

Abstract
Aptamers are single-stranded nucleic acid molecules that bind to and inhibit proteins and are commonly produced by systematic evolution of ligands by exponential enrichment (SELEX). Aptamers undergo extensive pharmacological revision, which alters affinity, specificity, and therapeutic half-life, tailoring each drug for a specific clinical need. The first therapeutic aptamer was described 25 years ago. Thus far, one aptamer has been approved for clinical use, and numerous others are in preclinical or clinical development. This review presents a short history of aptamers and SELEX, describes their pharmacological development and optimization, and reviews potential treatment of diseases including visual disorders, thrombosis, and cancer.
Keywords: aptamer, SELEX, antidote, therapeutics, pharmacology
INTRODUCTION
Aptamers are single-stranded deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) molecules that bind to protein targets by folding into a three-dimensional conformation, similar to antibodies. The word aptamer comes from the Latin aptus, meaning fit, and the Greek meros, meaning part or region. Aptamers are most often isolated by a method called systematic evolution of ligands by exponential enrichment (SELEX), first described 25 years ago. Since then, one aptamer has received approval from the US Food and Drug Administration (FDA) to treat macular degeneration of the eye, and numerous others are in preclinical or clinical trials. This review covers a brief history of aptamers and SELEX, describes postselection optimization providing pharmacokinetic flexibility, and includes antidote oligonucleotide reversal of aptamers to regulate aptamer activity rapidly. Finally, this review summarizes a list of therapeutic aptamer targets that are currently in preclinical or clinical studies and offers optimism that the future potential of aptamer therapeutics remains bright.
A BRIEF HISTORY OF APTAMERS
The idea that nucleic acids, whether DNA or RNA, could function as ligands and modulate the activity of target proteins was the result of research performed on viruses. Specifically, studies into the molecular biology of human immunodeficiency virus (HIV) and adenovirus demonstrated that these viruses encoded small, structured RNA that bind to endogenous proteins. These RNA ligands either facilitated viral replication or mitigated antiviral activity by the host (1, 2). A specific illustration is that HIV evolved a short, structured RNA sequence called trans-activation response or TAR. The TAR RNA binds to the cellular and viral proteins cyclin T1 and Tat, which are both involved in controlling gene expression and viral replication (1) (Figure 1). These findings suggested to virologists that nucleic acid molecules that bind proteins have the potential to become therapeutic drug agents.
Figure 1.
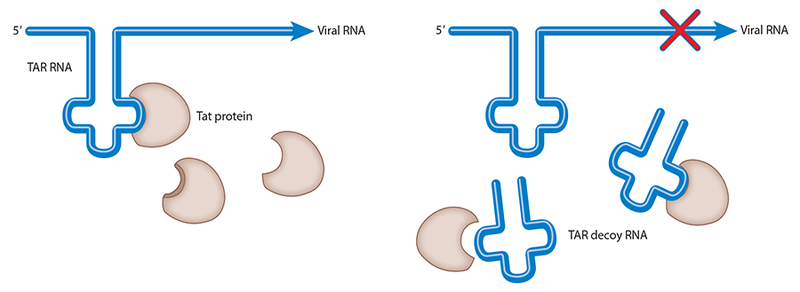
The TAR aptamer serves as a decoy, preventing the Tat protein from binding the endogenous TAR RNA, thereby inhibiting HIV gene expression and replication. Modified with permission from the Annual Review of Medicine. Abbreviations: HIV, human immunodeficiency virus; RNA, ribonucleic acid; TAR, trans-activation response.
In 1990, Sullenger et al. (3) published the first study demonstrating that an RNA aptamer designed to bind a viral protein prevented viral RNA-protein binding, thereby preventing viral replication. A TAR RNA-derived aptamer was expressed from a promoter to serve as a decoy, binding to Tat and cyclin T1 proteins in CD4+ T cells. Cells that expressed the TAR aptamer were made resistant to viral replication, demonstrating that RNA ligands represented potential therapeutic agents (3, 4).
Systematic Evolution of Ligands by Exponential Enrichment
Two groundbreaking studies were performed simultaneously and independently that demonstrated that nucleic acid ligands could be isolated against virtually any protein (5, 6). Tuerk & Gold (6) demonstrated that a large library of RNA molecules could be screened to find ligands that bound to T4 DNA polymerase with high affinity. Ellington & Szostak (5) demonstrated that a large library of RNA molecules could be screened, and they isolated RNA molecules that bound to organic dyes. Moreover, they stated that these RNA molecules folded into specific confirmations to bind to these dyes. They subsequently used this technique, known as SELEX, to demonstrate that this could also be performed using DNA ligands (7) (Figure 2). SELEX is an iterative process that starts with a large library of randomized nucleic acid sequences. In theory, the library contains two fixed ends to allow for reverse transcription PCR (RT-PCR) with a randomized region in the middle, typically up to 40 nucleotides. This yields a theoretical library of 440 different sequences. In practice, a portion of this large library, approximately 1015 different molecules, is then incubated with a target protein, and the nucleic acid molecules that bind to the protein are separated. To select RNA aptamers, the bound RNAs are amplified by RT-PCR and the resulting DNA templates transcribed subsequently. This new pool of nucleic acids is enriched for the target protein, and the process is repeated 8–12 times until an RNA pool with high affinity for the target protein is isolated. The pool is then sequenced and characterized to identify the aptamers with the highest affinity (Figure 2).
Figure 2.
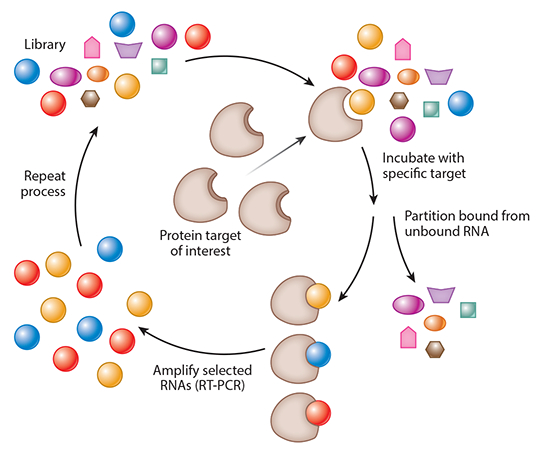
Systematic evolution of ligands by exponential enrichment (SELEX). Modified with permission from the Annual Review of Medicine.
The groundbreaking work performed by Sullenger et al. (3) demonstrated that nucleic acids that bind proteins could serve potentially as therapeutic agents. The simultaneous discovery of SELEX by Tuerk, Gold, Ellington, and Szostak enabled researchers to isolate aptamers to virtually any target (5, 6). The subsequent work on manipulating the structure and formulation of aptamers has expanded their pharmacological properties to make them a versatile class of compounds that can be tailored to specific clinical needs.
PHARMACOLOGICAL OPTIMIZATION OF APTAMERS
As with antibodies and peptide ligands, aptamers bind to their target by folding into a specific three-dimensional conformation that is dictated largely by the nucleic acid sequence. Similar to antibodies, nucleic acid aptamers bind to their targets with high affinity, demonstrating dissociation constants (Kd) from the low nanomolar to high picomolar range. In addition to high-affinity binding to their targets, aptamers also bind with high specificity, much like small molecules, discriminating between target proteins that share similar structural epitopes (8–11).
Aptamers can undergo chemical modification to modulate their pharmacokinetic profile. First, the internal structure of the molecule can be modified. The 2’-hydroxyl position on both purines and pyrimidines of RNA is a major target for nucleases. To protect against degradation, selections are often performed using an oligonucleotide library with a fluoro modification at the 2’-position of the pyrimidines (12, 13). To facilitate transcription of RNA aptamer pools, a mutant T7 RNA polymerase is used that demonstrates superior synthesis of modified oligonucleotides compared to conventional polymerase (14). With respect to the 2’-hydroxyl on purine nucleotides, performing postselection sequential replacement of hydroxyl groups with O-methyl residues can confer additional nuclease resistance (15). In addition to conferring nuclease resistance, modifications can also increase circulating half-life (t1/2). For example, carrier molecules such as polyethylene glycol (PEG) or cholesterol can be added to the ends of the aptamers and increase t1/2 to several hours (16).
Preclinical evaluation of drug compounds is an essential part of drug development. High- specificity binding of aptamers to their targets can result in poor or no cross-reactivity to homologs from other species. To solve this, a process called toggle SELEX was developed whereby the RNA pool is exposed to human protein target followed by reverse transcription and amplification. In the following round, the RNA pool is then exposed to the animal ortholog of the protein of interest. This iterative exposure, first to human then to preclinical species, continues sequentially until aptamers that bind to both the human and animal protein are identified (17) (Figure 3).
Figure 3.
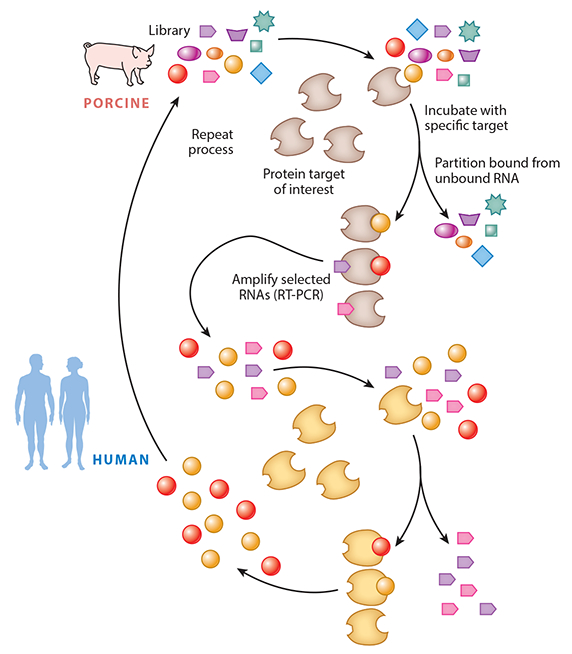
Toggle SELEX, whereby the pool is toggled between orthologs of the target protein to select an aptamer that binds to both targets. This facilitates preclinical evaluation of the aptamer. Modified with permission from the Annual Review of Medicine.
Clinical circumstances exist in which drugs need to be cleared from the system rapidly. The three main roots of clearance rely on the kidney, liver, or saturable mechanisms. This poses a significant problem in clinically ill patients with multisystem organ dysfunction. In order to avoid relying on the patient’s own clearance mechanisms, Rusconi et al. (10, 18) developed the concept of antidote oligonucleotides that bind to the aptamer by Watson-Crick base pairing, preventing it from inhibiting its target protein (Figure 4). In preclinical studies, the oligonucleotide antidote to coagulation factor IXa (FIXa) demonstrated complete reversal less than 5 min after administration (18, 19).
Figure 4.
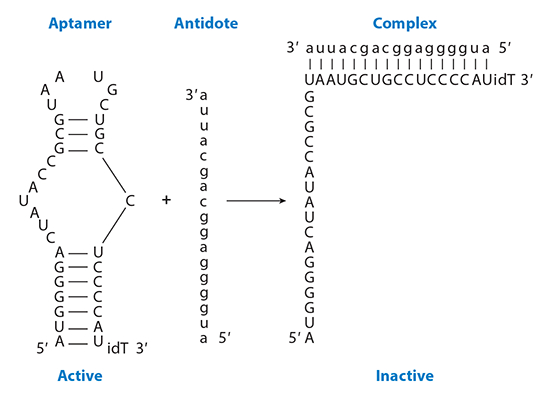
Antidote oligonucleotide control of aptamer activity. By binding to the aptamer using Watson-Crick base pairing, the aptamer cannot form its three-dimensional structure, thereby preventing it from binding to its target. Modified with permission from the Annual Review of Medicine.
In the course of selecting aptamers to a protein target, a single ligand can predominate early, possibly excluding other aptamers from binding to other sites that may be better diagnostic or therapeutic agents. To enrich the pool of binding oligonucleotides, researchers used complex protein mixes to enrich the initial SELEX pool, followed by a subsequent step of deconvolution SELEX to refine aptamers that bind to specific targets from the enriched pool (20). This concept was used to isolate therapeutic aptamers to both coagulation factors and von Willebrand factor (VWF) (21, 22). Complex SELEX is particularly attractive in oncology in which the aptamer chooses the targets. With this strategy, aptamer targets are not limited to specific proteins and might include specific posttranslational modifications, combinations of proteins, or even nonprotein targets that are unique to cancer cells. DNA aptamers were selected against several leukemia and solid tumor cell lines using in vitro selection against whole cells in culture (23–29). The concept was expanded further with in vivo selection against intrahepatic colorectal cancer in mice by administering the RNA library and subsequent enriched pools by intravenous injection. This resulted in isolation of an aptamer that bound to p68, an RNA helicase that is upregulated in colorectal cancer (30). A complete list of potential pharmacological advantages of aptamers is found in Table 1.
Table 1.
Properties of aptamers versus antibodies
| Aptamers | Antibodies |
|---|---|
| Binding affinity in low nanomolar to picomolar range | Binding affinity in low nanomolar to picomolar range |
| Entire selection is a chemical process carried out in vitro and can therefore target any protein | Selection requires a biological system, therefore difficult to raise antibodies to toxins (not tolerated by animal) or nonimmunogenic targets |
| Can select for ligands under a variety of conditions for in vitro diagnostics | Limited to physiological conditions for optimizing antibodies for diagnostics |
| Iterative rounds against known target limit screening processes | Screening monoclonal antibodies is time-consuming and expensive |
| Uniform activity regardless of batch synthesis | Activity of antibodies varies from batch to batch |
| Pharmacokinetic parameters can be changed on demand | Difficult to modify pharmacokinetic parameters |
| Investigator determines target site of protein | Immune system determines target site of protein |
| Wide variety of chemical modifications to molecule for diverse functions | Limited modifications to molecule |
| Return to original conformation after temperature insult | Temperature sensitive and undergo irreversible denaturation |
| Unlimited shelflife | Limited shelf life |
| No evidence of immunogenicity | Significant immunogenicity |
| Cross-reactive compounds can be isolated using toggle strategy to facilitate preclinical studies | No method for isolating cross-reactive compound |
| Aptamer-specific antidote can be developed to reverse the inhibitory activity of the drug | No rational method to reverse molecules |
Slow Off-Rate Modified Aptamers
Aptamers are negatively charged molecules, and as such, isolating an aptamer to a negatively charged protein or a protein that lacks a three-dimensional binding epitope owing to heavy glycosylation can be difficult (31). To circumvent these challenges, Gold et al. (32) developed slow off-rate modified aptamers (SOMAmers). The initial premise of SOMAmers was to add functional groups that mimic amino-acid side chains. SELEX libraries have a vast structural diversity based on 440 different sequences that result in a multitude of potential shapes. These amino acid-like side chains increase the chemical diversity of aptamers. By replacing the nucleotide at the 5’-position of uridine with hydrocarbons such as benzene or methyl groups, modified nucleic acid libraries were generated. When compared to nonmodified SELEX libraries, only the modified or SOMAmer libraries isolated high-affinity aptamers (32). The second pharmacological parameter that Gold et al. (32) sought to optimize was the off rate. To minimize nonspecific binding of SOMAmers to their target proteins, aptamers were isolated that had dissociation rates greater than 30 min (32).
Since 1990, research has focused on the flexibility of aptamers as drug agents, making them customizable, reversible, and capable of serving as both diagnostic and therapeutic agents of disease. Their potential has been fully realized in ophthalmology, for which the first aptamer was approved by the FDA to treat macular degeneration. Other promising therapeutic areas include thrombosis and cancer. We present a list of aptamers that have been developed with the potential capability for diagnosis and treatment of disease in cerebrovascular, cardiovascular, and peripheral vascular disease and oncology.
EYE DISORDERS
Vascular Endothelial Growth Factor
Neovascularization is a pathological process seen in age-related macular degeneration (AMD), retinopathy of prematurity, and diabetic retinopathy (33–35). Vascular endothelial growth factor (VEGF) is upregulated in all three diseases. Three RNA aptamers were isolated against VEGF165 with binding affinities (Kd) between 5 and 50 pM (36). In vivo experiments in adult guinea pigs demonstrated that the aptamer could inhibit VEGF-mediated capillary leakage by 58% (36). The aptamer was conjugated to a 40-kD PEG molecule at the 5’-end, and pharmacokinetic experiments were performed in rhesus monkeys. This optimized aptamer, renamed NX-1838, demonstrated a t1/2 of 9.3 h with peak concentrations of 4.9 μg/ml at 8–12 h (16). In a rat model of retinopathy of prematurity, NX-1838 reduced retinal neovascularization by 80% (37). Phase Ia studies demonstrated no significant risks with a single intravitreal administration of the aptamer. Furthermore, evaluation of patients with diabetic retinopathy 3 months after injection revealed no deterioration in vision and, in fact, a 27% improvement in vision of 3 lines or greater when examined by the Early Treatment of Diabetic Retinopathy Study (ETDRS) chart (37). The Phase II clinical trial was a multi-injection study with or without photodynamic therapy, which was the standard of care treatment at the time. Of those treated with NX-1838, now called pegaptanib, 87.5% had stable vision or improved vision and 25% demonstrated 3 lines or greater improvement by ETDRS. Of those patients who received both pegaptanib and photodynamic therapy, 60% had a 3 lines or greater visual improvement by ETDRS at 3 months (38).
Two concurrent, prospective, randomized, double-blinded, multicenter, dose-ranging Phase III controlled clinical trials were conducted comparing pegaptanib at three different doses in one eye versus sham injections in the other eye in patients with AMD. The aptamer was administered every 6 weeks for 48 weeks. The primary endpoint was the proportion of patients who lost fewer than 15 letters of visual acuity at 54 weeks. The study demonstrated that even at the lowest intravitreal injection dose of 0.3 mg, 70% of patients lost fewer than 15 letters of visual acuity, compared to 55% of the negative control group (P < 0.0001). The beneficial effect of pegaptanib was seen as early as 6 weeks after treatment compared to negative controls (39). Side effects of pegaptanib noted in the clinical trials included endophthalmitis, increased intraocular pressure, and rare cases of anaphylaxis and anaphylactoid reactions (40). These results led to pegaptanib being the first aptamer-based drug to receive approval from the FDA to treat macular degeneration in 2004. Since then, pegaptanib has been largely replaced by the antibody-based therapy ranibizumab that recognizes more isoforms of VEGF. However, it is important to recognize that pegaptanib paved the way for intraocular antiangiogenesis treatment and validated aptamer-based drug therapy.
More recently, two additional targets have been validated to treat AMD. Pegpleranib is a platelet-derived growth factor DNA aptamer that has been tested clinically with ranibizumab, an anti-VEGF antibody. A Phase I study demonstrated improved vision compared to ranibizumab alone (40). A Phase II dose escalation study of the two drugs showed similar results (40).
The second target for AMD therapy is complement component C5. Avacincaptad pegol is a modified RNA aptamer with a 40-kD PEG moiety on the 5’-end. An open-label Phase IIa study in patients with AMD demonstrated that avacincaptad pegol coadministered with ranibizumab demonstrated improved visual acuity compared to ranibizumab alone (40).
THROMBOSIS AND VASCULAR DISEASE
Developing antithrombotic aptamers takes advantage of targets that are extracellular and uses antidote oligonucleotides that reverse aptamer activity in the setting of hemorrhage. Several coagulation factors and a platelet protein have been targeted for aptamer selection and show promise to threat thrombosis.
Coagulation Factor IXa
In the classical cascade model of coagulation, FIXa becomes activated and, after binding to FVIIIa, ultimately leads to the prodigious amount of thrombin production necessary for thrombosis. To this end, a FIXa aptamer was selected using a 2’-fluoropyrimidine library that inhibited the activated partial thromboplastin time (aPTT) in a dose-dependent manner. An antidote oligonucleotide reversed the aptamer’s activity (10). Appending a cholesterol moiety to its 5’-end optimized the aptamer further. Now named Ch-9.3t, it was tested in vivo with antidote oligonucleotide 5–2C. The aptamer increased the aPTT reliably in a porcine model of anticoagulation. The antidote reversed that activity rapidly and sustainably. The aptamer also inhibited coagulation in a murine model of thrombosis, and antidote addition to aptamer-treated mice prevented hemorrhage (18). The FIXa aptamer/antidote pair was then compared to heparin and protamine in a porcine model of cardiopulmonary bypass. The results from this study demonstrated that the aptamer maintained patency of the circuit for 60 min, equal to the time for heparin. The antidote reversed the activity of the aptamer rapidly and sustainably. Finally, compared to heparin/protamine, animals treated with aptamer/antidote had a lower production of thrombin and lower inflammatory marker profile. Physiologically, the animals also demonstrated a lower change in cardiac output after antidote administration compared to protamine.
These latter studies led to the initial clinical trials of the aptamer/antidote pair (19). A Phase I study tested the aptamer, renamed RB006, and antidote, RB007, in healthy human subjects. Patients who received RB006 had a dose-dependent increase in aPTT. Patients who did not receive RB007 subsequently had sustained anticoagulation. Patients who received RB007 without RB006 had no physiological change and no quantifiable biological change. Those who received RB007 after RB006 administration had rapid reversal of aPTT (41). This study was then repeated in patients with coronary artery disease on antiplatelet therapy, with similar results (42). A Phase IIa trial studied RB006 and RB007 in patients undergoing percutaneous coronary intervention (PCI). Named REVERSAL-PCI, the study initially evaluated RB006 with a glycoprotein IIb/IIIa inhibitor as an additional antithrombotic agent, followed by aptamer administration alone. Antidote RB007 was administrated at a reduced dose to prevent in-stent thrombosis that could develop from complete reversal of RB007. Patients underwent PCI with RB006 alone successfully and tolerated partial and full reversal without any clinical evidence of in-stent thrombosis (43).
The RADAR trial was a partially blinded study that evaluated the aptamer/antidote pair in patients with acute coronary syndrome (ACS) and PCI. In addition to evaluating whether the aptamer could serve as a stand-alone anticoagulant in ACS and PCI, it also evaluated the percentage of antidote reversal of the aptamer that would prevent groin hemorrhages in the setting of immediate sheath removal. The aptamer provided sufficient anticoagulation in both clinical settings, and 50% antidote reversal was adequate to remove the sheath safely immediately after the procedure (44). These results paved the way for the Phase III trial, called REGULATE-PCI. It was a randomized, open-label design that compared aptamer to bivalirudin in PCI. The open- label design was to facilitate immediate sheath removal after reversal. The primary endpoint was a composite of ischemic events that included heart attack, stroke, in-stent thrombosis, and death up to 3 days after PCI. The trial ended before completion as 10 out of 1,600 patients had a serious allergic reaction within minutes of aptamer administration (45). In RADAR, 3 females also experienced a similar allergic reaction (44). Further analysis of these patients led to the identification of the PEG moiety, not the nucleic acid, as the causative agent of this reaction (46). A full study into the REGULATE-PCI patients is currently being performed. Despite the allergic reaction to PEG, these studies validated FIXa as a target in PCI. The aptamer prevented ischemia as well as bivalirudin, and there was a trend toward reduced thrombosis, which was not statistically powered as the trial was terminated at 25% percent of enrollment (45).
Thrombin
The conversion of prothrombin to thrombin is an essential step in thrombus formation. The first aptamer isolated to thrombin was a DNA aptamer (47). It bound to and inhibited exocite 1 on thrombin. Robust in vivo studies demonstrated anticoagulation in canine cardiopulmonary bypass as well as sheep and monkey hemofiltration (48, 49). The optimized version of the aptamer called NU172 is currently in human Phase IIa clinical trials (50).
von Willebrand Factor
Platelet adhesion occurs when platelet glycoprotein Ib/IX binds VWF that has been exposed on the vessel surface as a result of endothelial injury such as atherosclerotic disease. It is the first of two nonredundant pathways of platelet aggregation and an attractive target for aptamer selection. The first of two aptamers selected against VWF was ARC1772. This DNA aptamer bound to the gpIb domain of VWF, also known as the A-1 domain (51). A DNA/RNA hybrid aptamer with a 20-kD PEG moiety on the 5’-end was engineered subsequently and named ARC1779. ARC1779 showed promise in patients undergoing carotid endarterectomy by reducing thromboembolic events as measured by transcranial ultrasound but was also associated with hemorrhage. Furthermore, some patients had injection site reactions, echoing the concern of potential reactions to PEGylated aptamers (52).
More recently, an RNA aptamer was selected against VWF that bound to the A-1 domain and inhibited platelet aggregation in vitro and ex vivo (22). To mitigate the risk of hemorrhage, an antidote oligonucleotide was designed that reversed aptamer activity rapidly. A truncated version of this aptamer was tested in vivo in a murine model of carotid thrombosis and tail-bleeding model of hemorrhage. The aptamer prevented carotid occlusion for 60 min, the measured end of the experiment, compared to the negative control animals that occluded in approximately 10 min. In the tail-bleeding model, the aptamer-treated animals exhibited robust bleeding, approximately 4-fold above baseline, whereas mice treated with aptamer and antidote exhibited a bleeding profile similar to that of the negative control (53).
CANCER
Targeted therapy is a buzzword in cancer therapy, and numerous specific inhibitors, mostly small molecules and antibodies, are now in clinical use for various cancers. Aptamers are the ultimate in targeted therapy with respect to specificity. In the early years of aptamers, few aptamers were generated against cell surface proteins. Several aptamers that have shown promise as cancer therapeutics are discussed below.
Nucleolin
Ironically, the first aptamer to enter clinical trials for cancer therapy was not generated by SELEX but was discovered serendipitously based on its ability to be taken up by cancer cells (54). Its target was later determined to be nucleolin, a protein that is expressed in the nuclei of all cells but is overexpressed in the cytoplasm and on the plasma membrane of cancer cells relative to normal cells (55). At relatively high doses (micromolar), the 26-nucleotide G-rich DNA nucleolin aptamer (AS1411) has direct antiproliferative effects in vitro but is internalized efficiently even at nanomolar doses (56). Phase I studies in a variety of tumor types demonstrated that AS1411 was safe when delivered as a continuous infusion at doses up to 40 mg/kg/day. A subsequent Phase II study in renal cell carcinoma demonstrated a similarly low incidence of adverse events attributable to AS1411 (57). Only one patient had an adverse response, but it was dramatic and sustained. Dose- limiting toxicity was not reached in the AS1411 Phase I trials, presumably because drug costs limited escalation; therefore, higher response rates may have been achieved with higher doses.
The nucleolin aptamer is internalized and shuttled to the nucleus at a much lower concentration than that required for direct effects. Furthermore, owing its small size and ease of synthesis, it has received attention as a delivery agent for a variety of interesting types of cargo. AS1411 was conjugated to cobalt-ferrite nanoparticles for magnetic resonance imaging and concurrently labeled with 67Ga-citrate for radionuclide imaging (58). Studies in rat glioma xenografts demonstrated that the conjugates cleared the bloodstream rapidly and targeted tumor cells specifically and could therefore be used for imaging and therapy in vivo. Human serum albumin nanoparticles loaded with paclitaxel were delivered with the nucleolin aptamer and shown to inhibit growth of glioma xenografts in mice relative to nontargeted nanoparticles (59). AS1411 conjugated directly to doxorubicin (Dox) was delivered intravenously in mice that had hepatocellular cancer xenografts, resulting in inhibition of tumor growth (60). Other groups have functionalized different drug carriers with AS1411 and demonstrated targeted delivery of chemotherapeutic agents in vitro (61, 62). AS1411 was complexed noncovalently with multiple molecules of the porphyrin derivative TMPyP4 and used to deliver the molecules into breast cancer cells in vitro for photodynamic therapy (63). As a different type of photodynamic therapy, photosensitive gold nanorods were decorated with AS1411 and used to release Dox into cancer cells when stimulated with near- infrared light (64). Finally, as proof of concept that the nucleolin aptamer could actually deliver cargo to the nucleus, our group used AS1411 to deliver splice-switching oligonucleotides to cancer cells and mediate RNA splicing of a luciferase reporter construct in vitro (65).
Prostate-Specific Membrane Antigen
A desirable feature of a cancer target is specificity, and one of the best examples is prostate-specific membrane antigen (PSMA), a surface protein expressed in healthy prostate, prostate cancer, and the vasculature of a variety of solid tumors. PSMA has been studied extensively as a tumor marker as well as a target for imaging and therapy. Two nuclease-resistant RNA aptamers (A9 and A10) against PSMA were identified and have now been studied extensively in preclinical models (66). PSMA, also known as folate hydrolase, is typically considered a prostate cancer marker, but a recent study demonstrated that an optimized, truncated version of the A9 aptamer inhibited prostate cancer invasion and migration in vitro (67). When delivered daily as an intravenous injection, the aptamer inhibited the development of bone metastases in a mouse model in which PSMA- expressing cells were delivered by intracardiac injection. Most studies using the PSMA aptamer, however, have exploited the fact that PSMA is internalized. The PSMA aptamer has been conjugated directly to the peptide toxin gelonin (68) and to Dox, which is known to intercalate into DNA (69) for delivery and targeted killing of PSMA-expressing cells. To increase the amount of payload that can be delivered, PSMA aptamers have been conjugated to various nanomaterials, including quantum dots (70), iron oxide nanoparticles (71), and polymeric (PLGA-block-PEG) nanoparticles (72). The polymeric nanoparticles were loaded with the chemotherapeutic drug docetaxel, and the aptamer-targeted nanoparticles were shown to eradicate prostate cancer xenografts after a single intratumoral dose. This study suggests that the aptamer promotes intracellular delivery of the nanoparticles, but this study—like many other first-in-animal aptamer studies—is limited in that data for systemic delivery were not provided.
As one of the first aptamers used for drug delivery, the PSMA aptamer was also used for intracellular delivery of small interfering RNAs (siRNAs). siRNAs against a critical gene for cell survival, polo-like kinase-1 (Plk1), were appended to the 3’ end of a PSMA aptamer to target PSMA-expressing cells. In vitro experiments demonstrated specific silencing of Plk1 mRNA and induction of apoptosis. We observed that intratumoral injections of the PSMA aptamer-Plk1 siRNA chimera into prostate cancer xenografts resulted in tumor regression (73). This chimera was optimized and modified with PEG to enhance plasma stability. Systemic delivery of this construct induced tumor regression in prostate cancer xenografts (74). The PSMA aptamer was also used to deliver sRNA targeting nonsense-mediated messenger RNA decay to enhance antigen expression and presentation in prostate cancer cells and inhibit tumor growth in immunocompetent mice (75).
Human Epidermal Growth Factor Receptor 2
Another well-known cancer target is the EGF receptor family member human epidermal growth factor receptor 2 [HER-2 (erbB2)], which is amplified and overexpressed in a significant proportion of breast cancers as well as other epithelial tumors. HER-2 monoclonal antibodies, such as trastuzumab, are widely used clinically. The better characterized family member EGFR is overexpressed in many solid tumors and has also been targeted by monoclonal antibodies, such as cetuximab. As prototypes for receptor-mediated endocytosis, both HER-2 and EGFR have also been exploited for antibody-drug conjugates. A DNA aptamer generated to bind HER-2 inhibits growth of gastric cancer cells in vitro, and intraperitoneal delivery in mice of a trimeric version of the same aptamer (but not the monomer) inhibited growth of the same cells as xenografts (76). This improved effect is at least partly due to increased avidity (77) but may also be partly due to the increased t1/2 of the trimer compared to the monomer. Cell-based SELEX was used to identify an RNA aptamer that binds and internalizes into HER-2-expressing cells (78). Using a similar approach to that used for the PSMA aptamer, the aptamer was linked to siRNA against the antiapoptotic gene Bcl-2 and demonstrated that treatment of HER-2-overexpressing breast cancer cells in vitro improved sensitivity to chemotherapy. Although animal studies have not yet been reported, a natural RNA aptamer was generated against EGFR that was able to deliver gold nanoparticles into EGFR-expressing cancer cells (79). Subsequently, nuclease-resistant EGFR aptamer (E07) was internalized by and able to inhibit growth of EGFR-expressing epidermoid carcinoma cells in vitro (80). The EGFR aptamer was also used to deliver oligonucleotides incorporating gemcitabine, a nucleoside analog and chemotherapeutic drug, targeted for in vitro killing of pancreatic cancer cells (81).
AXL
Although not as well known as the EGF receptor family, the AXL tyrosine kinase receptor was identified as a transforming gene in myeloid leukemia and is overexpressed in several solid tumor types. A nuclease-resistant aptamer against AXL was isolated using cell-based SELEX that inhibited AXL signaling in vitro as well as growth of lung cancer xenografts in mice when injected directly into the tumor (82). An all-nucleic acid conjugate of the AXL aptamer was developed subsequently with let-7g, a well-characterized antitumor microRNA (83). In vitro, the conjugate not only inhibited AXL signaling but also decreased expression of genes regulated by let-7g. Administration of the conjugate to mice by tail vein injection increased let-7g levels and decreased growth in AXL-expressing xenografts.
MUC1
Aberrantly glycosylated forms of the mucin glycoprotein family are an important class of tumor surface markers, and MUC1 in particular is overexpressed on several epithelial cancer cells but not normal cells (84, 85). DNA aptamers have been developed that bind MUC1 glycoforms, one of which was conjugated to the photodynamic therapy agent chlorin e6. The aptamer delivered the agent to a variety of MUC1 + cancer cell lines, including breast, colon, lung, ovarian, and pancreatic cancers, which were killed by light activation in vitro (86). MUC1 has also been labeled with 99mTc for molecular imaging (87–89). When imaged by autoradiography, the MUC1 aptamers penetrated xenograft tissue quickly and uniformly but had relatively short t1/2 values, which will need to be improved if this approach is used for radionuclide therapy.
Carcinoembryonic Antigen
Carcinoembryonic antigen (CEA) is a tumor-associated antigen and one of the best-known markers for tumors of the gastrointestinal tract. Clinically, circulating levels of CEA are used for assessing prognosis and monitoring response to therapy. CEA is a glycosyl phosphatidyl inositol- linked cell surface protein, and radiolabeled CEA antibodies have been studied in animals and humans in molecular imaging and radionuclide therapy. A CEA-specific DNA aptamer has been shown to bind to CEA-expressing cancer cell lines but not CEA-negative cell lines in vitro (90, 91). A nuclease-resistant RNA aptamer against CEA was shown to inhibit invasion and migration of colorectal cancer cells in vitro (92). Aversion of this aptamer, modified with PEG, was able to inhibit liver metastasis in a mouse model in which the cells were incubated ex vivo with the CEA aptamer prior to injection into the spleen.
Protein Tyrosine Kinase 7
Using complex SELEX strategies, DNA aptamers have been isolated against several leukemia and solid tumor cell lines using in vitro selection against whole cells in culture (23–29). The aptamer’s specific protein target was identified in some but not all cases. One of the most promising aptamers was generated against a T cell acute leukemia cell line, and the target was determined to be protein tyrosine kinase 7 (PTK7), a cell surface receptor. The PTK7 aptamer was conjugated to Dox using an acid-labile hydrazone linkage (93). The aptamer-Dox conjugate bound and was internalized by leukemia cells but not normal cells. Dox is membrane permeable and is taken up by cells through passive diffusion. However, conjugating Dox with the aptamer restricted entry to cells that express PTK7, creating a sieve mechanism that should curb nonspecific uptake and minimize the toxic effects of Dox on normal cells. Researchers also functionalized gold nanoparticles with the PTK7 aptamer for delivery of additional Dox (94) and the photodynamic therapy agent chlorin e6 (95). This aptamer and others identified by cell-based SELEX have been used for in vitro delivery in a variety of creative ways, but unfortunately none have made significant progress in preclinical studies.
p68
An in vivo selection technique to identify an RNA aptamer that targets mouse liver tumors was performed by injection of the murine colon carcinoma cell line CT26 (30). The target was determined by affinity purification to be p68, an RNA helicase that is overexpressed in colon cancer and believed to shuttle between the cell surface and nucleus (96, 97). The aptamer was fluorescently labeled and injected intravenously into tumor-bearing mice. After the tumor was harvested, fluorescence microscopy visualized the aptamer in the nucleus and cytoplasm of tumor cells but not in normal tissue. This was the first report of this method, and the aptamer has now been used for delivery of therapeutic cargo. The study suggests that in vivo selection has the potential to identify novel cancer targets and target reagents simultaneously to specific cells.
Cytotoxic T Cell Antigen-4
Aptamers have potential roles in immunotherapy as well. Our group developed a nuclease-resistant RNA aptamer against cytotoxic T cell antigen-4 (CTLA-4), a cell surface receptor that downreg- ulates the immune response to tumors. Two aptamers that inhibited CTLA-4 function in vitro were truncated for large-scale synthesis. The ability of the CTLA-4-binding aptamers to enhance the potency of an antitumor vaccine was tested in an immunocompetent mouse melanoma allograft model (98). Aptamers were injected intraperitoneally at the time of tumor implantation and inhibited tumor growth to an extent comparable to that seen in mice treated with anti-CTLA-4 antibody but required a larger dose of aptamer. Potency was improved significantly with a tetramer of the same aptamer, presumably due to improved avidity as well as bioavailability.
T Cell Receptors OX40 and 4–1BB
Aptamers have been selected against 0X40 (99, 100) and 4–1BB (75), both of which are costimulatory receptors that function to promote T cell responses. When assembled as bivalent structures (using an all-nucleic acid scaffold), these aptamers were shown to have agonistic activity on their receptors, stimulating T cell activation in vitro. The bivalent aptamer against murine 4–1BB aptamer mediated tumor rejection when injected intratumorally in a mouse allograft model (75). The bivalent aptamer against murine 0X40 was delivered systemically to enhance the response of a dendritic cell-based vaccine in mice (99). No significant toxicities, such as autoimmunity, were observed in any of these in vivo studies. However, one of the challenges of translating aptamers is that aptamers against specific proteins from one species may not cross-react with homologs from another species, as in this case. As mentioned above, toggle SELEX can circumvent this potential challenge. With respect to 0X40, a new bivalent aptamer against human 0X40 had to be developed, which has been confirmed to activate human T cells ex vivo but has not yet been studied in human patients (100).
Angiopoietin-2
Inhibition of angiogenesis by anti-VEGF agents, such as bevacizumab, has been shown to improve response rates and survival in many types of cancer. However, the process of tumor angiogenesis is regulated by many factors, and inhibition of VEGF alone may not be enough. Tie2 is an endothelial transmembrane receptor tyrosine kinase that is expressed in vascular endothelium and is critical for embryonic blood vessel formation. Tie2 is activated by angiopoietin-1 (Ang1) (101). Angiopoietin-2 (Ang2) is a naturally occurring antagonist of Tie2 and is expressed at sites of active angiogenesis, such as tumors, and believed to potentiate the actions of other growth factors by destabilizing the vasculature (102). An aptamer was selected against Ang2 that binds with high affinity yet high specificity, as it does not bind Ang1. In vivo activity of the aptamer demonstrated inhibition of basic fibroblast growth factor-mediated neovascularization in a rat cornea model (103). A truncated version of the aptamer was conjugated to PEG and given systemically by continuous infusion via an osmotic pump in a murine colon carcinoma model. The aptamer decreased intratumoral vascular density as well as tumor growth (104). An Fc-peptide fusion protein (AMG 386) that inhibits both Ang1 and Ang2 has also shown safety and activity in Phase I trials (105). However, there are theoretical advantages to selective inhibition of Ang2 alone.
THE FUTURE OF APTAMER THERAPEUTICS
The unique characteristics of nucleic acid aptamers that represent a combination of the best features of small molecules and antibodies, including high binding affinity and specificity, provide vast potential for future applications. Many in the field believe that the next generation of aptamers will be particularly useful for diagnostic purposes, including molecular probes, biosensors, aptamer-based magnetic cell sorting, and immunoassays. There is a similar level of enthusiasm for their therapeutic potential in several areas, including but not limited to infectious disease and its complications, oncology, inflammatory disorders and conditions, and vascular disease. In addition to chemical manipulations that allow for heightened bioavailability and, when needed, antidote control, the emergence of varying formulations provides diversity for numerous therapeutic applications, including inhibition of coagulation proteases, antifibrinolytic proteins, proinflammatory proteins, and proangiogenesis factors that facilitate proinflammatory and prothrombotic environments at the vascular and tissue levels. The field of aptamer-based drug delivery to primary and metastatic tumor sites; infectious materials and localized safe havens, as encountered in tuberculosis and HIV; and diseased organs, including the brain with slow virus encephalopathy, as well as the modulation of systemic disease through delivery of miRNAs, is also emerging rapidly.
Finally, aptamer technology could be used as a platform for the development of a new generation of reversal agents for new oral anticoagulant drugs. Their target specificity and reversibility would allow an anticoagulant or platelet antagonist to be turned off for a brief period of time, followed by antidote reversal of the inhibitory effect and restoration of anticoagulation as the underlying condition and clinical circumstances dictate.
DISCLOSURE STATEMENT
S.M.N. is a coinventor of and holds patents describing aptamers as reversible antithrombotic agents. R.R.W. is a coinventor of and holds a patent on the use of aptamers as anticoagulants and antiangiogenesis agents. B.A.S. is a coinventor of and holds intellectual property on using aptamers as therapeutic agents.
ACKNOWLEDGMENTS
The authors would like to thank Debra G. Wheeler for her help in preparing the manuscript. S.M.N. is funded by NIH 5K12NS080223–3. R.C.B. and B.A.S. are funded by NIH 5U54HL112307–03.
LITERATURE CITED
- 1.Cullen BR, Greene WC. 1989. Regulatory pathways governing HIV-1 replication. Cell 58:423–26 [DOI] [PubMed] [Google Scholar]
- 2.Marciniak RA, Garcia-Blanco MA, Sharp PA. 1990. Identification and characterization of a HeL anuclear protein that specifically binds to the trans-activation-response (TAR) element of human immunodeficiency virus. PNAS 87:3624–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullenger BA, Gallardo HF, Ungers GE, Gilboa E. 1990. Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell 63:601–8 [DOI] [PubMed] [Google Scholar]
- 4.Sullenger BA, Gallardo HF, Ungers GE, Gilboa E. 1991. Analysis of trans-acting response decoy RNA- mediated inhibition of human immunodeficiency virus type 1 transactivation. J. Virol . 65:6811–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellington AD, Szostak JW. 1990. In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–22 [DOI] [PubMed] [Google Scholar]
- 6.Tuerk C, Gold L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–10 [DOI] [PubMed] [Google Scholar]
- 7.Ellington AD, Szostak JW. 1992. Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 355:850–52 [DOI] [PubMed] [Google Scholar]
- 8.Doudna JA, Cech TR, Sullenger BA. 1995. Selection of an RNA molecule that mimics a major autoanti- genic epitope of human insulin receptor. PNAS 92:2355–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SW, Sullenger BA. 1997. Isolation of a nuclease-resistant decoy RNA that can protect human acetylcholine receptors from myasthenic antibodies. Nat. Biotechnol 15:41–45 [DOI] [PubMed] [Google Scholar]
- 10.Rusconi CP, Scardino E, Layzer J, Pitoc GA, Ortel TL, et al. 2002. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 419:90–94 [DOI] [PubMed] [Google Scholar]
- 11.Rusconi CP, Yeh A, Lyerly HK, Lawson JH, Sullenger BA. 2000. Blocking the initiation of coagulation by RNA aptamers to factor VIIa. Thromb. Haemost 84:841–48 [PubMed] [Google Scholar]
- 12.Jellinek D, Green LS, Bell C, Lynott CK, Gill N, et al. 1995. Potent 2/-amino-2/-deoxypyrimidine RNA inhibitors of basic fibroblast growth factor. Biochemistry 34:11363–72 [DOI] [PubMed] [Google Scholar]
- 13.Willis MC, Collins BD, Zhang T, Green LS, Sebesta DP, et al. 1998. Liposome-anchored vascular endothelial growth factor aptamers. Bioconjug. Chem 9:573–82 [DOI] [PubMed] [Google Scholar]
- 14.Padilla R, Sousa R. 2002. AY639F/H784A T7 RNA polymerase double mutant displays superior properties for synthesizing RNAs with non-canonical NTPs. Nucleic Acids Res. 30:e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beigelman L, McSwiggen JA, Draper KG, Gonzalez C, Jensen K, et al. 1995. Chemical modification of hammerhead ribozymes: catalytic activity and nuclease resistance. J. Biol. Chem 270:25702–8 [DOI] [PubMed] [Google Scholar]
- 16.Tucker CE, Chen LS, Judkins MB, Farmer JA, Gill SC, Drolet DW. 1999. Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. J. Chromatogr. B Biomed. Sci. Appl 732:203–12 [DOI] [PubMed] [Google Scholar]
- 17.White R, Rusconi C, Scardino E, Wolberg A, Lawson J, et al. 2001. Generation of species cross-reactive aptamers using “toggle” SELEX. Mol. Ther 4:567–73 [DOI] [PubMed] [Google Scholar]
- 18.Rusconi CP, Roberts JD, Pitoc GA, Nimjee SM, White RR, et al. 2004. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat. Biotechnol . 22:1423–28 [DOI] [PubMed] [Google Scholar]
- 19.Nimjee SM, Keys JR, Pitoc GA, Quick G, Rusconi CP, Sullenger BA. 2006Anovel antidote-controlled anticoagulant reduces thrombin generation and inflammation and improves cardiac function in cardiopulmonary bypass surgery. Mol. Ther 14:408–15 [DOI] [PubMed] [Google Scholar]
- 20.Morris KN, Jensen KB,Julin CM, Weil M, Gold L. 1998. High affinity ligands from in vitro selection: complex targets. PNAS 95:2902–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Layzer JM, Sullenger BA. 2007. Simultaneous generation of aptamers to multiple gamma- carboxyglutamic acid proteins from a focused aptamer library using DeSELEX and convergent selection. Oligonucleotides 17:1–11 [DOI] [PubMed] [Google Scholar]
- 22.Oney S, Nimjee SM, Layzer J, Que-Gewirth N, Ginsburg D, et al. 2007. Antidote-controlled platelet inhibition targeting von Willebrand factor with aptamers. Oligonucleotides 17:265–74 [DOI] [PubMed] [Google Scholar]
- 23.Shangguan D,Li Y,Tang Z, Cao ZC, Chen HW, et al. 2006. Aptamers evolved from live cells as effective molecular probes for cancer study. PNAS 103:11838–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shangguan D, Meng L, Cao ZC, Xiao Z, Fang X, et al. 2008. Identification of liver cancer-specific aptamers using whole live cells. Anal. Chem . 80:721–28 [DOI] [PubMed] [Google Scholar]
- 25.Zhao Z, Xu L, Shi X, Tan W, Fang X, Shangguan D. 2009. Recognition of subtype non-small cell lung cancer by DNA aptamers selected from living cells. Analyst 134:1808–14 [DOI] [PubMed] [Google Scholar]
- 26.Mallikaratchy P, Tang Z, Kwame S, Meng L, Shangguan D, Tan W. 2007. Aptamer directly evolved from live cells recognizes membrane bound immunoglobin heavy mu chain in Burkitt’s lymphoma cells. Mol. Cell. Proteom 6:2230–38 [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Zhao Z, Bai H, Fu T, Yang C, et al. 2015. DNA aptamer selected against pancreatic ductal adenocarcinoma for in vivo imaging and clinical tissue recognition. Theranostics 5:985–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Zhang J, Ma Y, Pei X, Liu Q, et al. 2013. A cell-based single-stranded DNA aptamer specifically targets gastric cancer. Int. J. Biochem. Cell Biol 46:1–8 [PubMed] [Google Scholar]
- 29.Chen HW, Medley CD, Sefah K, Shangguan D, Tang Z, et al. 2008. Molecular recognition of small-cell lung cancer cells using aptamers. Chem. Med. Chem 3:991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mi J, Liu Y, Rabbani ZN, Yang Z, Urban JH, et al. 2010. In vivo selection of tumor-targeting RNA motifs. Nat. Chem. Biol 6:22–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rohloff JC, Gelinas AD, Jarvis TC, Ochsner UA, Schneider DJ, et al. 2014. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol. Ther. Nucleic Acids 3:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gold L, Ayers D, Bertino J, Bock C, Bock A, et al. 2010. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLOS ONE 5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamis AP, Miller JW, Bernal MT, D’Amico DJ, Folkman J, et al. 1994. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am. J. Ophthalmol 118:445–50 [DOI] [PubMed] [Google Scholar]
- 34.Kvanta A, Algvere PV, Berglin L, Seregard S. 1996. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Investig. Ophthalmol. Vis. Sci 37:1929–34 [PubMed] [Google Scholar]
- 35.Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE. 1995. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. PNAS 92:905–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruckman J, Green LS, Beeson J, Waugh S, Gillette WL, et al. 1998. 2/-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165): inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem 273:20556–67 [DOI] [PubMed] [Google Scholar]
- 37.Eyetech Study Group. 2002. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina 22:143–52 [DOI] [PubMed] [Google Scholar]
- 38.Eyetech Study Group. 2003. Anti-vascular endothelial growth factor therapy for subfoveal choroidal neovascularization secondary to age-related macular degeneration: phase II study results. Ophthalmology 110:979–86 [DOI] [PubMed] [Google Scholar]
- 39.Gragoudas ES,Adamis AP, Cunningham ETJ, Feinsod M, Guyer DR. 2004. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med 351:2805–16 [DOI] [PubMed] [Google Scholar]
- 40.Drolet DW, Green LS, Gold L, Janjic N. 2016. Fit for the eye: aptamers in ocular disorders. Nucleic Acid Ther 26:127–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dyke CK, Steinhubl SR, Kleiman NS, Cannon RO, Aberle LG, et al. 2006. First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation ofa drug-antidote pair for the controlled regulation offactor IXa activity. Circulation 114:2490–97 [DOI] [PubMed] [Google Scholar]
- 42.Chan MY, Cohen MG, Dyke CK, Myles SK, Aberle LG, et al. 2008. Phase 1b randomized study of antidote-controlled modulation of factor IXa activity in patients with stable coronary artery disease. Circulation 117:2865–74 [DOI] [PubMed] [Google Scholar]
- 43.Cohen MG, Purdy DA, Rossi JS, Grinfeld LR, Myles SK, et al. 2010. First clinical application of an actively reversible direct factor IXa inhibitor as an anticoagulation strategy in patients undergoing percutaneous coronary intervention. Circulation 122:614–22 [DOI] [PubMed] [Google Scholar]
- 44.Povsic TJ, Vavalle JP, Aberle LH, Kasprzak JD, Cohen MG, et al. 2013A Phase 2, randomized, partially blinded, active-controlled study assessing the efficacy and safety ofvariable anticoagulation reversal using the REG1 system in patients with acute coronary syndromes: results of the RADAR trial. Eur. Heart J 34:2481–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lincoff AM, Mehran R, Povsic TJ, Zelenkofske SL, Huang Z, et al. 2016. Effect of the REG1 anticoagulation system versus bivalirudin on outcomes after percutaneous coronary intervention (REGULATE- PCI): a randomised clinical trial. Lancet 387:349–56 [DOI] [PubMed] [Google Scholar]
- 46.Ganson NJ, Povsic TJ, Sullenger BA, Alexander JH, Zelenkofske SL, et al. 2016. Pre-existing antipolyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J. Allergy Clin. Immunol 137:1610–13.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. 1992. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 355:564–66 [DOI] [PubMed] [Google Scholar]
- 48.DeAnda A Jr., Coutre SE, Moon MR, Vial CM, Griffin LC, et al. 1994. Pilot study of the efficacy of a thrombin inhibitor for use during cardiopulmonary bypass. Ann. Thorac. Surg 58:344–50 [DOI] [PubMed] [Google Scholar]
- 49.Griffin LC,Tidmarsh GF, Bock LC,Toole JJ, Leung LL. 1993. Invivo anticoagulantproperties ofanovel nucleotide-based thrombin inhibitor and demonstration of regional anticoagulation in extracorporeal circuits. Blood 81:3271–76 [PubMed] [Google Scholar]
- 50.Buff MC, Schafer F, Wulffen B, Muller J, Potzsch B, et al. 2010. Dependence of aptamer activity on opposed terminal extensions: improvement of light-regulation efficiency. Nucleic Acids Res 38:2111–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang RH, Fremont DH, Diener JL, Schaub RG, Sadler JE. 2009. A structural explanation for the antithrombotic activity of ARC1172, a DNA aptamer that binds von Willebrand factor domain A1. Structure 17:1476–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Markus HS, McCollum C, Imray C, Goulder MA, Gilbert J, King A. 2011. The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy: a randomized trial. Stroke 42:2149–53 [DOI] [PubMed] [Google Scholar]
- 53.Nimjee SM, Lohrmann JD, Wang H, Snyder DJ, Cummings TJ, et al. 2012. Rapidly regulating platelet activity in vivo with an antidote controlled platelet inhibitor. Mol. Ther 20:391–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bates PJ, Laber DA, Miller DM, Thomas SD, Trent JO. 2009. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol 86:151–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soundararajan S, Wang L, Sridharan V, Chen W, Courtenay-Luck N, et al. 2009. Plasma membrane nucleolin is a receptor for the anticancer aptamer AS1411 in MV4–11 leukemia cells. Mol. Pharmacol 76:984–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soundararajan S, Chen W, Spicer EK, Courtenay-Luck N, Fernandes DJ. 2008. The nucleolin targeting aptamerAS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. CancerRes. 68:2358–65 [DOI] [PubMed] [Google Scholar]
- 57.Rosenberg JE, Bambury RM, Van Allen EM, Drabkin HA, Lara PN Jr., et al. 2014. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investig. New Drugs 32:178–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hwang DW, Ko HY, Lee JH, Kang H, Ryu SH, et al. 2010. Anucleolin-targeted multimodal nanoparticle imaging probe for tracking cancer cells using an aptamer. J. Nuclear Med 51:98–105 [DOI] [PubMed] [Google Scholar]
- 59.Guo J, Gao X, Su L, Xia H, Gu G, et al. 2011. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 32:8010–20 [DOI] [PubMed] [Google Scholar]
- 60.Trinh TL, Zhu G,Xiao X, Puszyk W, Sefah K,et al. 2015A synthetic aptamer-drug adduct for targeted liver cancer therapy. PLOS ONE 10:e0136673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aravind A, Jeyamohan P, Nair R, Veeranarayanan S, Nagaoka Y, et al. 2012. AS1411 aptamer tagged PLGA-lecithin-PEG nanoparticles for tumor cell targeting and drug delivery. Biotechnol. Bioeng 109:2920–31 [DOI] [PubMed] [Google Scholar]
- 62.Wu J, Song C, Jiang C, Shen X, Qiao Q, Hu Y. 2013. Nucleolin targeting AS1411 modified protein nanoparticle for antitumor drugs delivery. Mol. Pharm 10:3555–63 [DOI] [PubMed] [Google Scholar]
- 63.Shieh YA, Yang SJ, Wei MF, Shieh MJ. 2010. Aptamer-based tumor-targeted drug delivery for photodynamic therapy. ACS Nano 4:1433–42 [DOI] [PubMed] [Google Scholar]
- 64.Yang X, Liu X, Liu Z, Pu F, Ren J, Qu X. 2012. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer gated nanovehicles. Adv. Mater 24:2890–95 [DOI] [PubMed] [Google Scholar]
- 65.Kotula JW, Pratico ED, Ming X, Nakagawa O, Juliano RL, Sullenger BA. 2012. Aptamer-mediated delivery of splice-switching oligonucleotides to the nuclei of cancer cells. Nucleic Acid Ther 22:187–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lupold SE, Hicke BJ, Lin Y, Coffey DS. 2002. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 62:4029–33 [PubMed] [Google Scholar]
- 67.Dassie JP, Hernandez LI, Thomas GS, Long ME, Rockey WM, et al. 2014. Targeted inhibition of prostate cancer metastases with an RNA aptamer to prostate-specific membrane antigen. Mol. Ther 22:1910–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chu TC, Marks JWIII, Lavery LA, Faulkner S, Rosenblum MG, et al. 2006. Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res 66:5989–92 [DOI] [PubMed] [Google Scholar]
- 69.Bagalkot V, Farokhzad OC, Langer R, Jon S. 2006. An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew. Chem 45:8149–52 [DOI] [PubMed] [Google Scholar]
- 70.Bagalkot V, Zhang L, Levy-Nissenbaum E, Jon S, Kantoff PW, et al. 2007. Quantum dot-aptamer conjugates for synchronous cancer imaging, therapy, and sensing of drug delivery based on bi-fluorescence resonance energy transfer. Nano Lett 7:3065–70 [DOI] [PubMed] [Google Scholar]
- 71.Wang AZ, Bagalkot V, Vasilliou CC, Gu F, Alexis F, et al. 2008. Superparamagnetic iron oxide nanoparticle-aptamer bioconjugates for combined prostate cancer imaging and therapy. Chem. Med. Chem. 3:1311–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, et al. 2006. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. PNAS 103:6315–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McNamara JO II, Andrechek ER, Wang Y, Viles KD, Rempel RE, et al. 2006. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol 24:1005–15 [DOI] [PubMed] [Google Scholar]
- 74.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, et al. 2009. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol 27:839–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McNamara JOII, Kolonias D, Pastor F, Mittler RS, Chen L, et al. 2008. Multivalent 4–1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Investig 118:376–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mahlknecht G, Maron R, Mancini M, Schechter B, Sela M, Yarden Y. 2013. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. PNAS 110:8170–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mahlknecht G, Maron R, Schechter B, Yarden Y, Sela M. 2015. Multimerization of ERBB2/HER2 specific aptamer leads to improved receptor binding. Biochem. Biophys. Res. Commun 465:218–24 [DOI] [PubMed] [Google Scholar]
- 78.Thiel KW, Hernandez LI, Dassie JP, Thiel WH, Liu X, et al. 2012. Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res 40:6319–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li N, Larson T, Nguyen HH, Sokolov KV, Ellington AD. 2010. Directed evolution of gold nanoparticle delivery to cells. Chem. Commun 46:392–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li N, Nguyen HH, Byrom M, Ellington AD. 2011. Inhibition of cell proliferation by an anti-EGFR aptamer. PLOS ONE 6:e20299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ray P, Cheek MA, Sharaf ML, Li N, Ellington AD, et al. 2012. Aptamer-mediated delivery of chemotherapy to pancreatic cancer cells. Nucleic Acid Ther 22:295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cerchia L, Esposito CL, Camorani S, Rienzo A, Stasio L, et al. 2012. Targeting Axl with an high-affinity inhibitory aptamer. Mol. Ther 20:2291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Esposito CL, Cerchia L, Catuogno S, De Vita G, Dassie JP, et al. 2014. Multifunctional aptamer-miRNA conjugates for targeted cancer therapy. Mol. Ther 22:1151–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brayman M, Thathiah A, Carson DD. 2004. MUC1: a multifunctional cell surface component of reproductive tissue epithelia. Reprod. Biol. Endocrinol 2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gendler SJ. 2001. MUC1, the renaissance molecule. J. Mammary GlandBiol. Neoplasia 6:339–53 [DOI] [PubMed] [Google Scholar]
- 86.Ferreira CS, Cheung MC, Missailidis S, Bisland S, Gariepy J. 2009. Phototoxic aptamers selectively enter and kill epithelial cancer cells. Nucleic Acids Res 37:866–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Perkins AC, Missailidis S. 2007. Radiolabelled aptamers for tumour imaging and therapy. Q. J. Nuclear Med. Mol. Imaging 51:292–96 [PubMed] [Google Scholar]
- 88.Da Pieve C, Perkins AC, Missailidis S. 2009. Anti-MUC1 aptamers: radiolabelling with 99mTc and biodistribution in MCF-7 tumour-bearing mice. Nuclear Med. Biol 36:703–10 [DOI] [PubMed] [Google Scholar]
- 89.Borbas KE, Ferreira CS, Perkins A, Bruce JI, Missailidis S. 2007. Design and synthesis of mono- and multimeric targeted radiopharmaceuticals based on novel cyclen ligands coupled to anti-MUCl aptamers for the diagnostic imaging and targeted radiotherapy of cancer. Bioconjug. Chem 18:1205–12 [DOI] [PubMed] [Google Scholar]
- 90.Wang L, Liu B, Yin H, Wei J, Qian X, Yu L. 2007. Selection of DNA aptamer that specific binding human carcinoembryonic antigen in vitro. J. Nanjing Med. Univ 21:277–81 [Google Scholar]
- 91.Hashemi Tabar GR, Smith LC. 2010. DNA aptamers selected as molecular probes for diagnosis of cancereous cells. World Appl. Sci. J 8:16–21 [Google Scholar]
- 92.Lee YJ, Han SR, Kim NY, Lee SH, Jeong JS, Lee SW. 2012. An RNA aptamer that binds carcinoem- bryonic antigen inhibits hepatic metastasis of colon cancer cells in mice. Gastroenterology 143:155–65 e8 [DOI] [PubMed] [Google Scholar]
- 93.Huang YF, Shangguan D, Liu H, Phillips JA, Zhang X, et al. 2009. Molecular assembly of an aptamer- drug conjugate for targeted drug delivery to tumor cells. Chem. Biochem 10:862–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo YL, Shiao YS, Huang YF. 2011. Release of photoactivatable drugs from plasmonic nanoparticles for targeted cancer therapy. ACS Nano 5:7796–804 [DOI] [PubMed] [Google Scholar]
- 95.Wang J, Zhu G, You M, Song E, Shukoor MI, et al. 2012. Assembly of aptamer switch probes and photosensitizer on gold nanorods for targeted photothermal and photodynamic cancer therapy. ACS Nano 6:5070–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rossow KL, Janknecht R. 2003. Synergism between p68 RNA helicase and the transcriptional coactivators CBP and p300. Oncogene 22:151–56 [DOI] [PubMed] [Google Scholar]
- 97.Shin S, Rossow KL, Grande JP, Janknecht R. 2007. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res 67:7572–78 [DOI] [PubMed] [Google Scholar]
- 98.Santulli-Marotto S, Nair SK, Rusconi C, Sullenger B, Gilboa E. 2003. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. CancerRes. 63:7483–89 [PubMed] [Google Scholar]
- 99.Dollins CM, Nair S, Boczkowski D, Lee J, Layzer JM, et al. 2008. Assembling 0X40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol 15:675–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pratico ED, Sullenger BA, Nair SK. 2013. Identification and characterization of an agonistic aptamer against the T cell costimulatory receptor, 0X40. Nucleic Acid Ther. 23:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, et al. 1995. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 376:70–74 [DOI] [PubMed] [Google Scholar]
- 102.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, et al. 1999. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284:1994–98 [DOI] [PubMed] [Google Scholar]
- 103.White RR, Shan S, Rusconi CP, Shetty G, Dewhirst MW, et al. 2003. Inhibition of rat corneal angio- genesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. PNAS 100:5028–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sarraf-Yazdi S, Mi J, Moeller BJ, Niu X, White RR, et al. 2008. Inhibition of in vivo tumor angiogenesis and growth via systemic delivery of an angiopoietin 2-specific RNA aptamer. J. Surg. Res 146:16–23 [DOI] [PubMed] [Google Scholar]
- 105.Herbst RS, Hong D, Chap L, Kurzrock R, Jackson E, et al. 2009. Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. J. Clin. Oncol 27:3557–65 [DOI] [PubMed] [Google Scholar]