

Abstract
Cellular components of solid tumors including DNA are released into the bloodstream, but data on circulating-free DNA (cfDNA) in hepatocellular carcinoma (HCC) are still scarce. This study aimed at analyzing mutations in cfDNA and their correlation with tissue mutations in patients with HCC. We included 8 HCC patients treated with surgical resection for whom we collected paired tissue and plasma/serum samples. We analyzed 45 specimens, including multiregional tumor tissue sampling (n=24), peripheral blood mononuclear cells (PMBC, n=8), plasma (n=8) and serum (n=5). Ultra-deep sequencing (5,500x) of all exons was performed in a target panel of 58 genes, including frequent HCC driver genes and druggable mutations. Mutations detected in plasma included known HCC oncogenes and tumor suppressors (e.g., TERT promoter, TP53, NTRK3) as well as a candidate druggable mutation (JAK1). This approach increased the detection rates previously reported for mutations in plasma of HCC patients. A thorough characterization of cis mutations found in plasma confirmed their tumoral origin, which provides definitive evidence of the release of HCC-derived DNA fragments into the bloodstream. This study demonstrates that ultra-deep sequencing of cfDNA is feasible and can confidently detect somatic mutations found in tissue; these data reinforce the role of plasma DNA as a promising minimally invasive tool to interrogate HCC genetics.
Keywords: Liquid Biopsy, Liver Cancer, Cell-free DNA, Biomarker, Precision medicine
INTRODUCTION
The field of liquid biopsy has significantly grown in the last 5 years, and it is likely to play an important role in precision medicine, particularly in oncology. The main advantage of this technology is that it facilitates obtaining molecular information of the tumor using data from a blood draw1–4. This would result in a drastic decrease in the need of tissue biopsies, and allow for easy monitoring of cancer genetics. The release of tumor components such as circulating-free DNA (cfDNA) into the bloodstream has been known for decades1, 5. Despite the mechanisms of how DNA is released are still unclear, robust data suggest that tumor DNA fragments found in plasma (approximately 160 bp long) are derived mostly from apoptotic cancer cells6. Recent improvements in DNA sequencing technologies and digital PCR have enabled incorporating mutation analysis of cfDNA as a novel diagnostic tool in some malignancies like lung cancer. Potential applications include cancer surveillance7, identification of druggable mutations8, and detection of minimal residual disease after tumor surgical resection9. Analysis of cfDNA could be particularly useful to identify and monitor drug targets for molecular therapies. The basis for effectively inhibiting oncogene addiction relies on the identification of druggable oncodrivers, which are somatic mutations that predict response to ad hoc therapies. Many examples demonstrate the value of this approach, including the remarkable response to erlotinib in lung cancer patients with EGFR mutations10. As an example of the potential of liquid biopsy to identify mutations, the FDA recently approved the first blood-based diagnostic tool to detect a specific actionable EGFR mutation (L858R) in lung cancer patients11.
There are few reports on cfDNA in hepatocellular carcinoma (HCC), the most frequent form of primary liver cancer6, 12–14. Liver cancer is the second cause of cancer-related mortality worldwide, and it ranks first among tumors that increased mortality in the last 20 years15. Patients with HCC are typically diagnosed at intermediate or advanced stages, where potential curative treatments (e.g., surgical resection or liver transplantation) are not recommended16. Unlike most solid tumors, HCC can be confidently diagnosed with imaging techniques16, which has limited the access to tissue biopsies for retrospective biomarker studies. Thus, it is critical to develop new strategies to access HCC molecular information and facilitate biomarker discovery in HCC patients. Recent publications on mutation analysis of cfDNA in HCC reported variable detection rates that could be as low as 5% for certain genes such as TP53 or CTNNB112. A limitation of these studies is their focus on a limited number of specific hotspot mutations12, or a relatively low coverage given their low-stringent thresholds for mutations calling leading to the identification of a high number of plasma mutations with indeterminate confidence13, 14. Here, we report results of a new approach of targeted sequencing using ultra-deep coverage (5,500x) of all exons of the 58 most frequently mutated genes in HCC, in paired plasma and tissue samples of 8 patients. To assert the impact on intra-tumoral heterogeneity in detection rate, we conducted multi-regional sampling in tumor tissue. We also compared the performance of cfDNA detection rate in plasma versus serum in a subset of patients (n=5/8). Overall, we confirm the feasibility of this approach to detect mutations in plasma and provide the proof-of-principle to further develop this technology to monitor mutant DNA in HCC patients.
RESULTS
Ultra-deep sequencing of cfDNA detect tissue mutations in HCC patients
This study included 8 patients undergoing surgical resection for HCC with available paired tissue (i.e., multi-regional sampling in 5 patients) and blood samples. Patient demographics and tumors characteristics are summarized in Table 1. Briefly, all patients were male with a median age of 59 years. Cirrhosis was present in 3/8 (38%) patients, with viral hepatitis B (HBV) as the main etiologies for the underlying liver disease in 4/8 (50%) cases. Most patients had a single nodule (7/8, 88%) with a median tumor size of 4.6 cm. Vascular invasion or satellites were observed in 2/8 (25%) surgical specimens. Blood was collected preoperatively and further processed into plasma (n=8 patients), serum (n=5 patients), and PBMC (n=8 patients). The latter was used as germline DNA to call somatic events. We performed multi-regional sampling in some tumor specimens (total of 24 regions analyzed), which allowed interrogating different geographic locations of the same nodule. Altogether, DNA was extracted from 45 samples and submitted for next generation sequencing (NGS).
Table 1.
Clinical characteristics of the cohort
| Patient | Age | Gender | Etiology | Background liver |
Size (cm) |
Tissue samples analyzed (n) |
Plasma/ Serum |
Number of nodules |
Micro- vascular invasion |
Satellites | Degree of cell differentiation (WHO) |
AFP (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 74 | Male | Cryptogenic | Non-cirrhotic | 9.8 | 2 | Yes/Yes | 1 | Yes | No | Poor | 31 |
| 2 | 58 | Male | HBV | Cirrhotic | 3.5 | 5 | Yes/Yes | 1 | No | No | Well | 2 |
| 3 | 51 | Male | HCV | Cirrhotic | 6.5 | 3 | Yes/Yes | 4* | Yes | Yes | Moderate | 235 |
| 4 | 79 | Male | Cryptogenic | Non-cirrhotic | 13.5 | 6 | Yes/No | 1 | No | Yes | Well | 4 |
| 5 | 75 | Male | HBV | Non-cirrhotic | 4.7 | 5 | Yes/No | 1 | No | No | Poor | 2 |
| 6 | 60 | Male | Cryptogenic | Non-cirrhotic | 4.5 | 1 | Yes/Yes | 1 | No | No | Moderate | 51 |
| 7 | 38 | Male | HBV | Cirrhotic | 2.1 | 1 | Yes/Yes | 1 | No | No | Poor | 9 |
| 8 | 52 | Male | HBV | Non-cirrhotic | 2.1 | 1 | Yes/No | 1 | No | No | Moderate | 30 |
HBV: Hepatitis B virus; HCV: Hepatitis C Virus; NA: Not available
Multiregional samples and molecular analysis were performed on the largest nodule. Patient had multiple satellites in the main nodule
DNA extracted from plasma and serum yielded a median concentration of 12.5 ng/mL and 14.6 ng/mL, respectively. There were no differences in the rate of DNA recovery from both sources (p=0.42). Median DNA fragment size was 171 base pairs, which is consistent with previously reported fragment size of cfDNA in HCC6, and significantly lower than average fragment size for tissue extractions. Despite DNA fragments tend to be slightly larger in serum compared to plasma, the difference was not statistically significant (Fig. 1a). Average sequencing coverage was 1,500X and 5,500X for tissue and plasma, respectively (Fig. 1b). We leveraged ultra-deep coverage to detect rare somatic mutations that may be present in very small cell fractions of the tumor or mutations at extremely low allele frequencies in cfDNA. The sequencing panel included 58 genes comprising 943 regions and 3,808 probes (total region size of 193,000 bp), with an estimated coverage of the selected targets of 99.3%.
Figure 1.
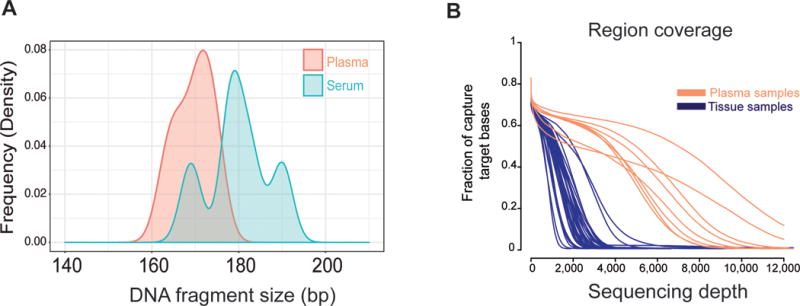
a) Distribution of DNA length fragments in plasma and serum samples.
b) Differential coverage for next-generation sequencing in tissue and plasma. Sequencing depth was significant higher in plasma to identify low-frequency mutations.
Targeted deep sequencing identified 21 somatic mutations in the tumor tissue of 6 out of the 8 patients analyzed (75%) (Table 2). These included known mutations in HCC such as TERT promoter (5/8), TP53 (3/8), CTNNB1 (2/8), JAK1 (1/8) or AXIN1 (1/8). We then looked for these same mutations in corresponding plasma and serum samples to assess their concordance with tissue (Fig. 2a). Among the 21 mutations identified in tissue with deep sequencing, reads harboring the same alternative allele were also detected in plasma or serum in 15/21 (71%) mutations (Fig. 2b). Among these 15 mutations, 9 (43%) were supported by enough reads of the mutant allele to be confidently called in cfDNA (p<0.05). As predicted, these 9 mutations displayed high variant allele fraction (VAF) based on the range of cfDNA previously described in cancer patients6. Considering a potential role of our gene panel for early HCC detection, our approach identified at least one mutation in the blood of 4/6 (67%) patients with tissue mutations (Fig. 2b). We selected a subset of tissue mutations (14/22, 66%) for orthogonal validation with Sanger sequencing and/or Droplet Digital PCR (ddPCR). All mutations tested for validation were confirmed (Table 2).
Table 2.
Mutations Identified in HCC-tissue and in Plasma
| Patient ID | Size (cm) |
Mutation location (hg.38) |
Gene | Tissue mutation validated by Sanger |
Tissue mutation validated by ddPCR |
Druggable | Sub- clonal |
Ref Allele |
Alt Allele |
VAF (%, Tissue) |
bVAF (%, plasma/serum) |
Plasma mutation validated by ddPCR |
Detection in plasma/serum (Hypergeometric test p-value) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 9.8 | 5:1295113 | TERT Prom | Not tested | Not tested | No | No | G | A | 23 | 0 | Not tested | Not detected | |
| 2 | 3.5 | 5:1295135 | TERT Prom | Yes | Not tested | No | No | G | A | 29 | 0.21 | Not tested | Detected (low confidence, p>0.05) | |
| 1:64839716 | JAK1 | Yes | Yes | Yes | No | A | G | 58 | 2.47 | Yes | Detected (high confidence, p<0.05) | |||
| 3:41224610 | CTNNB1 | Yes | Not tested | No | No | C | A | 32 | 0.02 | Not tested | Detected (low confidence, p>0.05) | |||
| 3 | 6.5 | 5:1295113 | TERT Prom | Yes | Not tested | No | No | G | A | 26 | 0.9 | Not tested | Detected (low confidence, p>0.05) | |
| 17:7675223 | TP53 | Yes | Yes | No | No | A | C | 74 | 0.4 | Yes | Detected (high confidence, p<0.05) | |||
| 2:21013095 | APOB | Not tested | Not tested | No | Yes | G | C | 25 | 0 | Not tested | Not detected | |||
| 5:150117795 | PDGFRB | Yes | Yes | Yes | No | C | T | 31 | 0.72 | Yes | Detected (high confidence, p<0.05) | |||
| 7:140800180 | BRAF | Not tested | Not tested | No | Yes | C | G | 21 | 0 | Not tested | Not detected | |||
| 4 | 13.5 | 5:1295113 | TERT Prom | Yes | Not tested | No | No | G | A | 36 | 0 | Not tested | Not detected | |
| 2:21029497 | APOB | Not tested | Not tested | No | Yes | A | G | 86 | 0 | Not tested | Not detected | |||
| 5 | 4.7 | 5:1295113 | TERT Prom | Yes | Not tested | No | No | G | A | 41 | 2.65 | Not tested | Detected (high confidence, p<0.05) | |
| 15:88183508 | NTRK3 | Not tested | Yes | No | No | A | T | 45 | 3.25 | Yes | Detected (high confidence, p<0.05) | |||
| 17:7674885 | TP53 | Yes | Yes | No | No | C | A | 80 | 4.12 | Yes | Detected (high confidence, p<0.05) | |||
| 17:7674888 | TP53 | Yes | Yes | No | No | T | G | 80 | 3.92 | Yes | Detected (high confidence, p<0.05) | |||
| 3:41224607 | CTNNB1 | Yes | Yes | No | No | A | C | 45 | 0.04 | Yes | Detected (low confidence, p>0.05 | |||
| 4:73417849 | ALB | Not tested | Not tested | No | Yes | A | G | 23 | 0 | Not tested | Not detected | |||
| 6 | 4.5 | No mutation identified in tissue | ||||||||||||
| 7 | 2.1 | 16:346446 | AXIN* | Not tested | Not tested | No | NA | C | A | 33 | 0.03 | Not tested | Detected (low confidence, p>0.05 | |
| 16:346447 | C | A | 33 | 0.02 | ||||||||||
| 17:7674216 | TP53 | Not tested | Yes | No | NA | C | A | 46 | 0.09 | Yes | Detected (high confidence, | |||
| p<0.05) | ||||||||||||||
| 7:116783444 | MET | Not tested | Not tested | Yes | NA | A | T | 28 | 0.05 | Not tested | Detected (low confidence, p>0.05) | |||
| 7:140777205 | BRAF | Yes | Yes | No | NA | T | A | 30 | 8.93 | Yes | Detected (high confidence, p<0.05) | |||
| 8 | 2.1 | No mutation identified in tissue | ||||||||||||
Ref All: Reference allele; Alt All: Alternative allele; tVAF : Variant allele frequency in tissue; bVAF : Variant allele frequency in blood; HCC: Hepatocellular carcinoma.
Dinucleotide mutation; Draggable is defined as per http://dgidb.genome.wustl.edu/
Figure 2.
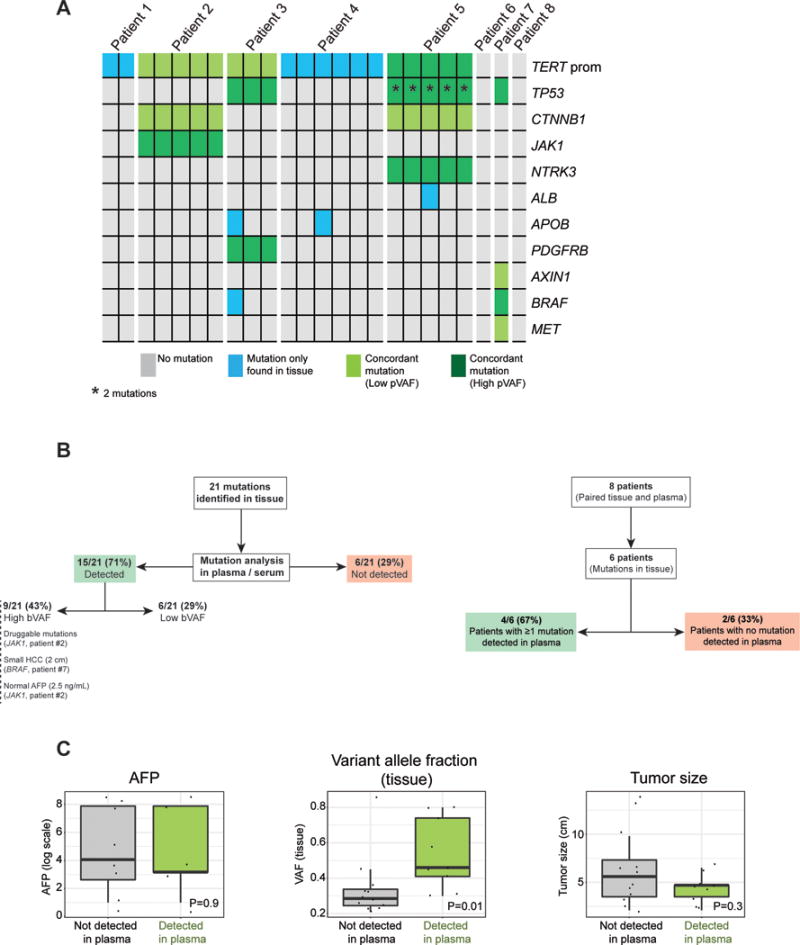
a) Heat-map of detected mutations with their concordance in tissue and plasma. Each column represents one tumor region and each row is a mutated gene found in tissue.
b) Performance of ultra-deep sequencing to detect plasma mutations, on a mutation-basis and on a patient-basis.
c) Impact of tumor size, AFP levels and variant allele fraction of the mutation in tissue in detection rate of cfDNA.
We next sought to orthogonally validate the mutations detected in plasma using Droplet Digital PCR (ddPCR). This technology allows for a highly sensitive quantitation of nucleic acids by partitioning a given sample into several thousand discrete droplets, and performing subsequent PCR amplification and fluorescence-based readout in each one of them17. Unlike conventional Sanger sequencing, ddPCR allows detecting mutations with a VAF as low as 0.01%. In addition to plasma DNA, we included paired tumor tissue DNA as a positive control. In our samples, median droplet count per reaction was 13,800, which allowed us to confidently detect mutations with very low VAF. All the plasma mutations tested were validated with ddPCR: JAK1 (VAF 1.22%, patient #2), TP53 (VAF 1.4%) and PDGFRB (VAF 1.5%, patient #3), NTKR3 (VAF 1.82%), CTNNB1 (VAF 1.57%) and the two TP53 mutations in cis (VAF 2.12%, patient #5), TP53 (VAF 0.3%) and BRAF (VAF 0.3%, patient #7, Fig. 4). In summary, we confirmed the detection performance of our ultra-deep targeted sequencing approach using ddPCR.
Figure 4.
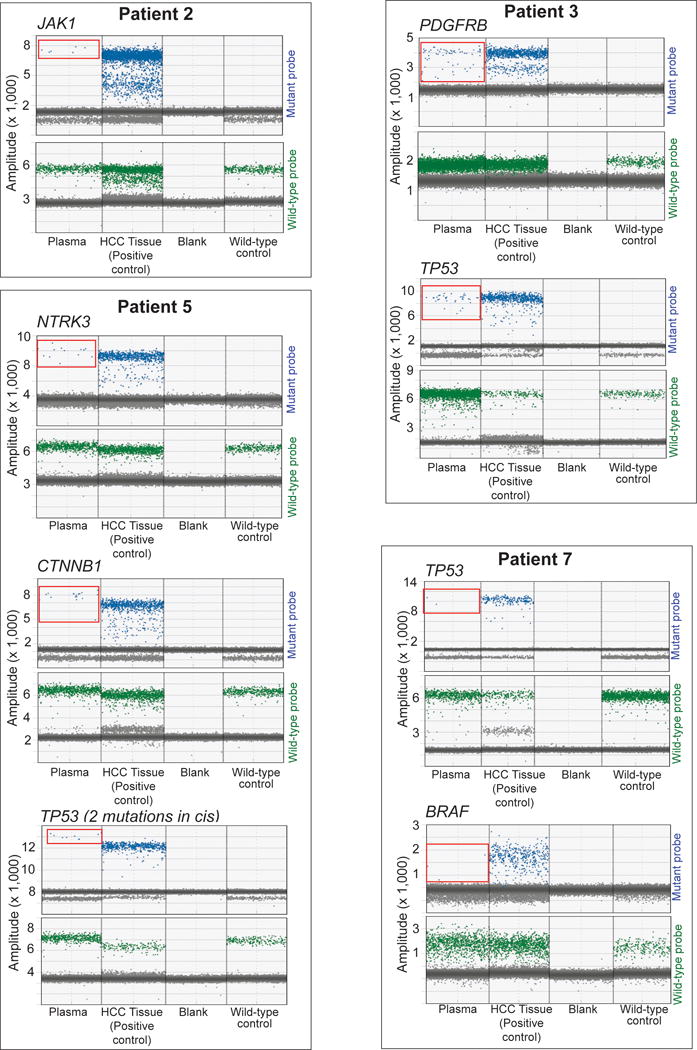
Mutation assay with ddPCR. Displayed are 1D amplitudes for mutant probe (upper lane) and wild-type probe (lower lane) for each mutation assay. Panels from left to right: Plasma and corresponding HCC tissue of same patient, blank (negative control), wild-type DNA (negative control). Blue: single positive droplets for mutant, green: single positive droplets for wild-type, grey: double negative droplets. Detected positive droplets for mutant alleles in plasma are circled in red.
Mutations in cfDNA are detected in small tumors and include a candidate druggable gene
We next sought to better characterize the mutations detected in cfDNA. Well-established oncogenes and tumor suppressors in HCC were confidently detected in plasma (e.g., TERT promoter in patient #5, TP53 in patients #3 and #5). Interestingly, we found two TP53 mutations in cis in patient #5 located 3 base-pairs apart (chromosome 17:7674885 and 17:7674888). We also found a somatic mutation in JAK1 (patient #2) (Fig. 1b), at the same hotspot as recently described18. This is particularly relevant because JAK1 mutations are druggable (e.g., ruxolitinib), and there is evidence demonstrating the oncogenic potential of this mutation in experimental models of HCC18. However, it is unknown if selective blockade of this mutation has anti-tumoral effects in HCC patients. These data support the use of mutant cfDNA as a tool to track candidate oncogene addiction loops on an individual basis. Notably, the BRAF mutation found in the plasma of patient #7 indicates that mutations in cfDNA can also be detected in small tumors. Patient #7 had a single nodule HCC of 2 cm in size without vascular invasion or satellites. Likewise, mutant cfDNA was also detected in patients with normal levels of the prognostic marker AFP, as exemplified by the JAK1 mutation found in the plasma of patient #2 (AFP 2 ng/mL), or the NTRK3, CTNNB1 and TP53 mutations detected in patient #5 (AFP 2 ng/mL).
A frequent criticism of studies detecting mutations in cfDNA is the lack of definitive evidence that the mutated reads are derived from the tumor. In other words, how can we be confident about a tumor origin of a given mutation in plasma? Our study provides definitive evidence of the release of HCC-derived DNA fragments into the bloodstream. Specifically, patient #5 had 2 distinct TP53 mutations in tissue located 3 base pairs apart, both detected with NGS and validated with Sanger sequencing and ddPCR. These mutations were also found in plasma, at a VAF of 4.12% and 3.92%, respectively, and validated with ddPCR (Fig. 3). Strikingly, when visualizing the mutant reads in plasma we found that both mutations were present in cis in the same reads while being absent in PMBC, which confirms their shared origin from malignant hepatocytes.
Figure 3.
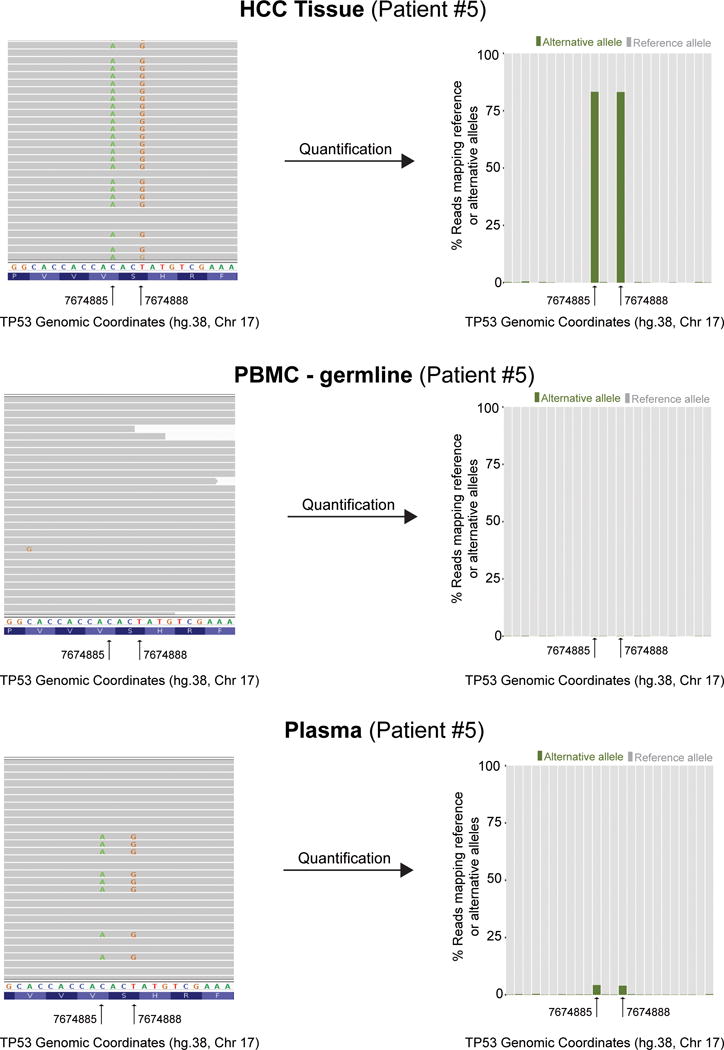
TP53 mutations in patient #5: Left panels illustrate read alignments using Integrative Genomics Viewer. Right panels show the percentage of alternative reads found in each compartment [i.e., tissue (top), PBMC (middle), plasma (bottom)]. Two adjacent mutations of TP53, located 3 base pairs apart, with a variant allele fraction of 80% were detected in tissue. The corresponding alternative alleles of these 2 mutations were not detected in PBMC, thus excluding germline events. Ultra-deep sequencing of plasma DNA detected the same 2 mutations in approximately 4% of circulating DNA fragments. Mutations in plasma are found in the exact same reads, identifying their shared origin from their tissue counterparts.
Detection rate in plasma and serum and impact of variant allele fraction
We next explored whether the mutation detection performance was influenced by the source of cfDNA (i.e., plasma or serum). This was not previously reported in HCC patients and could facilitate the retrospective analysis of samples stored in serum banks. We performed a paired analysis on the 5 patients for which both data points were available. In every case, plasma and serum were collected from the same blood draw, they were processed at the same time and under the same laboratory conditions. We did not find significant differences in the detection rate in plasma or serum. Among the 13 tissue mutations detected in these 5 patients, reads harboring the alternative allele were captured for 10 mutations in both plasma and serum, while 5 mutations were detected with high confidence (p<0.005) in both plasma and serum samples.
We explored whether any tumor characteristics could impact the detection rate for mutations in cfDNA. We failed to find differences in detection rate based on tumor size, vascular invasion, etiology of the underlying liver disease, cirrhosis or degree of cell differentiation (Figure 2c). However, the low sample size of our study does not allow us to definitively rule out a contribution of any of these factors in cfDNA mutation detection rate. Of note, we found significant differences in detection rate when considering the VAF of the mutation in tissue, with higher likelihood for detection in those mutations with higher VAF in tissue (Mann-Whitney P=0.02). Lastly, we evaluated the impact of intra-tumor genetic heterogeneity in plasma detection. Previous studies suggested the ability of mutation analysis of cfDNA to detect sub-clonal mutations19. Multi-regional sequencing data was available in 5 patients, with a total of 21 tumor regions analyzed. Similarly to recent data20, mutations in HCC driver genes such as TP53, CTNNB1 or TERT had a clonal distribution among all regions sampled from the same patient. However, our targeted gene panel did not allow us to extensively evaluate sub-clonal non-driver mutations. We found sub-clonal mutations such as APOB in patients #3 and #4, ALB in patient #5, and an intronic mutation in BRAF in patient #3. None of these sub-clonal mutations was detected in the blood.
DISCUSSION
The use of cfDNA as a novel source for cancer biomarkers will likely revolutionize personalized medicine. Its convenience and diagnostic power is well established in non-invasive prenatal testing21, and its application in oncology is rapidly evolving. In this study, we applied ultra-deep coverage NGS to the analysis of circulating DNA in HCC patients. In this pilot study, we demonstrate its ability to detect somatic mutations including known driver genes in HCC and a candidate druggable mutation (JAK1). In a clinical setting, this could allow monitoring response to selective oncogenic addition blockade using a minimally invasive approach.
There are previous reports of targeted sequencing of cfDNA in other tumor types. For instance, a recent study evaluated 568 mutations in six genes across samples from different tumor types, and confirmed its value to track emerging mutations upon tumor progression22 . Another study compared the mutation detection rate in tissue and plasma of a panel of 54 genes in 26 patients with pancreatobiliary malignancies23, further confirming the accuracy of this technology as a proxy to detect mutations in tissue. However, there are few reports that studied DNA mutations in plasma of HCC patients12–14, 24–26. Most of them analyzed a small number of genes (e.g., TERT promoter, TP53 and CTNNB1) and did not evaluate druggable mutations12, 24. Some studies reported an overall detection rate for tissue mutations in plasma as low as 12% (8/67), on a mutation-basis12. Our approach improves this rate to 43% (9/21), likely due to an increased sequencing effort (i.e., ultra-deep coverage), and a more comprehensive gene panel that included all exons of the 58 most frequently mutated genes in HCC.
Our analysis also detects mutant reads in plasma for 6 additional loci, but their VAF is too low to provide a confident call as per our pre-specified hyper-geometric test threshold. This limitation has been reported in other malignancies such as lung cancer27. Potential improvements could result from the use of molecular barcoding or an increase in the input cfDNA. Among these 6 mutations detected at low frequency in blood, there were 2 mutations in TERT promoter and CTNNB1. The former is widely known to be of particular challenge in term of sequencing because the 2 described hotspots are located within GC-rich regions. As a consequence, the detection of TERT promoter mutation in NGS studies was circumvent by alternative technique such as Sanger sequencing, even when using tissue samples28. Regarding CTNNB1, a recent study also failed to detect in blood any of the CTNNB1 mutations detected in tissue24. Interestingly, we were able to confirm the CTNNB1 mutation in patient #5 using ddPCR.
Ultra-deep targeted sequencing of cfDNA is still a technology under development, and considering its high financial cost and analytical requirements, it is unlikely to be widely available for routine testing. In addition, clinical implementation of the cfDNA analysis in HCC faces different challenges. First, the variant allele fraction (i.e., percentage of mutant DNA) of tumor cfDNA is generally beneath 2%, which is below threshold for minimal detection in most sequencing technologies at the usual sequencing depth rates (80-200x). Two recent studies reported whole exome as well as targeted sequencing of cfDNA in small series of 4 and 5 HCC patients, respectively13, 14. The sequencing coverage was lower than in our study, and the number of called SNV was significantly higher (with an average of 172-253 SNV per patient) after whole exome sequencing of cfDNA. Not surprisingly, the validation rate for these mutations using digital PCR was 53%. Our approach to address these limitations included increasing sequencing depth up to 5,500x, and integrating a hypergeometric test that enables to calibrate the confidence of the SNV called, based on predicted error distribution. Nevertheless, other technologies such as digital PCR are better suited to detect very low frequency variants. The improved performance of digital PCR for low VAF variants comes at a cost of limiting the number of mutation hotspots interrogated in each reaction. This may not be critical in tumors with well-defined recurrent hotspot mutations (e.g., EGFR in lung cancer29). However, in HCC most mutations affect different locations in a given gene, which complicates the use of digital PCR for multiple mutation monitoring. More recent analytical approaches have tried to reduce error rates without substantially sacrificing mutation calling in plasma by incorporating molecular barcoding and background polishing30, which could significantly reduce the amount of input DNA needed for this analyses. Notably, our detection performance was better for truncal mutations, which suggest a direct correlation between tumor burden and detection rate. Indeed, high VAF of a given mutation in tissue was significantly associated with higher odds for detection in blood. This suggests that the number of input DNA molecules in the plasma (i.e., genomic equivalents) is still a limiting factor to maximize mutation calling from cfDNA in plasma. Similar results have been recently reported in patients with lung cancer31. This may affect the ability of this technology to monitor sub-clonal mutations, which could be clinically relevant for those that are druggable.
Another challenge of this technology is its inability to identify the tumor site based on the mutations called in blood. This is key when considering mutation analysis of cfDNA as a cancer surveillance tool, but it is not so relevant when looking for tools to monitor response to oncogene addiction loops linked to actionable mutations. To a certain extent, mutation analysis on ctDNA could complement imaging techniques by tracking cancer clone evolution in response to therapies. For instance, the JAK1 mutation we found in tissue and blood in patient #2 could be easily monitored to potentially evaluate response to JAK1 inhibition. The advantages of monitoring tumor clone composition with cfDNA to overcome treatment resistance were recently confirmed in colorectal cancer32. Sequential analysis of KRAS mutations in cfDNA was instrumental to assess clonal competition over time. The relevance of this approach is that it demonstrated re-emergence of a KRAS wild-type clone, which provided the rationale to re-challenge the patient with an EGFR monoclonal antibody, despite initial progression on this therapy. Without mutation analysis of ctDNA, this case would have required more than 4 tumor biopsies, which is unfeasible in routine clinical conditions and emphasizes the power of this technology for precision medicine.
Despite JAK1 could be considered a druggable mutation, the overall detection rate of druggable mutations in plasma was low. Despite BRAF mutations are predictive biomarkers of response to vemurafenib in melanoma33, but there are no robust data to suggest a similar behavior in HCC. The lack of druggable mutations is an inherent limitation in HCC. Indeed, a recent report that evaluated druggable mutations in 10,000 cancer samples from different tumor types ranked HCC as second-to-last in terms of prevalence of druggable mutations34. However, there are other potential clinical applications of mutation analysis in cfDNA beyond the study of druggable mutations. For example, the detection of driver genes and monitoring of their allele frequency could be critical tools to trace cancer progression8, and to detect minimal residual disease9.
The main limitation of our study is related to its sample size. Also, all patients included were male and had early stage HCC (i.e., surgical resection candidates). As per study design, we wanted to ensure that we could recapitulate the mutational landscape of tumor tissue on the blood, and this required early stages with paired multi-regional tissue and blood samples. Hence, we cannot rule out different detection performance in patients with advanced HCC, but based on studies in other malignancies, detection rate scales up in parallel with tumor burden4. Also, we only performed mutation analysis in plasma at one time point, which did not allow us to monitor mutations at clinically significant events such as tumor recurrence.
A major strength of our study is the confirmation of the tumor origin of the mutant reads found in plasma, which has not yet been demonstrated in depth for HCC patients. The fact that the exact same reads harbored the two nearby TP53 mutations found in patient #5 supports the validity of this approach to investigate HCC genetics in blood. The small sample size precluded a comprehensive evaluation of how detection rate is influenced by clinical or pathological variables such as tumor size or presence of vascular invasion. We were able to confidently detect mutations in at least one small tumor (patient 7) and in 2 patients with normal levels of the prognostic biomarker AFP (patient 2 and 5). In summary, this pilot study demonstrates how ultra-deep targeted sequencing of cfDNA in the plasma of HCC patients is a feasible, reliable and minimally invasive approach to interrogate HCC genetics. It confidently identifies driver mutations in plasma and emerges as a promising tool for predictive biomarker development in HCC.
MATERIALS AND METHODS
Study participants
All patients were enrolled at a single institution (Icahn School of Medicine at Mount Sinai (ISMMS), New York, (US)) and provided informed consent for tissue/blood biobanking. Samples were provided by the ISMMS Biorepository (Project number BRC#228) and the study was approved by ISMMS IRB (#14-01011). All patients were diagnosed with hepatocellular carcinoma (HCC) based on the EASL/AASLD guidelines16, and they were all treated by surgical resection. Patients did not receive any other treatment prior resection.
Sample Collection
Blood
Peripheral venous blood was drawn prior surgery, and it was processed within 3 hours of collection. Plasma was collected in EDTA-containing tubes (BD Vacutainer) whereas serum was collected using clot activator tubes (BD Vacutainer). Plasma was separated by 2 centrifugation steps. The interphase layer containing peripheral blood mononuclear cells (PBMC), namely ‘buffy coat’ (germline DNA), was collected after a first centrifugation at 1,600g for 10 min at 4°C. The obtained plasma was further centrifuged at 16,000g for 10 min at 4°C to remove any cellular debris. For serum, whole blood was stored at room temperature (RT) for 30-60 minutes to allow clotting, and further centrifuged at 1,200g for 20 min at RT. Plasma, serum and PBMC were immediately aliquoted and stored at −80°C.
Tissue
Multi-regional tissue sampling of the surgical specimens was performed within 90 min of resection. The surgical specimen was examined and sectioned by a liver pathologist. Different regions within the same tumor nodule were sampled allowing for at least 1 cm of distance between each other. The final number of regions sampled per tumor varied according to tissue availability and presence of intra-tumoral necrosis. Tissue samples were immediately stored in RNA-later (Qiagen) for 24 hours at 4°C, and subsequently at −80°C.
DNA extraction, quality control and quantification
Circulating DNA was isolated from 5 mL and 1 mL of plasma and serum, respectively, using the QIAamp Circulating Nucleic Acid Kit (Qiagen). Germline DNA was isolated from PBMC using the QIAamp DNA Mini Kit (Qiagen). DNeasy Blood & Tissue kit (Qiagen) was used to extract DNA from fresh frozen tissue, following the recommendations from the manufacturer. Extracted DNA was immediately aliquoted and stored at −20°C. The purified plasma DNA was run on a 2100 Bioanalyzer Instrument (Agilent) for size estimation, and its concentration was measured by fluorometric quantitation using Qubit (ThermoFisher).
Next-Generation Library Preparation and Sequencing
Ultra-deep DNA sequencing was performed for all exons of a panel of 58 genes (Suppl. Table 1) selected based on 2 main criteria: 1) high prevalence of mutations in previous HCC studies28, 35 and 2) evidence of druggability36. Indexed Illumina NGS libraries were prepared from plasma, serum, tumor tissue and PBMC (germline DNA). Sequence captures of 58-gene-exons were carried out using the Biotinylated custom baits of Agilent SureSelect oligo pool (Agilent Cat # 5190-4808). All the procedures were carried out using vendor recommended protocols. 25-50 ng of plasma DNA were used for library construction without fragmentation using KAPA Hyper Prep Kit. Targeted enrichment was carried out using custom SureSelect oligo pool (Agilent). For tumor tissue and germline DNA, 50 ng DNA was used for tagmentation based SureSelect QXT library preparation (Agilent) followed by targeted custom sequence capture (Agilent); mean fragment size was ~250 bp. High fidelity Herculese II Fusion DNA polymerase supplied by Agilent (Cal # 600677) was utilized to prevent PCR errors. To minimize the risk of sequence error rate, we made sure that our Q30 of sequencing data was ≥90%. All samples were sequenced on HiSeq2500 sequencing platform, carrying out 100 bp single-end sequencing and pair-end sequencing for plasma/serum and tissue samples, respectively.
Sanger Sequencing
Sanger sequencing after PCR amplification using the primers listed in Suppl. Table 2 was used to further validate tissue mutations. Each PCR product was assessed on a 1.5% agarose gel, sequenced in both directions using Big Dye Terminator reactions and loaded on an ABI PRISM 3730xl DNA analyzer. Sequences were analyzed using the Applied Biosystems’s sequencing analysis software with the KB base-caller.
Droplet Digital PCR (ddPCR)
Droplets were generated using a QX100 Droplet generator (QuantaLife/BioRad) in a 20 ul reaction mix containing 2x ddPCR Supermix, 20x target/mutant (FAM) and wild-type (HEX) probes, 0.5 ul of restriction enzyme, and DNA sample or water. For each experiment, corresponding tumor tissue with the known mutation (detected by NGS and validated by Sanger) was used as a positive control, while a blank and wild-type DNA (adjacent liver tissue without mutation) served as two separate negative controls. DNA input for tissue was between 28-31 ng, where for plasma was 2.5 ng (Patient #2, JAK1 mutation) to 13.2 ng (Pat #3, TP53 and PDGFRB mutations) per reaction. Subsequent reading was performed with the QX100 Droplet reader (QuantaLife/BioRad) in “rare event detection” mode. Analysis was done with QuantaSoft Analysis Pro Software version 1.0.596. All the probes except for BRAF (BioRad #10031276/-9) were custom-made by BioRad. Sequences of the probes are listed in Suppl. Table 2.
Data Analysis
General quality control of the samples was assessed by descriptive statistics and exploratory plots with FastQC (Babraham Institute), including quality score and sequence content per base. The coverage for the targeted region was also investigated. Based on the mentioned metrics, we used a quality threshold of 20 for trimming read ends. Reads left at length smaller than 40 bp after quality trimming were filtered out. We mapped the reads to the reference genome GRCh38 (ENSEMBL) using BWA37 with the BWA-MEM algorithm. Alignments were filtered to include only the targeted region with an interval of 50 bp. PCR duplicates were tagged with GATK38 to assess their effect on variant calling. To avoid misdetection of sequencing- or PCR errors, we followed the best-practice standards of GATK39, and reads were re-aligned near indels. Subsequently, the mapping quality scores were re-calibrated according to BQSR (Base Quality Score Recalibration). VarScan2 was used for variant calling40. Filtering was done based on allele frequency (a threshold of 0.1 for solid tumors, where expected average VAF is centered around 0.5) and Fisher p-value of somatic variant of 0.001. Germ line variants were identified and filtered out with the standard germ line consensus calling in VarScan2. Because of the reported difficulty to sequence the two mutation hotspots located in the GC-rich TERT promoter, we assessed the presence and clonality of somatic mutations at these sites by manual inspection of the alignment files. Selected TERT promoter variants were further validated with Sanger. Annotation and effect prediction was performed with Variant Effect Predictor (Ensembl release 85), including SIFT and Polyphen scores of impact on protein function. For those tumors where multi-regional tissue sequencing data was available, we define clonal mutations as those detected in all regions of the tumor whereas sub-clonal were those not detected in all regions.
We assessed whether the somatic mutations called in solid tissue were detected in blood, for those with alternative allele frequencies that are beyond what we would expect in a random choice. For this purpose, and for each mutation found in tissue in each patient, we performed a hypergeometric test calculating the probability of observing a somatic mutation in blood (out of n mutational events tested for a patient) as compared to those having equal or higher read support (coverage of the alternative allele) under the null hypothesis of random selection. For those mutations with a hypergeometric p-value lower than 0.05, we rejected the null hypothesis and concluded that such somatic mutations were successfully detected in blood. For the purpose of significance testing, each SNP was considered as an independent event and the dinucleotide mutation in Patient #7 at gene AXIN1 at position 16:346446-346447 was counted as a single genomic event as per previous publications41.
Supplementary Material
Acknowledgments
The authors thank the office of Scientific Computing at the Icahn School of Medicine at Mount Sinai (ISMMS) for providing computational resources and staff expertise, as well as the ISMMS Tissue Biorepository for providing the samples. Sequencing was performed at the Genomics Core Facility at ISMMS.
Grant Support: IL is supported by a grant from the Swiss National Science Foundation, from Foundation Roberto & Gianna Gonella and Foundation SICPA. DD is supported by the Grant for Studies Broadening from the Spanish Association for the Study of the Liver (Asociación Española para el Estudio del Hígado, AEEH) and the Cancer Research Grant from Nuovo Soldati Foundation. SNM is supported by grants from the Brazilian National Council for Scientific and Technological Development, and “Associacao Piauiense de Combate ao Cancer”. JML is supported by grants from the European Commission (Heptromic, proposal number 259744; HEPCAR, proposal number 667273-2), the Samuel Waxman Cancer Research Foundation, the Spanish National Health Institute (J.M.L: SAF-2010-16055), the Asociación Española para el Estudio del Cáncer and the U.S. Department of Defense (CA150272P1). AV is supported by the U.S. Department of Defense (CA150272P3) and the Tisch Cancer Institute (Cancer Center Grant P30 CA196521). SLF is supported by NIH grant DK56621 and the U.S. Department of Defense (Contract No. W81XWH-16-1-0455). This study was funded by the American Association for the Study of Liver Diseases Foundation (AASLDF) Alan Hofmann Clinical and Translational Award to AV.
Abbreviations
- AFP
Alpha-fetoprotein
- BCLC
Barcelona Clinic Liver Cancer
- cfDNA
Circulating-free DNA
- HBV
Hepatitis B virus
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C virus
- IQR
interquartile range
- NGS
Next-generation sequencing
- PBMC
Peripheral blood mononuclear cell
- VAF
Variant allele frequency
Footnotes
Conflicts of interests: The authors declare no potential conflicts of interest.
Raw sequencing data: Sequence data is publicly available through ArrayExpress repository (accession number: E-MTAB-5123).
- Study concept and design: IL, CVM, AV
- Acquisition of data: IL, DD, AC, JvF, SNM, DS, AS, SW, MIF, SNM, MM, SNT, PT, CA, MS, AV
- Analysis and interpretation of data: IL, CVM, AS, JvF, SNM, PT, SNT, SLF, JML, MS, AV
- Drafting of the manuscript: IL, CVM, AV
- Critical revision of the manuscript for important intellectual content: IL, CVM, DD, AC, JvF, AS, DS, SNM, SW, MIF, SNM, MM, PT, SNT, SLF, JML, CA, MS, AV
References
- 1.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 2.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 3.Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 4.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mandel P, Metais P. C R Seances Soc Biol Fil. 1948;142:241–243. [Not Available] [PubMed] [Google Scholar]
- 6.Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin XJ, Chong Y, Guo ZW, Xie C, Yang XJ, Zhang Q, et al. A serum microRNA classifier for early detection of hepatocellular carcinoma: a multicentre, retrospective, longitudinal biomarker identification study with a nested case-control study. Lancet Oncol. 2015;16:804–815. doi: 10.1016/S1470-2045(15)00048-0. [DOI] [PubMed] [Google Scholar]
- 8.Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016;6:147–153. doi: 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra392. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 11.FDA approves first blood test to detect gene mutation associated with non-small cell lung cancer [news release] Silver Spring MD: U.S. Food and Drug Administration; Jun 1, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm504488.htm Accessed June 1, 2016. [Google Scholar]
- 12.Liao W, Yang H, Xu H, Wang Y, Ge P, Ren J, et al. Noninvasive detection of tumor-associated mutations from circulating cell-free DNA in hepatocellular carcinoma patients by targeted deep sequencing. Oncotarget. 2016;7:40481–40490. doi: 10.18632/oncotarget.9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang A, Zhao X, Yang XR, Li FQ, Zhou XL, Wu K, et al. Circumventing intratumoral heterogeneity to identify potential therapeutic targets in hepatocellular carcinoma. J Hepatol. 2017 doi: 10.1016/j.jhep.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Cai ZX, Chen G, Zeng YY, Dong XQ, Lin MJ, Huang XH, et al. Circulating tumor DNA profiling reveals clonal evolution and real-time disease progression in advanced hepatocellular carcinoma. Int J Cancer. 2017 doi: 10.1002/ijc.30798. [DOI] [PubMed] [Google Scholar]
- 15.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 16.European Association For The Study Of The L, European Organisation For R, Treatment Of C. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–943. doi: 10.1016/j.jhep.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014;25:1729–1735. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai W, Lim TK, Zhang T, Phang ST, Tiang Z, Guan P, et al. The spatial organization of intra-tumour heterogeneity and evolutionary trajectories of metastases in hepatocellular carcinoma. Nat Commun. 2017;8:4565. doi: 10.1038/ncomms14565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norton ME, Jacobsson B, Swamy GK, Laurent LC, Ranzini AC, Brar H, et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med. 2015;372:1589–1597. doi: 10.1056/NEJMoa1407349. [DOI] [PubMed] [Google Scholar]
- 22.Malapelle U, Mayo de-Las-Casas C, Rocco D, Garzon M, Pisapia P, Jordana-Ariza N, et al. Development of a gene panel for next-generation sequencing of clinically relevant mutations in cell-free DNA from cancer patients. Br J Cancer. 2017;116:802–810. doi: 10.1038/bjc.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov. 2015;5:1040–1048. doi: 10.1158/2159-8290.CD-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang A, Zhang X, Zhou SL, Cao Y, Huang XW, Fan J, et al. Detecting Circulating Tumor DNA in Hepatocellular Carcinoma Patients Using Droplet Digital PCR Is Feasible and Reflects Intratumoral Heterogeneity. J Cancer. 2016;7:1907–1914. doi: 10.7150/jca.15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ono A. Circulating Tumor DNA Analysis for Liver Cancers and Its Usefulness as a Liquid Biopsy. Cellular and Molecular Gastroenterology and Hepatology. 2015 doi: 10.1016/j.jcmgh.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Labgaa I, Villanueva A. Liquid biopsy in liver cancer. Discov Med. 2015;19:263–273. [PubMed] [Google Scholar]
- 27.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Izumchenko E, Chang X, Brait M, Fertig E, Kagohara LT, Bedi A, et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat Commun. 2015;6:8258. doi: 10.1038/ncomms9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–451. doi: 10.1038/nature22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 36.The Drug Gene Interaction Database
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–163. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.