

Abstract
Glycogen storage diseases (GSDs) of the liver are devastating disorders presenting with fasting hypoglycemia as well as hepatic glycogen and lipid accumulation, which could lead to long-term liver damage. Diet control is frequently utilized to manage the potentially dangerous hypoglycemia, but there is currently no effective pharmacological treatment for preventing hepatomegaly and concurrent liver metabolic abnormalities, which could lead to fibrosis, cirrhosis, and hepatocellular adenoma or carcinoma. In this study, we demonstrate that inhibition of glycogen synthesis using an RNAi approach to silence hepatic Gys2 expression effectively prevents glycogen synthesis, glycogen accumulation, hepatomegaly, fibrosis, and nodule development in a mouse model of GSD III. Mechanistically, reduction of accumulated abnormally structured glycogen prevents proliferation of hepatocytes and activation of myofibroblasts as well as infiltration of mononuclear cells. Additionally, we show that silencing Gys2 expression reduces hepatic steatosis in a mouse model of GSD type Ia, where we hypothesize that the reduction of glycogen also reduces the production of excess glucose-6-phosphate and its subsequent diversion to lipid synthesis. Our results support therapeutic silencing of GYS2 expression to prevent glycogen and lipid accumulation, which mediate initial signals that subsequently trigger cascades of long-term liver injury in GSDs.
Keywords: hepatomegaly, steatosis, fibrosis, cirrhosis, neoplasia
Graphical Abstract
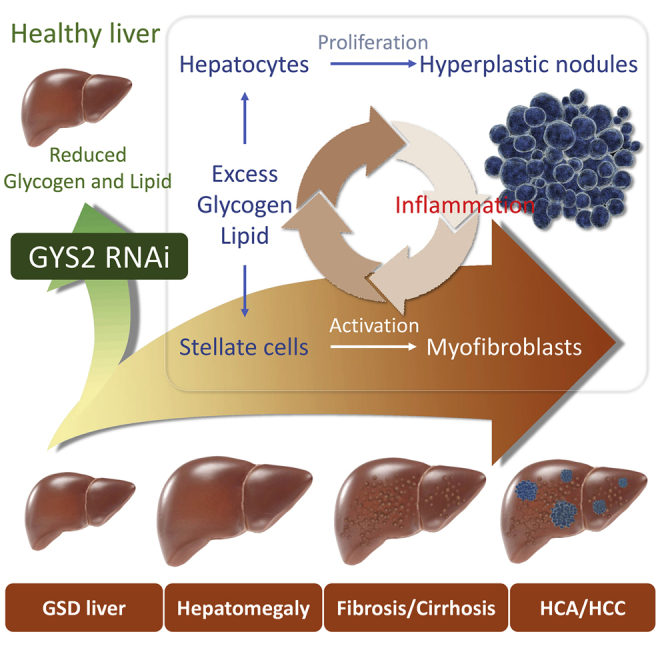
Pursell et al. demonstrate that abnormally structured glycogen and excess lipid accumulation trigger cascades of liver injury in glycogen storage diseases. Reduction of glycogen synthesis through an RNAi approach reverses liver injury, suggesting a potential therapeutic strategy to treat liver complications associated with hepatic glycogen storage diseases.
Introduction
The hepatic glycogen storage diseases (GSDs) are inherited disorders of glycogen metabolism that present clinically with hepatomegaly, liver injury, failure to thrive, and/or fasting hypoglycemia.1 These GSDs, including types 0, I, III, IV, VI, and IX, are caused by deficiencies in enzymes or transporters involved in glycogen synthesis or breakdown.1 In a healthy individual, excess postprandial glucose is converted via a multi-enzyme process into uridine diphosphate (UDP)-glucose, from which an initial glycogen polymer is formed on the glycogenin 2 (GYG2) dimer scaffold via α-1,4 glycosidic linkages using its autoglycosylation activity (Figure S1).2, 3 Glycogen synthase 2 (GYS2) continues to catalyze the addition of UDP-glucose onto existing glucose molecules to increase the length of linear glucose polymer chain. Subsequently, glycogen branching enzyme (GBE) is able to attach a new branch point with α-1,6 glycosidic linkage that serves as a new initial point for another cycle of linear glucose chain elongation. Working together, GYS2 and GBE build a complicated tree-like structured glycogen molecule with a GYG2 dimer in the center. During periods of fasting, glycogen phosphorylase (GLGP) starts to release glucose-1-phosphate from glycogen until a branching point is encountered. Glycogen debranching enzyme (GDE) activity then is required for transferring and liberating glucose molecules near the branching point before GLGP is able to further process glycogen and release glucose. Glucose-1-phosphate released from glycogen is then converted into glucose-6-phosphate (G6P) and subsequently to glucose by glucose 6-phosphatase (G6Pase) in the final step of glycogenolysis to maintain glucose homeostasis in times of fasting.4
GSD type Ia (GSD Ia) is caused by a deficiency of G6Pase and is characterized by the most severe hypoglycemia among all types of GSDs due to redundant role of G6Pase in converting G6P to glucose in the final step of both gluconeogenesis (GNG) and glycogenolysis pathways.5, 6, 7, 8, 9, 10 G6Pase is primarily expressed in the liver, kidneys, and intestine.10, 11 Liver and kidney G6Pase deficiency leads to the accumulation of G6P and subsequent metabolic complications, such as hypertriglyceridemia, hyperlactacidemia, and hyperuricemia.12, 13 The accumulation of glycogen and lipid in the liver leads to hepatomegaly and may contribute to the development of hepatocellular adenoma (HCA) or carcinoma (HCC) in some patients.14, 15, 16, 17, 18
The majority of the remaining hepatic GSDs, including types 0, III, VI, and IX, are associated with a milder, fasting, ketotic hypoglycemia due to an active GNG pathway in these patients capable of partially maintaining blood glucose homeostasis and compensating for the defective glycogenolysis pathway.1, 19 The increased activity of the GNG pathway from lipid and protein sources in these patients results in increased ketone body levels. GSD III is the most common among all ketotic GSDs.20 GSD III is caused by the deficiency of GDE (encoded by AGL) and is characterized by hepatomegaly and hyperlipidemia, with some patients developing liver damage, such as elevated circulating liver enzymes, fibrosis, cirrhosis, HCA, or HCC.20, 21, 22
To dissect the mechanisms underlying liver injury associated with hepatic GSDs, we investigated the effect of RNAi-mediated GYS2 enzyme reduction in GSD Ia and GSD III mouse models. We observed that accumulation of abnormally structured glycogen was accompanied by early damage signals, such as hepatocyte proliferation, myofibroblast activation, and mononuclear cell infiltration, leading to the subsequent development of fibrosis and nodule formation in GSD III mice. Silencing of Gys2 expression effectively reduced glycogen accumulation and prevented all assessed markers of early liver injury and progressive liver damage. Additionally, Gys2 reduction in a GSD Ia mouse model resulted in decreased glycogen and lipid levels and prevented subsequent apoptosis and cell proliferation.
Results
Gys2 mRNA Knockdown Inhibits Glycogen Synthesis in Wild-Type Mice
We identified Gys2 small interfering RNAs (siRNAs) by sequential screening in vitro (Figure S2). Potent siRNAs were then conjugated with N-acetylgalactosamine (GalNAc) sugar residues (designated as GYS2-1) or formulated in lipid nanoparticle (designated as GYS2-2) for specific reduction of Gys2 mRNA and GYS2 protein expression in liver (Figure S2). We then explored the therapeutic potential of Gys2 silencing in GSD mouse models (experimental designs summarized in Figure 1). GYS2-1 potently silences Gys2 mRNA (Figure 2A) and GYS2 protein levels (Figures 2B and 2C) in wild-type mice. Additionally, GYS2-1 and GYS2-2 treatment results in reduction of GYS2 protein and glycogen levels in wild-type mice in nearly all hepatocytes (Figures 2D, 2E, S2G, and S2H), suggesting that both GalNAc and lipid nanoparticle (LNP)-mediated siRNA delivery mechanisms are able to achieve homogeneous delivery of siRNA to hepatocytes and are suitable for further evaluation of the disease mechanism.
Figure 1.

Schematic Representation of the Experimental Designs for All Presented Studies
Wild-type (WT), GSD III (Agl−/−), and GSD Ia (L-G6pc−/−) mice were injected (downward open arrows) with either GYS2-1 or GYS2-2 with different dose levels, dosing regimens, and study durations (takedown denoted with upward filled arrows) as indicated. Age of mice is indicated within horizontal arrows. Results obtained from each experimental protocol are presented in their corresponding figures and supplemental figures as indicated.
Figure 2.

GYS2-1 Treatment Reduces Gys2 mRNA, GYS2 Protein Expression, Glycogen Synthesis, and Hepatomegaly in GSD III, Agl−/− Mice
The lead Gys2 siRNA conjugate, GYS2-1, was subcutaneously injected into WT and Agl−/− mice at a 10 mg/kg dose. Wild-type and Agl−/− mice were given 10 weekly doses of GYS2-1 starting at 4 weeks of age and sacrificed at 13 weeks of age after a 6-hr fast. In a second experiment, Agl−/− mice were given 18 weekly doses of GYS2-1 starting at 8 weeks of age and sacrificed at 26 weeks of age. Liver samples were collected 24 hr following the final dose. (A) Results of RT-PCR analysis on the effect of GYS2-1 on Gys2 mRNA expression in both wild-type (****p < 0.0001) and Agl−/− (**p ≤ 0.01) mice are shown. (B and C) Western blot analysis (B) and quantification (C) to measure GYS2 protein expression in both wild-type (p < 0.0001) and Agl−/− (**p ≤ 0.01) mice treated with GYS2-1 is shown. (D) Measurement of glycogen in prepared liver extracts in wild-type (p < 0.0001) and Agl−/− (**p ≤ 0.01 for 13 weeks and *p ≤ 0.05 for 26 weeks) mice treated with GYS2-1. (E) Detection of glycogen levels in formalin-fixed liver tissues in wild-type mice using PAS staining. (F) Liver to body weight ratio was measured in Agl−/− mice (***p ≤ 0.001 for 13 weeks and ****p < 0.0001 for 26 weeks) treated with GYS2-1. Data are presented as mean ± SD. Unpaired t test for statistical significance relative to PBS treatment group at each time point. (G) Representative images of livers from each group as indicated. The scale bar represents 1 cm. WT, wild-type littermate.
Gys2 mRNA Knockdown Prevents Glycogen Accumulation and Hepatomegaly in the Agl−/− GSD III Mouse Model
To evaluate whether accumulated glycogen molecules trigger liver abnormalities in GSDs, we employed a murine model of GSD III. Agl−/− mice have a germline mutation in the gene encoding GDE, resulting in accumulation of abnormally structured glycogen that is resistant to efficient breakdown by GLGP during fasting (Figure 2D).23, 24 As reported by others, we also observed that these mice develop hepatomegaly with abnormal histomorphology by four weeks of age (Figure S3),23 followed by an elevation of circulating liver enzymes and progression to periportal fibrosis by approximately eighteen weeks of age.23, 24 Agl−/− mice were injected with GYS2-1 weekly from four to thirteen weeks of age or from eight to twenty-six weeks of age (Figure 1). GYS2-1 potently suppressed hepatic Gys2 mRNA (Figure 2A) and GYS2 protein (Figures 2B and 2C) levels, resulting in a substantial reduction of hepatic glycogen levels in the Agl−/− mice (Figure 2D). Blockage of glycogen synthesis with chronic dosing of GYS2-1 reverses the hepatomegaly that is characteristic of Agl−/− mice (Figures 2F and 2G). Chronic dosing of GYS2-1 also reverses histomorphological characteristics of Agl−/− mice as evidenced by H&E and periodic acid-Schiff (PAS) staining (Figures 3A, S3A, and S3B) until they are similar to their wild-type littermates.
Figure 3.

GYS2-1 Treatment Results in a Normalization of Liver Pathology in an Agl−/− GSD III Mouse Model
(A) Agl−/− mice treated with PBS or GYS2-1 weekly for 18 doses starting at 8 weeks of age. Histological analysis was performed using H&E and PAS staining as indicated. Ki67-positive cells and infiltrated mononuclear cells (H&E:M) were noted in Agl−/− mice. Immunohistochemistry for α-smooth muscle actin (α-SMA) and Sirius Red staining were performed to detect stellate cell activation and fibrosis. Representative images of four individual animals from PBS- or GYS2-1-treated groups are shown. The scale bars represent 100 μm. (B) Double staining of Ki67 (dark brown) and CYP3A (cytochrome P450 3A, magenta) or α-SMA (magenta) was performed to detect cell proliferation in both hepatocytes and non-hepatocytes in livers of Agl−/− mice. (C) Quantitative analysis of immunohistochemistry of Ki67, α-SMA, and Sirius Red (*p ≤ 0.05) staining shown in (A) is shown. Detailed methodology of quantitative analysis can be found in Supplemental Information and Figures S4–S6. Data are presented as mean ± SD. Unpaired t test for statistical significance relative to PBS treatment group or wild-type animals (*p ≤ 0.05) is shown.
Interestingly, we observed that Gys2 mRNA and GYS2 protein levels were downregulated in Agl−/− mice in the absence of treatment when compared with their wild-type littermates (Figures 2A–2C). This observation suggests there may be physiological feedback in Agl−/− mice to mitigate the accumulation of excess glycogen by downregulation of Gys2 expression. However, despite this downregulation of Gys2 expression, Agl−/− mice still exhibit hepatomegaly and histomorphological characteristics of GSD III, suggesting that the remaining levels of GYS2 enzyme are sufficient to cause accumulation of glycogen in this GDE-deficient background. Importantly, GYS2-1 was able to further reduce the expression of Gys2 mRNA and GYS2 protein in Agl−/− mice, resulting in a reversal of the phenotypic characteristics of GSD III (Figures 1, 2, S3A, and S3B). Analysis of four-week-old Agl−/− mice sacrificed in the absence of treatment indicates that hepatomegaly and abnormal liver morphology occur early in life (Figure S3A),23 suggesting that the GSD III liver phenotype of Agl−/− mice is highly reversible with GYS2 reduction.
Silencing of Gys2 Protects against Initial and Progressive Liver Injury in the Agl−/− GSD III Mouse Model
As the life expectancy of patients with GSD III improves with diet control, cases of long-term liver complications continue to be reported.25, 26, 27 However, the mechanism remains unclear as to how liver damage in Agl-deficient patients leads to long-term complications, such as fibrosis, cirrhosis, HCA, and HCC. Immunohistochemistry of the livers of untreated Agl−/− mice for the cellular proliferation marker Ki67 shows increased clusters of positively stained cells surrounded by hepatocytes containing large vacuoles (Figures 3A, S3B, and S4). The increased Ki67-positive cells were detected on hepatocytes and non-hepatocytes, identified based on morphology and by immunohistochemistry for expression of cell-type-specific markers in the PBS-treated Agl−/− mice (Figure 3B). One hypothesis is that liver damage caused by glycogen accumulation induces neighboring cells (hepatocyte or non-hepatocyte) to proliferate, leading to myofibroblast activation in non-hepatocytes and the development of neoplasia in hepatocytes. Livers of Agl−/− mice did not have elevated activity of apoptotic caspases 3 and 7 (Figure S3C), supporting the hypothesis that slowly progressing liver damage, mediated by a non-apoptotic mechanism, induces cell proliferation in the GSD III mouse model. Notably, in Agl−/− mice injected with GYS2-1, cellular proliferation was reduced to the levels near wild-type littermates as evidenced by immunohistochemistry for Ki67 (Figures 3A, 3C, S3, and S4). We also observed that GYS2-1 prevents infiltration of mononuclear cells (Figure 3A). Importantly, we observed that Gys2 silencing greatly impeded the development of stellate cell activation and fibrosis in the livers of Agl−/− mice (Figures 3A–3C and S3–S6). These results suggest that reduction of GYS2 enzyme in the livers of Agl−/− mice can prevent hepatocyte damage from excess glycogen accumulation and the resulting cell proliferation, myofibroblast differentiation, inflammation, and development of fibrosis.
When compared to their wild-type littermates, Agl−/− mice also display signs of liver damage with elevated levels of circulating alanine aminotransferase (ALT), aspartate transaminase (AST), and alkaline phosphatase (ALP) that were exacerbated by fasting (Figures 4, 6, and S3D).23, 24 Gys2 mRNA silencing with GYS2-1 significantly reduces levels of ALT, AST, and ALP in Agl−/− mice to near wild-type baseline levels during both fasting and non-fasting conditions (Figures 4, 6, and S3D).
Figure 4.

GYS2-1 Treatment Reduces Circulating Liver Enzymes in an Agl−/− GSD III Mouse Model
GYS2-1 was subcutaneously injected into WT or Agl−/− mice weekly at a 10 mg/kg dose starting at 4 or 8 weeks of age and sacrificed at 13 or 26 weeks of age, respectively, as indicated. Analysis of serum samples was performed to measure circulating liver enzymes. ALT, alanine transaminase; AST, aspartate aminotransferase; ALP, alkaline phosphatase. Data are presented as mean ± SD. Unpaired t test for statistical significance relative to PBS treatment group. WKS, weeks. ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05.
Figure 6.

GYS2-1 Treatment Prevents Fibrosis in the Agl−/− GSD III Mouse Model
Agl−/− mice were treated with PBS or GYS2-1 monthly for 12 doses starting at 1 month of age. (A) Liver samples were collected 72 hr following the final dose and analyzed for collagen deposition using Sirius Red staining. Lower panel represents higher magnification of images in upper panel. Representative images of individual animals from PBS- or GYS2-1-treated groups are shown. The scale bars represent 1 mm or 200 μm in the upper or lower panel, respectively. (B) Analysis of Ki67 staining to detect hepatocellular proliferation in Agl−/− mice. Representative images of nodule and non-nodule areas of individual animals are shown. The scale bars represent 200 μm. (C) Quantitative analysis of immunohistochemistry of Ki67 and Sirius Red staining shown in (A) and (B), respectively, is shown. Detailed methodology of quantitative analysis can be found in Supplemental Information and Figures S4 and S8. (D) Analysis of serum samples was performed to measure circulating liver enzymes. Data are presented as mean ± SD.
Silencing of Gys2 Prevents Development of Hepatic Nodules in the Agl−/− GSD III Mouse Model
To investigate the long-term effect of inhibiting glycogen synthesis in Agl−/− mice, animals were injected with GYS2-1 monthly from 1 to 12 months of age (Figure 1). Repeatedly, silencing of Gys2 shows a tendency toward reducing glycogen synthesis and preventing hepatomegaly, liver toxicity, and fibrosis in Agl−/− mice (Figures 5, 6, S7, and S8). Importantly, small, sporadic nodules ranging from <1 to 3 mm in diameter were noted in the livers of Agl−/− mice, but not in GYS2-1-treated group. Histopathological analysis of the nodules identified hepatocytes with varying sizes, shapes, and irregular nuclei with a decrease in the number of vacuoles present (Figures 5 and S9). The largest nodule displays the “pushing border” characteristic indicative of an expanding cell mass that is adjacent to the parenchyma (Figure S9); however, no incidences of the development of hepatocellular carcinoma were observed by histopathology or immunohistochemistry analysis at one year of age (Figures S9 and S10). Increased cellular proliferation was detected in PBS-treated mice in both the parenchymal and nodule compartments at one year of age (Figure 6B), as it was in the younger mice (Figure 3). In contrast, silencing of Gys2 shows a tendency toward reduction of the proliferative index (Figure 6C). These results suggest that Gys2 silencing effectively prevents long-term liver injury and hyperplasia in the GSD III mouse model. Whereas small numbers of animals were included in this study, normalization of the liver phenotype with a high degree of homogeneous morphology throughout the entire liver was achieved with GYS2-1 treatment that was comparable to that of their wild-type littermates (Figures 5, 6, S7, and S8), suggesting that a GalNAc-mediated RNAi approach to prevent the accumulation of abnormally structured glycogen is effective in unbiasedly resolving the underlying disorder of all hepatocytes (Figure S11).
Figure 5.

GYS2-1 Treatment Prevents Nodule Development in the Agl−/− GSD III Mouse Model
Agl−/− mice were treated with PBS or GYS2-1 monthly for 12 doses starting at one month of age. Liver samples were collected 72 hr following the final dose. (A) Histological analysis was performed in prepared liver samples. The scale bar represents 100 μm. Each liver image shows a representative section from an individual animal. (B) Higher magnification images of sections of the nodules are shown. The scale bars represent 500 or 100 mm in the upper or lower panel, respectively.
Reduction of GYS2 Prevents Lipid Deposition in Hepatocytes in a GSD Ia Mouse Model
In GSD Ia, the loss of G6Pase activity leads to the accumulation of glycogen and lipid in hepatocytes. Here, we evaluated the effect of reducing Gys2 expression in a GSD Ia mouse model with liver-specific deletion of G6pc (L-G6pc−/−). The L-G6pc−/− mice develop liver injury representative of GSD Ia but with less severe hypoglycemia than patients.28 L-G6pc−/− mice were injected with the LNP-mediated GYS2-2 siRNA weekly for 5 weeks (Figure 1). Similar to our observations in Agl−/− mice, we demonstrate that Gys2 silencing greatly reduced hepatic Gys2 mRNA expression (Figure 7A), resulting in a reduction of hepatic glycogen accumulation (Figures 7B and 7E). In addition, L-G6pc−/− mice treated with GYS2-2 displayed similar liver morphology to wild-type controls whereas untreated L-G6pc−/− mice showed glycogen and lipid accumulation (Figure 7E). In this short-term treatment experiment, we observed a trend of decreased hepatic G6P accumulation with reduced Gys2 expression (Figure 7C), corresponding to the observed reduction in hepatic glycogen levels (Figure 7B), which serves as one of the main sources of hepatic G6P production from glycogenolysis. Reduction of elevated circulating ALT and AST was also observed in L-G6pc−/− mice with Gys2 silencing (Figure 8A). The activity of apoptotic caspases 3 and 7 and the proliferative index were also increased in L-G6pc−/− mice compared to wild-type controls (Figures 7D, 7E, and 8B), suggesting that lipid deposition and damage-induced cellular proliferation in the liver are linked to apoptosis in the L-G6pc−/− GSD Ia mouse model. A tendency toward decreased caspase 3 and caspase 7 activity (not statistically significantly; p = 0.24) was observed in livers of L-G6pc−/− mice after Gys2 silencing (Figure 8B). All L-G6pc−/− animals (both PBS and GYS2-2-treated groups) exhibit nearly undetectable G6pc mRNA expression levels (Figure S12) and severe fasting hypoglycemia (Figure S13), suggesting that the observed efficacy is specific to Gys2 silencing and not to random variation in recombination efficiency.28 Our data thus suggest that reduction of hepatic glycogen levels by Gys2 silencing results in the decline of hepatic G6P buildup and lipid deposition, thereby preventing damage-induced cell proliferation in the livers of L-G6pc−/− mice (Figures 7 and 8).
Figure 7.

Silencing of Gys2 mRNA Expression Reduces Glycogen Accumulation and Restores Normal Liver Morphology in a L-G6pc−/− GSD Ia Mouse Model
L-G6pc−/− mice were dosed intravenously with PBS or GYS2-2 (Gys2 siRNA formulated in lipid nanoparticle) weekly for 5 weeks. Liver samples were collected 48 hr following the final dose. (A) RT-PCR analysis was performed to measure Gys2 mRNA expression in L-G6pc−/− mice (****p < 0.0001). (B) Measurement of glycogen content in liver extracts was performed in L-G6pc−/− mice (**p ≤ 0.01). (C) Glucose-6-phosphate levels were measured in L-G6pc−/− mice treated with PBS or GYS2-2. (D) Quantitative analysis of Ki67-positive liver cells in L-G6pc−/− mice. Numbers (N) of male (M) and female (F) animals in each group are indicated. (E) Histological analysis to characterize histopathology of L-G6pc−/− mice using H&E, Oil Red O staining, and Ki67 immunohistochemistry. Liver samples stained with both Oil Red O and hematoxylin (Oil Red O + H) are indicated. Representative images of three individual animals from PBS- or GYS2-2-treated groups are shown. The scale bar represents 100 μm. Data are presented as mean ± SD. Unpaired t test for statistical significance relative to PBS control group.
Figure 8.

Silencing of Gys2 mRNA Expression Reduces Liver Toxicities in a L-G6pc−/− GSD Ia Mouse Model
L-G6pc−/− mice were dosed intravenously with PBS or GYS2-2 weekly for 5 weeks. Liver and blood samples were collected 48 hr following the final dose. (A) Analysis of serum samples was performed to measure circulating liver enzymes. (B) Measurement of caspase-3/-7 activity was performed on liver extracts to detect apoptosis. Numbers (N) of male (M) versus female (F) animals in each group are indicated. Unpaired t test for statistical significance relative to PBS control group or wild-type animal (****p < 0.0001; ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05). Data are presented as mean ± SD.
In summary, our results suggest that glycogen accumulation and lipid deposition are able to initiate liver damage cascades, resulting in increased cellular proliferation of hepatocytes and non-hepatocytes in both GSD Ia and GSD III mouse models. Suppression of Gys2 mRNA expression through an RNAi approach reduces glycogen and fatty acid accumulation homogenously and prevents subsequent liver damage of nearly all hepatocytes in mouse models of GSD Ia and GSD III.
Discussion
Short- and long-term liver complications occur in many types of GSDs, but there is currently no clear understanding of the mechanisms underlying disease progression. We demonstrate that accumulation of glycogen triggers the initial damage cascade, and this injury can be effectively reversed by Gys2 silencing in animal models of GSD Ia and III. However, Gys2 silencing has no effect on preventing fasting hypoglycemia (Figure S13). Whereas Gys2-deficient patients (GSD 0) have mild hypoglycemia and liver complications,29, 30, 31 hypoglycemia was not exacerbated by Gys2 silencing (Figure S13). Thus, targeting GYS2 could provide a potentially safe therapeutic approach for preventing liver complications, such as HCA and HCC in GSD Ia and III.
Mechanisms Underlying Progression of Liver Injury in Hepatic GSDs
Our data suggest that aberrant accumulation of glycogen initiates sequential events that are responsible for long-term liver complications. However, each hepatic GSD also exhibits unique phenotype. Indeed, GSD III hepatomegaly is characterized by the accumulation of abnormal glycogen (called phosphorylase limit dextrin), whereas glycogen molecule is normal in GSD I. Moreover, neutral lipids are accumulated in GSD Ia and lead to the development of steatosis. Whereas HCC development generally occurs on fibrotic or cirrhotic liver, HCC develops from an HCA transformation event in GSD Ia in the absence of fibrosis. In addition, hepatic diseases of GSD Ia and GSD III are characterized by different metabolic remodeling events that could play distinguished roles in liver tumorigenesis.13 For GSD III, increased cell proliferation was observed long before hyperplastic nodule formation. Similar to other fibrotic chronic liver diseases, we observed that proliferation and differentiation of stellate cells proceed the development of fibrosis. Our histology data suggest that inflammation may also play an important role for liver disease progression. In GSD Ia mice, our results suggest that reduction of glycogen content by Gys2 silencing led to a decrease of lipid accumulation, where modifications of metabolism subsequently prevent liver injury. Our understanding of mechanisms and development of liver injury of GSDs can further provide insight into potential intervention approaches, an area where the field has been progressing slowly, with the exception of advances in dietary control and gene therapy.32
GSD III patients develop fasting, ketogenic hypoglycemia as a result of fatty acid oxidation during gluconeogenesis as a means to compensate for the deficiency of glycogenolysis.19 Thus, we hypothesized that gluconeogenesis activity would be increased in GSD III mice during fasting in order to maintain blood glucose levels. As hypothesized, we detected an elevation of ketone bodies in Agl−/− GSD III mice during fasting (Figure S13C). We did not detect any significant difference in the elevation of ketone bodies following treatment of GYS2-1, suggesting Gys2 silencing has no effect on gluconeogenesis (Figure S13C). This result also suggests that abnormally structured glycogen, and not ketone bodies, causes liver toxicity and subsequent complications in GSD III. Interestingly, D,L-3-hydroxybutyrate, a synthetic ketone, has been used as an alternative energy source for treating a few GSD III patients with cardiomyopathy where no aggravated liver phenotype was reported, suggesting elevated ketone bodies will not cause liver complications in GSD III patients.33, 34
Silencing Hepatic GYS2 as a Potential Therapeutic Approach: Advantages and Limitations
We demonstrate that Gys2 silencing effectively and rapidly reversed the disease phenotype in GSD Ia and GSD III animal models, supporting its therapeutic potential for preventing liver complications. Accumulation of abnormally structured glycogen in hepatocytes directly or indirectly causes stress; however, reversal can be achieved rapidly by reducing glycogen synthesis. A similar but less specific approach has been reported with rapamycin, an mTOR inhibitor, which moderately reduced GYS2 expression, glycogen accumulation, and liver fibrosis in a canine model of GSD III with a range of other mTOR targets also affected.35 These results further support the role of accumulated glycogen in the disease pathology of GSD III and the therapeutic potential of GYS2 silencing. In addition to GSD Ia and III, liver complications are also associated with other types of GSDs, including types VI and IX.36, 37 Mechanistically, reduction of GYS2 expression with siRNA treatment should also reduce glycogen accumulation of GSD VI and IX. However, the effect of Gys2 silencing has yet to be confirmed in animal models of these GSDs.
RNAi-mediated inhibition of glycogen synthesis has the advantage to unvaryingly reach and correct glycogen accumulation in nearly all hepatocytes (Figures 5, 6, S2G, S2H, S8, and S11), subsequently eliminating abnormal cell proliferation throughout. This is particularly important for designing therapeutics aimed at preventing hepatic malignancies in livers where widespread and sustained cell proliferation has been observed (Figures 3, 6, and S4). Tumorigenesis can theoretically initiate from any small population of damaged hepatocytes that are actively proliferating, making the homogeneous silencing of Gys2 and the resulting prevention of hepatocyte damage particularly valuable for preventing disease progression.
As a potential therapeutic approach, GYS2 silencing may improve liver complications; however, it will not prevent hypoglycemia (Figure S13). Nevertheless, it has the potential to complement a gene therapy approach by ameliorating the cellular abnormalities associated with specific GSDs. In addition to liver complications, a high percentage of GSD III patients also eventually develop muscle weakness or heart complications.37, 38, 39 As expected, GYS2-1 treatment does not reduce glycogen accumulation in skeletal muscle due to the liver specificity of the GalNAc conjugation delivery approach (Figure S14). Theoretically, GYS1, which is predominantly expressed in both muscle and heart tissue, where it serves as the main enzyme for glycogen synthesis, can be targeted to improve myopathy and cardiomyopathy in affected patients. An alternative tissue-specific ligand-receptor approach other than the GalNAc-asialoglycoprotein receptor system utilized here will need to be identified for skeletal and cardiac muscle-specific delivery40 before future investigation of RNAi-mediated silencing of Gys1. Similarly, this current limitation of liver-specific delivery of RNAi will need to be resolved before we could prevent against kidney damage that is associated with high percentage of GSD I patients.41
In summary, our preclinical studies suggest that glycogen accumulation directly or indirectly induces a liver injury cascade in GSD animal models, supporting further exploration of applying RNAi to inhibit glycogen synthesis as a universal therapeutic approach for GSD-related liver disease.
Materials and Methods
Selection of Lead Gys2 siRNAs
GYS2-1 and GYS2-2 siRNAs were identified through a process of large-scale screening of siRNAs for mRNA knockdown activity in vitro in HEK293 cells stably expressing mouse Gys2 (Figure S2). RNA strands for siRNA duplexes were synthesized purified at Integrated DNA Technologies (Coralville, IA). The GYS2-1 siRNA had a 36-mer and 22-mer duplex RNA structure, with a 36-nt sense strand composed of modified RNA and GalNAc conjugation that was annealed to a 22-nt modified RNA antisense strand. The GYS2-2 had a 25-mer and 27-mer duplex RNA structure with a 25-nt modified sense strand annealed to a 27-nt modified RNA antisense strand. The GYS2-2 siRNA duplex was formulated in lipid nanoparticles. The siRNAs were modified with either 2′-OMe or 2′-F on their sugar moieties.
Generation of GSD Mouse Models and In Vivo Testing
All animal experiments complied with the animal protocols approved by Dicerna’s Institutional Animal Care and Use Committees (IACUCs). Mice were kept in a pathogen-free facility, housed using an Innovive disposable caging system with corn cob bedding (Innovive, San Diego, CA) with free access to Picolab diet for research animals by Purina (Scott Pharma, Marlborough, MA) and water unless otherwise noted. All experiments were done during the 12-hr light cycle. CD-1 mice (Charles River Laboratories, Wilmington, MA) were used for in vivo screening of Gys2 siRNA activity. C57BL/6 mice (Harlan Laboratories, Indianapolis, IN) were used for testing the efficacy of the lead GYS2-1 and GYS2-2 siRNA.
Agl−/− mice were produced from embryonic stem cells (ESCs) with a knockout of the Agl gene, which were acquired from the European Conditional Mouse Mutagenesis Program (EUCOMM, Germany). ESC injections and the generation of heterozygous Agl+/− mice were performed by geneOway (Lyon, France). Heterozygous Agl+/− mice were bred to homozygous for all studies. Wild-type and heterozygous littermates were used as controls.
L-G6pc−/− mice were generated and maintained as described previously.11, 28
Agl−/− and L-G6pc−/− animals were used to evaluate the efficacy of GYS2-1 and GYS2-2, respectively. GalNac-conjugated siRNA (GYS2-1) was injected subcutaneously, and LNP-formulated siRNA (GYS2-2) was administered intravenously. Blood samples were collected via tail vein (interim measurement) or cardiac puncture (terminal measurement) and used either as whole blood or processed to serum and plasma. Agl−/− mice were subjected to six hours of fasting prior to study termination except where fed state indicated. L-G6pc−/− mice were subjected to six hours of fasting during week four, after which blood samples were taken. At study termination, mice were euthanized according to IACUC guidelines and liver tissue samples were collected. Liver samples were stored in RNAlater (Thermo Fisher Scientific, Waltham, MA) for RNA preparation, fresh frozen in liquid nitrogen, or fixed in 10% neutral-buffered formalin (VWR International, Radnor, PA).
Analysis of Blood Chemistry Parameters
Interim blood glucose measurements were made on whole blood using a glucometer. Terminal blood collections were processed to serum and plasma for measurement of blood chemistry parameters. Serum chemistry and blood glucose levels were measured by IDEXX BioResearch Laboratories (Grafton, MA). Blood glucose measurements were confirmed using a glucose assay kit (Abcam, Cambridge, MA) according to the manufacturer’s instructions. ALT activity assay kit (Abcam, Cambridge, MA) was used for time course measurement of ALT according to the manufacturer’s instructions. For L-G6pc−/− mice, plasma glucose, cholesterol, and triglycerides were analyzed as described.11, 28
RNA Preparation and Real-Time PCR
Tissue samples were homogenized in QIAzol Lysis Reagent using TissueLyser II (QIAGEN, Valencia, CA). RNA was then purified using MagMAX Technology according to manufacturer instructions (Thermo Fisher Scientific, Waltham, MA). High-capacity cDNA reverse transcription kit (Thermo Fisher Scientific, Waltham, MA) was used to prepare cDNA. Mouse-specific Gys2 and Hprt1 primers (Integrated DNA Technology, Coralville, IA) were used for PCR on a CFX96 or CFX384 Real-Time PCR Detection System (Bio-Rad, Hercules, CA).
Western Blot
Tissue lysates were prepared using TissueLyser II (QIAGEN, Valencia, CA) with T-PER Tissue Protein Extraction Reagent and protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA). Total protein concentration was measured by BCA Protein Assay (Thermo Fisher Scientific, Waltham, MA), and equal protein concentrations were resolved by NuPAGE 4%–12% Bis-Tris SDS-PAGE (Thermo Fisher Scientific, Waltham, MA). Electrophoresed proteins were transferred to nitrocellulose membranes using the iBlot Dry Blotting System (Thermo Fisher Scientific, Waltham, MA) and blocked with Odyssey Blocking Buffer (Li-Cor Biosciences, Lincoln, NE). Membranes were then incubated with rabbit anti-glycogen synthase antibody (Cell Signaling Technology, Danvers, MA) and with mouse anti-glyceraldehyde 3-phosphate dehydrogenase antibody (Abcam, Cambridge, MA). Anti-rabbit IRDye 680 and anti-mouse IRDye 800 secondary antibodies (Li-Cor Biosciences, Lincoln, NE) were used for detection, and signal intensity was measured using the Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE).
Measurement of Liver Glycogen and Glucose-6-Phosphate Levels
Tissue lysates were prepared in water using TissueLyser II (QIAGEN, Valencia, CA). Tissue homogenate was split into two samples of equal volume. The first sample (Glycogen) was incubated at 95°C for 10 min and then centrifuged to isolate the supernatant. The supernatant was diluted in water and analyzed by a Glycogen Assay Kit (Abcam, Cambridge, MA). The second sample was immediately centrifuged to isolate the supernatant, which was used to measure total protein concentration by BCA Protein Assay (Thermo Fisher Scientific, Waltham, MA). Measured glycogen levels were normalized to total protein concentration.
Concentrations of G6P in L-G6pc−/− mouse livers were measured as described previously.28
Histological and Immunohistochemistry Analysis
Tissues were fixed in 10% neutral-buffered formalin overnight and then transferred to 70% ethanol. Embedding in paraffin, preparation of slides, and H&E staining was done at Mass Histology Service (Worcester, MA). PAS (Sigma-Aldrich, St. Louis, MO) and Sirius Red (Abcam, Cambridge, MA) staining were performed according to the manufacturer’s instructions. For immunohistochemistry experiments, paraffin sections were deparaffinized and rehydrated. Heat-mediated antigen retrieval (citrate buffer [pH 6.0]) was performed for Ki67 immunohistochemistry samples. Endogenous peroxidases and alkaline phosphatase were blocked with BLOXALL solution (Vector Laboratories, Burlingame, CA). Rabbit monoclonal anti-Ki67 antibody (1:100 dilution; Abcam, Cambridge, MA), rabbit monoclonal anti-alpha smooth muscle actin antibody (1:200 dilution; Abcam, Cambridge, MA), and mouse monoclonal anti-CYP3A antibody (1:50 dilution; Santa Cruz Biotechnology, Dallas, TX) were diluted in SignalStain Antibody Diluent (Cell Signaling Technology, Danvers, MA) and incubated overnight at 4°C. Binding of the primary antibody was detected using a goat anti-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP) antibody (Antibodies-online, Atlanta, GA) and SignalStain DAB Substrate Kit (Cell Signaling Technology, Danvers, MA). Oil Red O Lipid Stain was performed on frozen tissue samples according to the manufacturer’s instructions (Abcam, Cambridge, MA) in the presence or absence of hematoxylin. PAS staining was performed on formalin-fixed tissue samples according to the manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO). Results were visualized using an Olympus BX51, a Nikon Eclipse Ti microscope, and an Olympus BX61VS slide scanner using Image Pro Premier 9.1, NIS-Elements BR3.2, and Olympus VS-ASW image analysis software, respectively. Quantitative analysis of immunohistochemistry was performed using Olympus cellSens software. Briefly, for each tissue section, five regions of interest (ROIs) were selected, and an appropriate intensity threshold to specifically identify stained areas was set. Object counts and intensity were measured using an algorithm for each ROI. Importantly, the size of the ROI and the intensity threshold were kept constant for all tissue sections to be compared directly.
Assessment of Caspase Activity in Mouse Liver Tissue
Detection of caspase 3 and caspase 7 activity in mouse livers was performed essentially as described.42 100 μg/mL extract was mixed with an equal volume of Caspase-Glo Reagent (Promega, Madison, WI) and incubated at room temperature before reading on a SpectraMax M5 Plate Reader (Molecular Devices, Sunnyvale, CA).
Measurement of Ketone Body Levels
Ketone body levels were measured with Ketone Body Assay kit in terminal serum samples from wild-type and Agl−/− mice according to the manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO).
Author Contributions
Conceived and Designed Experiments, N.P., B.D.B., and C.L.; Performed the Experiments, N.P., J.G., W.Z., M.D., R.D., M.G., U.S., J.-S.Y., A.S., N.V., M.R., and G.M.; Analyzed the Data, N.P., J.G., F.R., and H.D.; Contributed Reagents/Material/Analysis Tools, R.S., B.K., and W.W.; Wrote the Manuscript, N.P., F.R., B.D.B., and C.L.
Conflicts of Interest
N.P., J.G., W.Z., R.D., M.D., U.S., J.-S.Y., N.V., R.S., B.K., A.S., W.W., M.A., H.D., B.D.B., and C.L. are or were employees of Dicerna Pharmaceuticals, which is developing siRNAs as therapeutics. M.G., M.R., G.M., and F.R. have received research grant support from Dicerna Pharmaceuticals.
Acknowledgments
We thank Doug Fambrough, David Miller, Jennifer Lockridge, and Jim Weissman for their review of this manuscript. We thank Geremew Desta for helping with animal care.
Footnotes
Supplemental Information includes fourteen figures and can be found with this article online at https://doi.org/10.1016/j.ymthe.2018.04.023.
Contributor Information
Bob D. Brown, Email: bbrown@dicerna.com.
Chengjung Lai, Email: clai@dicerna.com.
Supplemental Information
References
- 1.Wolfsdorf J.I., Weinstein D.A. Glycogen storage diseases. Rev. Endocr. Metab. Disord. 2003;4:95–102. doi: 10.1023/a:1021831621210. [DOI] [PubMed] [Google Scholar]
- 2.Akman H.O., Raghavan A., Craigen W.J. Animal models of glycogen storage disorders. Prog. Mol. Biol. Transl. Sci. 2011;100:369–388. doi: 10.1016/B978-0-12-384878-9.00009-1. [DOI] [PubMed] [Google Scholar]
- 3.Han H.S., Kang G., Kim J.S., Choi B.H., Koo S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016;48:e218. doi: 10.1038/emm.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adeva-Andany M.M., González-Lucán M., Donapetry-García C., Fernández-Fernández C., Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85–100. doi: 10.1016/j.bbacli.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou J.Y., Mansfield B.C. Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease. Hum. Mutat. 2008;29:921–930. doi: 10.1002/humu.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chou J.Y., Matern D., Mansfield B.C., Chen Y.T. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr. Mol. Med. 2002;2:121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- 7.Moses S.W. Historical highlights and unsolved problems in glycogen storage disease type 1. Eur. J. Pediatr. 2002;161(Suppl 1):S2–S9. doi: 10.1007/s00431-002-0997-6. [DOI] [PubMed] [Google Scholar]
- 8.Ozen H. Glycogen storage diseases: new perspectives. World J. Gastroenterol. 2007;13:2541–2553. doi: 10.3748/wjg.v13.i18.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kishnani P.S., Austin S.L., Abdenur J.E., Arn P., Bali D.S., Boney A., Chung W.K., Dagli A.I., Dale D., Koeberl D., American College of Medical Genetics and Genomics Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014;16:e1. doi: 10.1038/gim.2014.128. [DOI] [PubMed] [Google Scholar]
- 10.Soty M., Gautier-Stein A., Rajas F., Mithieux G. Gut-brain glucose signaling in energy homeostasis. Cell Metab. 2017;25:1231–1242. doi: 10.1016/j.cmet.2017.04.032. [DOI] [PubMed] [Google Scholar]
- 11.Mutel E., Gautier-Stein A., Abdul-Wahed A., Amigó-Correig M., Zitoun C., Stefanutti A., Houberdon I., Tourette J.A., Mithieux G., Rajas F. Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice: induction of renal and intestinal gluconeogenesis by glucagon. Diabetes. 2011;60:3121–3131. doi: 10.2337/db11-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sever S., Weinstein D.A., Wolfsdorf J.I., Gedik R., Schaefer E.J. Glycogen storage disease type Ia: linkage of glucose, glycogen, lactic acid, triglyceride, and uric acid metabolism. J. Clin. Lipidol. 2012;6:596–600. doi: 10.1016/j.jacl.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gjorgjieva M., Oosterveer M.H., Mithieux G., Rajas F. Mechanisms by which metabolic reprogramming in GSD1 liver generates a favorable tumorigenic environment. J. Inborn Errors Metab. Screen. 2016;4 2326409816679429. [Google Scholar]
- 14.Franco L.M., Krishnamurthy V., Bali D., Weinstein D.A., Arn P., Clary B., Boney A., Sullivan J., Frush D.P., Chen Y.T., Kishnani P.S. Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J. Inherit. Metab. Dis. 2005;28:153–162. doi: 10.1007/s10545-005-7500-2. [DOI] [PubMed] [Google Scholar]
- 15.Wang D.Q., Fiske L.M., Carreras C.T., Weinstein D.A. Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J. Pediatr. 2011;159:442–446. doi: 10.1016/j.jpeds.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iguchi T., Yamagata M., Sonoda T., Yanagita K., Fukahori T., Tsujita E., Aishima S., Oda Y., Maehara Y. Malignant transformation of hepatocellular adenoma with bone marrow metaplasia arising in glycogen storage disease type I: a case report. Mol. Clin. Oncol. 2016;5:599–603. doi: 10.3892/mco.2016.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassiman D., Libbrecht L., Verslype C., Meersseman W., Troisi R., Zucman-Rossi J., Van Vlierberghe H. An adult male patient with multiple adenomas and a hepatocellular carcinoma: mild glycogen storage disease type Ia. J. Hepatol. 2010;53:213–217. doi: 10.1016/j.jhep.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Calderaro J., Labrune P., Morcrette G., Rebouissou S., Franco D., Prévot S., Quaglia A., Bedossa P., Libbrecht L., Terracciano L. Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. J. Hepatol. 2013;58:350–357. doi: 10.1016/j.jhep.2012.09.030. [DOI] [PubMed] [Google Scholar]
- 19.Sentner C.P., Hoogeveen I.J., Weinstein D.A., Santer R., Murphy E., McKiernan P.J., Steuerwald U., Beauchamp N.J., Taybert J., Laforêt P. Glycogen storage disease type III: diagnosis, genotype, management, clinical course and outcome. J. Inherit. Metab. Dis. 2016;39:697–704. doi: 10.1007/s10545-016-9932-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kishnani P.S., Austin S.L., Arn P., Bali D.S., Boney A., Case L.E., Chung W.K., Desai D.M., El-Gharbawy A., Haller R., ACMG Glycogen storage disease type III diagnosis and management guidelines. Genet. Med. 2010;12:446–463. doi: 10.1097/GIM.0b013e3181e655b6. [DOI] [PubMed] [Google Scholar]
- 21.Shen J.J., Chen Y.T. Molecular characterization of glycogen storage disease type III. Curr. Mol. Med. 2002;2:167–175. doi: 10.2174/1566524024605752. [DOI] [PubMed] [Google Scholar]
- 22.Bernier A.V., Sentner C.P., Correia C.E., Theriaque D.W., Shuster J.J., Smit G.P., Weinstein D.A. Hyperlipidemia in glycogen storage disease type III: effect of age and metabolic control. J. Inherit. Metab. Dis. 2008;31:729–732. doi: 10.1007/s10545-008-0919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu K.M., Wu J.Y., Chen Y.T. Mouse model of glycogen storage disease type III. Mol. Genet. Metab. 2014;111:467–476. doi: 10.1016/j.ymgme.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Pagliarani S., Lucchiari S., Ulzi G., Violano R., Ripolone M., Bordoni A., Nizzardo M., Gatti S., Corti S., Moggio M. Glycogen storage disease type III: a novel Agl knockout mouse model. Biochim. Biophys. Acta. 2014;1842:2318–2328. doi: 10.1016/j.bbadis.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 25.Haagsma E.B., Smit G.P., Niezen-Koning K.E., Gouw A.S., Meerman L., Slooff M.J., The Liver Transplant Group Type IIIb glycogen storage disease associated with end-stage cirrhosis and hepatocellular carcinoma. Hepatology. 1997;25:537–540. doi: 10.1002/hep.510250307. [DOI] [PubMed] [Google Scholar]
- 26.Siciliano M., De Candia E., Ballarin S., Vecchio F.M., Servidei S., Annese R., Landolfi R., Rossi L. Hepatocellular carcinoma complicating liver cirrhosis in type IIIa glycogen storage disease. J. Clin. Gastroenterol. 2000;31:80–82. doi: 10.1097/00004836-200007000-00020. [DOI] [PubMed] [Google Scholar]
- 27.Demo E., Frush D., Gottfried M., Koepke J., Boney A., Bali D., Chen Y.T., Kishnani P.S. Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? J. Hepatol. 2007;46:492–498. doi: 10.1016/j.jhep.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mutel E., Abdul-Wahed A., Ramamonjisoa N., Stefanutti A., Houberdon I., Cavassila S., Pilleul F., Beuf O., Gautier-Stein A., Penhoat A. Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J. Hepatol. 2011;54:529–537. doi: 10.1016/j.jhep.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Bachrach B.E., Weinstein D.A., Orho-Melander M., Burgess A., Wolfsdorf J.I. Glycogen synthase deficiency (glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. J. Pediatr. 2002;140:781–783. doi: 10.1067/mpd.2002.124317. [DOI] [PubMed] [Google Scholar]
- 30.Nessa A., Kumaran A., Kirk R., Dalton A., Ismail D., Hussain K. Mutational analysis of the GYS2 gene in patients diagnosed with ketotic hypoglycaemia. J. Pediatr. Endocrinol. Metab. 2012;25:963–967. doi: 10.1515/jpem-2012-0165. [DOI] [PubMed] [Google Scholar]
- 31.Kasapkara C.S., Aycan Z., Açoğlu E., Senel S., Oguz M.M., Ceylaner S. The variable clinical phenotype of three patients with hepatic glycogen synthase deficiency. J. Pediatr. Endocrinol. Metab. 2017;30:459–462. doi: 10.1515/jpem-2016-0317. [DOI] [PubMed] [Google Scholar]
- 32.Koeberl D.D., Kishnani P.S., Bali D., Chen Y.T. Emerging therapies for glycogen storage disease type I. Trends Endocrinol. Metab. 2009;20:252–258. doi: 10.1016/j.tem.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharya K. Investigation and management of the hepatic glycogen storage diseases. Transl. Pediatr. 2015;4:240–248. doi: 10.3978/j.issn.2224-4336.2015.04.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valayannopoulos V., Bajolle F., Arnoux J.B., Dubois S., Sannier N., Baussan C., Petit F., Labrune P., Rabier D., Ottolenghi C. Successful treatment of severe cardiomyopathy in glycogen storage disease type III With D,L-3-hydroxybutyrate, ketogenic and high-protein diet. Pediatr. Res. 2011;70:638–641. doi: 10.1203/PDR.0b013e318232154f. [DOI] [PubMed] [Google Scholar]
- 35.Yi H., Brooks E.D., Thurberg B.L., Fyfe J.C., Kishnani P.S., Sun B. Correction of glycogen storage disease type III with rapamycin in a canine model. J. Mol. Med. (Berl.) 2014;92:641–650. doi: 10.1007/s00109-014-1127-4. [DOI] [PubMed] [Google Scholar]
- 36.Dagli A.I., Weinstein D.A. Glycogen storage disease type VI. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., Amemiya A., editors. GeneReviews. University of Washington, Seattle; 2009. https://www.ncbi.nlm.nih.gov/books/NBK5941/ [Google Scholar]
- 37.Roscher A., Patel J., Hewson S., Nagy L., Feigenbaum A., Kronick J., Raiman J., Schulze A., Siriwardena K., Mercimek-Mahmutoglu S. The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada. Mol. Genet. Metab. 2014;113:171–176. doi: 10.1016/j.ymgme.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 38.Austin S.L., Proia A.D., Spencer-Manzon M.J., Butany J., Wechsler S.B., Kishnani P.S. Cardiac pathology in glycogen storage disease type III. JIMD Rep. 2012;6:65–72. doi: 10.1007/8904_2011_118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mogahed E.A., Girgis M.Y., Sobhy R., Elhabashy H., Abdelaziz O.M., El-Karaksy H. Skeletal and cardiac muscle involvement in children with glycogen storage disease type III. Eur. J. Pediatr. 2015;174:1545–1548. doi: 10.1007/s00431-015-2546-0. [DOI] [PubMed] [Google Scholar]
- 40.Khan T., Weber H., DiMuzio J., Matter A., Dogdas B., Shah T., Thankappan A., Disa J., Jadhav V., Lubbers L. Silencing myostatin using cholesterol-conjugated siRNAs induces muscle growth. Mol. Ther. Nucleic Acids. 2016;5:e342. doi: 10.1038/mtna.2016.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y.T. Type I glycogen storage disease: kidney involvement, pathogenesis and its treatment. Pediatr. Nephrol. 1991;5:71–76. doi: 10.1007/BF00852851. [DOI] [PubMed] [Google Scholar]
- 42.Liu D., Li C., Chen Y., Burnett C., Liu X.Y., Downs S., Collins R.D., Hawiger J. Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J. Biol. Chem. 2004;279:48434–48442. doi: 10.1074/jbc.M407190200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.