMR (126 MHz, C6D6) δ 214.5, 162.6, 92.0, 76.8, 65.7, 64.9, 64.5; IR (Neat Film NaCl) 3344, 2049, 1971, 1666, 1449, 1368, 1304, 1288 cm−1; HRMS (FAB+) m/z: [M – CO]+ calc′d for C9H6Cl3FeNO3 336.8763, found 336.8769.
Model cycloaddition substrate 20
To a round-bottom flask charged with Zn(OTf)2 (14.9 mg, 0.041 mmol, 0.05 equiv) and toluene (0.2 mL) at 0 °C was added aryl cyclopentenol 1822 (151.3 mg, 0.868 mmol, 1.0 equiv) by syringe. To this suspension was added a solution of 19 (370 mg, 1.01 mmol, 1.2 equiv) in toluene (0.2 mL) by cannula transfer, with further washing by additional toluene (0.2 mL). A yellow precipitate was observed at the beginning of the addition, and this turned into a thick slurry upon completion of the addition. The ice bath was allowed to expire over 1.5 h and the reaction was stirred for an additional 6 h. The crude reaction mixture was transferred directly onto a 5 g silica gel loading cartridge and purified by automated flash chromatography using a 40 g silica column (1:0 → 9:1 hexanes/EtOAc) to afford 20 (250.8 mg, 0.663 mmol, 76% yield) as a pale yellow oil. Rf = 0.56 (4:1 hexanes/EtOAc); 1H NMR (300 MHz, C6D6) δ 6.97 (ABq, ΔδAB = 0.06, JAB = 7.8 Hz, 4H), 5.88–5.85 (m, 1H), 4.45–4.44 (m, 1H), 3.96–3.94 (m, 1H), 3.55 (ABq, ΔδAB = 0.01, JAB = 9.2 Hz, 2H), 3.46 (s, 2H), 3.33 (s, 1H), 2.28 (ddd, J = 13.8, 6.9, 5.4 Hz, 1H), 2.14 (s, 3H), 1.86 (ddd, J = 13.7, 6.9, 5.4 Hz, 1H); 13C{1H} NMR (126 MHz, C6D6) δ 215.0, 142.3, 140.1, 135.9, 131.6, 129.5, 127.4, 84.9, 82.5, 64.65, 64.64, 64.0, 62.3, 50.1, 41.2, 21.0; IR (Neat Film NaCl) 2863, 2044, 1959, 1513, 1075, 1048, 613 cm−1; HRMS (FAB+) m/z: [M]+ calc′d for C20H18O4Fe 378.0554, found 378.0551.
Model cycloadduct 2129
To a vigorously stirring solution of cycloaddition substrate 20 (683 mg, 1.806 mmol, 1.0 equiv) in acetone (1.81 mL, 1 mM) was added CAN (1.98 g, 3.61 mmol, 2.0 equiv) under ambient atmosphere. After 15 min, a second portion of CAN (1.98 g, 3.61 mmol, 2.0 equiv) was added. After an additional 5 min, consumption of 20 by TLC (4:1 hexanes/Et2O, developed twice) was observed and the reaction was quenched by addition of sat aq NaHCO3 (50 mL). Stirring was ceased after 15 min, the solids were allowed to settle and the supernatant was decanted into a flask (to prevent bumping) and concentrated in vacuo to ca. 50 mL. This slurry and the remnants of the flask were transferred to a separatory funnel with minimal acetone, diluted with brine (10 mL) and pentane (200 mL). The layers were separated, the organic phase was washed with water (2 × 100 mL), and the combined aq layers were extracted with 1:1 hexanes/Et2O (200 mL). The combined organic layers were concentrated to ca. 25 mL, transferred to a sep funnel and diluted with CH2Cl2 (30 mL) and brine (25 mL). The layers were separated, the aq was extracted with CH2Cl2 (2 × 30 mL), the organics were dried over MgSO4, filtered, and concentrated to a dark orange oil. The crude material was purified by flash chromatography on SiO2 (2.5 × 21 cm, 15:1 → 9:1 hexanes/Et2O, slow gradient) to afford 21 (326.2 mg, 1.37 mmol, 76% yield) as a colorless oil that solidified in a −20 °C freezer. Rf = 0.54 (4:1 hexanes/Et2O, developed twice); 1H NMR (500 MHz, CDCl3) δ 7.01 (ABq, ΔδAB = 0.03, JAB = 8.2 Hz, 4H), 6.35 (d, J = 2.1 Hz, 1H), 6.31 (d, J = 2.3 Hz, 1H), 4.71–4.69 (m, 1H), 3.89 (ABq, ΔδAB = 0.04, JAB = 9.8 Hz, 2H), 3.27 (td, J = 7.8, 3.4 Hz, 1H), 3.06 (app t, J = 6.1 Hz, 1H), 2.94 (s, 1H), 2.46 (ddd, J = 13.8, 7.0, 2.4 Hz, 1H), 2.35 (app t, J = 4.8 Hz, 1H), 2.32 (s, 3H), 2.03 (ddd, J = 13.8, 8.9, 4.9 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 142.9, 140.3, 138.4, 135.6, 129.3, 127.2, 84.1, 71.2, 59.6, 54.9, 52.6, 50.3, 46.5, 44.6, 21.1; IR (Neat Film NaCl) 3025, 2949, 1514, 1074, 1041, 1025, 811, 744 cm−1; HRMS (FAB+) m/z: [M + H – H2]+ calc′d for C17H17O 237.1279, found 237.1271.
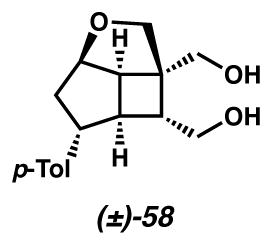
Diol 58
To a solution of cycloadduct 21 (18.0 mg, 75.5 μmol, 1.0 equiv) in a 2:1 mixture of CH2Cl2 (1.0 mL) and MeOH (0.5 mL) was added a solution of Sudan Red 7b (25 μL of a 0.05 wt % in MeOH) and cooled to −78 °C. The resulting pink solution was sparged with a gentle stream of oxygen for ca. 1 min, then ozonolyzed until consumption of 21 by TLC (indicator typically turned colorless just prior to completion). The solution was sparged with oxygen for another 1 min, and NaBH4 (28.8 mg, 0.76 mmol, 10 equiv) was added and the bath was removed. When the reaction reached room temperature, CH2Cl2 (2 mL) was added followed by quenching with 10% HCl (1 mL). The layers were separated, the aq extracted with CH2Cl2 (3 × 2 mL), the organics were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by flash chromatography on SiO2 to afford 58 (18.2 mg, 66.3 μmol, 88% yield) as a colorless oil. Rf = 0.31 (3:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) δ 7.04 (ABq, ΔδAB = 0.08, JAB = 8.1 Hz, 4H), 4.60 (app t, J = 4.3 Hz, 1H), 4.23 (d, J = 11.5 Hz, 1H), 4.09 (d, J = 9.2 Hz, 1H), 3.87–3.74 (comp m, 2H), 3.69 (d, J = 9.2 Hz, 1H), 3.55 (d, J = 11.5 Hz, 1H), 3.42 (ddd, J = 10.5, 7.0, 3.7 Hz, 1H), 3.09 (br s, 2H), 2.82 (dd, J = 8.5, 5.3 Hz, 1H), 2.58 (dd, J = 14.0, 7.2 Hz, 1H), 2.36 (dd, J = 11.0, 5.4 Hz, 1H), 2.31 (s, 3H), 1.99 (app dt, J = 8.7, 4.4 Hz, 1H), 1.73 (ddd, J = 14.0, 10.5, 3.5 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3) δ 143.1, 135.7, 129.3, 127.0, 86.2, 80.6, 63.5, 63.4, 51.4, 50.8, 50.7, 49.1, 44.8, 42.2, 21.1; IR (Neat Film NaCl) 3332 (br), 2922, 1514, 1436, 1100, 1037, 811 cm−1; HRMS (FAB+) m/z: [M + H – H2]+ calc′d for C17H21O3 273.1491, found 273.1485.
Optimized preparation of model olefin 25
The following procedure represents a telescoped preparation of olefin 25. Individual optimization procedures can be found in the supporting information. A solution of cyclobutene 21 (326.2 mg, 1.369 mmol, 1.0 equiv) in CH2Cl2/MeOH (5:1, 27.4 mL, 0.05 M) containing NaHCO3 (34.5 mg, 0.411 mmol, 0.3 equiv) and Sudan Red 7b (150 μL of a 0.05 wt % solution in MeOH) at −78 °C was sparged with a stream of oxygen for ca. 1 min and ozonolyzed until consumption by TLC analysis (typically as indicator turned colorless). After sparging the solution with oxygen for an additional 3 min, the reaction was capped with a drying tube and warmed to room temperature. The solution was filtered through a cotton plug, washing with benzene (3 mL). The reaction was concentrated in vacuo to ca. 2 mL, and to this flask was added a stir bar, septum, and the flask was evacuated/purged briefly (3x). The crude was dissolved in CH2Cl2 (13.7 mL), cooled to 0 °C, and to this was added Ac2O (387 μL, 4.11 mmol, 3.0 equiv) and Et3N (286 μL, 2.05 mmol, 1.5 equiv). The bath was removed and the reaction was stirred at room temperature for 8 h, diluted with CH2Cl2 (25 mL), washed with 2% HCl (10 mL), then 10% NaOH (10 mL), dried over MgSO4, filtered, and concentrated in vacuo to a pale yellow oil. The crude oil was purified by flash chromatography on SiO2 (2.5 × 8 cm, 4:1 → 1:1 hexanes/Et2O) to afford acetal 22 (61.5 g, 0.205 mmol, 15% yield) and a mixture of aldehyde 24 and acetals 23 and 32 (293.5 mg, 0.977 mmol, 71% yield).
The mixture of 23, 24 and 32 (293.5 mg) was dissolved in MeOH (19.5 mL, 0.05 M) and to this was added oven-dried 4 Å MS (489 mg, 0.5 g/mmol). After 24 h at room temperature the reaction was diluted with EtOAc (20 mL), filtered through a plug of Celite, and concentrated to a turbid yellow oil. This was dissolved in CH2Cl2 and passed through a small SiO2 plug and concentrated in vacuo to afford a pale yellow oil (312.1 mg).
To a solution of Ph3P•CH3Br (390 mg, 1.09 mmol, 1.05 equiv) in THF (3 mL) at 0 °C was added KOt-Bu (105 mg, 0.935 mmol, 0.9 equiv). The resulting bright yellow solution was stirred for 1 h, and a solution of aldehyde 24 and acetals 23 and 32 (312.1 mg) in THF (2 mL, 0.2 M total) was quantitatively transferred via cannulation. The bath was removed after 5 min and at 5 h the reaction was quenched with water (5 mL) and diluted with Et2O (5 mL). The layers were separated, the aq was extracted with Et2O (2 × 10 mL), the combined organics were dried over Na2SO4, filtered, and concentrated in vacuo to a pale yellow oil. The crude material was purified by flash chromatography on SiO2 (2.5 × 15 cm, 9:1 → 1:1 hexanes/Et2O) to afford olefin 25 (162.2 mg, 0.544 mmol, 40% yield over three steps) and ca. 7:3 mixture of acetals 23 and 32 (59.5 mg, 0.198 mmol, 14% yield over three steps).
Model acetal 2229
Rf = 0.52 (1:2 hexanes/Et2O); 1H NMR (500 MHz, CDCl3) δ 7.13 (d, J = 7.8 Hz, 2H), 7.05 (d, J = 8.1 Hz, 2H), 5.37 (s, 1H), 4.77 (dd, J = 5.2, 4.0 Hz, 1H), 4.14 (d, J = 9.7 Hz, 1H), 4.03 (d, J = 9.7 Hz, 1H), 3.57 (app t, J = 6.4z Hz, 1H), 3.50 (s, 3H), 3.49 (ddd, J = 10.8, 6.8, 4.3, 1H), 2.57 (dd, J = 14.2, 7.0 Hz, 1H), 2.55–2.53 (comp m, 2H), 2.33 (s, 3H), 1.80 (ddd, J = 14.4, 10.8, 3.6 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 175.5, 141.3, 136.3, 129.5, 127.2, 109.0, 85.9, 73.0, 56.8, 55.5, 51.9, 51.3, 51.2, 44.5, 44.2, 21.1; IR (Neat Film NaCl) 2927, 1773, 1515, 1353, 1182, 1143, 1118, 1018, 990, 930, 814 cm−1; HRMS (FAB+) m/z: [M + H]+ calc′d for C18H21O4 301.1440, found 301.1448.
Model desired acetal 2329
Rf = 0.33 (1:2 hexanes/Et2O); 1H NMR (500 MHz, CDCl3) δ 7.08 (ABq, ΔδAB = 0.04, JAB = 8.0 Hz, 4H), 5.40 (s, 1H), 4.71 (dd, J = 5.4, 3.1 Hz, 1H), 4.08 (d, J = 10.5 Hz, 1H), 3.78 (d, J = 10.5 Hz, 1H), 3.55 (dt, J = 11.6, 6.1 Hz, 1H), 3.48 (s, 3H), 3.31 (dd, J = 7.4, 5.7 Hz, 1H), 2.89 (d, J = 2.4 Hz, 1H), 2.66 (ddd, J = 7.6, 5.2, 2.4 Hz, 1H), 2.53 (dd, J = 13.8, 6.7 Hz, 1H), 2.31 (s, 3H), 1.76 (ddd, J = 13.8, 11.7, 3.1 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 177.7, 140.1, 136.1, 129.4, 127.1, 106.3, 85.7, 71.8, 56.8, 55.0, 51.1, 50.0, 47.9, 46.2, 43.9, 21.1; IR (Neat Film NaCl) 2925, 2847, 1772, 1516, 1352, 1207, 1168, 1144, 1108, 1042, 943, 814, 729, 705 cm−1; HRMS (FAB+) m/z: [M + H]+ calc′d for C18H21O4 301.1440, found 301.1444.
Model aldehyde 24
Aldehyde 24 was difficult to isolate as a pure compound, as it usually contained varying quantities of acetal 23. It has the following spectral properties: 1H NMR (300 MHz, CDCl3) δ 9.76 (s, 1H), 7.07 (ABq, ΔδAB = 0.05, JAB = 8.3 Hz, 4H), 4.69 (dd, J = 4.6, 3.9 Hz, 1H), 4.10 (d, J = 9.6 Hz, 1H), 3.91 (d, J = 9.7 Hz, 1H), 3.70 (s, 3H), 3.59 (dd, J = 8.3, 5.3 Hz, 1H), 3.45 (ddd, J = 10.7, 7.1, 3.7 Hz, 1H), 3.06–2.97 (comp m, 2H), 2.63 (dd, J = 14.2, 7.2 Hz, 1H), 2.31 (s, 3H), 1.81 (ddd, J = 14.1, 10.7, 3.4 Hz, 1H).
Model olefin 25
Rf = 0.62 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.07 (ABq, ΔδAB = 0.04, JAB = 8.0 Hz, 4H), 5.94 (dd, J = 17.5, 10.8 Hz, 1H), 5.18 (dd, J = 10.8, 1.1 Hz, 1H), 5.14 (dd, J = 17.5, 1.1 Hz, 1H), 4.65–4.63 (m, 1H), 4.03 (d, J = 9.2 Hz, 1H), 3.62 (s, 3H), 3.57 (d, J = 9.2 Hz, 1H), 3.40 (ddd, J = 10.6, 7.1, 3.6 Hz, 1H), 3.26 (dd, J = 8.5, 5.2 Hz, 1H), 3.03 (ddd, J = 8.7, 5.2, 3.6 Hz, 1H), 2.98 (d, J = 5.5 Hz, 1H), 2.64 (dd, J = 14.1, 7.3 Hz, 1H), 2.31 (s, 3H), 1.78 (ddd, J = 14.0, 10.6, 3.4 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 173.3, 142.6, 135.6, 134.7, 129.2, 127.1, 116.3, 86.5, 78.9, 55.2, 53.0, 51.6, 51.2, 51.1, 45.0, 40.8, 21.1; IR (Neat Film NaCl) 2952, 2922, 1733, 1515, 1435, 1210, 1158, 1055, 1037, 919, 812 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C19H22O3 298.1569, found 298.1580.
Model desired acetal 3229
Rf = 0.19 (1:2 hexanes/Et2O); 1H NMR (500 MHz, CDCl3) δ 7.08 (ABq, ΔδAB = 0.04, JAB = 8.0 Hz, 4H), 5.30 (s, 1H), 4.68 (dd, J = 5.6, 3.3 Hz, 1H), 4.07 (d, J = 9.5 Hz, 1H), 3.72 (dd, 7.2, 6.2 Hz, 1H), 3.69 (d, J = 9.5 Hz, 1H), 3.63 (s, 3H), 3.50 (dt, J = 11.4, 5.8 Hz, 1H), 2.85 (d, J = 2.7 Hz, 1H), 2.66 (ddd, J = 7.6, 4.9, 2.7 Hz, 1H), 2.54 (dd, J = 13.9, 6.8 Hz, 1H), 2.31 (s, 3H), 1.79 (ddd, J = 13.9, 11.4, 3.3 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 175.8, 140.8, 136.1, 129.4, 127.1, 104.0, 86.0, 72.7, 58.7, 54.5, 50.23, 50.20, 47.7, 44.8, 44.0, 21.1; IR (Neat Film NaCl) 2922, 1770, 1515, 1450, 1386, 1209, 1170, 1106, 1041, 995, 942, 813 cm−1; HRMS (MM: ESI/APCI) m/z: [M + H]+ calc′d for C18H21O4 301.1434, found 301.1432.
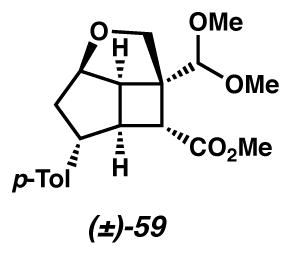
Model dimethyl acetal 59
To a solution of acetal 23 (1.0 equiv) in MeOH (25 mM) was added either La(OTf)3 or Sm(OTf)3 (0.20 equiv). The reaction was stirred until complete conversion by TLC, diluted with Et2O, filtered through a plug of Celite, and concentrated in vacuo. The crude 1H NMR showed 59 as the exclusive product. Rf = 0.36 (1:2 hexanes/Et2O); 1H NMR (600 MHz, CDCl3) δ 7.06 (ABq, ΔδAB = 0.04, JAB = 7.9 Hz, 4H), 4.69 (s, 1H), 4.58 (dd, J = 4.8, 3.8 Hz, 1H), 3.87 (ABq, ΔδAB = 0.01, JAB = 9.2 Hz, 2H), 3.67 (s, 3H), 3.51 (s, 3H), 3.39 (s, 3H), 3.36 (ddd, J = 10.7, 7.4, 3.8 Hz, 1H), 3.23 (dd, J = 8.6, 5.3 Hz, 1H), 2.93 (ddd, J = 8.8, 5.4, 3.6 Hz, 1H), 2.82 (d, J = 5.7 Hz, 1H), 2.60 (dd, J = 14.1, 7.4 Hz, 1H), 2.30 (s, 3H), 1.75 (ddd, J = 14.1, 10.4, 3.6 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 174.1, 142.7, 135.6, 129.2, 127.1, 105.0, 85.8, 75.0, 58.3, 56.6, 54.5, 52.1, 51.7, 51.4, 49.4, 45.0, 42.4, 21.1; IR (Neat Film NaCl) 2952, 1727, 1515, 1435, 1362, 1210, 1069, 1042, 977, 813 cm−1; HRMS (FAB+) m/z: [M + H – H2]+ calc′d for C20H25O5 345.1702, found 345.1701.
Model acid 33
To a solution of olefin 25 (162.2 mg, 0.544 mmol, 1.0 equiv) in THF (10.9 mL, 0.05 M) at 0 °C was added KOTMS (698 mg, 5.44 mmol, 10 equiv) in one portion. The cooling bath was removed and the reaction was stirred until consumption by TLC analysis (typically 5–6 h). The reaction was then cooled to 0 °C and slowly quenched with 1 N HCl (10 mL), diluted with EtOAc (20 mL) and brine (5 mL). The layers were separated, the aq was extracted with EtOAc (3 × 20 mL), the combined organics were dried over Na2SO4, filtered, and concentrated to a pale yellow semisolid. The crude material was purified by flash chromatography on SiO2 (1:1 hexanes/EtOAc) to give 33 (148.7 mg, 0.523 mmol, 96% yield) as a white solid. Rf = 0.23 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.06 (ABq, ΔδAB = 0.04, JAB = 8.0 Hz, 4H), 6.00 (dd, J = 17.5, 10.8 Hz, 1H), 5.21 (dd, J = 10.8, 1.1 Hz, 1H), 5.17 (dd, J = 17.6, 1.2 Hz, 1H), 4.64 (dd, J = 4.8, 3.6 Hz, 1H), 4.04 (d, J = 9.3 Hz, 1H), 3.57 (d, J = 9.3 Hz, 1H), 3.40 (ddd, J = 10.5, 7.2, 3.5 Hz, 1H), 3.26 (dd, J = 8.2, 5.2 Hz, 1H), 3.00–2.95 (comp m, 2H), 2.64 (d, J = 14.2, 7.3 Hz, 1H), 2.31 (s, 3H), 1.78 (ddd, J = 14.0, 10.6, 3.4 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 177.7, 142.6, 135.7, 134.4, 139.3, 127.7, 116.6, 86.6, 79.0, 54.9, 53.2, 51.14, 51.10, 44.9, 40.8, 21.1; IR (Neat Film NaCl) 2923, 1729, 1700, 1515, 1418, 1223, 1053, 992, 918, 812 cm−1; HRMS (FAB+) m/z: [M + H]+ calc′d for C18H21O3 285.1491, found 285.1495.
Model α-diazo cyclobutyl ketone 34
To a solution of acid 33 (14.7 mg, 51.7 μmol, 1.0 equiv) in CH2Cl2 (2 mL, 0.025 M) at 0 °C was added a solution of oxalyl chloride (107 μL of a 1.45 M solution in CH2Cl2, 155 μmol, 3.0 equiv), followed by 1 drop of DMF. The reaction was stirred for 1 h, at which point the stir bar was removed and the volatiles were removed on a rotovap purged with argon. The septum and stir bar were replaced and the crude material was further dried under high vacuum for 10 min. The resulting crude semisolid was partially dissolved in CH2Cl2 (1 mL) and transferred quantitatively via Teflon cannula to a vigorously stirring solution of excess diazomethane (5–8 mL) at 0 °C. After 30 min the cooling bath was removed, and after a further hour the diazomethane was pulled off via water aspirator. The pale yellow solution was filtered through a small SiO2 plug (Et2O) and concentrated in vacuo. The crude material was purified by flash chromatography on SiO2 (9:1 → 3:1 hexanes/EtOAc) to afford 34 (13.2 mg, 42.8 μmol, 83% yield) as a bright yellow oil that solidified in a −20 °C freezer. Rf = 0.31 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.05 (ABq, ΔδAB = 0.04, JAB = 7.9 Hz, 4H), 6.01 (dd, 17.5, 10.8 Hz, 1H), 5.17 (d, J = 10.8 Hz, 1H), 5.11–5.06 (br m, 2H), 4.64 (app t, J = 4.0 Hz, 1H), 3.97 (br d, J = 9.1 Hz, 1H), 3.66 (br d, J = 8.6 Hz, 1H), 3.36 (ddd, J = 10.6, 7.7, 3.5 Hz, 1H), 3.19 (br s, 1H), 3.11 (br s, 1H), 2.92 (br s, 1H), 2.64 (dd, J = 14.1, 7.3 Hz, 1H), 2.30 (s, 3H), 1.78 (ddd, J = 13.7, 10.5, 3.0 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 193.6, 142.5, 135.7, 134.9, 129.3, 127.1, 116.0, 86.5, 78.6, 60.8, 55.3, 53.6, 51.4, 44.9, 40.5, 30.4, 21.1; IR (Neat Film NaCl) 2955, 2921, 2100, 1633, 1514, 1370, 1352, 1048, 812 cm−1; HRMS (FAB+) m/z: [M + H]+ calc′d for C19H21O2N2 309.1603, found 309.1619.
Model homologated ester 37
A solution of α-diazo cyclobutyl ketone 34 (3.2 mg, 104 μmol) in MeOH (5.2 mL, 2 mM) in a dried quartz tube was irradiated in a Luzchem rayonette (λ = 350 nm) for 1.5 h. The solution was concentrated in vacuo and revealed homologated ester 37 as the sole product by crude 1H NMR. An analytical sample was obtained from purification by preparative TLC on SiO2 (2:1 hexanes/EtOAc) to give 37 (2.4 mg, 7.7 μmol, 74% yield) as a colorless oil. Rf = 0.43 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.04 (ABq, ΔδAB = 0.04, JAB = 5 Hz, 4H), 5.89 (dd, J = 16.9, 10.2 Hz, 1H), 5.22 (d, J = 10.7 Hz, 1H), 5.13 (d, J = 17.5 Hz, 1H), 4.64 (app t, J = 3.9 Hz, 1H), 3.97 (d, J = 8.7 Hz, 1H), 3.51 (s, 3H), 3.50 (d, J = 8.7 Hz, 1H), 3.46–3.42 (m, 1H), 3.23 (dd, J = 7.9, 5.8 Hz, 1H), 2.61 (dd, J = 14.1, 7.3 Hz, 1H), 2.54–2.36 (comp m, 3H), 2.30 (s, 3H), 2.11 (app dt, J = 7.7, 3.8 Hz, 1H), 1.78 (ddd, J = 12.7, 10.4, 2.1 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 173.1, 143.6, 135.5, 135.3, 129.2, 127.1, 116.4, 86.7, 79.2, 52.4, 51.9, 51.6, 50.1, 47.0, 46.9, 45.0, 37.2, 21.1; IR (Neat Film NaCl) 2951, 2922, 1736, 1514, 1435, 1207, 1163, 1041, 916, 808 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C20H24O3 312.1726, found 312.1725.
Wolff/Cope rearrangement to cyclooctadienone 35
Photochemical/Thermal
A solution of α-diazo cyclobutyl ketone 34 (3.5 mg, 11.4 μmol) in benzene (5.7 mL, 2 mM) in a dried quartz tube was irradiated in a Luzchem photochemical reactor (λ = 310 nm) for 10 min, and then the lamp was turned off and the quartz tube was placed in an 80 °C oil bath for 2 h. The reaction was concentrated in vacuo and purified by preparative TLC on SiO2 (1:1 hexanes/Et2O, developed twice) to give model cyclooctadienone 35 (1.9 mg, 6.8 μmol, 59% yield) as a colorless oil.
Microwave (thermal)
A solution of α-diazo cyclobutyl ketone 34 (2.3 mg, 7.5 μmol) in toluene (1.5 mL, 5 mM) was prepared in a non-dried microwave vial containing a stir bar under ambient atmosphere. The vial was sealed and irradiated in a Biotage Initiator microwave reactor at 400 W until the temperature reached 140 °C, and the temperature was maintained for 20 min. The vial was cooled to room temperature, the seal was removed, and the contents were concentrated in vacuo. Reaction conversion was monitored by 1H NMR analysis (C6D6). The crude material was purified by preparative TLC on SiO2 (2:1 hexanes/EtOAc) to give 35 (2.0 mg, 7.1 μmol, 95% yield) as a colorless oil that solidified in a −20 °C freezer. Rf = 0.32 (3:1 hexanes/EtOAc); 1H NMR (CDCl3, 500 MHz) δ 7.15 (ABq, ΔδAB = 0.02, JAB = 8.3 Hz, 4H), 5.80–5.73 (comp m, 2H), 5.63–5.59 (m, 1H), 4.76 (app t, J = 5.2 Hz, 1H), 4.49 (dd, J = 13.1, 1.5 Hz, 1H), 4.40 (dd, J = 13.2, 1.0 Hz, 1H), 3.66 (td, J = 6.8, 4.0 Hz, 1H), 3.41 (dd, J = 14.7, 8.1 Hz, 1H), 3.23 (app t, J = 11.3 Hz, 1H), 3.13–3.05 (comp m, 2H), 2.34 (s, 3H), 2.30 (dd, J = 13.8, 5.9 Hz, 1H), 1.93 (ddd, J = 13.8, 12.1, 4.6 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 203.3, 146.8, 139.7, 138.4, 136.9, 129.7, 129.2, 127.9, 144.2, 85.3, 72.2, 53.9, 50.6, 48.1, 45.4, 41.4, 21.2; 1H NMR (500 MHz, C6D6) δ 6.99 (d, J = 7.7 Hz, 2H), 6.86 (d, J = 6.9 Hz, 2H), 5.77 (dt, J = 13.5, 1.3 Hz, 1H), 5.56 (d, J = 13.6 Hz, 1H), 5.11–5.08 (m, 1H), 4.35 (app t, J = 5.1 Hz, 1H), 4.24 (d, J = 13.1 Hz, 1H), 4.11 (d, J = 13.2 Hz, 1H), 3.04–2.89 (comp m, 4H), 2.70 (app t, J = 11.3 Hz, 1H), 2.20 (dd, J = 13.6, 5.9 Hz, 1H), 2.15 (s, 3H), 1.53–1.46 (m, 1H); 13C{1H} NMR (126 MHz, C6D6) δ 201.9, 147.2, 139.2, 138.6, 136.8, 130.1, 130.0, 128.7, 114.4, 85.5, 72.5, 53.8, 51.2, 48.3, 45.9, 41.9, 21.3; IR (Neat Film NaCl) 3015, 2922, 1693, 1661, 1516, 1435, 1318, 1208, 1062, 1030, 817 cm−1; HRMS (MM: ESI/APCI) m/z: [M – H]− calc′d for C19H19O2 279.1391, found 279.1384.
Anti-cyclopentenol 38
A 2 L, 3-neck flask was charged with CuCN (2.01 g, 112.2 mmol. 0.2 equiv) and THF (280 mL), and the suspension was cooled to ca. −20 °C (internal) using a cryocool. To this was added a solution of MeMgCl (109 mL, 337 mmol of a 3.1 M solution in THF) and the internal temperature warmed to −14 °C. After 30 min at −20 °C, a solution of monoacetate (+)-1735 (15.95 g, 112.2 mmol, 1.0 equiv) in THF (40 mL) was slowly transferred (quantitative) via cannulation at such a rate that the internal temperature does not rise above −10 °C (required ca. 1 h). After 30 min the reaction was slowly quenched with sat. aq NH4Cl (100 mL), 50% sat. brine (100 mL), the cooling bath was removed and the viscous suspension was stirred vigorously for several hours. Additional water (200 mL) and 3% HCl (100 mL) was added and the layers were separated. The aq layer was extracted with Et2O (3 × 200 mL), the combined organics were dried over MgSO4, filtered, and concentrated carefully (water bath = 5 °C, down to 30 torr) to a pale yellow oil. The crude material was purified by short path distillation (bp = 88–92 °C, 40 torr) to afford a 95:5 mixture of 38 and 39 (8.847 g, 90.2 mmol, 80% yield). The early distillation fractions and Et2O washings from the apparatus were combined and purified by flash chromatography on SiO2 (2.5 × 27 cm, 6:1 → 1:1 pentane/Et2O) to provide another 1.049 g of 38 and 39 (combined 9.996 g, 101.9 mmol, 91% yield). Rf (38) = 0.35 (1:1 hexanes/EtOAc); Rf (39) = 0.29 (1:1 hexanes/EtOAc); bp = 88–92 °C (40 torr). An analytical sample of 38 was obtained from the column conditions above. 1H NMR (500 MHz, CDCl3) δ 5.89 (dd, J = 5.5, 1.9 Hz, 1H), 5.79 (dt, J = 4.8, 2.3 Hz, 1H), 4.87 (dt, J = 4.8, 2.3 Hz, 1H), 2.95 (dqdt, J = 9.5, 7.2, 4.9, 2.3 Hz, 1H), 1.96 (ddd, J = 14.0, 7.5, 2.6 Hz, 1H), 1.71 (ddd, J = 14.0, 7.1, 5.2 Hz, 1H), 1.48 (br s, 1H), 1.03 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 142.0, 132.1, 77.6, 42.7, 38.5, 21.0; IR (Neat Film NaCl) 3338 (br), 2956, 2870, 1354, 1088, 1017, 982, 742 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C6H10O 98.07317, found 98.07171; [α]D21 −272.2 (c 0.39, CHCl3, 99% ee). GC conditions: 45 °C isothermal, GTA column, tR (min): major = 37.7, minor = 36.7.
Syn-cyclopentenol 16
To a suspension of Ph3P (13.41 g, 51.12 mmol, 1.2 equiv) and benzoic acid (6.243 g, 51.12 mmol, 1.2 equiv) in toluene (237 mL) at −75 °C (internal) was added DIAD (10.1 mL, 51.12 mmol, 1.2 equiv) dropwise, neat over 15 min. The resulting yellow suspension was stirred vigorously for 30 min, at which point a solution of 38 and 39 (4.1806 g, 42.60 mmol, 1.0 equiv) in toluene (47 mL, 0.15 M total) was transferred via cannula quantitatively over 30 min (observed maximum temperature increase to −70 °C). When ca. 1/3 of this solution was added, the reaction mixture turned homogeneous. After complete addition of 38 and 39, the reaction was stirred for an additional 30 min (white precipitate had formed) and quenched with sat. aq NaHCO3 (100 mL) and water (100 mL) and the contents were warmed to room temperature. The layers were separated, the aq was extracted with Et2O (2 × 50 mL), and the combined organics were shaken with 3% aq H2O2 until TLC showed disappearance of Ph3P. The layers were separated, the aq was extracted with Et2O (1 × 50 mL), and the combined organics were dried over MgSO4, filtered, and concentrated to a pale yellow solid. The crude material was purified by flash chromatography on SiO2 (7 × 7.5 cm, 1:0 → 24:1 hexanes/Et2O, dry loaded onto SiO2) to give the expected syn-benzoate (60, 7.792 g, 38.53 mmol, 90% yield) as a pale yellow oil. Rf = 0.57 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 8.05-8.03 (m, 2H), 7.56–7.53 (m, 1H), 7.44–7.41 (m, 2H), 6.03 (dd, J = 4.4, 2.0 Hz, 1H), 5.90–5.86 (comp m, 2H), 2.80–2.73 (m, 1H), 2.65 (dt, J = 14.0, 7.8 Hz, 1H), 1.51 (dq, J = 12.5, 3.7 Hz, 1H), 1.16 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 166.6, 142.9, 132.9, 130.8, 129.7, 128.6, 128.4, 80.9, 38.9, 38.7, 21.7; IR (Neat Film NaCl) 2961, 1716, 1451, 1340, 1315, 1272, 1110, 711 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C13H14O2 202.0994, found 202.1019; [α]D25.7 +123.8 (c 1.175, CHCl3, 98–99% ee).
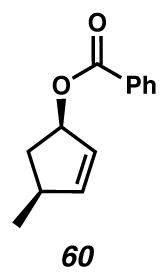
To a solution of syn-benzoate 60 (6.255g, 30.93 mmol, 1.0 equiv) in MeOH (62 mL, 0.5 M) was added K2CO3 (8.549 g, 61.85 mmol, 2.0 equiv) in one portion. After completion as judged by TLC analysis (3 h, 3:1 hexanes/EtOAc), the reaction was concentrated carefully in vacuo to a slurry (ca. 5–10 mL). The white slurry was diluted with brine (25 mL) and extracted with Et2O (4 × 25 mL, follow by TLC), the organics were dried over MgSO4, filtered, and concentrated carefully in vacuo. The crude material was purified by flash chromatography on SiO2 (5 × 12 cm, 6:1 → 1:1 pentane/Et2O) and concentrated down to 100 torr until 1H NMR analysis revealed the absence of solvent to afford 16 (2.728 g, 27.79 mmol, 90% yield) as a colorless oil. Rf = 0.25 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.82 (dt, J = 5.5, 1.5 Hz, 1H), 5.73 (dt, J = 5.5, 2.0 Hz, 1H), 4.79 (br s, 1H), 2.66–2.59 (m, 1H), 2.52 (dt, J = 13.4, 7.6 Hz, 1H), 1.79 (br s, 1H), 1.17 (dt, 13.4, 5.4 Hz, 1H), 1.09 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 140.4, 132.8, 77.8, 42.6, 39.0, 22.0; IR (Neat Film NaCl) 3338 (br), 3048, 2959, 2870, 1456, 1356, 1322, 1115, 1051, 755 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C6H10O 98.07317, found 98.07135; [α]D27 −23.0 (c 0.475, CHCl3, 98.2% ee). GC conditions: 50 °C isothermal, GTA column, tR (min): major = 21.2, minor = 20.7.
Cycloaddition substrate 40
To a round-bottom flask containing Zn(OTf)2 (271 mg, 0.745 mmol, 0.05 equiv) and toluene (3.7 mL) at 0 °C was added cyclopentenol 16 (1.45 g, 14.8 mmol, 1.0 equiv) by syringe. To this suspension was added a solution of cyclobutadiene trichloroacetimidate 19 (6.38 g, 17.4 mmol, 1.2 equiv) in toluene (2.0 mL) by cannula transfer, with further washing by toluene (1.7 mL). A yellow precipitate was observed at the beginning of the addition, and this turned into a viscous slurry upon completion of the addition. The ice bath was allowed to expire over 1.5 h and the reaction was stirred for an additional 0.5 h at ambient temperature. The crude reaction mixture was transferred directly onto a 25 g silica gel loading cartridge and purified by automated flash chromatography using a 125 g silica column (1:0 → 19:1 hexanes/EtOAc) to afford 40 (3.65 g, 12.2 mmol, 82% yield) as a pale yellow oil. Rf = 0.73 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, C6D6) δ 5.72 (app dt, J = 5.6, 1.9 Hz, 1H), 5.66 (app dt, J = 5.6, 1.6 Hz, 1H), 4.27–4.24 (m, 1H), 3.53 (ABq, ΔδAB = 0.01, JAB = 9.1 Hz, 2H), 3.48 (s, 2H), 3.32 (s, 1H), 2.43–2.39 (m, 1H), 2.15 (app dt, J = 13.3, 7.6 Hz, 1H), 1.26 (ddd, J = 17.0, 11.2, 6.9 Hz, 1H), 0.98 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, C6D6) δ 215.0, 140.5, 130.5, 85.2, 82.6, 64.64, 64.58, 64.0, 62.2, 39.1, 38.9, 21.6; IR (Neat Film NaCl) 2961, 2871, 2046, 1965, 1359, 1076, 1055, 757 cm−1; HRMS (FAB+) m/z: [M]+calc′d for C14H14FeO4 302.0242, found 302.0244; [α]D25 +22.7 (c 0.86, hexane, 98% ee).
Cycloadduct 4129
A solution of cycloaddition substrate 40 (2.3582 g, 7.806 mmol, 1.0 equiv) dissolved in acetone (780 mL, 10 mM) in a 1 L round-bottom flask fitted with a reflux condenser was warmed in a 70 °C oil bath. When the solution approached reflux, the condenser was momentarily removed and Me3NO•2H2O (8.77 g, 78.9 mmol, 10 equiv) was added in a single portion. The solution was allowed to reflux and within 10 min the reaction vessel was filled with a rust colored precipitate. After 4 h a second portion of Me3NO•2H2O (4.35 g, 45.9 mmol, 5.8 equiv) was added. The solution was heated at reflux for an additional 17 h after which the reaction was judged to be complete by TLC analysis (4:1 hexanes/EtOAc). The solution was cooled to room temperature and poured directly onto a SiO2 column (25 × 5 cm) packed in pentane. The column was washed with 0 → 10% Et2O in pentane, and all fractions containing cyclobutene 41 were combined and concentrated carefully to a volume of ca. 30 mL by atmospheric pressure distillation. This solution was purified by flash chromatography on SiO2 (packed with pentane, eluted with 20:1 pentane/Et2O). The fractions containing 41 were combined and concentrated to a volume of ca. 10 mL by atmospheric pressure distillation. This pale yellow cyclobutene solution in pentane was used directly in the following reaction. An analytical sample of cyclobutene 41 could be prepared by further chromatography and exhaustive distillation of solvent. Rf = 0.39 (3:1 hexanes/Et2O); 1H NMR (500 MHz, CDCl3) δ 6.27 (d, J = 2.2 Hz, 1H), 6.22 (s, 1H), 4.84 (app q, J = 7.2 Hz, 1H), 4.04 (d, J = 9.1 Hz, 1H), 3.94 (d, J = 8.6 Hz, 1H), 3.00 (s, 1H), 2.89 (app t, J = 5.8 Hz, 1H), 2.23 (tq, J = 12.7, 6.4 Hz, 1H), 2.10–2.05 (comp m, 2H), 1.37 (ddd, J = 13.0, 13.0, 7.0 Hz, 1H), 0.97 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 140.2, 138.4, 84.7, 70.3, 57.9, 52.0, 47.8, 44.8, 39.0, 37.3, 14.3; IR (Neat Film NaCl) 2955, 2865, 1458, 1334, 1089, 1075, 1057, 1032, 931, 740 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C11H14O 162.1045, found 162.1026; an optical rotation was not obtained due to the volatility of this compound. Cyclobutene 41 was found to possess an optical purity (ee) of 98% by chiral GC analysis; GC conditions: 110 °C isothermal, GTA column, tR (min): major = 13.6, minor = 13.3.
Oxidative cleavage of 41, equilibration and olefination
In a 250 mL round-bottom flask, the cyclobutene 41 solution prepared above was diluted with CH2Cl2 (130 mL) and methanol (26 mL, 5:1, 0.05 M total). To this was added NaHCO3 (205.2 mg, 2.44 mmol, 0.3 equiv) and a few drops of Sudan Red (0.05 wt % in MeOH) until the solution became a pale pink color (ca. 10 drops). The reaction vessel was cooled to −78 °C and the solution was sparged with O2 gas (0.5 L/min) for 2 min. The reaction was then ozonolyzed (setting the ozone generator to “5” with an O2 flow rate of 0.5 L/min) for 60 min, at which point the pink color of the solution had disappeared and the reaction was judged to be complete by TLC analysis. The ozone was sparged with O2 gas (1 L/min) through the solution for 2 min, and the pale yellow solution was warmed to room temperature and filtered through a cotton plug to remove the solid NaHCO3. The cotton plug was washed with benzene (10 mL) and the filtrate was concentrated to a small volume (ca. 3–4 mL). The resulting crude yellow oil was dissolved in CH2Cl2 (78 mL), cooled to 0 °C, and to this was added Et3N (1.63 mL, 11.7 mmol, 1.5 equiv) and Ac2O (2.21 mL, 23.4 mmol, 3.0 equiv) dropwise via syringe. After 6 h, the reaction was quenched by the addition of 2 M HCl (25 mL), the organic layer was separated and washed with 2 M NaOH (25 mL), and the combined aqueous layers were extracted with CH2Cl2 (5 × 25 mL). The organics were dried over MgSO4, filtered, and concentrated to afford a pale brown oil which was passed through a SiO2 plug eluting with EtOAc, and concentrated to afford a pale yellow oil (0.8504 g, 3.8 mmol, three steps, 48% crude yield) containing mostly acetals 42 and 43.
The crude pale yellow oil prepared above was azeotroped from benzene (2 × 10 mL) in a 250 mL round-bottom flask and dissolved in MeOH (76 mL, 0.05 M). To this was added oven dried 4 Å MS (1.90 g, 0.5 g/mmol) and the flask was fitted with a reflux condenser and heated to reflux using an 80 °C oil bath. After 6 h, the reaction was judged complete by 1H NMR analysis of a reaction aliquot and the reaction was cooled to room temperature. Most of the 4 Å MS were removed by filtration through Celite eluting with EtOAc. The filtrate was concentrated and the resultant turbid oil was further purified by filtration through a SiO2 plug with EtOAc. This filtrate was concentrated to afford a yellow oil (0.8420 g) containing mostly aldehydes derived from 42 and 43 with the requisite acetals. This mixture was used directly in the following reaction.
A flask containing Ph3P•CH3Br (1.62 g, 4.54 mmol, 1.2 equiv) was partially dissolved with THF (15 mL) and cooled to 0 °C. To this was added KOt-Bu (423 mg, 3.77 mmol, 1.0 equiv) in one portion, and the solution immediately displayed a bright yellow color. The crude yellow oil of aldehydes/acetals (0.8420 g, ca. 3.7 mmol) prepared above was azeotroped from benzene (2 × 10 mL), dissolved in THF (7.5 mL), cooled to 0 °C, and transferred dropwise via positive pressure cannulation into the solution of phosphorane over ca. 10 min. The flask was then washed with a second portion of THF (7.5 mL) to ensure quantitative transfer. The reaction was gradually allowed to warm to room temperature. After 18 h the reaction was quenched by the addition of H2O (25 mL) and extracted with Et2O (4 × 20 mL) then EtOAc (2 × 20 mL). The combined organics were dried with MgSO4, filtered and concentrated in vacuo. The crude yellow residue was purified flash chromatography on SiO2 (15 × 2 cm, 20:1 → 4:1 hexanes/EtOAc) to afford olefins 44 and 45 (384.4 mg, 1.729 mmol, 2.7:1 ratio, 22.2% yield over four steps from cycloaddition substrate 40) as a colorless oil and acetals 42 and 43 (400.5 mg, 1.786 mmol; 2.7:1 ratio, 22.9% yield over four steps from 40) as pale yellow oil. Olefins 44 and 45 could be separated by further flash chromatography on SiO2 (20:1 → 9:1 hexanes/EtOAc), and acetals 42 and 43 could be separated by further flash chromatography on SiO2 (3:1 → 1:1 hexanes/EtOAc).
Olefin 44
Rf = 0.46 (9:1 hexanes/EtOAc, developed thrice); 1H NMR (500 MHz, CDCl3) δ 5.95 (dd, J = 17.5, 10.8 Hz, 1H), 5.15 (dd, J = 10.8, 1.2 Hz, 1H), 5.11 (dd, J = 17.5, 1.2 Hz, 1H), 4.59 (ddd, J = 6.3, 6.3, 1.5 Hz, 1H), 3.99 (d, J = 9.0 Hz, 1H), 3.64 (s, 3H), 3.54 (d, J = 9.0 Hz, 1H), 3.16 (d, J = 7.1 Hz, 1H), 3.02 (app t, 7.1 Hz, 1H), 2.95 (app q, J = 7.4 Hz, 1H). 2.39 (ddq, J = 14.0, 10.4, 7.0 Hz, 1H), 2.18 (ddd, J = 14.6, 10.4, 5.7 Hz, 1H), 1.71 (ddd, J = 14.6, 6.3, 1.7 Hz, 1H), 1.02 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 173.6, 134.9, 115.9, 87.1, 78.7, 52.6, 51.6, 51.4, 44.8, 42.5, 38.9, 37.2, 17.2; IR (Neat Film NaCl) 2954, 1736, 1436, 1363, 1236, 1206, 1162, 1042, 920 cm−1; HRMS (MM: ESI/APCI) m/z: [M + H]+ calc′d for C13H19O3 223.13287, found 223.13255; [α]D16.8 −4.73 (c 1.18, CH2Cl2, 98% ee).
Olefin 45
Rf = 0.39 (9:1 hexanes/EtOAc, developed thrice); 1H NMR (500 MHz, CDCl3) δ 5.86 (ddd, J = 17.3, 10.3, 7.5 Hz, 1H), 5.05 (ddd, J = 17.1, 1.5, 1.5 Hz, 1H), 5.02 (ddd, J = 10.3, 1.4, 1.4 Hz, 1H), 4.67 (td, J = 6.6, 2.8 Hz, 1H), 4.02 (d, J = 9.4 Hz, 1H), 3.88 (d, J = 9.4 Hz, 1H), 3.68 (s, 3H), 3.30 (app t, J = 7.2 Hz, 1H), 3.00 (app t, J = 7.2 Hz, 1H), 2.54 (app q, 7.2 Hz, 1H), 2.35 (d sextets, J = 9.6, 7.2 Hz, 1H), 2.18 (ddd, J = 14.5, 9.7, 6.3 Hz, 1H), 1.67 (ddd, J = 14.5, 7.7, 2.8 Hz, 1H), 1.01 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 172.3, 137.9, 115.9, 87.1, 76.0, 55.8, 51.7, 48.9, 44.9, 43.6, 41.8, 38.0, 16.2; IR (Neat Film NaCl) 2953, 5873, 1731, 1436, 1295, 1207, 1041, 917 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C13H18O3 222.1256, found 222.1267; [α]D15.1 −0.49 (c 0.72, CH2Cl2, 98% ee).
Acetal 4229
Rf = 0.29 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 1H), 4.80 (ddd, J = 6.5, 6.5, 4.5 Hz, 1H), 3.99 (ABq, ΔδAB = 0.01, JAB = 10.5 Hz, 2H), 3.48 (s, 3H), 3.13 (app t, J = 6.7 Hz, 1H), 2.89 (d, J = 3.3 Hz, 1H), 2.61 (ddd, J = 6.9, 6.9, 3.3 Hz, 1H), 2.39–2.30 (m, 1H), 2.06 (ddd, J = 14.4, 8.4, 6.3 Hz, 1H), 1.60 (ddd, J = 13.3, 8.5, 4.5 Hz, 1H), 1.11 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 179.0, 107.4, 86.7, 70.9, 56.8, 52.4, 51.5, 44.7, 40.8, 38.6, 37.2, 16.9; IR (Neat Film NaCl) 2961, 2877, 1772, 1353, 1150, 1128, 1100, 1062, 936, 710 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C12H16O4 224.1049, found 224.1052; [α]D18.3 +73.0 (c 1.13, CH2Cl2, 98% ee).
Acetal 4329
Rf = 0.19 (2:1 hexanes/EtOAc); mp = 151.5–153 °C (Et2O); 1H NMR (600 MHz, CDCl3) δ 5.30 (s, 1H), 4.89 (td, J = 7.2, 5.8 Hz, 1H), 4.19 (d, J = 9.6 Hz, 1H), 4.11 (d, J = 9.6 Hz, 1H), 3.49 (s, 3H), 3.33 (dd, J = 7.0, 6.3 Hz, 1H), 2.58 (d, J = 4.0 Hz, 1H), 2.40 (td, J = 6.0, 4.0, Hz, 1H), 2.26 (ddq, J = 13.7, 11.1, 6.8 Hz, 1H), 2.16 (dt, J = 14.1, 7.1 Hz, 1H), 1.55 (ddd, J = 14.0, 11.1, 5.7 Hz, 1H), 1.03 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 177.2, 108.4, 86.6, 73.2, 56.5, 55.5, 50.1, 42.8, 41.2, 38.7, 38.2, 15.3; IR (Neat Film NaCl) 2934, 1766, 1460, 1359, 1199, 1171, 1143, 1130, 1115, 1063, 1045, 916, 904, 691 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C12H16O4 224.1049, found 224.1044; [α]D17.9 −56.7 (c 0.62, CH2Cl2, 98% ee). Crystals suitable for X-ray analysis were obtained by slow evaporation from Et2O.
Acid 46
To a solution of olefin 44 (41.0 mg, 0.184 mmol, 1.0 equiv) in THF (3.7 mL, 0.05 M) cooled to 0 °C was added KOTMS (236 mg, 1.84 mmol, 10 equiv) in one portion. After 5 min the reaction was warmed to room temperature. At 12 h the reaction was cooled to 0 °C and slowly quenched with 10% HCl (4 mL) and diluted with brine (4 mL) and EtOAc (10 mL). The layers were separated and the aq layer was extracted with EtOAc (3 × 10 mL), the combined organics were dried over Na2SO4, filtered, and concentrated to a pale yellow oil. The crude was purified by flash chromatography on SiO2 (6:1 → 3:1 hexanes/EtOAc, CH2Cl2 load) to afford 46 (35.2 mg, 0.169 mmol, 92% yield) as a white solid. Rf = 0.21 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 6.04 (dd, J = 17.5, 10.8 Hz, 1H), 5.20 (dd, J = 10.8, 1.2 Hz, 1H), 5.17 (dd, J = 17.5, 1.2 Hz, 1H), 4.61 (td, J = 6.4, 1.6 Hz, 1H), 4.02 (d, J = 9.1 Hz, 1H), 3.54 (d, J = 9.1 Hz, 1H), 3.22 (d, J = 7.3 Hz, 1H), 3.05 (app t, J = 7.1 Hz, 1H), 2.91 (app q, J = 7.6 Hz, 1H), 2.40 (d sextets, J = 10.4, 7.0 Hz, 1H), 2.20 (ddd, J = 14.7, 10.4, 5.8 Hz, 1H), 1.73 (ddd, J = 14.7, 6.3, 1.7 Hz, 1H), 1.05 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 178.6, 134.5, 116.2, 87.2, 78.8, 52.8, 51.2, 44.7, 42.5, 39.0, 37.2, 17.2; IR (Neat Film NaCl) 3085 (br), 2958, 2930, 1731, 1704, 1418, 1283, 1241, 1086, 1041, 996, 921 cm−1; HRMS (EI+) m/z: [M]+ calc′d for C12H16O3 208.1100, found 208.1094; [α]D15.3 +28.3 (c 0.97, CH2Cl2, 98% ee).
α-Diazo cyclobutyl ketone 47
To a solution of acid 46 (62.5 mg, 0.300 mmol, 1.0 equiv) in CH2Cl2 (6.0 mL, 0.05 M) at 0 °C was added a solution of oxalyl chloride (353 μL of a 1.7 M solution in CH2Cl2, 0.600 mmol, 2.0 equiv), followed by 1 drop of DMF. The reaction was stirred for 45 min at 0 °C, at which point the stir bar was removed, toluene was added (6 mL), and the volatiles were removed on a rotovap purged with argon. The septum and stir bar were replaced and the crude material was further dried under high vacuum for 10 min. The resulting crude semisolid was partially dissolved in CH2Cl2 (2 mL) and THF (4 mL) and transferred quantitatively via Teflon cannula to a vigorously stirring solution of excess diazomethane (ca. 30 mL) containing IRA-67 (161 mg, ca. 0.9 mmol, 3.0 equiv) at 0 °C. The flask was further washed with CH2Cl2 (4 mL) and THF (2 mL) and quantitatively transferred. After 3.5 h the cooling bath was removed and the diazomethane was pulled off via water aspirator. The pale yellow solution was filtered through a small SiO2 plug (Et2O) and concentrated in vacuo. The crude material was purified by flash chromatography on SiO2 (6:1 → 2:1 hexanes/Et2O) to afford 47 (63.2 mg, 0.272 mmol, 91% yield) as a bright yellow oil that solidifies in a −20 °C freezer. Rf = 0.24 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.01 (dd, J = 17.5, 10.8 Hz, 1H), 5.15 (dt, J = 10.9, 1.1 Hz, 1H), 5.10 (br s, 1H), 5.05 (d, J = 17.5 Hz, 1H), 4.59 (app t, J = 6.1 Hz, 1H), 3.92 (d, J = 9.0 Hz, 1H), 3.63 (d, J = 9.0 Hz, 1H), 3.11 (br d, J = 14.8, 2H), 2.96 (app t, J = 6.9 Hz, 1H), 2.44–2.35 (m, 1H), 2.20 (td, J = 12.6, 5.5 Hz, 1H), 1.70 (dd, J = 14.7, 6.3 Hz, 1H), 0.99 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 193.7, 134.9, 115.7, 87.2, 78.3, 54.8, 53.3, 51.9, 50.4, 42.6, 38.3, 37.3, 17.2; IR (Neat Film NaCl) 3081, 2956, 2100, 1635, 1373, 1047, 919 cm−1; HRMS (FAB+) m/z: [M + H]+ calc′d for C13H17N2O2 233.1290, found 233.1296; [α]D20.2 −66.3 (c 0.99, CH2Cl2, 98% ee).
α-Diazo cyclobutyl ketone 48
α-Diazoketone 48 was prepared by the same procedure as described for 47 using acid 46 (17.3 mg, 83.1 μmol, 1.0 equiv), but with freshly prepared and KOH-dried diazoethane (ca. 20 mL). After 4 h at 0 °C the excess diazoethane was removed via water aspirator. The pale orange solution was filtered through a small SiO2 plug (Et2O) and concentrated in vacuo. The crude material was purified by flash chromatography on SiO2 (6:1 → 4:1 hexanes/Et2O, CH2Cl2 load) to afford 48 (13.0 mg, 52.8 μmol, 64% yield) as a bright yellow oil. Rf = 0.38 (2:1 hexanes/Et2O); 1H NMR (500 MHz, CDCl3) δ 5.95 (dd, J = 17.5, 10.9 Hz, 1H), 5.10 (d, J = 10.8 Hz, 1H), 5.05 (d, J = 17.6 Hz, 1H), 4.57 (app t, J = 5.7 Hz, 1H), 3.86 (d, J = 9.3 Hz, 1H), 3.66 (d, J = 9.3 Hz, 1H), 3.34 (d, J = 6.8 Hz, 1H), 3.28 (app q, J = 7.5 Hz, 1H), 2.93 (app t, J = 6.9 Hz, 1H), 2.45–2.36 (m, 1H), 2.16 (ddd, J = 14.9, 10.1, 5.2 Hz, 1H), 1.94 (s, 3H), 1.70 (dd, J = 14.5, 5.9 Hz, 1H), 0.98 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 14.3, 134.8, 115.5, 87.0, 77.3, 53.0, 52.5, 47.7, 42.3, 36.7, 36.3, 17.3, 8.3; IR (Neat Film NaCl) 2957, 2926, 2064, 1631, 1286, 1050 cm−1; HRMS (FAB+) m/z: [M + H]+ calc′d for C14H19O2N2 247.1447, found 247.1457; [α]D19.9 +71.6 (c 0.57, CH2Cl2, 98% ee).
Cyclooctadienone 49
A solution of α-diazo cyclobutyl ketone 47 (69.2 mg, 0.271 mmol) in toluene (54 mL, 5 mM) was partitioned equally into three non-dried 20 mL microwave reaction vessels containing a stir bar under ambient atmosphere. Each vial was sealed and irradiated in a Biotage Initiator microwave reactor at 400 W until the temperature reached 160 °C, and the temperature was maintained for 15 min. The vial was cooled to room temperature, the seal was removed, and the contents were concentrated in vacuo. Reaction conversion was monitored by crude 1H NMR analysis (CDCl3). The crude material was purified by flash chromatography on SiO2 (9:1 → 6:1 → 3:1 hexanes/EtOAc) to give 49 (43.9 mg, 0.215 mmol, 79% yield) as a colorless oil that solidifies in a −20 °C freezer. Rf = 0.35 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.08 (dd, J = 12.4, 6.5 Hz, 1H), 5.93 (app dt, J = 12.4, 1.9 Hz, 1H), 5.56–5.51 (m, 1H), 4.59 (dd, J = 14.1, 6.9 Hz, 1H), 4.38 (d, = 12.1 Hz, 1H), 4.32 (m, 1H), 3.44 (m, 1H), 3.28 (dd, J = 14.3, 9.8 Hz, 1H), 3.18–3.13 (m, 1H), 2.99 (ddd, J = 14.3, 6.2, 1.1 Hz, 1H), 2.36 (tq, J = 12.9, 6.5 Hz 1H), 2.18 (dtd, J = 13.2, 6.5, 1.1 Hz, 1H), 1.44 (td, J = 13.2, 5.9 Hz, 1H), 1.11 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 204.4, 146.1, 139.2, 131.6, 114.2, 85.1, 74.6, 52.9, 47.8, 40.1, 38.9, 15.0; IR (Neat Film NaCl) 2958, 2874, 1691, 1666, 1116, 1064, 1032, 974, 867 cm−1; HRMS (FAB+) m/z: [M + H]+ calc′d for C13H17O2 205.1229, found 205.1223; [α]D20.4 −642 (c 1.38, CH2Cl2, 98% ee).
Wolff/Cope rearrangement of 48
A solution of α-diazo cyclobutyl ketone 48 (17.7 mg, 71.9 μmol) in heptane (14.4 mL, 5 mM) was prepared in a non-dried 20 mL microwave reaction vessel under ambient atmosphere and sealed. The contents were irradiated in a Biotage Initiator microwave reactor at 400 W until the temperature reached 150 °C, and the temperature was maintained for 10 min. The reaction was cooled to room temperature and TLC analysis showed consumption of 221. The solution was concentrated in vacuo and purified by preparative TLC on SiO2 (3:1 hexanes/EtOAc, develop twice) to afford α-methyl cyclooctadienone 50 (6.6 mg, 30.2 μmol, 42% yield) as a colorless oil and cyclopropane 51 as a single diastereomer.
Cyclooctadienone 50
Rf = 0.41 (3:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 5.80 (app dq, J = 7.2, 1.3 Hz, 1H), 5.52–5.48 (m, 1H), 4.58 (td, J = 7.3, 6.2 Hz, 1H), 4.38 (d, J = 12.1 Hz, 1H), 4.31 (m, 1H), 3.36 (td, J = 8.2, 1.2 Hz, 1H), 3.24 (dd, J = 15.6, 9.5 Hz, 1H), 3.02–2.98 (comp m, 2H), 2.32 (td, J = 13.0, 6.2 Hz, 1H), 2.16 (ddd, J = 13.2, 7.1, 1.0 Hz, 1H), 1.84 (t, J = 1.5 Hz, 3H), 1.47 (td, J = 13.3, 5.9 Hz, 1H), 1.08 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 207.6, 146.6, 137.2, 132.5, 113.7, 85.1, 74.5, 53.3, 47.0, 46.8, 40.3, 38.9, 20.8, 15.0; IR (Neat Film NaCl) 2956, 2923, 2874, 1693, 1667, 1452, 1375, 1076, 1045, 1020, 973, 873, 838 cm−1; HRMS (MM: ESI/APCI) m/z: [M + H]+ calc′d for C14H19O2 219.1380, found 219.1379; [α]D25 −573 (c 0.35, CHCl3, 98% ee)
Cyclopropane 5129
Rf = 0.36 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 4.80 (dt, J = 7.2, 6.0 Hz, 1H), 3.94 (d, J = 9.3 Hz, 1H), 3.84 (d, J = 9.3 Hz, 1H), 2.86 (d, J = 1.8 Hz, 1H), 2.84 (dd, J = 7.4, 5.8 Hz, 1H), 2.25 (d, J = 2.9 Hz, 1H), 2.16 (dddd, J = 10.5, 10.5, 8.6, 4.1 Hz, 1H), 2.12–2.07 (m, 1H), 2.04 (ddd, J = 8.7, 5.8, 2.8 Hz, 1H), 1.56–1.54 (comp m, 2H), 1.49 (dddd, J = 13.6, 10.2, 5.5, 4.1 Hz, 1H), 1.22 (s, 3H), 0.96 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 198.7, 86.3, 74.3, 67.1, 57.9, 51.0, 49.7, 46.7, 43.1, 38.8, 37.4, 32.7, 15.2, 9.4; IR (Neat Film NaCl) 2953, 2923, 2868, 1776, 1449, 1073, 1015, 937 cm−1; HRMS (MM: ESI/APCI) m/z: [M + H]+ calc′d for C14H19O2 219.1380, found 219.1382; [α]D25 +46.1 (c 0.38, CHCl3, 98% ee)
Supplementary Material
Acknowledgments
The authors thank the NIH-NIGMS (R01GM080269), Eli Lilly (predoctoral fellowship to M.R.K.), the Danish Council for Independent Research/Natural Sciences (postdoctoral fellowship to T.J.), Amgen, AbbVie, Bristol-Myers Squibb, Boehringer-Ingelheim, Merck and Caltech for generous financial support. Dr. Takeharu Toyoshima and Angela Guerrero are acknowledged for contributions to substrate preparation and reaction scouting. Drs. David VanderVelde and Scott Ross of the Caltech NMR facility are thanked for invaluable assistance with NMR experiments and helpful discussions. Lawrence Henling and Michael Day are gratefully acknowledged for X-ray crystallographic structural determination. Dr. Mona Shahgholi and Naseem Torian are acknowledged for assistance with high-resolution mass spectrometry.
Footnotes
Additional experimental procedures, X-ray data and new compound spectra (1H, 13C, IR and other NMR) are available in the Supporting Information.
References and Endnotes
- 1.Hensens OD, Zink D, Williamson JM, Lotti VJ, Chang RSL, Goetz MA. J Org Chem. 1991;56:3399–3403. [Google Scholar]
- 2.Yoganathan K, Rossant C, Glover RP, Cao S, Vittal JJ, Ng S, Huang Y, Buss AD, Butler MS. J Nat Prod. 2004;67:1681–1684. doi: 10.1021/np049844c. [DOI] [PubMed] [Google Scholar]
- 3.Fujimoto H, Nakamura E, Okuyama E, Ishibashi M. Chem Pharm Bull. 2000;48:1436–1441. doi: 10.1248/cpb.48.1436. [DOI] [PubMed] [Google Scholar]
- 4.Tezuka Y, Takahashi A, Maruyama M, Tamamura T, Kutsuma S, Naganawa H, Takeuchi T. Novel Antibiotics, AB5362-A, B, and C, Their Manufacture, Their Use as Fungicides, and Phoma Species AB5362. 10045662. Japan Patent JP. 1998
- 5.Takahashi H, Hosoe T, Nozawa K, Kawai K-i. J Nat Prod. 1999;62:1712–1713. doi: 10.1021/np990146f. [DOI] [PubMed] [Google Scholar]
- 6.Sato A, Morishita T, Hosoya T. S-19777 as Endothelin Antagonist, Its Manufacture with Emericella Aurantiobrunnea, and Its Use as Pharmaceutical. 10306087. Japan Patent JP. 1998
- 7.For reviews of the preparation of eight-membered carbocycles and their use in the synthesis of complex molecules, see: Petasis NA, Patane MA. Tetrahedron. 1992;48:5757–5821.Mehta G, Singh V. Chem Rev. 1999;99:881–930. doi: 10.1021/cr9800356.Maier ME. Angew Chem, Int Ed. 2000;39:2073–2077. doi: 10.1002/1521-3773(20000616)39:12<2073::aid-anie2073>3.0.co;2-0.Yet L. Chem Rev. 2000;100:2963–3007. doi: 10.1021/cr990407q.Michaut A, Rodriguez J. Angew Chem, Int Ed. 2006;45:5740–5750. doi: 10.1002/anie.200600787.Yu ZX, Wang Y, Wang Y. Chem – Asian J. 2010;5:1072–1088. doi: 10.1002/asia.200900712.Hog DT, Webster R, Trauner D. Nat Prod Rep. 2012;29:752–779. doi: 10.1039/c2np20005h.Urabe D, Asaba T, Inoue M. Chem Rev. 2015;115:9207–9231. doi: 10.1021/cr500716f.
- 8.For recent advancements, see: Watson IDG, Ritter S, Toste FD. J Am Chem Soc. 2009;131:2056–2057. doi: 10.1021/ja8085005.Zhu C, Zhang X, Lian X, Ma S. Angew Chem, Int Ed. 2012;51:7817–7820. doi: 10.1002/anie.201202971.Iwai T, Okochi H, Ito H, Sawamura M. Angew Chem, Int Ed. 2013;52:4239–4242. doi: 10.1002/anie.201300265.Arichi N, Yamada K-i, Yamaoka Y, Takasu K. J Am Chem Soc. 2015;137:9579–9582. doi: 10.1021/jacs.5b06576.
- 9.(a) Piers E, Boulet SL. Tetrahedron Lett. 1997;38:8815–8818. [Google Scholar]; (b) Walker SD. PhD Thesis. University of British Columbia; Vancouver, Canada: 2002. A Synthetic Approach to the Variecolin Class of Sesterterpenoids: Total Synthesis of (±)-5-Deoxyvariecolin, (±)-5-Deoxyvariecolol and (±)-5-Deoxyvariecolactone. A New Cycloheptenone Annulation Method Employing the Bifunctional Reagent (Z)-5-Iodo-1-tributylstannylpent-1-ene. [Google Scholar]
- 10.(a) Molander GA, Quirmbach MS, Silva LF, Jr, Spencer KC, Balsells J. Org Lett. 2001;3:2257–2260. doi: 10.1021/ol015763l. [DOI] [PubMed] [Google Scholar]; (b) George KM. PhD Thesis. University of Pennsylvania; Philadelphia, PA: 2005. Natural Product Total Synthesis: I. The Total Synthesis of (+)-Isoschizandrin. II. Progress Toward the Total Synthesis of Variecolin. [Google Scholar]
- 11.Li K, Wang C, Yin G, Gao S. Org Biomol Chem. 2013;11:7550–7558. doi: 10.1039/c3ob41693c. [DOI] [PubMed] [Google Scholar]
- 12.Hog DT, Mayer P, Trauner D. J Org Chem. 2012;77:5838–5843. doi: 10.1021/jo300726z. [DOI] [PubMed] [Google Scholar]
- 13.Brill ZG, Grover HK, Maimone TJ. Science. 2016;352:1078–1082. doi: 10.1126/science.aaf6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Hong AY, Krout MR, Jensen T, Bennett NB, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2011;50:2756–2760. doi: 10.1002/anie.201007814. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hong AY, Bennett NB, Krout MR, Jensen T, Harned AM, Stoltz BM. Tetrahedron. 2011;67:10234–10248. doi: 10.1016/j.tet.2011.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Sarpong R, Su JT, Stoltz BM. J Am Chem Soc. 2003;125:13624–13625. doi: 10.1021/ja037587c. [DOI] [PubMed] [Google Scholar]; (b) Su JT, Sarpong R, Stoltz BM, Goddard WA., III J Am Chem Soc. 2004;126:24–25. doi: 10.1021/ja037716p. [DOI] [PubMed] [Google Scholar]
- 16.(a) May JA, Stoltz BM. J Am Chem Soc. 2002;124:12426–12427. doi: 10.1021/ja028020j. [DOI] [PubMed] [Google Scholar]; (b) Tambar UK, Kano T, Stoltz BM. Org Lett. 2005;7:2413–2416. doi: 10.1021/ol050705b. [DOI] [PubMed] [Google Scholar]; (c) Tambar UK, Kano T, Zepernick JF, Stoltz BM. J Org Chem. 2006;71:8357–8364. doi: 10.1021/jo061236+. [DOI] [PubMed] [Google Scholar]; (d) Streuff J, White DE, Virgil SC, Stoltz BM. Nat Chem. 2010;2:192–196. doi: 10.1038/nchem.518. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nelson HM, Murakami K, Virgil SC, Stoltz BM. Angew Chem, Int Ed. 2011;50:3688–3691. doi: 10.1002/anie.201008003. [DOI] [PubMed] [Google Scholar]
- 17.Grubbs RH, Pancoast TA, Grey RA. Tetrahedron Lett. 1974;15:2425–2426. [Google Scholar]
- 18.Tallarico JA, Randall ML, Snapper ML. J Am Chem Soc. 1996;118:9196–9197. [Google Scholar]
- 19.Limanto J, Snapper ML. J Org Chem. 1998;63:6440–6441. [Google Scholar]
- 20.Our pursuit of an sp3-hybridized linker to join the cyclopentenol and cyclobutadiene was guided by Snapper’s observations: Limanto J, Snapper ML. J Am Chem Soc. 2000;122:8071–8072.
- 21.Deardorff DR, Myles DC. Org Synth. 1989;67:114–120. [Google Scholar]
- 22.Kobayashi Y, Nakata K, Ainai T. Org Lett. 2005;7:183–186. doi: 10.1021/ol047953c. [DOI] [PubMed] [Google Scholar]
- 23.The preparation of (cyclobutadiene)tricarbonyliron ether complexes from acid-sensitive allylic alcohols are typically accomplished from the reaction of potassium alkoxides with (bromomethyl cyclobutadiene)tricarbonyliron (see reference 19). In our hands, this procedure was variable and furnished only modest yields (41–69%) of desired ether 20.
- 24.Rai AN, Basu A. Tetrahedron Lett. 2003;44:2267–2269. [Google Scholar]
- 25.Preliminary efforts for the regioselective functionalization of diol 58 were unsuccessful.
- 26.Schreiber SL, Claus RE, Reagan J. Tetrahedron Lett. 1982;23:3867–3870. [Google Scholar]
- 27.For a review of the unsymmetrical ozonolysis of cycloalkenes see: Testero SA, Suárez AG, Spanevello RA, Mangione MI. Trends Org Chem. 2003;10:35–49.Testero SA, Mangione MI, Suárez AG, Spanevello RA. Eur J Org Chem. 2013:5236–5245.
- 28.For example see: Labadie GR, Luna LE, Gonzalez Sierra M, Cravero RM. Eur J Org Chem. 2003;2003:3429–3434.Breder A, Chinigo GM, Waltman AW, Carreira EM. Angew Chem, Int Ed. 2008;47:8514–8517. doi: 10.1002/anie.200803284.Xu Z, Wang Q, Zhu J. Angew Chem, Int Ed. 2013;52:3272–3276. doi: 10.1002/anie.201209970.Maehara T, Motoyama K, Toma T, Yokoshima S, Fukuyama T. Angew Chem, Int Ed. 2017;56:1549–1552. doi: 10.1002/anie.201611574.
- 29.The structural and relative stereochemical assignments were established by multiple methods that include 1D and 2D NMR analysis. See the supporting information for further details and tabulated structural data.
- 30.Kawamura S-i, Yamakoshi H, Nojima M. J Org Chem. 1996;61:5953–5958. [Google Scholar]
- 31.The Lewis acids La(OTf)3 or Sm(OTf)3 effect the conversion of acetal 23 to dimethyl acetal 58 as the sole product.
- 32.(a) Vogel E. Justus Liebigs Ann Chem. 1958;615:1–14. [Google Scholar]; (b) Hammond GS, DeBoer CD. J Am Chem Soc. 1964;86:899–902. [Google Scholar]; (c) Trecker DJ, Henry JP. J Am Chem Soc. 1964;86:902–905. [Google Scholar]; (d) Berson JA, Dervan PB. J Am Chem Soc. 1972;94:7597–7598. [Google Scholar]
- 33.(a) Schneider M. Angew Chem. 1975;87:717–718. [Google Scholar]; (b) Brown JM, Golding BT, Stofko JJ., Jr J Chem Soc, Perkin Trans. 2;1978:436–441. [Google Scholar]; (c) Schneider MP, Rau A. J Am Chem Soc. 1979;101:4426–4427. [Google Scholar]; (d) Gajewski JJ, Hawkins CM, Jimenez JL. J Org Chem. 1990;55:674–679. [Google Scholar]; (e) Özkan I, Zora M. J Org Chem. 2003;68:9635–9642. doi: 10.1021/jo035173w. [DOI] [PubMed] [Google Scholar]
- 34.(a) Sudrik SG, Chavan SP, Chandrakumar KRS, Pal S, Date SK, Chavan SP, Sonawane HR. J Org Chem. 2002;67:1574–1579. doi: 10.1021/jo010951a. [DOI] [PubMed] [Google Scholar]; (b) Presset M, Coquerel Y, Rodriguez J. J Org Chem. 2009;74:415–418. doi: 10.1021/jo8021567. [DOI] [PubMed] [Google Scholar]
- 35.(a) Reetz MT, Eipper A, Tielmann P, Mynott R. Adv Synth Catal. 2002;344:1008–1016. [Google Scholar]; (b) Tietze LF, Stadler C, Böhnke N, Brasche G, Grube A. Synlett. 2007:485–487. [Google Scholar]
- 36.Alcohol isomer 39 is not reactive under these Mitsunobu conditions.
- 37.Limanto J, Tallarico JA, Porter JR, Khuong KS, Houk KN, Snapper ML. J Am Chem Soc. 2002;124:14748–14758. doi: 10.1021/ja0203162. [DOI] [PubMed] [Google Scholar]
- 38.TLC analysis of the reaction progress indicated cycloadduct 41 as the major product, however, we are thus far unable to obtain consistent isolated yields due to the volatility of this compound and its challenging isolation from a large volume of acetone.
- 39.The reaction yield for the two steps post cycloaddition/ozonolysis is excellent (94%), indicating the low overall yield for the four steps is the result of either problematic cycloaddition or ozonolysis procedures. The challenges that we have encountered with the volatility and purification of cycloadduct 41 suggest a notable limitation of this reaction sequence.
- 40.The absolute stereochemistry of acetal 43 could not be determined from the X-ray diffraction data. See the supporting information for details.
- 41.(a) Wilds AL, Meader AL., Jr J Org Chem. 1948;13:763–779. doi: 10.1021/jo01163a024. [DOI] [PubMed] [Google Scholar]; (b) Kennedy M, McKervey MA. J Chem Soc, Perkin Trans. 1;1991:2565–2574. [Google Scholar]
- 42.For a recent review on common methods for the preparation of α-diazocarbonyl compounds see: Ford A, Miel H, Ring A, Slattery CN, Maguire AR, McKervey MA. Chem Rev. 2015;115:9981–10080. doi: 10.1021/acs.chemrev.5b00121.
- 43.Application of photochemical/thermal reaction conditions to α-diazo cyclobutyl ketone 48 produced similar results.
- 44.Kirmse W. Eur J Org Chem. 2002:2193–2256. [Google Scholar]
- 45.(a) Kaplan F, Meloy GK. J Am Chem Soc. 1966;88:950–956. [Google Scholar]; (b) Sorriso S, Piazza G, Foffani A. J Chem Soc B. 1971:805–809. [Google Scholar]; (c) Paliani G, Sorriso S, Cataliotti R. J Chem Soc, Perkin Trans. 1976;2:707–710. [Google Scholar]; (d) Wang Y, Toscano JP. J Am Chem Soc. 2000;122:4512–4513. [Google Scholar]; (e) Wang J, Burdzinski G, Kubicki J, Platz MS. J Am Chem Soc. 2008;130:11195–11209. doi: 10.1021/ja803096p. [DOI] [PubMed] [Google Scholar]
- 46.Variation of reaction temperature did not impact the ratio of products for the tandem rearrangement of 48.
- 47.Love BE, Jones EG. J Org Chem. 1999;64:3755–3756. doi: 10.1021/jo982433e. [DOI] [PubMed] [Google Scholar]
- 48.Werner EA. J Chem Soc Trans. 1919:1093–1102. [Google Scholar]
- 49.Williams MJ, Deak HL, Snapper ML. J Am Chem Soc. 2007;129:486–487. doi: 10.1021/ja0674340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.