

Abstract
Cytokines are signaling molecules secreted and sensed by immune and other cell types, enabling dynamic inter-cellular communication. Although a vast amount of data on these interactions exists, this information is not compiled, integrated or easily searchable. Here we report immuneXpresso, a text mining engine that structures and standardizes knowledge of immune inter-cellular communication. We applied immuneXpresso to PubMed to identify relationships between 340 cell types and 140 cytokines across thousands of diseases. The method distinguishes between incoming and outgoing interactions, and includes the effect of the interaction and the cellular function involved. These factors are assigned a confidence score and linked to the disease. Leveraging the breadth of this network, we predict and experimentally verify previously unappreciated cell-cytokine interactions. We also build a global immune-centric view of diseases and use it to predict cytokine-disease associations. This standardized knowledgebase (www.immunexpresso.org) opens up new directions for interpretation of immune data and model-driven systems immunology.
Protective immunity is mediated through a complex system of interacting cells whose communication network is primarily governed by secreted molecules, chiefly the cytokines and chemokine family proteins. Until recently, the high complexity of the immune system was approached by researchers using reductionist approaches, but technological advances now enable acquisition of large data sets, with broad enumeration of cell subset types and functions, protein, gene expression and more1. In addition, papers in immunology alone are being published at the rate of approximately one every 30 minutes. To maximize discovery, research results must transition to organized standardized models of knowledge, on which automated computational processing is deployed.
Biomedical text mining efforts have been an important means of grasping at the breadth and complexity of biological systems. With efforts invested into recognizing biologically relevant entities, such as genes, diseases, chemicals and genomic variants2–8, driven by gold-standards9,10 and community-wide efforts11,12,13,14, text mining is enabling automatic identification of complex biological relations15,16 and full-scale networks.. Recent research has expanded to additional types of molecular events17–19, with relation extraction methods ranging from co-occurrence15,19, pattern-matching and rule-based methods, to dependency parse graph analysis20,21 and machine learning21. However, to date, text mining approaches have not addressed large-scale inter-cellular communication networks and, in particular, those describing directional cell-cytokine interactions.
Biological literature mining has shown utility for hypothesis generation, particularly in disease contexts22–24. Similarly, data-driven disease classifications have shown benefit in understanding shared mechanisms, empowering target identification and drug repositioning choices25–28. Yet to date, such classifications have not addressed cellular cross-talk and how the immune system may impact disease.
To establish a foundation for systematic reasoning over the inter-cellular network, we built immuneXpresso (iX), a comprehensive high-resolution knowledgebase of directional inter-cellular interactions, text-mined from all available PubMed abstracts across a broad range of disease conditions. Interactions captured by iX include both direct cytokine binding/secretion events and more distant, indirect influencing relations, scored and filtered to emphasize precision. We use the resulting knowledge standardization to characterize the immune inter-cellular network and to predict and experimentally validate cell-cytokine interactions. Leveraging the breadth and context-awareness of the knowledgebase, we build an immune-centric view of diseases and explore its modularity to predict cytokine-disease associations.
RESULTS
A text mining pipeline to extract inter-cellular interactions
We designed a computational pipeline focused on mining the primary literature for identification of cells, inter-cellular signaling molecules (i.e., cytokines) and the directional relations between them (Fig. 1a, Online Methods) and applied it across the entire PubMed (approximately 16 million articles published electronically by July 2017). Briefly, for each individual sentence, the analysis pipeline tags cells, cytokines and diseases, as well as standardizes terminology through official ontologies to allow for hierarchical data analysis at multiple resolutions (Supplementary Tables 1-4). We examine sentence structure to identify syntactically related cell, verb and cytokine. From each such ‘evidence record’, we the relation’s directionality, polarity (representing its positive, negative or neutral effect) and when possible, the resulting cellular biological function (Supplementary Table 5). We distinguish between ‘outgoing’ relations, describing cytokine secretion by a given cell type, and ‘incoming’ relations, describing events in which a cytokine affects a cell type, either directly via binding or indirectly. Finally, for each unique triple of cell, cytokine and directionality, summarized across all its evidence records, we use a trained machine learning classifier to make a call on whether the collected evidence indeed describes an interaction (Online Methods). We assign confidence scores to these and link to the conditions (e.g., diseases) co-mentioned in the same abstracts. In addition, we annotate independent entity mentions, without interaction, of cells and cytokines to allow for entity co-occurrence and enrichment statistics.
Figure 1. immuneXpresso assembles a system level directional inter-cellular interaction network.
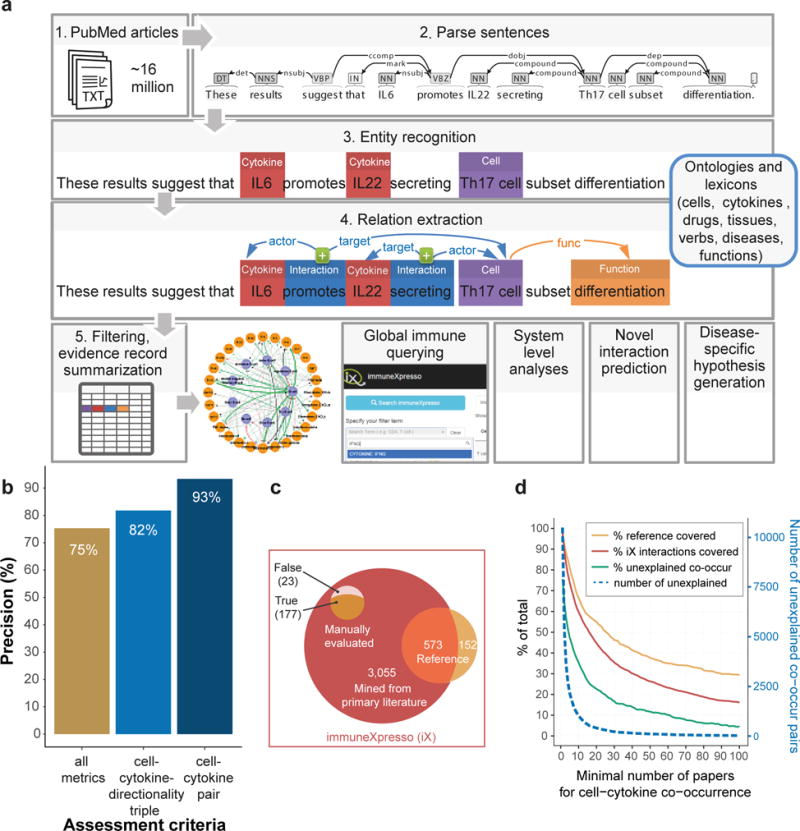
(a) Pubmed abstracts were mined to identify cell, cytokine and context entities and map them to ontologies. Semantically linked cells, cytokines and verbs were extracted and characterized to assign interaction directionality, polarity, and the biological function this interaction yields. Results were scored and filtered to yield a global machine-readable view of immune inter-cellular signaling across a large breadth of conditions. (b) Evaluation of cell-cytokine relation extraction precision,. 590 randomly selected evidence records (i.e., relations extracted from individual sentences) were evaluated by a human curator, post machine learning based filtering, to assess entity recognition performance, cell-cytokine relation extraction, including detection of verb, directionality, polarity and, where relevant. biological function. (c) Venn diagram depicting the precision (90%) of literature-derived cell-cytokine interactions (i.e. cell-cytokine-directionality triples) and coverage (76%) of the manually annotated reference book29. (d) Evaluation of precision and recall of co-occurrence based relation extraction. Cell-cytokine pairs, linked by co-occurrence within sentence boundaries, are filtered by their evidence strength (x-axis). Coverage of relations derived from the reference book and from the semantic parsing approach (“iX interactions”) is shown, as well as the percentage (or amount as a dotted line, right y-axis) of co-occurring pairs appearing in none of the resources above, likely reflecting false positive rate.
To assess the precision of entity recognition, expert human curators evaluated 100 randomly chosen annotations each for cells, cytokines and diseases and found the automatic annotation to be 91%, 96% and 93% correct, respectively (Online Methods, Supplementary Tables 6-8). Similarly, for precision of relation extraction, randomly chosen 590 interaction evidence records (i.e., particular sentence instances) and manually evaluated entity recognition, ontology mapping, verb and relation detection, directionality, polarity and cellular function identification (Online Methods, Supplementary Tables 9, 10). We observed a conservative true positives rate of 75% when all metrics were considered, 82% when assessing triple precision (cell-cytokine-directionality) and 93% when checking cell-cytokine relation pair extraction only, ignoring directionality, polarity and other relation characteristics (Fig. 1b).
To evaluate performance in interaction recall (i.e., identifying known interactions), we manually curated directional interactions from a reference book covering up-to-date knowledge of cytokines29, and used it as a gold standard (Online Methods). Our machine learning derived knowledgebase covered 79% of the interactions described in the reference book, yet was close to five times larger, containing an additional 3,055 directional interactions. Manual assessment of 200 of these yielded an 11.5% false positive rate, suggesting that a large amount of biologically meaningful interactions appear only in the primary literature.
Finally, we unified all identified interactions (manual and machine derived) into a single knowledgebase which we named immuneXpresso (iX) (Fig. 1c). To quantify the advantages of this semantic-based approach, we compared its precision and recall with an alternative of assuming cells and cytokines co-occurring in the same sentence interact, without sentence structure analysis (Online Methods). Though the full set of co-occurrence based relations showed 98% recall of both the reference book-curated and iX cell-cytokine pairs, 75.6% of these relations appeared in neither resource, suggesting a very high false positive rate for a co-occurrence approach, even when we threshold by a minimal number of repeat co-occurrences (Fig. 1d).
At present, iX contains a total of 4,118 directional cell-cytokine interactions (Supplementary Table 11), three times as many incoming interactions as outgoing ones, an enrichment qualitatively echoed in reference book annotated interactions. These interactions stem from more than 31,000 articles (Supplementary Table 12). In addition, using the iX pipeline, we collected annotations of thousands of diseases (11,260 distinct disease terms. 2,179 of them appearing in at least 100 papers) and identified mentions of 1,300 cell types, 360 of which are hematopoietic, and 170 cytokines in these disease contexts (Supplementary Table 11, 13). iX is freely accessible for querying through www.immunexpresso.org, as well as via the ImmPort web site30.
System-level characterization of inter-cellular interactions
iX offers a unique opportunity for a system-level view of inter-cellular information flow. Given the large number of cells and cytokines, many of which are poorly understood, we first grouped cells into 16 major categories based on the cell ontology hierarchy, and cytokines into families based on structure and function (Online Methods). This yielded a bi-partite inter-cellular interaction network showing information flow between cell types and cytokine families (Fig. 2a). We noted that cell-types, irrespective of the number of identified cell subsets in their lineage, interacted with a large number of cytokine families. Replotting the network using the highest cell and cytokine resolution in iX we observed an increase in distinct cell subset -cytokine profiles per cell-type, particularly for outgoing interactions (Supplementary Fig. 1a, shown for CD4+ T-cells). Yet, the bulk of interactions were still described solely at a low cellular resolution, indicating that the unique cytokine milieu profile of distinct cellular subsets is for the most part still lacking.
Figure 2. System level characteristic of inter-cellular information flow in the literature-derived network.
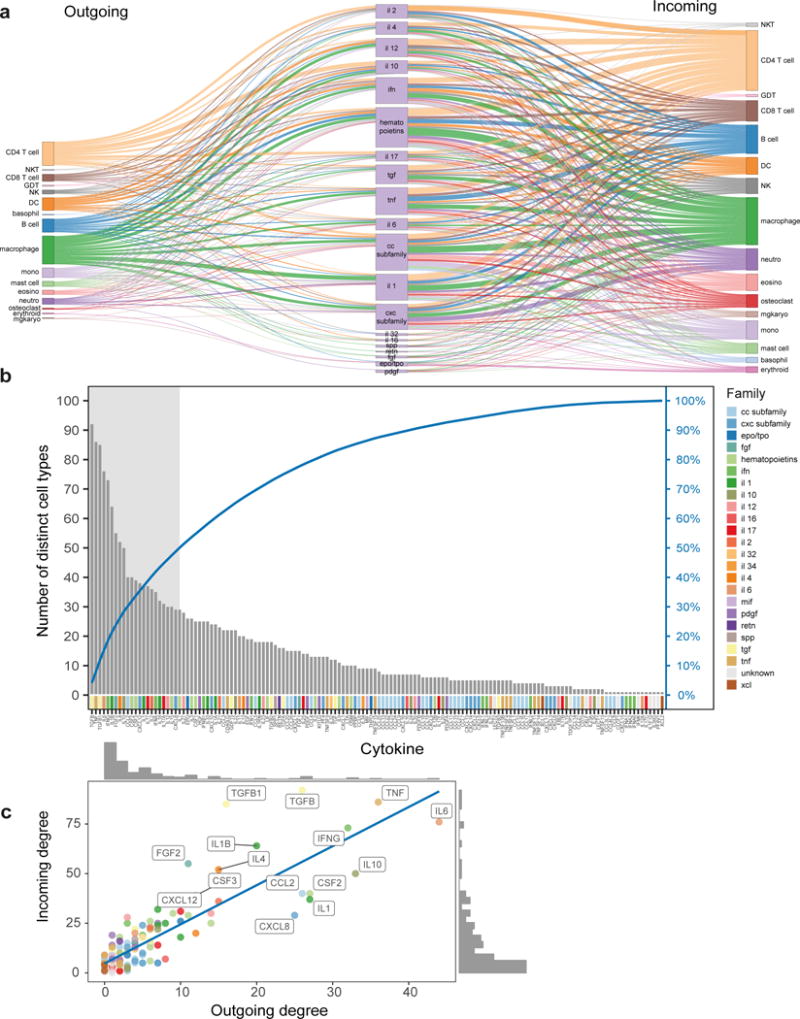
(a) Sankey plots showing bi-partite information flow between cellular secretion of cytokine families and those cytokine families affecting a variety of cell subsets. (b) A sorted histogram illustrating the number of unique cell types affected by each of the 144 cytokines (incoming interactions). Second y-axis displays the information as a cumulative sum (blue line). 50% of incoming edges are attributed to only 23 (16%) cytokines (grey area). Cytokine family classification appears as coloring of individual members along x-axis. (c) Scatter plot highlighting the strong correlation in cytokine degrees between incoming and outgoing directions (n=145 cytokines, r=0.86 pval <0.001 Pearson’s).
Signaling of some cytokines may be highly specific or broadly affecting multiple cell subsets, constituting hubs of the inter-cellular interaction network. iX enables to study global properties of the intercellular interaction network. For each interaction, we identified the highest cellular resolution it was reported for and calculated for each cytokine the number of cellular interactions it was associated with (i.e., its degree), covering both hematopoietic (HPC) and non-hematopoietic cell types. This demonstrated the existence of only a few hubs in the network, followed by a long tail of modestly and low interacting cytokines (Fig. 2b for incoming and Supplementary Fig. 1b for outgoing interactions). We noted that 50% of the incoming interactions in the network were formed by 23 (16% of total) cytokines. These included the top hubs TNF, TGFB, IL6 and IFNG. Similarly, in the reverse outgoing direction, we attributed 50% of edges to 17 (15% of total) cytokines. Cytokine degrees in incoming and outgoing directions showed high correlation (Fig. 2c, r=0.86 Pearson’s), as in the gold-standard reference book (Supplementary Fig. 1c, r=0.69 by Pearson). This correlation was lower yet still observed upon removal of autocrine interactions which may inflate similarity (r=0.73 and 0.36 for iX and the reference book respectively) and following removal of low degree cytokines (r=0.5 by Pearson), suggesting that hubs in the literature-derived inter-cellular interaction network appear to be bi-directional, both targeting and secreted by a large number of cell-types.
Immune inter-cellular network knowledge is biased
Analyzing the cytokine degree distribution, we could not reject power-law distribution for either incoming or outgoing interactions (Supplementary Fig. 2, p=0.73 and p=0.47 respectively for incoming and outgoing, Online Methods). Heavy-tailed network distributions may arise due to a research bias, yielding a “rich get richer” phenomenon31,32. Conversely, such degree distributions may arise naturally due to biological network structure33. Analysis of cytokine interaction knowledge accumulation showed the existence of one or two connection-rich leaders per cytokine family, with other family members trailing well behind (Fig. 3a, Supplementary Fig. 3). These were predominantly founding cytokine family members, such as IL6, IFNG, IL10 and TNF, and maintained their overwhelming dominance even when we discarded all explicit references to global family mentions in the text (e.g. TNF-family). We detected a low global correlation between a cytokine’s date of discovery and its degree, suggesting inter-cellular communication knowledge has not reached saturation, either for hubs or for less connected cytokines (Supplementary Fig. 4, r=−0.27, −0.26 Pearson’s for incoming and outgoing interactions respectively, driven by a few highly dominant hubs). Analysis of recent 5-year change in connectivity degree suggested for some hubs, such as FGF2, their iX degree likely reflects cellular interactivity potential, whereas others, such as IL6 and IL22, were still accumulating new connections at a high rate (Fig. 3b).
Figure 3. Immune inter-cellular network knowledge is biased and far from saturated.
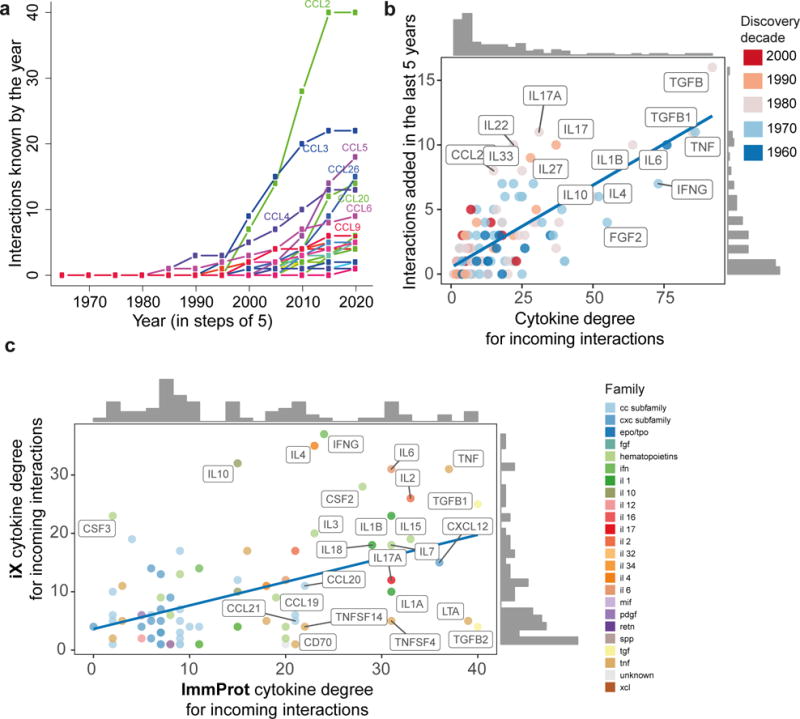
(a) Unequal information gain within cytokine families. Representative plot using the CC chemokine subfamily of the incoming interaction knowledge gain over time, in a 5-year window from initial date of the chemokine’s identification in the text. Colored lines represent individual chemokine family members and display the total number of distinct cell types known as interacting with the chemokine by the respective year (x-axis). (b) The number of distinct cell types a cytokine is known to act on (x-axis) is positively correlated with the number of new cytokine-cell interactions added in recent years (y-axis; n=134 cytokines, r=0.76 pval<0.001, Pearson’s), yet up-and-coming hubs, such as IL22, are identifiable as well. (c) Cytokine degrees in the iX knowledgebase (incoming interactions, restricted to hematopoietic cells) are positively correlated with those derived from the characterization of cytokine receptors on cells in the ImmProt project34 (n=82 cytokines present in both datasets, ρ=0.38 pval<0.001, spearman). Cytokines below the line suggest putative currently unknown interactions.
To assess how well the literature-derived knowledge represents experimental data, we compared iX cytokine degrees to those obtained from ImmProt, a high-resolution proteomics compendium quantifying proteins’ expression in rested and activated states in more than 20 sorted immune cell types34. We approximated outgoing and incoming interactions based on the expression of cytokine and cytokine receptor proteins in ImmProt cellular profiles. Comparison of incoming cytokine degrees in the resulting experimental network with the literature-derived ones (Fig. 3c) demonstrated significant correlation (ρ=0.38 by Spearman, p-val<0.001, based on 82 cytokines present in both datasets), with lower correlation for the opposite direction (ρ=0.26 by Spearman p-val=0.1, 41 shared cytokines), likely due to ImmProt secretion profiles measured in a single condition.
Prediction of cell-cytokine interactions
Using the iX knowledgebase up to 2014, we systematically predicted cell-cytokine interactions, using three orthogonal approaches (Fig. 4a, Online Methods): an unsupervised or supervised analysis of cytokine-cell interactions iX profiles independent of external data or by contrasting iX information versus receptor/cytokine gene expression data on cell subsets as reflected in the ImmProt34 and ImmGen databases35 (e.g., LTA in Fig. 3c). This systematic prediction process yielded 472 incoming and 367 outgoing ranked interaction candidates (Supplementary Tables 14, 15). Of these, we manually evaluated 78 predictions by extensive literature searches (Fig. 4b). This process identified 55% of candidates as already observed, true positive interactions, 3% with evidence published only following the prediction 2014 training set and 3% with evidence of cytokine receptor expression only. This high rate of recovery of known interactions was reassuring and suggested that the remaining 40% of candidate interactions were enriched for previously unrecognized interactions.
Figure 4. Prediction and validation of cell-cytokine interactions.
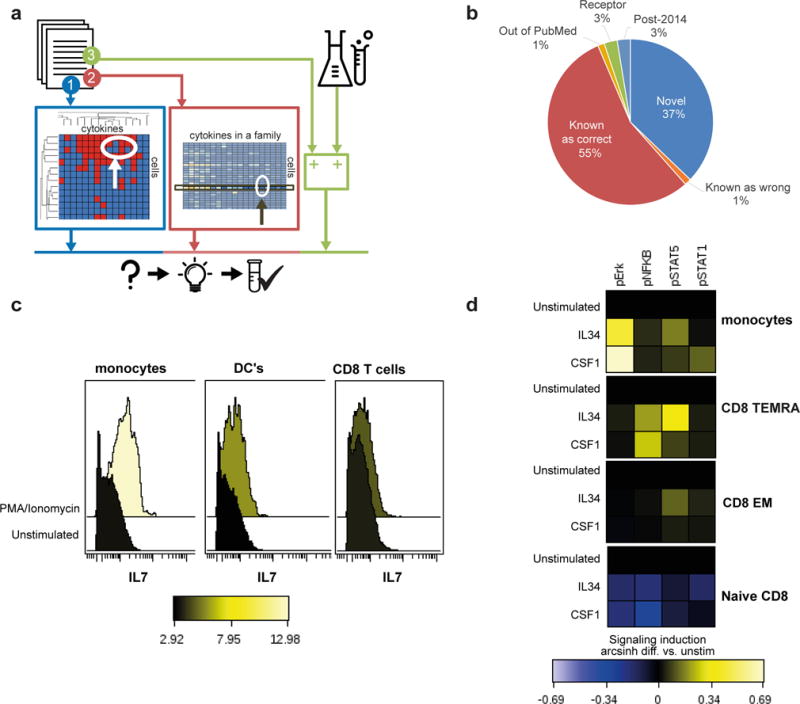
(a) Diagram illustrating three different methodologies for predicting novel cell-cytokine interactions: (1) by unsupervised clustering and filling in of missing ‘gaps’ (2) through supervised analysis of cytokine families (3) by comparison to measured external mRNA or proteome data. (b) Manual evaluation of the predicted high-confidence interactions using free literature search. (c) IL7 production by monocyte, dendritic cell and CD8 T cell subsets of freshly isolated PBMC’s from 2 healthy donors (PMA/ionomycin, 4hrs) by intra-cellular cytokine staining (CyTOF).. (d) Heatmap summary of pNFKB and pSTAT5 phosphorylation in CD8+ effector memory cell subsets, triggered by IL34 stimulation (100ng/ml, 15min) of freshly isolated human PBMCs.
We tested the validity of two top-rated candidate interaction predictions: For IL7, our supervised cytokine family prediction approach suggested an outgoing interaction (i.e. secretion) in monocytes (the literature currently describes the opposite interaction only, that is, IL7 affects monocytes36). In agreement with the prediction, we observed IL7 production by monocytes in activated human PBMC population. In addition, as expected, we detected dendritic cell production of IL7 and no IL7 production in CD8+ T cells, as reported in the literature (Fig. 4c). Similarly, our unsupervised prediction approach suggested that IL34 activates signaling in T-cells. We also detected expression of a corresponding receptor, CSF1R, on resting CD8+ T cell subsets in ImmProt34 (Supplementary Fig. 5). We stimulated human PBMCs with IL34 and observed robust phosphorylation of Erk in monocytes, as has previously been reported37. We also observed activation of pNFKB, pSTAT5 by IL34 in CD8+ effector memory T cells (Fig. 4e). Moreover, our prediction also supported our recent validation of CD4+ memory cells induction by IL34 signaling following upregulation of the CSF1R during activation34.
Immune-centric classification of diseases
We reasoned that the structured format and breadth of iX can be leveraged to obtain an immune-centered perspective on diseases and their relations. To do so, we picked a set of 188 broadly studied diseases whose associated abstracts we sampled to obtain a characteristic inter-cellular immune profile (Fig. 5a, Supplementary Fig. 6, Supplementary Table 16). These we clustered in an unsupervised manner to assemble an immune-centered map of disease similarities and differences (Online Methods).
Figure 5. Global interaction profile analysis across diseases.
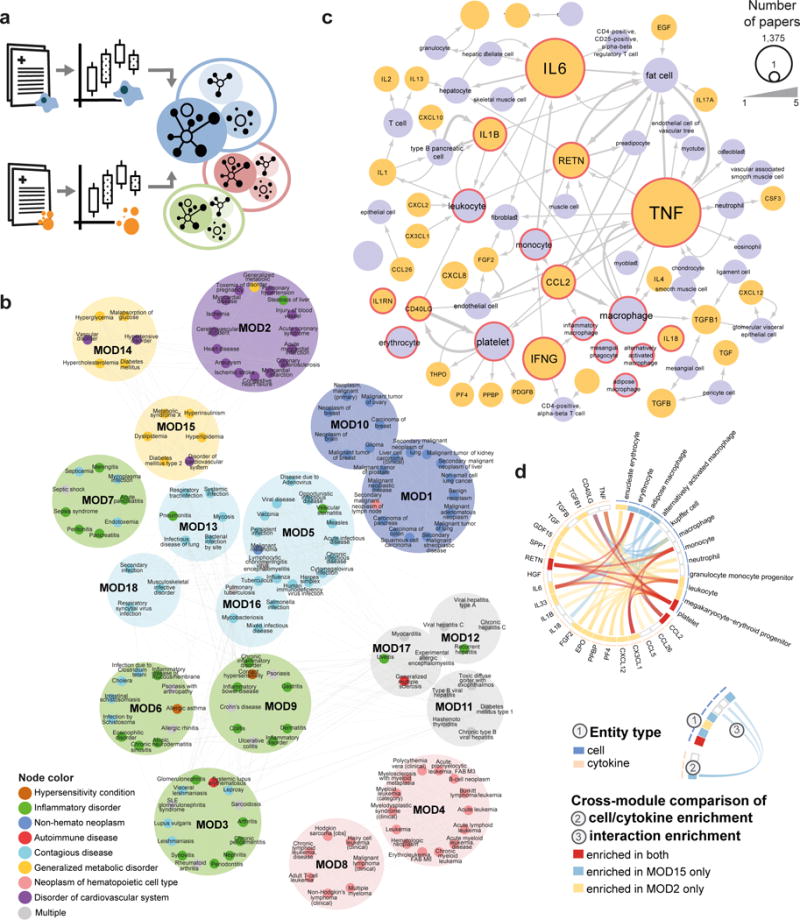
(a) Diagram illustrating the methodology of disease similarity module extraction: for 188 top-cited diseases, papers co-mentioning the disease either with cytokines or with hematopoietic cells were repeatedly sampled to identify cells and cytokines mentioned frequently in the particular context. Those entities, together with their potential interactions (as reported in the overall iX knowledgebase), form disease signaling profiles, used to build a global disease similarity map. (b) The immune-centered global map of disease similarities and differences, assembled in an unsupervised manner for 188 top-cited diseases. Nodes in graph correspond to diseases (colored by their known clinical disease categories), edges to significant correlation between diseases. Modules are colored by the most abundant SNOMED category, demonstrating large overlap between module separation and clinical classification. (c) iX extracts context-specific cells (purple), cytokines (orange) and interactions for a variety of conditions (here Diabetes mellitus type 2, as reported in the overall iX knowledgebase), providing a global view of the condition-specific immune signaling and emphasizing its key players. In red are cells and cytokines forming the sampling-based disease immune profile. (d) Circos plot showing enriched module signatures (cell, cytokine and interactions) compared to background for cardiovascular and metabolic syndrome diseases (Modules 2 and 15).
Analysis of this clustering outcome divided diseases into 18 modules based on associated cells, cytokines and interactions (Fig. 5b). We observed mixed agreement of this classification with a clinically based one (SNOMED): Some modules clustered clinically similar phenotypes: for example, Module 2 showed strong clustering of cardiovascular diseases, whereas Module 9 captured inflammatory bowel disorders and psoriasis. Similarly, cancers were grouped into four modules based on tissue type. In contrast, in some cases the immune-centered clustering yielded modules that included diseases from presumably unrelated clinical conditions: Modules 14 and 15 suggested a high degree of immune similarity between metabolic disorders and a subset of cardio-vascular diseases (e.g., Myocardial infarction and Acute coronary syndrome) - an observation also supported by high inter-connectivity of Modules 14 and 15 with Module 2.
We used iX to automatically generate an inter-cellular immune interaction map for type 2 diabetes, based on 5,484 abstracts (Fig. 5c, Online Methods). The network recapitulated the molecular basis of the disease, capturing the key role of tissue resident inflammatory macrophages, monocytes, fat cells and pre-adipocytes in secreting the pro-inflammatory cytokines TNF, IL6 and IL1B, triggering the release of adipokines that contribute to development of insulin resistance38. This profile co-clustered with other diseases in Module 15, consisting of metabolic and cardiovascular diseases, an appreciated link39. Enrichment analysis of Module 15, as well as its closely associated Module 2, compared to all others (Fig. 5d, Online Methods), showed a strong association with the pro-inflammatory adipokine RETN and its interaction with monocytes. RETN has been proposed to link the heightened inflammatory state in aged and obese individuals to insulin resistance, vascular inflammation and LDL cholesterol levels thus contributing to the risk of developing metabolic syndrome40. Moreover, elevated RETN directly induces IL6 secretion41, contributing to the constant low-grade inflammatory state associated with age and cardio-vascular conditions42,43. Thus, whereas phenotypically Modules 15 and 2 represent different pathological conditions, their molecular commonalities suggest consideration for common therapeutic interventions.
Prediction of cytokine-disease associations
Analysis of cytokine profiles across the 188 diseases revealed three predominant cytokine classes with respect to disease: disease-specific, module specific and backbone cytokines that are associated with the overwhelming majority of tested diseases (Fig. 6a, Supplementary Table 17, Online Methods). We noted a high overlap between backbone cytokines and those previously identified as hubs (Fig. 2b), suggesting that their pan-disease universality stems from the dominant role in the overall inter-cellular interaction network.
Figure 6. iX informs less studied diseases.
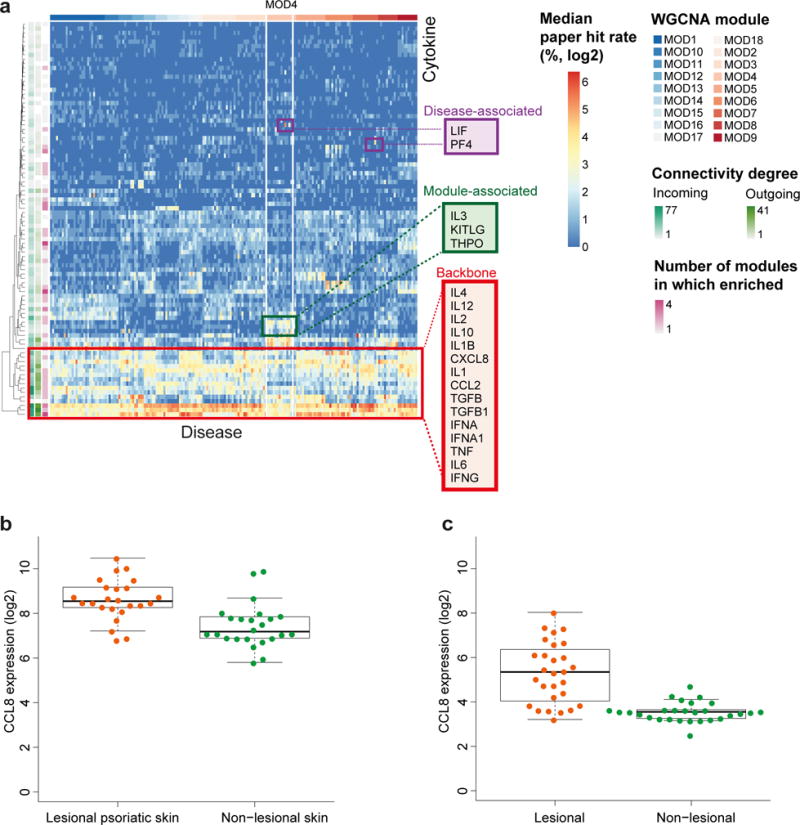
(a) Heatmap showing cytokine mention frequencies across 188 top-cited diseases. Color scale indicates the median percentage (log2-transformed) of paper hits for each cytokine (rows) in the context of each disease (columns, sorted by the unsupervised module classification), across 200 paper sampling iterations. An unequal immune system utilization is observed with respect to three cytokine classes: backbone cytokines highly observed across most modules, module specific and those specifically enriched in select diseases. Class examples highlighted in red. Annotation tracks on left indicate cytokine degrees. (b, c) CCL8 is differentially expressed in psoriasis. Shown is the expression of CCL8 in healthy skin versus lesion biopsies of psoriatic patients at baseline (paired two-sided Wilcoxon signed rank test, p-val=2.47e-05 and 1.04e-07, n=24 and n=28 independent samples). Box-plot elements: center line, median; box limits, first to third quartile (Q1 to Q3); whiskers, extend to the most extreme data point within 1.5 × IQR from the box; data points.
We hypothesized that given the unequal within-module knowledge per disease we may be able to predict de-novo cytokine-disease associations. For each module, we hierarchically clustered disease subsets across all cytokines and systematically predicted cytokine-disease associations (Online Methods). This yielded over 466 ranked candidates, stemming from all 18 modules (Supplementary Table 18). As we assembled the disease immune profiles by sampling, we checked the predicted cytokine-disease associations on the full, non-sampled, iX knowledgebase, which validated 81% of predicted interactions as true positives, suggesting a likely enrichment for unappreciated cytokine-disease associations in the remaining set (Supplementary Fig. 7).
To test this, we looked for experimental confirmation of two of the top-rated candidate associations, CCL8 and CCL24 in psoriasis, which to the best of our knowledge have not been reported. We analyzed two publicly available gene expression data sets for psoriasis44,45 and found CCL24 and CCL8 to be significantly upregulated in psoriatic lesions vs healthy skin in one45 and both datasets respectively (paired two-sided Wilcoxon signed rank test, p-val=0.0048, 2.47e-05 and 1.04e-07 respectively, Fig. 6b and Fig. 6c).
DISCUSSION
Knowledge of the immune inter-cellular network is crucial for understanding immune response in health and disease. However, the high system complexity leaves even expert researchers struggling to maintain a mental picture of the immune milieu and often leads to knowledge biases. We leverage a computational model of inter-cellular interaction network knowledge to accelerate discovery of novel interactions and the context in which they occur, identify disease-associated immune profiles and build an immune centric disease classification. Viewing diseases from such an angle yields a modular disease organization with partial overlap with clinical disease classification.
The knowledge we capture is unique in breadth and resolution, yet it can be expanded even further. Full-text article support, as well as identification of interactions described beyond sentence boundaries and using more complex sentence structures, would boost the volume of captured evidence and our ability to obtain more reliable and richer view of interactions, with less bias, particularly those that are understudied. We foresee extension of this approach to also include additional events such as direct cell-cell interactions and downstream inter-cellular signaling to capture complex cellular interaction cascades. The extensive meta-data we extract for each article, including MESH terms and bibliographic information, together with detailed characterization of the captured interactions, may be used for advanced filtering, allowing focus on the most authoritative knowledge. Beyond this, we envision that the structured formatting of knowledge we have achieved can be leveraged by machine learning applications, using statistical analyses of domain frequency and chronological pattern biases to identify potential discrepancies and erroneous claims in the published knowledge.
Technological advances now enable to step beyond assaying a narrow set of measures to high dimensional phenotyping across the breadth of the immune system at an unprecedented scale. This data is primarily analyzed by statistical analysis methods which are geared to identify differences and correlations, yet lacking any backend model of the immune system’s structure and function, lack in their ability to leverage domain knowledge or interpret what these differences mean. This results in an interpretation process which primarily manual, not systematized and relies strongly on investigator conjecture. Intelligent systematized interpretation requires having a machine-readable map of how immune components are connected and a formalized reasoning framework on which one could test hypotheses and refine knowledge. Here we built immuneXpresso, a framework that structures and standardizes our knowledge of immune intercellular interactions, under many conditions, and updates periodically. Its integration with high-dimensional immune data will enable paradigms of reasoning over heterogeneous cell populations, making first steps towards transforming immunology to systemized, model-based science; a true ‘systems immunology’.
ONLINE METHODS
iX pipeline execution environment
The computational pipeline assembling the iX knowledgebase was executed on a high-performance computing cluster, running a batch job scheduling system with up to 150 simultaneous jobs allowed. Details of the specific pipeline steps appear below. It typically takes roughly 2 weeks to generate the iX database from start to finish for the entire corpus.
Corpus selection, parsing and indexing
Abstracts of all English articles published electronically between 1960 and July 2017 (~16 million) were downloaded from PubMed using the EFetch utility. For each abstract, we extracted additional metadata, such as the article title, year of publication, whether it is a review or not and its assigned MESH terms. We focus on the mammalian immune system due to the rapid evolution of the immune system46,47, and in particular restrict our analysis to abstracts assigned a mouse and/or human MESH term, as these are the prime organisms relevant for biomedical research for which information is available. We used the Stanford Parser engine48 to split abstract text to sentences and words, perform part-of-speech tagging, extract sentence syntactic structure and grammatical relations (i.e. “typed dependencies”). This information was then indexed within the Elasticsearch engine (https://www.elastic.co/), to allow querying for sub-corpora potentially containing biological entities of interest. Moreover, preserving text processing products in the index eliminates the need for future time-consuming abstract reparsing, if entity recognition is expanded to support additional entity types (e.g., drugs, tissues or pathways).
Entity recognition and ontologies
To identify mentions of biological entities of interest (diseases, cells and cytokines) within article abstracts, we applied a dictionary-based approach with dictionaries either adapted from standard public knowledgebases or assembled from scratch. For diseases, we first queried elasticsearch for articles containing synonym phrases, listed by manually curated compilation of UMLS Metathesaurus49, and then post-processed the returned abstracts, sentence-by-sentence, to look for the matches and extract precise information for them. In particular, for each identified disease mention, we recorded its position in the sentence, the particular synonym used, as well as the public Concept Unique Identifier (CUI) and concept name, as defined by the SNOMED CT controlled vocabulary of the Metathesaurus (Supplementary Tables 8, 13). This post-processing stage drops conditions contained within longer disease entities (e.g., “deficiency”, “vitamin deficiency” are dropped, retaining only the most specific “vitamin A deficiency” term within the sentence “The essential role of vitamin A in kidney development has been demonstrated in vitamin A deficiency and gene targeting studies.”). To achieve this, we automatically examined all diseases with overlapping sentence positions to retain those with the longest position span and, among them, containing the highest number of words.
For cells, initial testing suggested that straightforward lookup of terms contained within the official Cell Ontology50 would miss a substantial fraction of cell occurrences in text due to the large number of possible forms of describing cell subsets. This pluralism in naming is hard to anticipate automatically, both when describing cells by name (e.g. “human CD8+ terminally differentiated memory cell” does not appear in the Cell Ontology and would not be captured by straightforward dictionary lookup) or by cell surface marker combination (e.g. “CD3+CD4+CD45RA+ cell”), whose delineation in the Cell Ontology is limited. To resolve this, we expanded the Cell Ontology with manually curated set of synonyms, and, more importantly, introduced a small lexicon of seed words that served a starting point, an anchor, for cell recognition in sentences (e.g., cell, lymphocyte, macrophage; see Supplementary Table 1 for the full list).
Cytokine dictionary was assembled manually, due to lack of an established lexicon (Supplementary Note 1).
For cell and cytokine entity recognition, we query elasticsearch for articles containing either a mention of a cytokine synonym or a cell “seed” word. The focus is on articles mentioning either or both cell and/or cytokine, to serve the basis for further relation extraction. Akin to diseases, we post-process candidate articles to better characterize the matches and, for cells, to expand from seeds to often multi-word hard-to-anticipate cell name phrases, using typed dependencies (Supplementary Note 2, Supplementary Fig. 8). In addition, the captured cell phrases are mapped to the Cell Ontology (Supplementary Note 3).
Last, following cell and cytokine mention candidate extraction, we analyzed their within-sentence positions to filter out erroneous identification of overlapping entities from different ontologies (e.g., erroneous “granulocyte” or “macrophage” cell matches within the cytokine entity “granulocyte-macrophage colony-stimulating factor”). For all remaining cell and cytokine mentions, we recorded their start and end positions in sentence, the particular phrase/synonym used, as well as the representative ID and concept name, assigned either by official Cell Ontology for cells or by our manually constructed dictionary for cytokines (Supplementary Tables 6, 7; see Supplementary Tables 1, 3 for frequencies of terms captured per each cell seed and cytokine lexicon concept respectively).
Relation extraction
Following extraction of articles containing either cytokine synonym or cell seed word mentions, we applied sentence-by-sentence post-processing to detect verbs, and when possible, link cells, cytokines and verbs into relations. We analyze sentence typed dependencies48 to detect semantically related cells and cytokines within sentence boundaries and identify the directionality (i.e., a cytokine acting on a cell, like “IL6 promotes Th17 cell differentiation”, or the opposite, a cell producing a cytokine, like “T cells secrete IL2”), polarity (i.e., positive, negative or neutral effect of the interaction, such as up-regulation, inhibition or just alteration of a cell function respectively), as well as the cellular biological function impacted by this relation (e.g., cell proliferation or apoptosis elicited by the acting cytokine).
Per-sentence relation extraction process included several steps: First, we found verbs in sentence by looking for words tagged as VB, VBD, VBG, VBN, VBP or VBZ by part-of-speech tagger. For each verb we examined all typed dependencies it was governing, attempting to resolve verb tense. In particular, we marked verbs tagged as VBN and governing either “passive nominal subject”, “passive auxiliary” or “agent” dependencies as passive. Second, to allow further relation directionality detection, we performed cell-cytokine semantic linking by examining all possible entity combinations with a verb located between a cell and a cytokine. We considered a candidate (verb, cell, cytokine) tuple as semantically related, if the sentence contained a directional dependency tree path from one of the elements to the other two. For example, in the sentence “These results suggest that IL6 promotes IL22 secreting Th17 subset differentiation” on Fig. 1a, there is a path from the verb “promotes” to the cytokine IL6 and to the Th17 cell, allowing (promote, IL6, Th17) relation identification. Third, we used the verb tense and the cell/cytokine order in sentence to resolve relation directionality. In the example above, since verb tense is active, and the cytokine precedes the cell entity, we identify the cytokine as acting on the cell. Finally, we applied a manually assembled verb classification lexicon (Supplementary Table 5) to assign relation polarity (i.e. positive in the example above). Verb classification lexicon lookup was performed using stemmed forms for both lexicon terms and the relation verb.
For the special case of cell-cytokine relations included within cell descriptions, “noun phrase-internal relations” (e.g. “IFNγ-producing CD4+ T cells”, “IL2-activated NK cells” or “NK cell IFN-gamma”), we applied a tailored rule-based approach (Supplementary Note 4).
If relation directionality could be deduced directly from the verb (e.g., express or stimulate), as marked by human curators of the verb lexicon, we preferred the directionality denoted by the lexicon to directionality-related decisions made by the algorithm above.
Finally, we applied cell entity-containing noun phrase analysis to detect cellular biological functions elicited by the interacting cytokine, such as “migration” in the “GM-CSF enhanced reactive oxygen species release and neutrophil migration in vitro”. We examined noun phrase words on right of the recognized cell entity to look for one or more matches from a manually curated cellular function list.
In terms of terminology, we refer to each detected (cell entity occurrence, cytokine entity occurrence, directionality, polarity, cellular function) relation in a sentence as a relation evidence record, while summarization of evidence records through representative labels and IDs of the entities produces unique candidate (cell, cytokine, directionality) interactions. Polarity and cellular function are ignored during summarization. We defined interaction context by the disease entities co-occurring in the same abstracts with the identified relation evidence records.
Filtering and scoring
We choose to put strong emphasis on precision over recall to ensure benefit and increase adoption by the community. To do so, we developed a confidence scoring methodology for both individual relation evidence records and summarized interactions, as well as performed four filtering stages as follows:
Baseline filtering: For each putative relation evidence record (i.e., a directional cell-cytokine relation extracted from an individual sentence), we compiled a set of sentence level features to capture the complexity of the sentence from which this evidence emerged. These included sentence length, number of typed dependencies, entities and relations detected, suspect for negation presence, cell ontology mapping score, as well as the distance between the cell and the cytokine occurrences. Next, we filtered evidence with potentially lower confidence, such as negated sentences or sentences with more than one non-noun phrase-internal relation detected or evidence with low cell ontology mapping score (Supplementary Note 3). This yielded a “baseline” subset of putative interaction evidence records used in the subsequent scoring and filtering stages.
Evidence record confidence scoring: all individual records were assigned confidence scores, based on sentence-level features (Supplementary Note 5) to allow focus on the highest confident evidence, both when queried by iX web-based interface users and during analyses, as well as serve a feature for further filtering, as detailed below.
Summary scoring: All summarized interaction (i.e., unique cell-cytokine-directionality triples summarized across the corpus) were assigned with summary and enrichment scores (Supplementary Note 6) to allow focusing on highly cited interactions.
Lasso-based filtering of summarized interactions: Last, to choose optimal parameters for classifying and further filtering of summarized interactions (i.e., unique cell-cytokine-directionality triples), we built a lasso logistic regression model51. Here, we summarized evidence level features into the interaction level across all evidence records, such as minimal/maximal sentence length, minimal/maximal cell-cytokine entity distance in sentence, the presence of mouse/human MESH term annotation in any of the evidence papers and minimal/maximal evidence confidence scores described in (2) above. Additionally, we defined interaction-level features, such as the overall number of evidence records, the summary and enrichment scores defined in (3) above. For all verbs in the training set, we added an interaction-level feature reflecting verb presence in any of the evidence records, producing 178 features in total. We trained the lasso regression model on a set of 203 randomly selected interactions, summarizing 788 baseline evidence records identified by iX. We labeled the summarized interaction as positive if an only if at least one of its evidence records was manually classified as having cell, cytokine and directionality identified correctly. The resulting set of manually labeled (cell-cytokine-directionality) triples was used for training the lasso model, separately for incoming and outgoing interactions, with leave-one-out cross-validation. This procedure identified a linear combination of sparse feature weightings which we then applied to all putative summarized interactions, classifying them as either “true” or “false”. Most prominent features included maximal evidence confidence score, having “mouse” MESH assigned for evidence record articles, as well as presence of several verbs, such as “produce”, “synthesize” and “affect”. We enforced classification of “true” for interactions having at least one noun phrase-internal evidence record, due to their very high identification precision. Filtering out summarized interactions classified as “false”, together with all their evidence records, yielded the resulting dataset which we used for further performance evaluation and system-wide analyses (Supplementary Table 11 for a breakdown of the articles, records, entities and interactions remaining at various stages; Supplementary Table 12 for PubMed articles for the resulting cell-cytokine relation evidence)
Named Entity Recognition performance evaluation
NER precision for all entity types was assessed by human curators. In addition, for cells, both precision and recall were examined by comparing to the gold standard Colorado Richly Annotated Full Text (CRAFT) corpus9 (Supplementary Note 7).
Relation precision evaluation
Relation extraction precision assessment was based on manual evaluation of randomly chosen relation evidence records (Supplementary Note 8).
Reference book network assembly and relation recall evaluation
To the best of our knowledge, no gold standard for directional cell-cytokine relations exist as of now. Thus, to assess iX text mining process performance in capturing existing knowledge in general, and, evaluate relation recall in particular, 11 human curators manually annotated interaction mentions, specifying the cell/cytokine terms and interaction direction from a reference book with encyclopedic display of up-to-date knowledge about cytokines and their interactions29. These we mapped to the Cell Ontology and the cytokine lexicon respectively to summarize and acquire reference IDs consistent with those used in iX. This process yielded 725 unique (cell, cytokine, directionality) interaction triples which we compared to those captured by iX, while allowing non-exact cell type match along the Cell Ontology hierarchy to account for the varying level of granularity of cell type reporting in literature. This comparison assessed the proportion of reference book triples captured by iX, as well as the number of triples unique to either the reference book or iX. For the latter, to estimate false positive proportion, we manually evaluated 200 (cell, cytokine, directionality) triples, randomly selected from the iX knowledgebase not covered by the reference book. An interaction was classified as false positive by the human curators, if none of the evidence records collected by iX for that triple reported the directional interaction in question. Following the assessment, we unified the list of interactions identified via literature text-mining and reference book annotation into a single knowledgebase.
Evaluation of co-occurrence based relation extraction performance
A co-occurrence based approach would not be able to capture interaction directionality, an essential characteristic of inter-cellular communication, as our typed dependency-based method inherently does. Still, we aimed to assess quality of those easier-to-extract relations. To the best of our knowledge, no gold standard for cell-cytokine relations exist as of now. Thus, to evaluate co-occurrence based relation extraction performance and contrast it with the typed dependency-based approach, we used the interaction network we manually assembled from the reference book (see “Reference book network assembly and relation recall evaluation” section above) and discarded interaction directionality for both reference-derived and typed dependency-based interactions (post-lasso filtering, hereafter referred to as iX interactions), producing two sets of cell-cytokine pairs to compare with. We linked cell and cytokine entities, previously recognized as co-appearing within sentence boundaries, into relation evidence records and summarized them into interactions, with the number of distinct articles mentioning the relation defining the strength of evidence.
Cytokine degrees and power law fit
The literature-derived nature of the iX network inherently reflects the fact that study of cellular interactions is performed at a varying level of granularity, rather than necessarily using the most specific cell type. As such, to avoid situations whereby the same interaction is counted multiple times when calculating degree distributions, we discarded interactions of less specific cell types, if a more specific cell type was known to interact with the same cytokine in the same direction. Cytokine degree counts, calculated separately per direction, took into account interactions with both hematopoietic and non-hematopoietic cells. Discrete power-law and log-normal distribution fit calculations were performed using the poweRlaw R package52.
Proteome comparison
We examined the similarity of iX and proteome interactions by inspecting each interaction direction separately and comparing cytokine degrees in these data sets. For outgoing interactions, we compared the number of distinct interacting cell types captured by iX and the number of those reported to express the particular cytokine in the ImmProt compendium34. For incoming interactions, we examined the proteomic profiles of cytokine receptor genes and approximated cytokine-cell interactions by mapping expressed receptors to the respective cytokines, based on the KEGG “Cytokine-cytokine receptor interaction - Homo sapiens (human)” (hsa04060) pathway entry53,54. We then compared the resulting proteome-derived degrees to those captured by iX for each cytokine. To avoid situations whereby the same interaction is counted multiple times due to literature reporting at varying levels of cell type granularity, we discarded iX interactions of less specific cell types, if a more specific cell type was known to interact with the same cytokine in the same direction. Moreover, to focus on similar cellular compartments, we discarded non-hematopoietic cell interactions from iX, thereby calculating cytokine degrees across hematopoietic cell types only in both data sets.
Novel cell-cytokine interaction prediction
To systematically predict novel interactions between immune cells and cytokines, we applied several strategies, separately for each interaction direction:
Based on similarity of global signaling profiles - we assembled a global literature-derived Boolean matrix indicating whether an interaction has been described for each cell-cytokine pair. We used hierarchical clustering to group together cytokines with similar interaction profiles across all cell types and hypothesized that non-interacting cell-cytokine pairs located within highly connected clusters might actually interact. Therefore, we scanned clusters, confined to all possible combinations of column and row dendrogram branches, and derived interaction candidates for “gaps” in clusters with at least 85% of interacting pairs. We scored the resulting candidates by counting the number of clusters that predicted the interaction to be novel.
Based on similarity of cytokine structure or function - we calculated the proportion of cytokine family members known from the literature to interact with each cell type and derived novel interaction candidates by hypothesizing that cells interacting with at least 30% of a cytokine family and with its most interactive member (i.e. usually the founding family member), might interact with other cytokines in the family as well. We used the aforementioned proportion to rank the resulting cell-cytokine interaction candidates.
Based on differences between literature-derived knowledge and experimental data (Supplementary Note 9)
Manual evaluation of novel cell-cytokine interaction candidates
We evaluated 78 interactions with the best overall scoring (Supplementary Tables 14, 15). Candidates were scored by manual evaluation (e.g. we considered predictions made by multiple methodologies stronger), however incorporated multiple other criteria in defining which candidate interaction to validate. For example, we considered a candidate interaction whose reverse directionality was not reported yet, stronger than the one for which it has been captured by iX (as that reverse directionality might result from erroneous identification, invalidating prediction novelty). A subset of candidate interactions with no evidence identified using manual search were then chosen for experimental validation.
Experimental validation of novel cell-cytokine interaction predictions
Whole blood was obtained by consent from healthy volunteers through venipuncture. PBMC fraction was separated by a standard centrifugation at 1500RPM on Ficoll gradient without brake. For phospho-flow, cells were washed twice with Dulbecco’s PBS and subjected to stimulation for 15min with IL34 and CSF1 (Peprotech, Asia) at 100ng/ml. Cells were fixed for 10min at RT with 1.6% PFA (Pierce) and stained for 1hr at RT with a mix of metal-tagged antibodies. Further, cells were permeabilized with ice-cold methanol, followed by intracellular staining with metal-tagged antibodies against a phosphorylated form of Erk-36/38, p-NFKB, pSTAT1 and pSTAT5. Antibodies for phosphorylated targets were obtained from Fluidigm.
For intracellular cytokine staining, PBMC’s were stimulated with PMA (150ng/ml)/ionomycin(1mM) (Sigma) for 4 hours at 37C in complete medium containing monensin and brefeldin-A at 1:1000 dilution (e-bioscience). Extracellular epitopes were stained for one hour with metal-tagged antibody mix, cells were fixed with PFA as described above and permeabilized for 1 hour on ice with saponin permeabilization buffer (e-Bioscience). Intracellular staining of IL7 was performed on ice in saponin-containing buffer. All extracellular and cytokine-specific antibodies were conjugated in-house using MaxPar kits from Fluidigm or pre-conjugates purchased from Fluidigm. Cells were stained with Cell-ID Ir191/193 for viability stain and acquired on a CyTOF1 (DVS, Fluidigm) instrument.
Assembling disease similarity modules
We defined a context for cells, cytokines and cell-cytokine interactions by diseases co-occurring in the same abstracts, while disease mentions were captured using manually curated compilation of the UMLS Metathesaurus49. To identify disease similarity modules, we focused on 188 top-cited diseases (co-mentioned in at least 500 papers with cytokines and at least 500 papers with hematopoietic cells), that we could classify as pertaining to at least one of the eight predefined clinical categories (e.g., disorder of cardiovascular system, generalized metabolic disorder, neoplasm of hematopoietic and non-hematopoietic cell types, autoimmune diseases and hypersensitivity conditions). To define clinical categories, we used SNOMED CT ontology available through UMLS Metathesaurus49 and manually expanded its autoimmune disease category with publicly available autoimmune-related disease list (http://www.aarda.org/autoimmune-information/list-of-diseases). As a preliminary step to module assembly, we extracted a non-specific across-disease control profile by repeated paper sampling from the entire corpus of 521,625 disease-HPC and 438,012 disease-cytokine co-occurrence papers respectively, without limiting to any particular context (200 iterations of 200 papers each). We examined cells and cytokines mentioned in the sampled papers, assembling the distribution of hits for each HPC and cytokine across sampling iterations, to serve a control for further disease-specific profile assembly.
Next, for each of the 188 diseases of interest, we assembled its underlying signaling profile by applying several steps: (1) performing 200 samplings of 200 random papers from the disease-specific sub-corpus, to control for differences in corpus size, followed by extraction of the distribution of hits for each cell and cytokine across sampling attempts (2) filtering out under-represented entities, that is, those with a hit proportion median lower in disease-specific than in the control sampling and (3) linking cells and cytokines co-occurring in the same disease profile to interacting pairs, using interaction potential captured in the overall iX network. Last, we performed unsupervised clustering of the resulting Boolean disease profiles (0/1 indicating whether the particular cell, cytokine or pair is a part of the profile) using WGCNA R package55,56 to assemble the set of immune-centric disease similarity modules based on the binary distance metric.
Disease module signature extraction
To extract features (i.e. cells, cytokines and their interacting pairs) characterizing disease modules, we applied a hypergeometric test, independently for each feature, examining whether the feature is over-represented in the particular module comparing to the entire set of 188 diseases. We corrected for multiple testing using Benjamini-Hochberg.
Novel cytokine-disease association prediction
To test cytokine utilization across conditions, we repeatedly sampled papers from disease-specific sub-corpora, separately for each of the 188 top-cited diseases (200 iterations of 200 papers each). We extracted the distribution of paper hit proportions for each cytokine-disease pair across iterations, and used the median proportion as the measure of cytokine mention frequency in that particular context.
To systematically predict novel cytokine-disease associations, we employed global within-module immune similarity and applied the following steps separately for each disease module: (1) assembly of literature-derived Boolean matrix indicating whether a cytokine was a part of the disease profile, for each disease in the module (2) hierarchical clustering to group together module diseases displaying similar profiles across all cytokines and hypothesizing that an association should exist between currently unlinked cytokine-disease pairs within highly linked clusters. Therefore, we scanned clusters, confined to all possible combinations of column and row dendrogram branches, and derived cytokine-disease association candidates for “gaps” in clusters with at least 30% of linked cytokine-disease pairs. We scored the resulting candidates by counting the number of clusters that predicted the particular association to be novel.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
Data of the inter-cellular communication network and disease context is hosted on ImmPort with periodic updates and available for query and download with standardization to ontologies at www.immunexpresso.org.
Code availability
Analysis code available upon request.
Supplementary Material
Acknowledgments
We thank A. Butte and M. Davis for fruitful discussions and advice, N. Geifman for assistance with cytokine ontology development, D. Dougall for contribution to the cell lexicon, members of the Shen-Orr lab for reference book curation, D. Cohen for the high-performance computing cluster support, R. Reichart for Text Mining insights, P. Dunn and S. Bhattacharya for the user interface development support. This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases [BISC contract number HHSN272201200028C] and the Rappaport Family Institute for Research in the Medical Sciences award to SSO.
Footnotes
AUTHOR CONTRIBUTIONS
K.K. designed, performed and interpreted the analyses, led design and development of the software, implemented relation extraction, filtering and network assembly, and wrote the manuscript, E.S. interpreted the analyses, performed quality control, designed and carried out the experimental validations and wrote the manuscript, A.Z.K. implemented indexing, ontology support, reference book annotation software and broadly contributed to the entire computational pipeline development, Y.K. conceived the pipeline architecture and broadly contributed to the software formation, Y.G. assisted with cell entity recognition, G.S.M. performed quality control on Text Mining output and assisted with cytokine ontology development, T.D. assisted with quality control on Text Mining output, M.B. contributed to quality control and prediction evaluation, N.A.R. wrote the software and implemented the website backend, J.C. and J.W. designed and developed the user interface, J.C.R. and F.M. provided and interpreted the proteomic data, N.A. assisted with machine learning for quality control, D.R. contributed to quality control and interpretation of disease profiles, S.S.O. conceived the idea, oversaw, designed and interpreted the analyses, and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
K.K. and Y.K are employees and co-founders of CytoReason. S.S.O. and E.S. are co-founders and serve as scientific advisors and/or consultants to CytoReason.
References
- 1.Maecker HT, et al. New tools for classification and monitoring of autoimmune diseases. Nat Rev Rheumatol. 2012;8:317–28. doi: 10.1038/nrrheum.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann R, Valencia A. Implementing the iHOP concept for navigation of biomedical literature. Bioinformatics. 2005;21:252–258. doi: 10.1093/bioinformatics/bti1142. [DOI] [PubMed] [Google Scholar]
- 3.Jimeno A, et al. Assessment of disease named entity recognition on a corpus of annotated sentences. BMC Bioinformatics. 2008;9:1–10. doi: 10.1186/1471-2105-9-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leaman R, Dogan RI, Lu Z. DNorm: Disease name normalization with pairwise learning to rank. Bioinformatics. 2013;29:2909–2917. doi: 10.1093/bioinformatics/btt474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald RT, et al. An entity tagger for recognizing acquired genomic variations in cancer literature. Bioinformatics. 2004;20:3249–3251. doi: 10.1093/bioinformatics/bth350. [DOI] [PubMed] [Google Scholar]
- 6.Tanenblatt M, Coden A, Sominsky I. The ConceptMapper Approach to Named Entity Recognition. Proc Seventh Conf Int Lang Resour Eval Lr. 2010:546–551. [Google Scholar]
- 7.Funk C, et al. Large-scale biomedical concept recognition: an evaluation of current automatic annotators and their parameters. BMC Bioinformatics. 2014;15:59. doi: 10.1186/1471-2105-15-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah NH, et al. Comparison of concept recognizers for building the Open Biomedical Annotator. BMC Bioinformatics. 2009;10:S14. doi: 10.1186/1471-2105-10-S9-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bada M, et al. Concept annotation in the CRAFT corpus. BMC Bioinformatics. 2012;13:161. doi: 10.1186/1471-2105-13-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JD, Ohta T, Tateisi Y, Tsujii J. GENIA corpus - A semantically annotated corpus for bio-textmining. Bioinformatics. 2003;19 doi: 10.1093/bioinformatics/btg1023. [DOI] [PubMed] [Google Scholar]
- 11.Arighi CN, et al. Overview of the BioCreative III Workshop. BMC Bioinformatics. 2011;12:1–9. doi: 10.1186/1471-2105-12-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Ohta T, Pyysalo S, Kano Y. Overview of BioNLP 2009 shared task on event extraction. Process Shar Task. 2009:1–9. [Google Scholar]
- 13.Kim JD, et al. Proceedings of the BioNLP Shared Task 2011 Workshop. Association for Computational Linguistics; 2011. Overview of BioNLP Shared Task 2011; pp. 1–6. [Google Scholar]
- 14.Kim JD, Ohta T, Tsujii J. Corpus annotation for mining biomedical events from literature. BMC Bioinformatics. 2008;9:1–25. doi: 10.1186/1471-2105-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ananiadou S, Pyysalo S, Tsujii J, Kell DB. Event extraction for systems biology by text mining the literature. Trends Biotechnol. 2010;28:381–390. doi: 10.1016/j.tibtech.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Pyysalo S, et al. Event extraction across multiple levels of biological organization. Bioinformatics. 2012;28:575–581. doi: 10.1093/bioinformatics/bts407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahmood ASMA, Wu TJ, Mazumder R, Vijay-Shanker K. DiMeX: A text mining system for mutation-disease association extraction. PLoS One. 2016;11:1–26. doi: 10.1371/journal.pone.0152725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee K, et al. BRONCO: Biomedical entity Relation ONcology COrpus for extracting gene-variant-disease-drug relations. Database. 2016;2016:1–13. doi: 10.1093/database/baw043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verspoor KM, Heo GE, Kang KY, Song M. Establishing a baseline for literature mining human genetic variants and their relationships to disease cohorts. BMC Med Inform Decis Mak. 2016;16 doi: 10.1186/s12911-016-0294-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Hunter L, Kešelj V, Verspoor K. Approximate Subgraph Matching-Based Literature Mining for Biomedical Events and Relations. PLoS One. 2013;8 doi: 10.1371/journal.pone.0060954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Björne J, et al. Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Association for Computational Linguistics; 2009. Extracting Complex Biological Events with Rich Graph-based Feature Sets; pp. 10–18. [Google Scholar]
- 22.Rzhetsky A, Seringhaus M, Gerstein MB. Getting started in text mining: part two. PLoS Comput Biol. 2009;5:e1000411. doi: 10.1371/journal.pcbi.1000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu F, et al. Biomedical text mining and its applications in cancer research. J Biomed Inform. 2013;46:200–11. doi: 10.1016/j.jbi.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Jensen LJ, Saric J, Bork P. Literature mining for the biologist: from information retrieval to biological discovery. Nat Rev Genet. 2006;7:119–129. doi: 10.1038/nrg1768. [DOI] [PubMed] [Google Scholar]
- 25.Goh KI, et al. The human disease network. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linghu B, Snitkin ES, Hu Z, Xia Y, Delisi C. Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network. Genome Biol. 2009;10:R91. doi: 10.1186/gb-2009-10-9-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu G, Agarwal P. Human disease-drug network based on genomic expression profiles. PLoS One. 2009;4:e6536. doi: 10.1371/journal.pone.0006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kilpinen S, et al. Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9,783 samples from 175 types of healthy and pathological tissues. Genome Biol. 2008;9:R139. doi: 10.1186/gb-2008-9-9-r139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dembic Z. The Cytokines of the Immune System: The Role of Cytokines in Disease Related to Immune Response. Elsevier Science; 2015. [Google Scholar]
- 30.Bhattacharya S, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58:234–9. doi: 10.1007/s12026-014-8516-1. [DOI] [PubMed] [Google Scholar]
- 31.Edwards AM, et al. Too many roads not taken. Nature. 2011;470:163–165. doi: 10.1038/470163a. [DOI] [PubMed] [Google Scholar]
- 32.Barabasi AL, Albert R. Emergence of scaling in random networks. 1999;509:11. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 33.Barabási AL. Scale-free networks: a decade and beyond. Science. 2009;325:412–413. doi: 10.1126/science.1173299. [DOI] [PubMed] [Google Scholar]
- 34.Rieckmann JC, et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol. 2017 doi: 10.1038/ni.3693. [DOI] [PubMed] [Google Scholar]
- 35.Heng TSP, Painter MW. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–4. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 36.Moller P, Bohm M, Czarnetszki BM, Schadendorf D. Interleukin-7. Biology and implications for dermatology. Exp Dermatol. 1996;5:129–137. doi: 10.1111/j.1600-0625.1996.tb00107.x. [DOI] [PubMed] [Google Scholar]
- 37.Lin H, et al. Discovery of a Cytokine and Its Receptor by Functional Screening of the Extracellular Proteome. Science (80-) 2008;320:807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 38.Asghar A, Sheikh N. Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell Immunol. 2017 doi: 10.1016/j.cellimm.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Peter G, et al. Multifactorial Intervention and Cardiovascular Disease in Patients with Type 2 Diabetes. N Engl J Med. 2003;348:383–393. doi: 10.1056/NEJMoa021778. [DOI] [PubMed] [Google Scholar]
- 40.Park HK, Kwak MK, Kim HJ, Ahima RS. Linking resistin, inflammation, and cardiometabolic diseases. Korean J Intern Med. 2017;32:239–247. doi: 10.3904/kjim.2016.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hillenbrand A, Weiss M, Knippschild U, Wolf AM, Huber-Lang M. Sepsis-Induced Adipokine Change with regard to Insulin Resistance. Int J Inflam. 2012;2012:972368. doi: 10.1155/2012/972368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen-Orr SS, et al. Defective Signaling in the JAK-STAT Pathway Tracks with Chronic Inflammation and Cardiovascular Risk in Aging Humans. Cell Syst. 2016;3:374–384.e4. doi: 10.1016/j.cels.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furman D, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. 2017;23:174–184. doi: 10.1038/nm.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell CB, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. 2014;192:3828–3836. doi: 10.4049/jimmunol.1301737. [DOI] [PubMed] [Google Scholar]
- 45.Yao Y, et al. Type I interferon: potential therapeutic target for psoriasis? PLoS One. 2008;3:e2737. doi: 10.1371/journal.pone.0002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes AL. eLS. John Wiley & Sons, Ltd; 2001. Vertebrate Immune System: Evolution. [DOI] [Google Scholar]
- 47.Du Pasquier L. The immune system of invertebrates and vertebrates. Comp Biochem Physiol Part B Biochem Mol Biol. 2001;129:1–15. doi: 10.1016/s1096-4959(01)00306-2. [DOI] [PubMed] [Google Scholar]
- 48.De Marneffe M-C, MacCartney B, Manning CD. Generating typed dependency parses from phrase structure parses. Proceedings of the 5th International Conference on Language Resources and Evaluation (LREC 2006) 2006 [Google Scholar]
- 49.Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004;32:D267–D270. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bard J, Rhee SY, Ashburner M. An ontology for cell types. Genome Biol. 2005;6:R21. doi: 10.1186/gb-2005-6-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tibshirani R. Regression Shrinkage and Selection Via the Lasso. J R Stat Soc Ser B. 1994;58:267–288. [Google Scholar]
- 52.Gillespie CS. Fitting Heavy Tailed Distributions: The {poweRlaw} Package. J Stat Softw. 2015;64:1–16. [Google Scholar]
- 53.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;559 doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langfelder P, Horvath S. Fast {R} Functions for Robust Correlations and Hierarchical Clustering. J Stat Softw. 2012;46:1–17. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data of the inter-cellular communication network and disease context is hosted on ImmPort with periodic updates and available for query and download with standardization to ontologies at www.immunexpresso.org.