

Abstract
The importance of understanding how interleukin-33 (IL-33) is regulated (particularly by miRs) is critical in IL-33 biology, and evidence of this in asthma pathology is limited. MicroRNA profiling of cells isolated from bronchoalveolar lavage of 14 asthmatic patients and 11 healthy controls revealed miR-200b and miR-200c were significantly reduced in asthmatic patients compared with healthy controls. The reduction was validated in two independent models of allergen-induced allergic airway inflammation and further demonstrated to be inversely correlated with asthma severity, as well as increased IL-33 production in asthmatic patients. In addition, the miR-200b and miR-200c binding sites in the 3′ UTR of IL-33 mRNA were identified by bioinformatics analysis and reporter gene assay. More importantly, introduction of miR-200b and miR-200c reduced, while inhibition of endogenous miR-200b and miR-200c increased, the induction of IL-33 expression in lung epithelial cells. Exogenous administration of miR-200b to lungs of mice with allergic inflammation resulted in a decrease in IL-33 levels and resolution of airway inflammation phenotype. In conclusion, miR-200b and miR-200c by regulating the expression of IL-33 have a role in bronchial asthma, and dysregulation of expression of miR-200b/c may be the underlying mechanism resulting in the asthmatic phenotype.
Keywords: asthma, microRNA, IL-33, allergic airway inflammation
The authors hypothesized that changes in the microRNA profile from the bronchoalveolar lavages of asthmatics and mouse airway inflammation models can be exploited for novel therapeutics to treat asthma. Their results demonstrated the role of miR-200 in regulation of interleukin-33 expression and also the alleviation of animal airway inflammation.
Introduction
Asthma is known as a common chronic inflammatory airway disease that is characterized by variable and recurring airflow obstruction, chronic airway inflammation, and bronchial hyper-responsiveness.1, 2 The incidence rate of asthma ranges from 2% to 18% of individuals, with approximately 300 million people affected, and accounts for 250,000 deaths each year.3 The pathophysiology of allergic asthma is currently known as maladaptive inflammatory responses to ubiquitous environmental stimuli in genetically susceptible people.4 More specifically, allergic asthma is defined as a chronic inflammatory disorder of the airways mediated by CD4+ T cells polarized to a type 2 helper (Th2) differentiation.5 Th2 cytokines, including interleukin-4 (IL-4), IL-5, and IL-13, drive the cardinal features of the disease: pulmonary eosinophilia, elevated concentrations of serum immunoglobulin E (IgE), airway hyper-responsiveness, and excessive production of mucus in the airways.5, 6 IL-33, previously known as a member of the IL-1 cytokine family, is an inducer of the Th2 branch of adaptive immunity and signals through the membrane-bound ST2 protein.7, 8 The IL-33/ST2 axis triggers the release of several proinflammatory mediators, such as chemokines and cytokines, and induces systemic Th2-type inflammation. Further, the IL-33/ST2 pathway also contributes to allergen-induced airway inflammation and hyper-responsiveness, both important features of asthma development.7, 9, 10 Elevated IL-33 mRNA levels were measured in the lung tissue from subjects with asthma compared with normal controls.11, 12 Until now, it remains unclear how IL-33 expression is regulated in vivo both at the transcriptional and the posttranscriptional levels. Especially, the posttranscriptional regulation of IL-33 expression is also unclear in vivo physiologically in bronchial asthma development.
MicroRNAs (miRNAs) are discovered as a type of non-coding RNA and proven to regulate gene expression via mRNA degradation and translational repression.13 Dysregulation of miRNA expression has been known to alter cellular pathways and disease development, including allergic asthma.14, 15, 16, 17 Downregulation of miR-133a contributed to upregulation of RhoA in bronchial smooth muscle cells of asthmatic patients.18 miR-192 expression was reduced in peripheral blood of asthmatic individuals undergoing an allergen inhalation challenge.19 miR-21 was upregulated in allergic airway inflammation and regulates IL-12p35 expression.20 Another miRNA molecule, let-7, could downregulate IL-13 production in allergic airway inflammation.21 Investigations of the role of altered miRNAs in bronchial asthma would reveal their relevance in the biologic and clinical behavior during the disease. In this study, we identified that miR-200b and miR-200c were downregulated in the airways of asthmatics. Furthermore, we used two different mouse allergen-induced airway inflammation models to investigate the contribution of these two miRNAs to the observed airway phenotype. Administration of miR-200b to the airways during allergen sensitization reverses airway inflammation. Using 3′ UTR-Luciferase reporter assays, we identified binding sites for the miRNAs and downregulation of luciferase activity, thus providing evidence for direct regulation of IL-33 induction.
Results
Reduced Expression of miR-200b/c in Asthmatic Patients and Its Correlation with Asthma Activity
In an initial effort to identify differentially expressed miRNAs in asthmatic patients, we profiled the expression of 366 miRNAs by using a TaqMan miRNA assay. Total RNA samples were obtained from the cells isolated from bronchoalveolar lavage of 14 asthmatic patients and 11 healthy controls. The age of asthmatic patients is 14 ± 5 years old compared with 15 ± 6 years old for healthy controls in the pilot study. The study revealed differential expression of 39 miRNAs in bronchoalveolar lavage fluid of patients with asthma as compared with healthy controls. Among them, we found that 10 miRNAs (let-7a/b, miR-30a, miR-99a, miR-27a, miR-133a, miR-155, miR-24, and miR-200b/c) were more than 3-fold lower in patients versus healthy controls; 3 miRNAs (miR-21, miR-1268, and miR-663a) were more than 3-fold higher in patients versus healthy controls (Table 1; Figure 1A). Among these miRNAs, miR-200b/c in particular has not been reported in allergic asthma pathogenesis. Because defects in the negative regulation system can cause unabated immune activation, even inflammatory diseases, we further explored the role of miR-200b/c in asthma development.
Table 1.
Selected Differentially Expressed miRNAs in Bronchoalveolar Lavage Cells from Bronchial Asthma
| miRbase-18 Name | Accession Number | Log2 Fold Difference (versus Healthy Control Subjects) | FDR Q Value |
|---|---|---|---|
| let-7a | MI0000060 | 0.25 | 1.3 × 10−3 |
| let-7b | MI0000063 | 0.34 | 3.2 × 10−4 |
| miR-30a | MI0000088 | 0.38 | 2.5 × 10−5 |
| miR-99a | MI0000101 | 0.36 | 1.9 × 10−4 |
| miR-27a | MI0000085 | 0.30 | 4.7 × 10−4 |
| miR-133a | MI0000450 | 0.15 | 5.2 × 10−5 |
| miR-155 | MI0000681 | 0.38 | 1.5 × 10−3 |
| miR-24 | MI0000080 | 0.20 | 6.2 × 10−3 |
| miR-200b | MI0000342 | 0.19 | 3.4 × 10−3 |
| miR-200c | MI0000650 | 0.25 | 1.2 × 10−2 |
| miR-21 | MI0000077 | 1.99 | 2.4 × 10−5 |
| miR-1268 | MI0006405 | 2.65 | 5.1 × 10−3 |
| miR-663a | MI0003672 | 3.05 | 4.0 × 10−3 |
Accession number and miRNA information are available from http://www.mirbase.org. FDR, false discovery rate.
Figure 1.

Reduced Expression of miR-200b/c in Asthmatic Patients and Its Correlation with Asthma Severity
(A) Heatmap of 13 differentially expressed miRNAs between 14 asthmatic patients and 11 normal controls. Relative expression is log2 transformed. A, asthma; C, control. (B–D) Real-time qPCR of miR-200a (B), miR-200b (C), and miR-200c (D) expression between 10 asthmatic patients and 10 normal controls. (E and F) miR-200b (E) and miR-200c (F) expression levels were compared among subjects with moderate asthma (n = 29) and severe asthma (n = 20) versus control subjects (n = 22). *p < 0.05 (Student’s unpaired and two-tailed t test).
To validate the results from the pilot array study, we compared between our miRNA array profiling and real-time RT-PCR data, and the results showed the strong correlation among these methods (Pearson correlation coefficient = 0.99; p = 0.001; data not shown). In addition, we examined the expression of miR-200 by the TaqMan PCR method described above in 10 asthmatic patients and 10 normal controls from the same subjects’ cohort (Figures 1B–1D). Similarly, there was a significant decreasing expression of miR-200b and miR-200c in the asthmatic patients compared with those of healthy controls (p = 0.015 and p = 0.022, respectively).
To examine the correlation between miR-200b/c expression and clinical asthma severity, we subsequently studied miR-200b/c expression in a larger group of samples: 29 patients with moderate asthma, 20 patients with severe asthma, and 22 healthy controls (Table 2). As shown in Figures 1E and 1F, the expression of miR-200b and miR-200c was significantly lower in moderate asthmatic patients compared with normal controls (all comparisons: p < 0.05). Furthermore, we detected reduced miR-200b/c levels (even lower, all comparisons: p < 0.05) in subjects with severe asthma compared with those with normal controls. Taken together, the results suggested that the miR-200 family is intrinsically reduced in asthma patients and further demonstrated that asthma disease levels affect the expression of the miR-200 family.
Table 2.
Clinical Characteristics of Asthmatic and Control Groups from Independent Cohorts
| Healthy Controls (n = 22) | Moderate Asthma (n = 29) | Severe Asthma (n = 20) | |
|---|---|---|---|
| Characteristics | |||
| Age (years) | 21 ± 5 | 17 ± 6 | 22 ± 3 |
| Male/female | 12/10 | 19/10 | 14/6 |
| Atopy | 4 (18%) | 25 (86%) | 20 (100%) |
| Age at first symptoms (years) | – | 5 ± 1 | 3 ± 1 |
| Duration of symptoms (years) | – | 11 ± 3 | 12 ± 3 |
| Parental smoking | 7 (21%) | 12 (41%) | 10 (50%) |
| FEV1 (% predicted) | 98 ± 11 | 71.5 ± 12 | 65.5 ± 9.5 |
| Bronchodilator reversibility (%) | 3.5 ± 1.5 | 20 ± 6 | 19 ± 7 |
| Total IgE (U/mL) | 56 (19–136) | 412 (145–1,096) | 412 (145–1,096) |
| Medications | |||
| Inhaled corticosteroid (mg/day) | – | 1.7 (0.7–1.6) | 1.7 (0.7–1.6) |
| Maintenance oral prednisolone | – | 5 | 8 |
| Long-acting β-agonist | – | 28 | 20 |
| Leukotriene receptor antagonist | – | 21 | 18 |
| Theophylline | – | 15 | 15 |
| ACT score (/25) | – | 14 (8–17) | 14 (8–17) |
| FENO50 (ppb; normal, <24 ppb) | – | 52 (27–67) | 55 (29–69) |
| Sputum eosinophil (%; normal, <2.5%) | – | 8 (3.5–28) | 9 (4–24) |
ACT, Asthma Control Test; FENO50, fraction of exhaled nitric oxide at 50 mL/s; FEV1, forced expiratory volume in 1 s; ppb, parts per billion.
miR-200b/c Reduction in Two Mouse Models of Allergic Airway Inflammation
To further examine the in vivo correlation between miR-200b/c expression and allergic asthma, we aimed to determine the levels of miR-200 using real-time RT-PCR analysis in two independent asthma models (ovalbumin [OVA] and A. fumigatus induced experimental asthma in mice). In the OVA model, mice were sensitized by two intraperitoneal (i.p.) injections of OVA and aluminum hydroxide. The A. fumigatus model involves a unique mucosal sensitization route (intranasal) compared with the OVA model. Although the methods of experimental asthma induction are different, both mouse models of allergic airway inflammation have similar phenotypes, including Th2-associated eosinophilic inflammation, mucous production, and airway hyper-responsiveness (Figure S1). First, we examined the OVA-induced model of allergic airway inflammation and demonstrated that OVA-challenged mice had a 2.2-fold (p < 0.01) reduction of miR-200b and a 1.8-fold repression of miR-200c (p < 0.01) in bronchoalveolar lavage cells compared with saline-challenged mice (Figure 2A). Second, we examined the A. fumigatus model of allergic airway inflammation and demonstrated that antigen-challenged mice have a 2.05-fold decrease in miR-200b level (p < 0.01) and a 2.15-fold repression of miR-200c (p < 0.01) compared with control mice (Figure 2B). These results indicated that miR-200b/c were downregulated in allergic airway inflammation.
Figure 2.
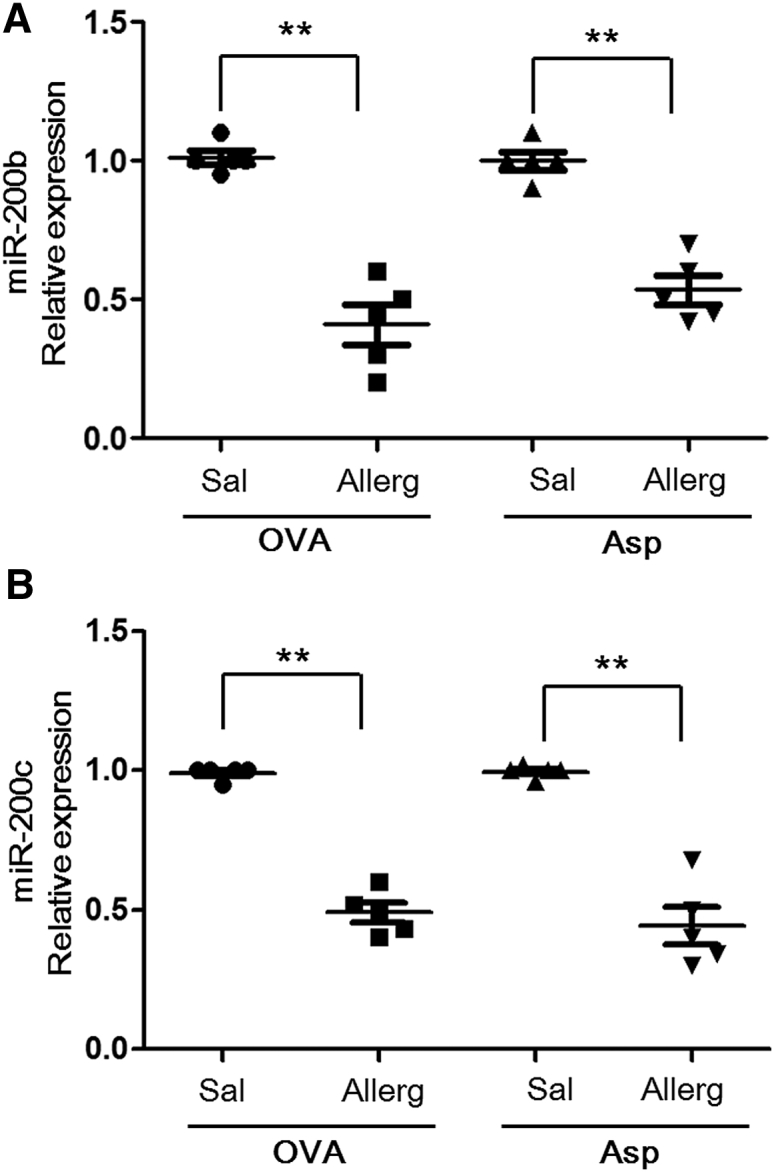
miR-200b/c Reduction in Two Mouse Models of Allergic Airway Inflammation
miR-200b (A) and miR-200c (B) expression were assessed in OVA and A. fumigatus-induced models of allergic airway inflammation. The relative expression levels were determined by qRT-PCR normalized to snoRNA135. Data are represented as mean ± SEM; n = 5 mice per group; data are representative of three experiments. **p < 0.01 (Student’s unpaired and two-tailed t test).
Confirmation of Target Site for miR-200b/c in IL-33 3′ UTR
Integration of miRNA target predictions from multiple algorithms has been reported to substantially increase the functional correlations and decrease the false-positive rate compared with single algorithms. Common predicted targets of miR-200b/c using miRanda and TargetScan algorithms arrived with a list of 13 predicted targets (data not shown), including IL-33, which was reported to be overexpressed in asthmatic patients. Our bioinformatics analysis predicted the binding of miR-200b/c to the 3′ UTR of IL-33 in a region encompassing 655–661 bases (Figure 3A). Therefore, IL-33 was predicted to have a potential miR-200 binding site conserved across species.
Figure 3.

Confirmation of Target Site for miR-200b/c in IL-33 3′ UTR
(A) Predicted binding site for miR-200b/c in 3′ UTR of IL-33. (B) Relative luciferase activity in Jurkat cells cotransfected with control firefly luciferase vector (pMIR-Report), or a firefly luciferase reporter vector containing the 3′ UTR of IL-33, or a firefly luciferase vector with perfect miR-200b/c binding site in the 3′ UTR (pMIR-200), and either the pre-miR-200 expression vector (pMIRNA1-Pre-miR-200) or control vector (pMIRNA1-Control). Firefly luciferase activity was normalized to the Renilla luciferase activity and then to the average of the control firefly luciferase reporter. n = 4 per group; data are representative of three experiments. An unpaired t test was used to compare reporter gene activity. (C) Jurkat, Raji, THP-1, and A549 cells were transfected with pMIR-REPORT-IL-33 3′ UTR to correlate the expression of miR-200b/c with IL-33 3′ UTR regulatory activity. (D) MRC5 cells were transfected with a miR-200b/c expression vector; 24 hr later, cells were stimulated with IL-13 to induce production of IL-33. Overexpression of miR-200b and miR-200c greatly reduced the induction of IL-33 production. (E) Silencing the endogenous miR-200b/c via transfection with inhibitory oligonucleotides increased IL-33 production. **p < 0.01 (Student’s unpaired and two-tailed t test).
We next performed reporter gene assay to confirm the bioinformatics prediction. When luciferase reporter vector containing the intact 3′ UTR was transfected in Jurkat cells, a significant downregulation in the luciferase expression in Jurkat cells with respect to control was observed (Figure 3B). This downregulation was not seen in mutant 3′ UTR construct lacking the predicted base miR-200b/c binding site. However, cotransfection with another non-specific miRNA, Cel-67, did not have any appreciable effect on luciferase expression. Most importantly, when miR-200b/c was cotransfected along with pMIR-REPORT-IL-33 3′ UTR (mutant), there was minimal reduction in the luciferase expression. Also, cotransfection of pMIR-REPORT-IL-33 3′ UTR (mutant) with Cel-67 had no effect on luciferase expression. These experiments indicate that miR-200b/c regulates IL-33 expression by interacting with its 3′ UTR.
To correlate the expression of miR-200b/c with IL-33 3′ UTR regulatory activity, Raji, THP-1, and A549 cells were transfected with pMIR-REPORT-IL-33 3′ UTR. A substantial fall in luciferase activity was observed in Jurkat, Raji, A549, and THP-1 cells (Figure 3C). Thus, the downregulatory effect of IL-33 3′ UTR was found to be correlated with the levels of mature miR-200b/c present in these cells. These results confirmed that expression of miR-200b/c and its involvement in IL-33 regulation were independent of specific cell lines.
To explore the association between miR-200b/c levels and activation of IL-33 production, we first sought to determine whether miR-200b/c could intrinsically modulate the onset and activation of the IL-33 pathway. The effects of miR-200b/c on IL-33 production were initially explored in lung epithelial cells. MRC5 cells were transduced with a miR-200b/c expression vector; 24 hr later, cells were stimulated with IL-13 to induce production of IL-33. As shown in Figure 3D, overexpression of miR-200b and miR-200c greatly reduced the induction of IL-33 production (both p < 0.01). Furthermore, silencing the endogenous miR-200b/c via transfection with inhibitory oligonucleotides increased IL-33 production (Figure 3E). Thus, the results suggested that miR-200b/c negatively regulates the production of IL-33.
Correlation between Reduced miR-200b/c Levels and Increased IL-33 Production in Asthmatic Patients
Because miR-200 family member deficiencies might reflect defects in negative regulation of the immune response, we explored whether sustained reduced expression of miR-200 affected, or was associated with, activation of the IL-33 biologic pathway in asthma patients. We next performed an analysis to determine whether there was any correlation between miR-200b/c levels and IL-33 production in asthmatic patients. It is interesting, although not surprising, that a negative correlation was found between reduced miR-200b and miR-200c levels and increased IL-33 production (r = −0.21, p = 0.04 and r = −0.32, p = 0.03, respectively) (Figure 4). These results confirmed the inverse correlation between reduced miR-200b/c levels and increased IL-33 production in asthma patients.
Figure 4.

Inverse Relationship between Reduced miR-200 Levels and Increased IL-33 Production in Asthmatic Patients
A negative correlation was performed between reduced miR-200b and miR-200c levels (qRT-PCR) and increased IL-33 production (ELISA) in asthmatic patients (r = −0.21, p = 0.04 and r = −0.32, p = 0.03, respectively).
Intranasal Delivery of miR-200 Reduces IL-33 Levels in Mice with Allergic Airway Inflammation
To determine the role of miR-200 in IL-33-mediated inflammation, we used the murine model of allergic airway inflammation (OVA) resembling allergic asthma. Two experiments were performed to confirm that the intranasal miRNA mimic was efficiently delivered to the lung and taken up by the lung tissue. First, delivery of exogenous mature miR-200b mimic to lungs through the intranasal route resulted in a 9-fold increase in miR-200b levels above the baseline value (Figure 5A). Second, intranasal administration of Dyomics 547 (DY547)-labeled miR-200 or Cel-67 oligonucleotides to OVA mice followed by measure of IL-33 production revealed that uptake of miR-200b mimic in the lungs of mice with allergic inflammation was associated with significant reductions in IL-33 levels in bronchoalveolar lavage fluid supernatants (Figure 5B) and sera (Figure 5C) of OVA mice compared with those seen in Cel-67 mice. We then examined mRNA expression of three representative IL-33-inducible genes to determine the coordinate activation of the IL-33 pathway in vivo. Uptake of miR-200 mimic in the lungs of mice with allergic inflammation led to significant reduction effects on the levels of inflammatory cytokines, such as IL-4, IL-5, and IL-13 (Figure 5D), compared with those seen in Cel-67 mice.
Figure 5.

Exogenous miR-200b Reduces IL-33 Levels in Mouse Airway Inflammation
(A) miR-200b levels after delivery of exogenous mature miR-200b mimic to lungs through the intranasal route. (B and C) IL-33 levels in bronchoalveolar lavage fluid supernatants (B) and sera (C) after intranasal administration of Dyomics 547 (DY547)-labeled miR-200 or Cel-67 oligonucleotides to OVA mice. (D) Inflammatory cytokines, including IL-4, IL-5, and IL-13, after uptake of miR-200 or Cel-67 in the lungs of mice. *p < 0.05; **p < 0.01 (Student’s unpaired and two-tailed t test).
We further examined the correlation with miR200b/c with full-length IL-33 or mature cleaved IL-33 in asthmatic lung tissue. The activities of full-length mouse (amino acids 1–266) and matured mouse (mm, 109–266) forms of IL-33 were analyzed by western blot by using in vivo lung tissues (Figure S2). Results showed that administration of miR-200b oligonucleotides in the mice lungs was associated with significant reductions in full-length and matured mouse IL-33 production.
Intranasal Delivery of miR-200 Mimic Alleviates Asthma Features
To determine whether miR-200-mediated reduction in IL-33 levels translated into improvement of the asthmatic condition, we determined the effects of intranasal miR-200 on asthma features, such as airway hyper-responsiveness and airway inflammation, in a murine model. Administration of miR-200b (n = 5), but not Cel-67 (n = 5), significantly reduced the infiltration of inflammatory cells in the peribronchial and perivascular regions (Figures 6A and 6B); in addition, administration of miR-200 mimic (n = 5) significantly reduced the increase in airway resistance in response to increasing concentrations of methacholine (Figure 6C). These data indicated that intranasal delivery of miR-200 mimic could alleviate asthma features.
Figure 6.

Exogenous miR-200b Attenuates Airway Inflammation and Airway Hyper-Responsiveness in Asthmatic Mice
(A) Lung sections stained with H&E in mice after administration of miR-200b (n = 5), but not Cel-67 (n = 5). Histology scale bar, 100 μM. (B) Inflammation scores in the lung tissue of miR-200b- or Cel67-administrated mice treated with OVA. Data are presented as the mean ± SD (n = 5). **p < 0.01 (Student’s unpaired and two-tailed t test). (C) Airway resistance with increasing concentrations of methacholine.
We further confirm the in vivo results by using small interfering RNA (siRNA) against mouse IL-33 (mIL-33). 1 × 106 IFU (infectious units/mL) of LentimiRa-GFP-siRNA mIL-33 virus and LentimiRa-GFP-negative control Scramble virus were administered intratracheally into the anesthetized animals 3 days before OVA administration. Real-time PCR showed that IL-33 mRNA was significantly reduced in LentimiRa-GFP-siRNA mIL-33 virus-infected animals compared with negative controls (Figure S3A). H&E staining demonstrated that LentimiRa-GFP-siRNA mIL-33 virus could significantly reduce the infiltration of inflammatory cells in the peribronchial and perivascular regions (Figure S3B).
Discussion
Dysregulation of miRNAs has been related with the pathogenesis in human bronchial asthma,13, 15 which further demonstrated the negative feedback regulation of allergic inflammation via both regulators of inflammatory miRNAs in asthmatic patients.18, 20, 21 Although our knowledge about the miRNA regulation of allergic inflammation has progressed in the last several years, multiple areas warrant future investigation. In particular, the polarized Th responses could be regulated by potential miRNAs targeting different components of the T cell polarization pathways. Upregulation of miR-21 appears to promote Th2 and attenuates Th1 responses by targeting IL-12 expression.20 Upregulation of miR-146a could potentially enhance the T regulatory (Treg) cell-mediated suppression of Th1 responses and result in unopposed Th2 activation.22 The let-7 family members appear to target IL-13 expression; downregulation of let-7 could enhance Th2 responses by upregulating IL-13 expression.21 The diverse role of miRNAs in disease processes may bring the fundamental changes to understanding the pathogenesis of asthma and to developing new therapeutic strategies. In this study, we performed an miRNA array of asthmatic cohorts and in vivo experimental allergic inflammation mice models, and found that the miR-200b/c could regulate IL-33 expression. Furthermore, exogenous administration of miR-200 mimic to lungs of OVA-dependent murine model of allergic airway inflammation resulted in a decrease in IL-33 levels, resolution of airway inflammation, and reduction in airway hyper-responsiveness. Our findings extend the role of miRNAs in the pathogenesis of allergic diseases and indicated the role and regulation of miR-200b/c in asthma to better understand its pathogenesis. To explore possible reasons for the reduced expression of miR-200 in asthmatic patients, we performed the bioinformatic analysis and identified a potential CpG island in the promoter of miR-200. This island corresponded exactly to the position of a fragment harboring the putative nuclear factor κB (NF-κB) binding sites. It would be interesting to explore whether DNA methylation changes caused by NF-κB signaling account for lowered expression of miR-200 in patients with asthma.
IL-33 is known as one of the earliest released signaling molecules following epithelial damage and can orchestrate the recruitment and activation of the cells responsible for allergic diseases. Unregulated IL-33 activity could activate T helper type 2 cells, mast cells, dendritic cells, eosinophils, and basophils, ultimately leading to increased expression of cytokines and chemokines that promotes the disease progression of asthma. The requirement for IL-33 in Th2 cell generation and activity was demonstrated in a pulmonary granuloma model driven by Schistosoma mansoni eggs and in a murine model of asthma driven by ovalbumin sensitization.23 Similarly, differentiation of human CD4+ cells in vitro in the presence of IL-33 enhanced antigen-dependent IL-5 and IL-13 production.24 In addition to influencing CD4 cellular differentiation, IL-33 works as a chemo-attractant for Th2 cells, recruiting Th2 cells to lymph nodes and tissue.25 IL-33 can influence dendritic cell maturation and activity, leading to their enhanced expression of major histocompatibility complex (MHC) class II and CD86. These activated dendritic cells (DCs), when cultured with naive CD4+ T cells, led to their differentiation in a fashion characterized by production of IL-5 and IL-13.26 Given the importance of IL-33 in the immune response and the consequences of IL-33 over-production in asthmatic patients, we propose that an approach involving replenishment of miR-200b/c would reduce IL-33 production. Interestingly, when the miR-200b/c mimic was introduced into lung cells of asthmatic patients, the expression of IL-33 was decreased. These results suggest that miR-200b/c levels could be manipulated to provide useful therapeutic interventions for asthma.
There are still some limitations in this study. First, the sample size in this study is small. Results have to be validated in an independent sample-size cohort including potential confounding clinical variables. The study participants are mostly adolescents, which could limit the generalizability of the study to younger and older populations. In addition, although we demonstrated downregulation of IL-33 by miR-200b/c overexpression, there may be other effects that have not been examined in this study. It is critical in terms of establishing potential off-target effects.
In conclusion, our results demonstrate that reduced expression of miR-200 is relevant to the biologic and clinical features of bronchial asthma development. Our findings suggest that miRNAs may serve as therapeutic targets for the treatment of asthma via regulation of the IL-33 signaling pathway.
Materials and Methods
Ethics Statement
The study was performed in accordance with relevant guidelines and regulations, following the approval of the licensing committee of Tongji Medical College, Huazhong University of Science and Technology. Written informed consent was obtained from asthmatic patients and healthy subjects, as well as their guardians. This work received approval from the institution ethics committee and conformed to the tenets of the Declaration of Helsinki.
Subject Collection
Patients and control subjects were recruited from Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. Among the subjects, there were 63 patients with asthma and 33 healthy controls. In the 63 patients, 40 cases are male and 23 cases are female. All asthmatic patients had currently one or more symptoms and physical examination results that were compatible with the asthma definition by the American Thoracic Society.27 Severe persistent asthma is defined by symptoms that occur daily and often. Lung function of severe asthma is less than 60% of the normal level without treatment. Bronchoalveolar lavage was performed during asthmatic patients’ visits (1–15 days post-exacerbations). Over-the-counter drugs were used in some patients before bronchoalveolar lavage; however, relevant information is not available. Healthy subjects were recruited from the general population who answered negatively to a screening questionnaire for respiratory symptoms and showed normal findings on a simple medical examination. Total serum IgE levels were determined with the Pharmacia CAP System.
RNA Processing
Bronchoalveolar lavage obtained from each study subject was collected into tubes, and cells were immediately separated using the Ficoll method (Amersham, Uppsala, Sweden). Bronchoalveolar lavage fluid cell counts and differential cell counts of subjects in the pilot study are displayed in Table S1. Alveolar macrophage accounted for more than 80% of cell types in bronchoalveolar lavage fluid in asthmatic patients and healthy controls. No significant differences in cell types (macrophages, neutrophils, and lymphocytes) were found between the two groups (Table S1). Total small RNAs were extracted using a mirVana miRNA isolation kit according to the manufacturer’s directions (Ambion, Austin, TX, USA). RNA was subjected to RT (reverse transcription) using a TaqMan miRNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) and Megaplex RT primers following the manufacturer’s protocol. For the pilot study, expression of the 366 miRNAs included in the TaqMan miRNA assays Human Panel-Early Access kit (Applied Biosystems) was examined according to the manufacturer’s protocol.
Real-Time PCR
In subsequent studies, the TaqMan kit specified for quantification of the miR-200 family was used, and the expression level of each sample was normalized to that of U6, a reference small nuclear RNA. TaqMan assays were performed in duplicate or in triplicate on a 7900HT real-time instrument (Applied Biosystems). Relative expression levels were calculated using the 2−ΔΔCt method.
Plasmid Construction
miR-200 expression vectors were constructed by inserting the designed primer pairs into the pSUPER basic vector (OligoEngine, Seattle, WA, USA). To create IL-33 3′ UTR luciferase reporter constructs, we cloned fragments of 3′ UTR from the IL-33 gene harboring the predicted miR-200 binding sites downstream of the firefly luciferase cassette in the pMIR-REPORT vector (Ambion). All constructs were sequenced, and expression vectors were prepared with the use of an Endofree plasmid kit (QIAGEN, Chatsworth, CA, USA). Following transfection, overexpression of miR-200 was confirmed by qPCR.
Cell Culture and Transfection
Jurkat, Raji, THP-1, A549, and MRC5 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS). Cell transfection was performed using Lipofectamine 2000 (Invitrogen, Foster City, CA, USA). For transfection of MRC5 cells, 3 × 106 cells were electroporated with 1.5 μg of an empty or miR-200b/c expression vector using Lipofectamine 2000 (Invitrogen).
Reporter Gene Assay
Cells were seeded in the wells of a 24-well plate and then transfected with a mixture of 250 ng of 3′ UTR luciferase reporter vector and 10 ng of pRL-TK vector, along with 400 ng of either an empty vector or the miR-200 expression plasmid. After 24 hr, cells were lysed and luciferase activity was measured with a luminometer by using a dual-luciferase reporter assay system (Promega, Madison, WI, USA). The ratio of firefly luciferase to Renilla luciferase was obtained for each well.
IL-4, IL-5, IL-13, and IL-33 Cytokine Release by ELISA
IL-33 levels were measured in serum by commercial sandwich ELISA (GenWay Biotech, San Diego, CA, USA). In summary, an ELISA plate was coated with capture antibody (affinity-purified chicken anti-human IL-33 Ab) in 0.05 M carbonate-bicarbonate followed by blocking. Serum samples were then added followed by horseradish peroxidase (HRP)-conjugated secondary monoclonal antibody (mAb). Tetramethyl benzidine was subsequently added to the reaction, which was stopped by applying 2 M H2SO4. Optical density was measured by microtiter plate reader at 450 nm. Serum level of IL-33 was read off from a standard curve according to the manufacturer’s instruction. Similarly, the supernatant of bronchoalveolar lavage fluid from mice was used to perform ELISA for IL-4, IL-5, and IL-13 (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol. Results were expressed in picograms and normalized by protein concentrations.
Experimental Asthma Induction
Experimental asthma was induced by injection with 100 μg of OVA and 1 mg of aluminum hydroxide as adjuvant twice, followed by two 50 μg OVA or saline intranasal challenges 3 days apart, starting least 10 days after the second sensitization. Mice were sacrificed 18–24 hr after the second challenge. Aspergillus fumigatus antigen-associated asthma was induced by challenging mice intranasally three times per week for 3 wk with 100 μg (50 μL) of A. fumigatus extract or 50 μL of saline each time. Mice were sacrificed 48 hr after the last challenge. Methods for performing bronchoalveolar lavage in mice were described in brief. After the tube and needle were put into the trachea, a 1-cc syringe with 0.9 cc sterile saline was loaded and saline was injected into the lungs. After aspirating saline by pulling the barrel of the syringe, the syringe was removed from the needle, and recovered lavage fluid was injected to a 15-mL Falcon tube on ice. The procedures were repeated for four washes per animal. LentimiRa-GFP-siRNA mIL-33 virus and LentimiRa-GFP-negative control Scramble virus were obtained from Applied Biological Materials (ABM) (Richmond, BC, Canada). 1 × 106 IFU were administered intratracheally into the anesthetized animals 3 days before OVA administration. All animals were housed under specific pathogen-free conditions in accordance with institutional guidelines, which were approved by the Institutional Animal Care and Use Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
miRNA Design and Delivery
miR-200 and Cel-67 oligonucleotides were designed in such a manner that the 3′ terminal in both strands was chemically modified with 2′-O-methoxy substitution to impart stability (Dharmacon, Lafayette, CO, USA). The oligonucleotides were dissolved in water, and the working dilution was prepared in PBS and administered to mice by using the InExpose inhalation exposure system (Scireq, Montreal, QC, Canada) on 3 consecutive days (days 24, 25, and 26) 30 min before OVA challenge. The mice were sacrificed the next day.
Measurement of Airway Hyper-Responsiveness
Airway hyper-responsiveness in the form of airway resistance was estimated in anesthetized mice by using the FlexiVent system (Scireq), which uses a computer-controlled murine ventilator and integrates with respiratory mechanics. Final results were expressed as airway resistance with increasing concentrations of methacholine.
Lung Histology
Formalin-fixed, paraffin-embedded lung tissue sections were examined for airway inflammation with H&E as described previously.21, 28 For the examination of bronchial inflammation, lung tissue sections were stained with H&E visualized with a DM LB microscope and scored using established criteria29 in a blinded manner by the pathologists, averaging their judgments for the categorization.
Statistical Analysis
Data were analyzed using GraphPad Prism 5 software. The nonparametric Mann-Whitney U test was used to draw comparisons between groups, with the exception that an unpaired t test was used to compare reporter gene activity. Spearman’s test was used for correlation studies. A p value (two-tailed) less than 0.05 was considered statistically significant.
Author Contributions
X.T. and G.Y. performed experiments and wrote the main manuscript text. F.W., J.F., and Y.J. performed statistical analysis. G.Y. and J.W. were responsible for conception of the project, design of the experiment, and writing of the manuscript. All authors reviewed the manuscript.
Conflicts of Interest
We declare no competing financial interests.
Acknowledgments
This study was supported by the Hubei Province Health and Family Planning Scientific Research Project (grant WJ2017M114 to G.Y.), the Fundamental Research Funds for the Central Universities (grant 2017KFYXJJ240 to G.Y.), and the Natural Science Foundation of China (NSFC grant 81470100 to X.T.).
Footnotes
Supplemental Information includes three figures and one table and can be found with this article online at https://doi.org/10.1016/j.ymthe.2018.04.016.
Supplemental Information
References
- 1.Elias J.A., Zhu Z., Chupp G., Homer R.J. Airway remodeling in asthma. J. Clin. Invest. 1999;104:1001–1006. doi: 10.1172/JCI8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bossé Y., Paré P.D., Seow C.Y. Airway wall remodeling in asthma: from the epithelial layer to the adventitia. Curr. Allergy Asthma Rep. 2008;8:357–366. doi: 10.1007/s11882-008-0056-0. [DOI] [PubMed] [Google Scholar]
- 3.Platts-Mills T.A., Carter M.C., Heymann P.W. Specific and nonspecific obstructive lung disease in childhood: causes of changes in the prevalence of asthma. Environ. Health Perspect. 2000;108(Suppl 4):725–731. doi: 10.1289/ehp.00108s4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fish J.E., Peters S.P. Airway remodeling and persistent airway obstruction in asthma. J. Allergy Clin. Immunol. 1999;104:509–516. doi: 10.1016/s0091-6749(99)70315-5. [DOI] [PubMed] [Google Scholar]
- 5.Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 1999;17:255–281. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- 6.Umetsu D.T., McIntire J.J., Akbari O., Macaubas C., DeKruyff R.H. Asthma: an epidemic of dysregulated immunity. Nat. Immunol. 2002;3:715–720. doi: 10.1038/ni0802-715. [DOI] [PubMed] [Google Scholar]
- 7.Schmitz J., Owyang A., Oldham E., Song Y., Murphy E., McClanahan T.K., Zurawski G., Moshrefi M., Qin J., Li X. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Smith D.E. IL-33: a tissue derived cytokine pathway involved in allergic inflammation and asthma. Clin. Exp. Allergy. 2010;40:200–208. doi: 10.1111/j.1365-2222.2009.03384.x. [DOI] [PubMed] [Google Scholar]
- 9.Lloyd C.M. IL-33 family members and asthma—bridging innate and adaptive immune responses. Curr. Opin. Immunol. 2010;22:800–806. doi: 10.1016/j.coi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oboki K., Ohno T., Kajiwara N., Arae K., Morita H., Ishii A., Nambu A., Abe T., Kiyonari H., Matsumoto K. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. USA. 2010;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mato N., Bando M., Yamasawa H., Hosono T., Mizushina Y., Sata M., Ohki G., Sugiyama Y. [Role of IL-33 in bronchial asthma] Nihon Kokyuki Gakkai Zasshi. 2010;48:419–425. [PubMed] [Google Scholar]
- 12.Préfontaine D., Nadigel J., Chouiali F., Audusseau S., Semlali A., Chakir J., Martin J.G., Hamid Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 13.Fabian M.R., Sonenberg N., Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 14.Foster P.S., Plank M., Collison A., Tay H.L., Kaiko G.E., Li J., Johnston S.L., Hansbro P.M., Kumar R.K., Yang M., Mattes J. The emerging role of microRNAs in regulating immune and inflammatory responses in the lung. Immunol. Rev. 2013;253:198–215. doi: 10.1111/imr.12058. [DOI] [PubMed] [Google Scholar]
- 15.Jiang X. The emerging role of microRNAs in asthma. Mol. Cell. Biochem. 2011;353:35–40. doi: 10.1007/s11010-011-0771-z. [DOI] [PubMed] [Google Scholar]
- 16.Ariel D., Upadhyay D. The role and regulation of microRNAs in asthma. Curr. Opin. Allergy Clin. Immunol. 2012;12:49–52. doi: 10.1097/ACI.0b013e32834ecb7f. [DOI] [PubMed] [Google Scholar]
- 17.Lu T.X., Rothenberg M.E. Diagnostic, functional, and therapeutic roles of microRNA in allergic diseases. J. Allergy Clin. Immunol. 2013;132:3–13. doi: 10.1016/j.jaci.2013.04.039. quiz 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiba Y., Tanabe M., Goto K., Sakai H., Misawa M. Down-regulation of miR-133a contributes to up-regulation of Rhoa in bronchial smooth muscle cells. Am. J. Respir. Crit. Care Med. 2009;180:713–719. doi: 10.1164/rccm.200903-0325OC. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto M., Singh A., Ruan J., Gauvreau G.M., O’Byrne P.M., Carlsten C.R., FitzGerald J.M., Boulet L.P., Tebbutt S.J. Decreased miR-192 expression in peripheral blood of asthmatic individuals undergoing an allergen inhalation challenge. BMC Genomics. 2012;13:655. doi: 10.1186/1471-2164-13-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu T.X., Munitz A., Rothenberg M.E. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J. Immunol. 2009;182:4994–5002. doi: 10.4049/jimmunol.0803560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar M., Ahmad T., Sharma A., Mabalirajan U., Kulshreshtha A., Agrawal A., Ghosh B. Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J. Allergy Clin. Immunol. 2011;128:1077–1085.e1–e10. doi: 10.1016/j.jaci.2011.04.034. [DOI] [PubMed] [Google Scholar]
- 22.Enesa K., Zakkar M., Chaudhury H., Luong L.A., Rawlinson L., Mason J.C., Haskard D.O., Dean J.L., Evans P.C. NF-κB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J. Biol. Chem. 2008;283:7036–7045. doi: 10.1074/jbc.M708690200. [DOI] [PubMed] [Google Scholar]
- 23.Löhning M., Stroehmann A., Coyle A.J., Grogan J.L., Lin S., Gutierrez-Ramos J.C., Levinson D., Radbruch A., Kamradt T. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc. Natl. Acad. Sci. USA. 1998;95:6930–6935. doi: 10.1073/pnas.95.12.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smithgall M.D., Comeau M.R., Yoon B.R., Kaufman D., Armitage R., Smith D.E. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008;20:1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 25.Komai-Koma M., Xu D., Li Y., McKenzie A.N., McInnes I.B., Liew F.Y. IL-33 is a chemoattractant for human Th2 cells. Eur. J. Immunol. 2007;37:2779–2786. doi: 10.1002/eji.200737547. [DOI] [PubMed] [Google Scholar]
- 26.Rank M.A., Kobayashi T., Kozaki H., Bartemes K.R., Squillace D.L., Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009;123:1047–1054. doi: 10.1016/j.jaci.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am. Rev. Respir. Dis. 1987;136:225–244. doi: 10.1164/ajrccm/136.1.225. [DOI] [PubMed] [Google Scholar]
- 28.Siddiqui S., Martin J.G. Structural aspects of airway remodeling in asthma. Curr. Allergy Asthma Rep. 2008;8:540–547. doi: 10.1007/s11882-008-0098-3. [DOI] [PubMed] [Google Scholar]
- 29.Myou S., Leff A.R., Myo S., Boetticher E., Tong J., Meliton A.Y., Liu J., Munoz N.M., Zhu X. Blockade of inflammation and airway hyperresponsiveness in immune-sensitized mice by dominant-negative phosphoinositide 3-kinase-TAT. J. Exp. Med. 2003;198:1573–1582. doi: 10.1084/jem.20030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.