

Abstract
While poly(lactic-co-glycolic acid)-block-polyethylene glycol (PLGA-PEG) nanoparticles (NPs) can encapsulate drug cargos and prolong circulation times, they show non-specific accumulation in off-target tissues. Targeted delivery of drugs to tumor tissue and tumor vasculature is a promising approach for treating solid tumors while enhancing specificity and reducing systemic toxicity. AXT050, a collagen-IV derived peptide with both antitumor and antiangiogenic properties, is shown to bind to tumor-associated integrins with high affinity, which leads to targeted accumulation in tumor tissue. AXT050 conjugated to PLGA-PEG NPs at precisely controlled surface density functions both as a targeting agent to human tumor cells and demonstrates potential for simultaneous antitumorigenic and antiangiogenic activity. These targeted NPs cause inhibition of adhesion and proliferation in vitro when added to human triple-negative breast cancer cells and microvascular endothelial cells through binding to integrin αVβ3. Furthermore, we find an in vivo biphasic relationship between tumor targeting and surface coating density of NPs coated with AXT050. NPs with an intermediate level of 10% peptide surface coating show approximately two-fold greater accumulation in tumors and lower accumulation in the liver compared to non-targeted PLGA-PEG NPs in a murine biodistribution model. Display of biomimetic peptides from NP surfaces to both target and inhibit cancer cells has the potential to enhance the activity of cancer nanomedicines.
Keywords: Drug delivery, cancer, nanoparticle, peptide, targeting
INTRODUCTION
Drug delivery of chemotherapeutic compounds to breast cancer and other solid tumors is often limited by short half-lives and systemic toxicity stemming from an inability to specifically target the tumor site.1–3 A commonly proposed solution to this problem is encapsulation of drugs within a biodegradable polymer nanoparticle (NP) with specifically altered surface chemistry to aid in targeting of the tumor tissue.4,5 For example, PEGylated poly(lactic-co-glycolic acid) (PLGA-PEG) NPs have been used to improve the blood circulation time and accumulation in tumors, thereby enhancing efficacy of doxorubicin,6 platinum prodrugs,5,7 docetaxel,8,9 paclitaxel10,11 and other common chemotherapeutics in animal models. However, compared to the number of preclinical studies reported, these PLGA-PEG NPs have had relatively little success achieving FDA approval due to insufficient drug efficacy in the tumor and potential accumulation in off-target tissue such as the liver.12 Protein and peptide-loaded NPs have been explored as well, but are often limited by packing density within the polymer matrix of the NPs and challenges with encapsulation.13
Early strategies for targeting PLGA-PEG and other NPs to tumors relied in large part on the passive enhanced permeability and retention (EPR) effect, which hypothesized that NPs can extravasate through leaky neovasculature near the tumor and are not easily drained by irregularly formed lymph vessels.14 Though this effect has been shown to contribute to NP accumulation in solid tumors, further targeting is needed to reduce non-specific accumulation and buildup in other healthy tissues. Growing effort has focused on developing active targeting methods using ligands that target receptors known to be upregulated on the surface of neovasculature and tumor cells, such as integrin αvβ35,15,16 and folate receptors.11,17,18 A common ligand used in conjunction with PLGA-PEG NPs to target integrin αvβ3 is the peptide sequence RGD, including cyclic-RGD peptide. The RGD motif is present in the extracellular matrix components such as fibronectin and vitronectin, aiding in the binding of these components to the cell surface. The use of cyclic-RGD ligands has been shown to increase tumor localization and improve efficacy of a number of chemotherapeutic drugs in pre-clinical models.19 However, despite promising preclinical studies, integrin facilitated drug delivery has not been validated in clinical applications to date. In addition, aside from cyclic-RGD ligands and RGD mimics, non-RGD-based αvβ3 integrin targeting systems for nanoparticles have not yet been developed.19 Alternative nanomedicine targeting strategies utilize other biological molecules to reach different cellular targets such as RNA aptamer ligands to prostate-specific membrane antigen to better target PLGA-PEG NPs to prostate cancer cells.20
The roles of particle physical properties, such as size and shape, are increasingly being investigated in polymeric and inorganic NP drug delivery systems.21–23 The size of particles can be controlled such that following systemic administration, particles are large enough to avoid filtration by the kidneys (particle diameter designed to be greater than ~10 nm) and small enough to enable enhanced diffusion to reach a target tissue (particle diameter designed to be less than ~100 nm). Discoidal and ellipsoidal particles can marginate to the side of the blood vessel, enhancing their possibility of extravasation at the tumor site. While the delivery of drugs by polymeric NPs can also be improved by targeting, encapsulation capacity of drugs in PLGA-PEG NPs is generally limited to a small percentage of the total weight of the particle.4 Targeted NP systems are canonically limited to drug loading within the particle while the targeting ligand decorates the outside. The hydrophobic nature of polymeric NPs makes them ideal delivery systems for hydrophobic drugs, but limits the ability to load many drugs into this system with high efficiency.24 This poses a problem when a relatively larger dose of drug is required for therapeutic efficacy or enhanced durability. If a drug, such as a peptide, were covalently coupled to the nanoparticle surface, the total loading could potentially be increased by an order of magnitude compared to loading by physical encapsulation alone. A surface tethered drug can target a receptor of interest, facilitate avidity, and potentiate a therapeutic effect. If the surface tethered drug contains degradable linkages, it could also be designed to enable controlled release from the NP. Further, the amount of the targeting ligand on the surface of NPs can be optimized to increase accumulation in the tumor and minimize off target delivery, avoid interaction and clearance via the reticuloendothelial system, and enhance targeted cellular uptake.25,26
Anti-angiogenesis is one of many key methods of cancer therapy, as tumor growth requires robust neovascularization in its microenvironment for increased oxygen and nutrient supply.27 E. Lee et al. showed that a collagen-IV derived 20-mer peptide, that we refer to here as AXT050, inhibits tumor growth in a number of models, including a metastatic tumor model, via its potent anti-angiogenic, anti-lymphangiogenic, and anti-tumorigenic activities.28–31 The naked peptide is also shown to bind to integrin αvβ3 and disrupt integrin-dependent protein signaling pathways, thereby expanding the arsenal of VEGF-independent, anti-angiogenesis based cancer therapies.28–31 By surface-functionalization and encapsulation of the AXT050 peptide in PLGA-PEG NPs, we hypothesize that targeted NPs with high loading capacity can be fabricated. As naked AXT050 biomimetic peptide can therapeutically modulate angiogenesis through extracellular interactions, we further hypothesize that presentation of AXT050 from the NP surface and soluble PEG-AXT050 conjugates released from the surface via hydrolysis can directly inhibit angiogenesis.
Herein, we design, characterize, and validate PLGA-PEG-AXT050 NPs, including in vitro adhesion and proliferation inhibition using both whole particles and the breakdown products of degraded particles. We also report the optimization of PLGA-PEG NPs functionalized with AXT050 peptide for in vivo biodistribution to tumors using an orthotopic human triple-negative breast cancer (MDA-MB-231) mouse model that is challenging to target through conventional approaches.
MATERIALS AND METHODS
Materials
Poly(D,L-lactide-co-glycolide) (65/35), dimethyl formamide (DMF), dimethyl sulfoxide (DMSO), di-isopropylethylamine (DIPEA), methanol, and ether were purchased from Sigma-Aldrich (St. Louis, MO). PVA [Poly(vinyl alcohol); Mw 25,000] was purchased from Polysciences (Warrington, PA). Poly(D,L-lactide-co-glycolide) (50/50) with terminal methoxy groups (PLGA-mPEG) or terminal n-hydroxysuccinimide groups (PLGA-PEG-NHS) (each with MW 20,000:5,000 Da, PLGA:PEG) were purchased from Akina (West Lafayette, IN). AXT050,28,29 AXT051 (AXT050 analog where the last amino acid residue is swapped from a F to a W to enable stronger intrinsic fluorescence), biotinylated AXT050, and AXT050-IRD800 were custom synthesized and ordered from New England Peptide (Gardner, MA). Human recombinant integrin was purchased from R&D Systems (Minneapolis, MN). IRDye 800 CW was purchased from LI-COR (Lincoln, NE). Tissue culture reagents and cell lines were purchased from Lonza (Portsmouth, NH). Biological buffers were purchased from Fisher Scientific (Hampton, NH).
Binding kinetics between AXT050 and integrin αvβ3
The ForteBio Octet RED96, capable of an automated high-throughput assay using 96-well plates, was used to investigate the binding kinetic profile of AXT050 to integrin αvβ3. 25 μM stock of biotinylated AXT050 in 5% DMSO / 95% water was diluted 100-fold using 0.05% Tween 20 in 1X PBS. The peptide was incubated with streptavidin biosensor probe from Pall ForteBio (Fremont, CA) for 300 seconds, flanked by 60 seconds of incubation in the same buffer without the peptide. Subsequently in the binding association step, the peptide-attached biosensor probe was incubated with various concentrations of integrin αvβ3 ranging from 10 – 300 nM in the same buffer solution for 600 seconds. The probe is further incubated in the buffer solution without integrin for 1800 seconds to observe the dissociation profile. Optical interference signal resulting from the binding of integrins to peptide-attached probe was exported for analysis in MATLAB (Mathworks 2015). A 1:1 binding model was implemented in MATLAB and the resulting differential equation describing the concentration changes in peptide and peptide-integrin binding complex was solved using ODE15s function. The fitting was performed using patternsearch algorithm as part of the MATLAB optimization toolbox to determine kon and koff of the reaction using the experimental time course data.
Formulation of spherical and ellipsoidal PLGA nanoparticles
PLGA NPs were formulated using an emulsion method. First, PLGA (65/35) was first dissolved into DCM at 20 mg/mL in a test tube, vortexed to fully dissolve, and mixed with 20 mg/mL AXT050 stock in DMSO at the desired mass ratio of peptide to PLGA; a common formulation is 1:50 peptide:PLGA. The mixture was sonicated with the test tube on ice. Sonication (Misonix) was performed with an amplitude of 30, which equals approximately 5-10 W, for 20 seconds. This primary emulsion was immediately poured into 50 mL of 1% PVA solution and sonicated at an amplitude of between 30 and 100 for 2 minutes on ice. The full volume was then transferred to 100 mL of 0.5% PVA solution and stirred in a chemical hood for 3.5 hours. Then, NPs were washed three times by centrifuging at 4°C, 17 krpm for 10 minutes, removing the supernatant, and resuspending NPs with 30 mL of refrigerated Milli-Q water. After the last wash, 5 mL of water was added to resuspend the sample, which was then snap-frozen with liquid nitrogen for lyophilization. NPs were stored at −20°C prior to use.
To generate ellipsoidal PLGA NPs, pre-made spherical PLGA NPs were stretched using a previously described method.32 Briefly, lyophilized spherical PLGA NPs were suspended in water and mixed with 10% PVA/2% glycerol solution at 5 mg/mL NP concentration. The solution was added to a mold (5 cm × 7 cm rectangular petri dishes) and allowed to dry into a film. The film was heated above the Tg of PLGA and then stretched via a custom made stretching device, after which the film was dissolved, NPs washed as described above, and stored for future use.
Synthesis of peptide-functionalized PLGA-PEG
PLGA-PEG-NHS was dissolved at 170 mg/mL in DMF, and AXT050 or AXT051 in DMSO at 100 mg/mL was added at a 1.2:1 molar ratio in excess of the peptide. A 40-fold molar excess of DIPEA was added to the mixture and stirred overnight at room temperature. The mixture was then added dropwise to a cold mixture of ether (50%) and methanol (50%) and spun down to produce a pellet. The supernatant was discarded and the pellet was disturbed and washed in 100% methanol, and spun down repeatedly to remove unreacted peptide. The pellet was left to dry under vacuum for several hours to yield solid PLGA-PEG-AXT050. Typical batches yield 10-100 mg, approximately 80% of the starting material. A similar protocol was used for the functionalization of PLGA-PEG-NHS polymer with IRDye 800 CW to prepare fluorescent NPs.
High performance liquid chromatography was used to confirm the conjugation. 500 μg of the AXT051 functionalized PLGA-PEG reaction mixture (prior to precipitation in methanol and ether) was diluted in acetonitrile and run through an Agilent Poroshell 300 column. AXT051 was utilized as an analog of AXT050 that allows improved quantification of intrinsic UV absorption/fluorescence via the tryptophan residue at the C-terminus of the peptide. The reaction mixture was compared to a control mixture using PLGA-mPEG in place of the reactive PLGA-PEG-NHS. AUCs of peaks from unreacted peptide were compared to determine the extent of the reaction, which was calculated according to signals at 220 nm (peptide backbone), 280 nm (trp absorption), and 295/348 excitation/emission (trp fluorescence).
Formulation of peptide-functionalized PLGA-PEG nanoparticles
The functionalized PLGA-PEG NPs were formulated following a nanoprecipitation method. PLGA-PEG-AXT050 and PLGA-mPEG were each dissolved in DMF in any desired mass ratio (i.e. 10% NP: 1 to 9 mass ratio of PLGA-PEG-AXT050 to PLGA-mPEG) at 10 mg/mL total polymer concentration. For encapsulation, this was then mixed with AXT050 in DMSO at 5% (w/w) and/or with a dye such as 5-carboxytetramethyl-rhodamine (TAMRA). The polymer/peptide mix was then added dropwise to a volume of deionized water ten times larger than the DMF solution under magnetic stirring. After 4-6 hours stirring in a chemical hood, the NPs were filtered and concentrated using ultra centrifugation columns (EMD Millipore, UFC810096) and GPC spin columns (Thermo Scientific) using S-400 HR media (Sephacryl) to ensure all free peptide was filtered. NPs were then either used immediately or stored overnight at 4°C before use.
Quantification of peptide content on functionalized nanoparticles
LavaPep peptide quantification kit (Gel Company, LP022010) was used to determine peptide content following NP formation. As per the LavaPep protocol, particles were incubated at 3 mg/mL in the dark in the LavaPep working solution for 1 hour. The epicocconone dye interacts with the arg residues in the peptide to become highly fluorescent. Fluorescence was read at 530/590 nm on a Biotek HT Synergy plate reader. The signal from the nanoparticles was compared to a standard dilution curve of known amounts of free peptide.
Characterization of nanoparticles
The particle size distributions were generally measured via dynamic light scattering (Malvern Zetasizer Nano ZS90) at 25°C at a scattering angle of 90° and at a concentration of approximately 1 mg/mL NP in water, PBS, or FBS. Nanoparticle tracking analysis (NTA) was performed using a Nanosight NS500 instrument and NP solutions were diluted so that the nanoparticle concentrations were appropriate for NTA analysis.33 NTA videos were captured for 60 seconds and analyzed using Nanosight NTA software. For TEM sizing, 10 μL of NP sample at 1 mg/mL was dropped onto carbon coated copper grids and left to dry in chemical hood for 2 hours. Unstained TEM imaging was then performed using the Philips CM120 system. Zeta potential was measured on a Malvern Zetasizer Nano ZS90 at 1 mg/mL in 10 mM NaCl.
In vitro targeting activity assay
MDA-MB-231 cells and MEC cells were trypsinized and suspended in appropriate media at 2,000 cells/mL. 100% AXT050 peptide coated NPs and non-coated PEG-PLGA NPs that encapsulated TAMRA dye were added at 5 mg/mL to the cells in suspension, and the mixture was incubated while shaking at 37°C for 1 hour to allow binding between cells and particles. After 1 hour, cells were spun down and supernatant was discarded. Cells were resuspended in PBS and spun down twice to wash away any non-bound particles or dye in solution. Cells were resuspended in 1 mL of PBS and 100 μL of that solution was assessed for TAMRA fluorescence using a Biotek Synergy Plate Reader at 530/590 nm excitation/emission.
Integrin αvβ3, α5β1¸and human serum albumin were labeled with AlexaFluor 488 Protein Labeling Kit (Thermo Scientific) according to the manufacturer’s instructions. Labeled proteins at 50 μg/mL were incubated with either peptide conjugated or non-conjugated NPs in PBS at concentrations of 5 mg/mL. After 1 hour shaking at 37°C, NPs were separated from free proteins in solution via GPC spin columns (Thermo Scientific) using S-400 HR media (Sephacryl) and the fluorescence of the filtrate was measured. For free peptide competition experiments, a similar protocol was followed, but a 100-fold excess of non-labeled AXT050 peptide or an inactive scrambled peptide control sequence known to have no activity in in vitro adhesion activity assays was added to the NP solution before the addition of labeled peptide.
In vitro anti-tumor functional activity assay
Prior to use in in vitro activity assays, particles were transferred from ultrapure water to appropriate media using the same ultracentrifugation columns used for purification after NP formation. To test activity in an adhesion assay, particles were concentrated to 20 mg/mL in media and added to 96 well plates. Regular media was used a negative control and media supplemented with 100 μM and 25 μM of free AXT050 were used as positive controls. MDA-MB-231 triple-negative breast cancer cells or primary microvascular endothelial cells (MECs) were added at 20,000 cells/well on top of the particles. The plate was then incubated for 2 hours at 37°C and 5% CO2. Wells were next washed twice with DPBS with Ca2+ and Mg2+ and then filled with media containing 4 μg/mL Calcein AM dye. Plates were then incubated for 30 minutes and washed again with DPBS with Ca2+ and Mg2+. Fluorescence was read on a Biotek Synergy HT at 485/528 nm excitation/emission to quantify the number of cells adhered to the surface of the well.
Particle breakdown products were assessed for their efficacy relative to fully intact particles and free peptide by allowing hydrolysis of 10% functionalized particles in PBS at 37°C for 5 days on a shaker. The resulting broken down particles were confirmed to have lost their structure via dynamic light scattering as described above, and were determined to be broken down when particles could no longer be detected at sufficient concentration to analyze particle size. The breakdown products were then added to MDA-MB-231 cells as above and compared to free peptide and to equal amounts of peptide included on intact particles at 10% functionalization.
To test activity on MEC cells in a proliferation assay, 2000 cells/well were plated in 96-well plates in phenol red-free ECM-2MV media and allowed to adhere over 18-20 hours. Media was replaced with particles suspended in media at 5 mg/mL or AXT050 peptide in media or media alone. After four days, media was replaced with 100 μl MTT reagent as per the manufacturer’s recommendations. After four hours, 100 μl of SDS solution was added to each well and incubated at 37°C for another four hours. Absorbance was read at 570 nm on a Biotek Synergy HT plate reader to capture the change from MTT to formazan by mitochondrial reductase in the living cells.
In vivo pharmacokinetics of functionalized PLGA-PEG nanoparticles
Animals were housed and treated in accordance with NIH and IACUC guidelines for the care and use of laboratory animals (NIH Publication #85-23 Rev. 1985), and used protocols approved by the Johns Hopkins University Animal Care and Use Committees. 2*106 human triple-negative breast cancer cells (MDA-MB-231) mixed 1:1 with matrigel (Corning) were implanted orthotopically of a 5-week old athymic nude mouse (Charles River Laboratories, Wilmington, MA). Tumors were then allowed to grow until they reached a volume of 100 mm3. Mice received tail-vein injection of PBS, naked peptide, spherical or ellipsoidal PLGA NPs, or PLGA-PEG NPs with 0%, 10%, and 100% peptide functionalization. In the non-targeted biodistribution experiment, peptide was labeled with IRD 800 CW dye. In the functionalized NP experiment, 1% of the polymer used to form particles was bound to IRD-800 CW dye by NHS chemistry described above. All NPs contained 1% (w/w) IRDye 800CW. Blood samples were taken via saphenous vein in heparin-coated glass capillary tubes at 5, 15, 30, 60, 120, and 300 minutes as well as at 24 hours. Fluorescence in capillary tubes were imaged with LI-COR Pearl Impulse NIR Imager and quantified by assigning ROI. For biodistribution, whole live-animals were imaged at 5 and 24 hours, and then the animals were sacrificed at 24 hours to harvest, weight, and image organs separately. For imaging, Xenogen IVIS Spectrum Imager (Living Image v4.1 software) was used for naked peptide and non-targeted NP studies, while LI-COR Pearl Impulse NIR Imager (Pearl Impulse v2.0 software) with higher resolution and sensitivity was used for targeted NP studies.
RESULTS
Peptide AXT050 has high molecular binding affinity to integrin αvβ3
αvβ3 is one of the most overexpressed integrin isoforms in endothelial cells that compose the leaky vasculature around tumor tissue as well one of the most overexpressed integrin isoforms in tumor and tumor progenitor cells.34,35 As our goal was surface engineering of nanoparticles to target tumors and their vasculature, we evaluated the binding affinity between AXT050 and recombinant integrin αvβ3. The ForteBio Octet RED96, a label-free molecular binding assay that employs bio-layer interferometry to detect specific, native interaction of ligand and receptor, was used. The binding of AXT050 to αvβ3 revealed a kinetic binding curve typical of 1:1 association and dissociation (Figure 1A). The reaction rate constants kon and koff were determined to be 1 μM−1s−1 and 0.0032 s−1, respectively, by fitting the raw binding data to simple single-binding site association model in MATLAB. The resulting equilibrium dissociation constant KD was determined to be 3.2 nM, which is a strong affinity with the same order of magnitude to that of antibodies and specific enzyme-substrate interactions. AXT050’s strong binding affinity to integrin αvβ3 measured here corroborates a recent report that the anti-angiogenic peptide’s mechanism of action is via downstream signaling from its specific binding to integrin isoforms.31
Figure 1.
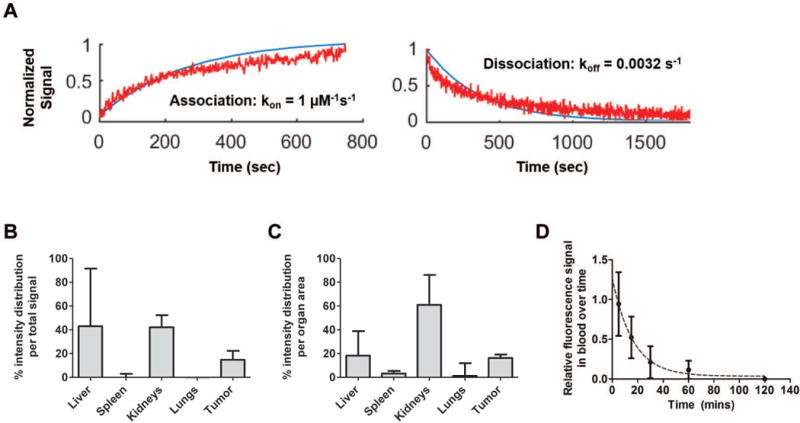
AXT050 binding and biodistribution. A AXT050 binds to αvβ3 with a kon of 1 μM−1s−1, koff of 0.0032 s−1, and KD of 3.2 nM. B-C AXT050 accumulated in MDA-MB-231 tumors and was cleared through the kidney with D a blood circulation half-life of ~11 min.
AXT050 localizes to tumor tissue following systemic delivery
Based on the biding affinity to integrin αvβ3, a biodistribution experiment of near-infrared fluorescence-tagged AXT050, AXT050-IRD800, injected intravenously was performed. An orthotopic human xenograft triple-negative breast cancer (MDA-MB-231) mouse model was used for this experiment with liver, kidneys, spleen, lungs, heart and the tumor harvested at 24 h post injection to detect fluorescence. Small biological molecules, such as a peptide composed of natural amino acids, are expected to degrade and clear from the blood quickly, and we find that AXT050 is cleared from the blood with a half-life of approximately 11 min. However, due to its binding affinity to integrin αvβ3, approximately ~15% of AXT050 was able to reach the tumor based on the fluorescence signal from tumor relative to the other organs within each mouse. As expected, kidneys were the main route of clearance for the small peptide, as seen by the highest level of its detection in the kidneys compared to other organs. Figure 1B-D illustrates these biodistribution findings.
Formulation and characterization of non-targeted nanoparticles
Three different NP systems were developed and investigated to enable improved delivery of the anti-angiogenic AXT050 peptide. For the first approach, PLGA NPs were fabricated using a double emulsion protocol to encapsulate peptide. The sonication settings and surfactant concentration were varied to determine their effect on particle size measured by Nanoparticle Tracking Analysis (NTA) as shown in Figure 2A. Increasing the sonication setting from 30 A to 60 A decreased hydrodynamic particle size from a micron scale to a size of approximately 200 nm, but further increases to the sonication power did not affect particle size. Increasing the PVA concentration from 2% to 3% also did not affect the NP size. TEM images of the NPs are shown in Figure 2A. Loading efficiency of the peptide into these PLGA NPs was found to be approximately 20% - 30%.
Figure 2.
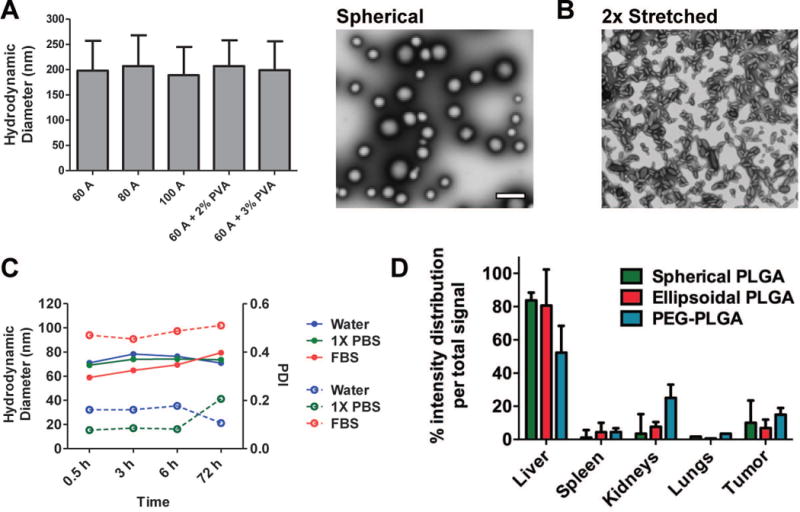
Biophysical characterization of non-targeted AXT050-containing PLGA NP. A Hydrodynamic diameter (NTA) of non-modified PLGA NPs formulated with different conditions and a representative TEM image of PLGA NPs from “60 A” condition, B TEM image of anisotropic PLGA NPs (“60 A” PLGA NPs stretched 2-fold), C Stability of PLGA-PEG NPs over 3 days in different media by DLS (closed circle/solid line: size, open circle/dashed line: polydispersity index), and D Biodistribution of non-targeted NPs. NTA: mean ± SD (n > 200 particles), TEM: scale bar = 1 μm.
To improve drug delivery, properties such as physical shape (higher aspect ratio) and chemical surface functionalization (PEGylation) have been previously shown to influence the biological response of drug delivery nanoparticles and enhance blood circulation and passive targeting.36,37 To explore the effect of shape on the NPs, a previously developed thin-film stretching particle method32 was utilized on the ~200 nm spherical PLGA NPs to create ellipsoidal PLGA NPs with aspect ratio of 2.8 by stretching 2-fold along one axis (Figure 2B). To explore the role of surface coating, PLGA-PEG NPs were also prepared to deliver the peptide. These NPs were made with PLGA-PEG block co-polymers using a nanoprecipitation method. PLGA-PEG NPs had a stable hydrodynamic particle of approximately ~70 nm in a range of media, including water, phosphate buffered saline (PBS), and 100% fetal bovine serum (FBS) (Figure 2C).
Biodistribution of non-targeted nanoparticles
A biodistribution experiment with the same design as with the naked peptide was performed using the non-targeted NP systems. The injected groups were PBS only, spherical PLGA NPs, ellipsoidal PLGA NPs, and PLGA-PEG NPs all loaded with AXT050-IRD800 labeled peptide. In all treated groups, the total amount of peptide injected was the same, at 20 μg per mouse. In Figure 2D and Figure S1, the relative distribution of the fluorescence across the different organs within each mouse is shown. By encapsulating AXT050 in nanoparticles, the liver became the primary organ of biodistribution for all three conditions (spherical PLGA NP, ellipsoidal PLGA NP, and PEGylated PLGA NP). There were no statistically significant differences between the groups in terms of accumulation in the tumor after 24 hours (7-15%).
Formation and characterization of targeted nanoparticles
In order to enhance accumulation of NPs in the tumor, we engineered targeted PLGA-PEG NPs using AXT050 peptide as the targeting ligand. We synthesized surface-functionalized PLGA-PEG NPs by conjugating AXT050 to PLGA-PEG-NHS and then forming NPs via nanoprecipitation such that AXT050 ligands coat the outer surface of the particles as illustrated in Scheme 1A and 1B. HPLC showed approximately 90% conjugation of PLGA-PEG-NHS to free peptide (Figure S2). Formation of functionalized PLGA-PEG copolymers prior to NP formation allowed control over the amount of peptide exposed on the surface of the NPs and optimization of surface ligand density. As physicochemical properties, such as ligand density, surface charge, and particle size, have a large effect on stealth properties and thus half-life of the particle,36 this synthesis method was important as it allowed us to mix functionalized and inert PLGA-PEG polymers in any ratio that we desired to tune the surface properties of the NPs.
Scheme 1.
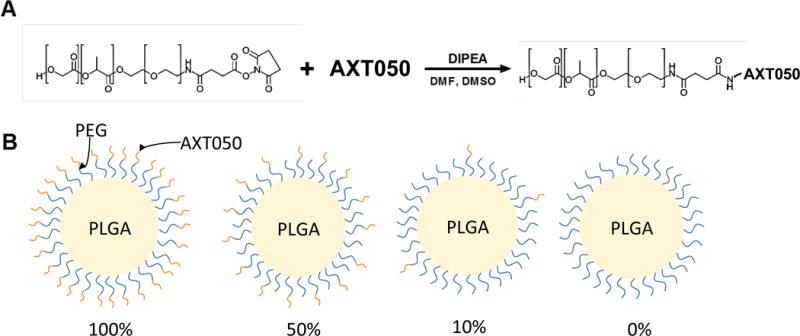
Fabrication of PLGA-PEG-AXT050 nanoparticles. (A) PLGA-PEG-NHS polymer is reacted with AXT050 to form PLGA-PEG-AXT050 conjugates. (B) Nanoparticles are formed by nanoprecipitation utilizing PLGA-PEG-AXT050 and PLGA-mPEG at varied mass ratios including 100%:0%, 50%:50%, 10%:90%, and 0%:100% PLGA-PEG-AXT050: PLGA-mPEG polymer.
NP size was found to be affected by functionalization, with a greater percentage of AXT050 peptide on the surface leading to greater particle size as seen in Figures 3A and 3B. The percentage (e.g. 100%, 50%) represents the percentage of conjugated polymer (PLGA-PEG-AXT050) by mass relative to the total polymer (conjugated polymer and methoxy-terminated PLGA-PEG) used in NP formulation. NP size, as measured by z-average hydrodynamic diameter, ranged from approximately 65 nm for 0% NP to 80 nm for 100% NP. A similar effect of increased size has been seen in other studies, which show that PEG length can have a significant effect on size.38 Increased peptide surface-functionalization also resulted in wider particle size distributions. Zeta potential was observed to be negative (approximately −20 mV) and not significantly affected by the surface peptide content, which may be explained by the overall neutral charge of the peptide (Figure 3C). LavaPep peptide quantification determined that approximately 80% of the theoretical peptide mass was exposed on the surface of the NP, regardless of the ratio of peptide-conjugated polymer to non-conjugated polymer used in the NP formulation.
Figure 3.
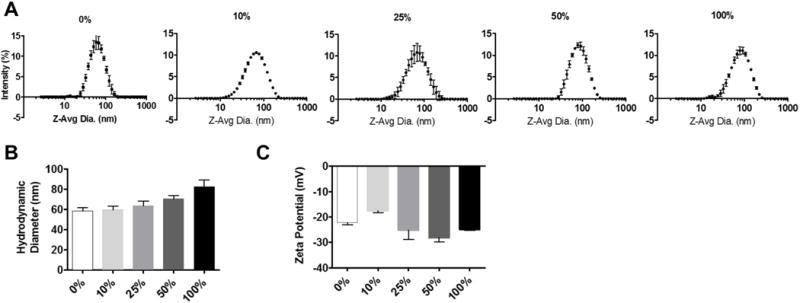
Biophysical characterization of PLGA-PEG-AXT050 nanoparticles. A The PLGA-PEG-AXT050 nanoparticles have a hydrodynamic particle diameter of approximately 65-80 nm. B Hydrodynamic particle diameter increased slightly as the percentage of AXT050 peptide conjugation increased. C Particle zeta potential was determined to be approximately -20 mV, without AXT050 conjugation content significantly affecting the surface charge.
AXT050 targeted nanoparticles bind to cells and recombinant integrin in vitro
In order to confirm the preferential homing of the targeted NPs to integrin, which we found to have a high molecular binding affinity, the NP binding to vascular cells expressing integrins, cancer cells expressing integrins, and free recombinant integrin was evaluated. First, 100% AXT050 peptide-coated PLGA-PEG NPs, which refers to the mass percentage of PLGA-PEG-AXT050 compared to PLGA-mPEG, encapsulated with TAMRA dye were incubated with MECs and triple-negative breast cancer cells, and then spun down to separate free NPs from those bound to cells. As depicted in Figure 4A, cells incubated with surface-functionalized NPs consistently showed approximately 4-fold and 2-fold higher signals to indicate binding compared to control PLGA-PEG NPs without peptide (0%) in MEC and MDA-MB-231 cells, respectively.
Figure 4.
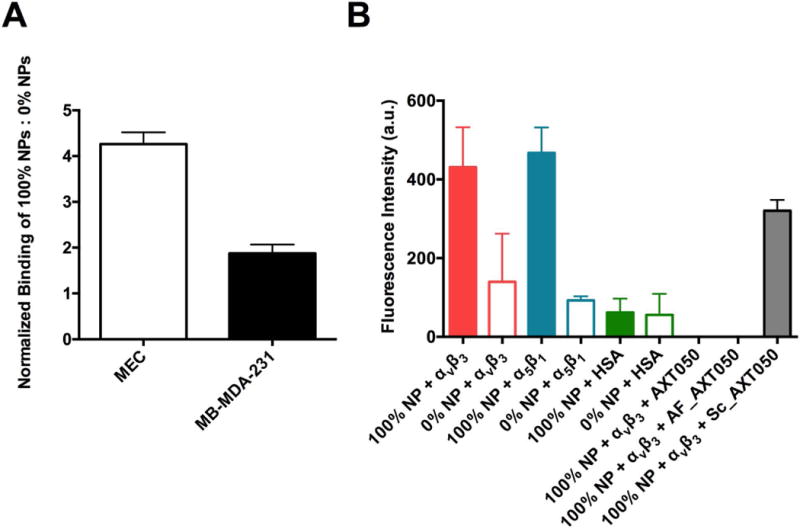
Targeted binding of nanoparticles to cells and integrin molecules. A Binding of targeted (PLGA-PEG-AXT050) 100% NPs to MEC cells and MB-MDA-231 cells in suspension normalized to the binding of untargeted (PLGA-mPEG) 0% NPs. B Binding of targeted 100% NPs and non-targeted 0% NPs to integrin αvβ3, α5β1, and to human serum albumin as a negative control. Binding of targeted 100% NPs to integrin αvβ3 in the presence of a 100-fold excess of AXT050 peptide, an active fragment of AXT050 peptide, and scrambled AXT050 peptide.
When NPs were incubated with labeled αvβ3, α5β1, and human serum albumin (HSA), 100% AXT050-functionalized targeted NPs showed significantly higher binding compared to non-targeted 0% AXT050 NPs with both integrin isoforms (Figure 4B). Critically, NPs were found to have higher binding to the integrins compared to HSA, indicating that the targeted NPs bind preferentially to these integrins with low non-specific interactions. Finally, when 100% NPs were incubated with labeled αvβ3 and a 100-fold excess of AXT050 or an active fragment of AXT050 (AF_AXT050) (Figure 4B), competition from the excess peptide completely prohibited binding of the targeted PLGA-PEG-AXT050 NPs to integrin. In contrast, when an inactive scrambled variant of AXT050 peptide was used as the free competitor rather than active AXT050, signal from the PLGA-PEG-AXT050 NPs binding to labeled integrin persisted. Although interactions between functionalized NPs and additional biomolecules may also be present, the specificity and high-affinity of the binding between the peptide-coated NPs and αvβ3 integrin can enable cellular targeting of the nanoparticles.
Efficacy of targeted nanoparticles in vitro
Human triple-negative breast cancer cells (MDA-MB-231) and primary microvascular endothelial cells (MEC) were used to demonstrate anti-tumorigenic and anti-angiogenic activity of peptide-functionalized and peptide-loaded NPs in vitro with dose response related to both the density of targeting ligand and the presence or absence of encapsulated peptides. Recently, we demonstrated that the naked form of an analog peptide could inhibit the proliferation and adhesion of MDA-MB-231 cells at concentrations of approximately 25-100 μM in vitro.39 At NP concentration of 10 mg/mL, particles showed a peptide dose-dependent inhibition of adhesion on both cell types. NPs were analyzed at varying levels of functionalization and encapsulation as shown in Figure 5A and 5B. Relative to control wells, wells with 100% PLGA-PEG-AXT050 NPs encapsulating 1% (w/w) free AXT050 consistently showed the highest adhesion inhibition on all cell types. This is expected as this condition represents the greatest amount of peptide delivered to the target cells. Increased levels of surface conjugated AXT050 and encapsulated AXT050 increased efficacy in a dose dependent fashion (full statistical results are available in the Supplementary Materials). Whether AXT050 was encapsulated or not, as AXT050 conjugation and display from the NP surface increased (100% > 50% > 10% > 0%), so did the biological activity of the NPs. When comparing surface conjugated AXT050 NPs to free bolus administration of AXT050, it was observed that unloaded 10% PLGA-PEG-AXT050 NPs, which correspond to approximately 40 μM of surface conjugated peptide, showed significantly less (p < 0.001) inhibition of adhesion on both cell types than 25 μM naked bolus AXT050 peptide, suggesting the availability and presentation of the peptide from the NP surface could affect its biological potency. Since the cells are plated in a monolayer, and the peptide is displayed from the nanoparticle’s three-dimensional surface, it is likely that more than half of the displayed peptide could be inaccessible to the cellular integrins due to the peptide’s orientation on the NPs in vitro. However, this effect may become less relevant in vivo as particle breakdown occurs over several days within a tumor microenvironment compared to several hours as measured in in vitro adhesion assays. Decorated particles with no encapsulated peptide that were subject to degradation in PBS solution at 37°C showed increased activity in in vitro adhesion assays using MDA-MB 231 cells compared to intact particles, but decreased activity compared to equal doses of free peptide (Figure S3). The increased efficacy after particle breakdown is likely due to PEG-peptide released from the surface as particles undergo hydrolysis. This is consistent with our findings that PEG-peptide shows adhesion activity against MDA-MB 231 cells, but with less activity as the length of the PEG increases (Figure S4). Degradation of particles in the days following initial administration could result in further efficacy when PEG-peptide fragments and peptide metabolites escape the NPs through hydrolysis and proteolysis respectively. In MDA-MB-231 cells, 50-100% PLGA-PEG-AXT050 NPs encapsulating 1% AXT050 reduced human cancer cell adhesion by 80-81%. In MECs, these two leading NP formulations reduced adhesion in these endothelial cells by 85-87% as well. These results demonstrate that these peptide coated nanoparticles have the biological capability to disrupt the endothelial cells that make up the leaky vasculature supplying oxygen and nutrients to tumors as well as disrupt the human breast cancer cells themselves.
Figure 5.
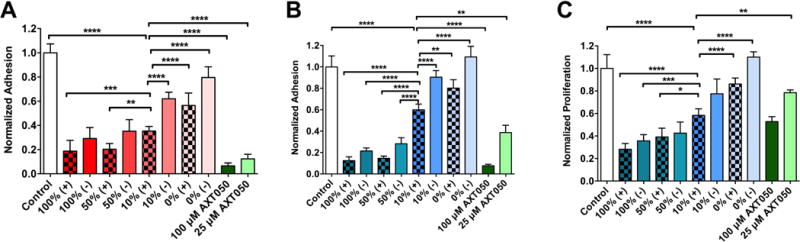
Functional biological activity of NPs composed of 0%-100% PLGA-PEG-AXT050 that contain (+) or do not contain (-) encapsulated AXT050. NPs inhibit adhesion of A MDA-MB231 cells and B MECs. (C) NPs also inhibit the proliferation of MECs normalized to untreated control. One-way ANOVA with Bonferroni post-tests comparing all pairs. Due to space limitations on the figures, representative comparisons with 10% PLGA-PEG-AXT050 encapsulating AXT050 found to be statistically significant are shown in the figures and all other comparisons are shown in the Supplementary Information, (α=0.05; *: p<0.05, **: p<0.01, ***: p<0.001, ****: p < 0.0001).
We also evaluated NP activity against MEC proliferation over 4 days. In addition to adhesion, NPs also inhibit proliferation of MECs with dose dependence of the peptide at the NP concentration of 5 mg/mL. Unlike the adhesion assay, where the effective peptide concentration appeared lower than surface conjugated peptide, likely to do orientation of immobilized peptide, proliferation inhibition more clearly followed the total amount of peptide present in the nanoparticle. As shown in Figure 5C, unloaded 10% and 50% NPs, which have equivalent total peptide concentration of 20 and 100 μM, show comparable levels of proliferation inhibition as 25 and 100 μM bolus naked peptide groups. One possible explanation is particle degradation over the 4-day experiment. Free, soluble peptide becomes available through release of encapsulated peptide, release of PEG-AXT050 via hydrolysis of PLGA,4 or protease-mediated degradation of AXT050 into active fragments. For example, AXT050 conjugated with PEG of two different molecular weights still resulted in adhesion inhibition of MECs at 25 and 100 μM concentrations (Figure S4). This suggests that the combination of PEG-AXT050 and AXT050 metabolites have activity in addition to the activity generated by PLGA-PEG-AXT050 in an intact particle. This provides a mechanism for sustained release of active AXT050 in the tumor microenvironment following initial binding of the PLGA-PEG-AXT050 NPs to overexpressed αvβ3 integrin on tumors and tumor vasculature. In this manner, the surface-conjugated peptide functions as a NP targeting agent, a multivalent surface bound drug agent, and a reservoir for controlled release of drug agent over time. This combined function, along with multimodality at affecting both tumors and tumor vasculature (endothelial cells), is promising for a cancer nanomedicine strategy.
Finally, we investigated the effect of AXT050 peptide surface coating to improve the targeting of PEG-PLGA NPs to mice inoculated with orthotopic human triple-negative breast cancer (MDA-MB-231) (Figure 6A-D). We evaluated the pharmacokinetics of the PLGA-PEG-AXT050 NPs in the blood and the effect of increasing AXT050 conjugation. Figure 6C shows the half-life curves for targeted NPs over approximately 5 hours, after which the signal reached baseline levels. We observed blood half-lives of approximately 110, 103, and 45 minutes for 0%, 10%, and 100% PLGA-PEG-AXT050 NPs, respectively. This extended half-life of nearly 2 hours for 0-10% PLGA-PEG-AXT050 (100-90% PEG-PLGA) NPs clearly demonstrates the beneficial effect of PEGylation at slowing the clearance of these particular NPs from the blood. Half-life was found to decrease with increasing amount of peptide conjugated to the surface of the NP. This decrease in half-life may be a result of increased peptide density obviating the stealth effect of PEG, leading to particle destabilization or increased opsonization and subsequent uptake by the immune system.40 Alternatively, it is possible that a very high density of the peptide on the surface of the NPs could lead to increased avidity and potential off-target binding of the NPs. Thus, maximizing the amount of peptide on the nanoparticle surface does not lead to optimal pharmacokinetics.
Figure 6.
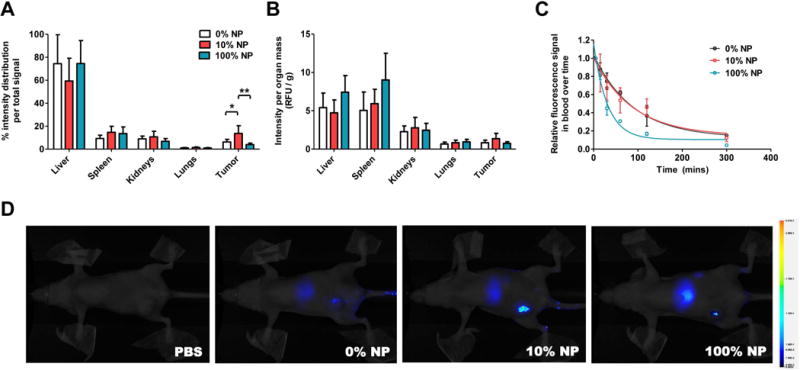
In vivo tumor targeting of PLGA-PEG-AXT050 NPs to human triple-negative breast cancer tumors. A-B PLGA-PEG-AXT050 NPs accumulate in human TNBC tumors and exhibit a biphasic relationship between the level of AXT050 conjugation and TNBC tumor accumulation with 10% PLGA-PEG-AXT050/90% PLGA-mPEG NPs being optimal. C The half-life of the 100% PLGA-PEG-AXT050 NPs was 45 minutes and longer half-lives were observed with 10% NPs (103 min) and 0% NPs (110 min), which were composed of increasing levels of PLGA-mPEG rather than PLGA-PEG-AXT050. D Animal imaging demonstrates the whole body biodistribution of the peptide nanoparticles, with the greatest tumor accumulation being with the 10% NPs.
Interestingly, biodistribution results showed a biphasic trend in tumor accumulation of targeted NPs (Figure 6A-B). Using labeled AXT050 peptide, the fluorescence signal in the tumor for 10% PLGA-PEG-AXT050 (90% PEG-PLGA) NP at 24 h post injection was 14% of the total fluorescence measured in all harvested organs, a 2.2-fold increase compared to non-targeted 0% PLGA-PEG-AXT050 (100% PEG-PLGA) NP and a 3.5-fold increase from 100% PLGA-PEG-AXT050 in this head to head study. Moreover, 10% NP showed 14% lower signal in liver than both 0% and 100% NP. This finding highlights the utility of combining the AXT050 targeting peptide to the surface of the PEGylated NPs. This finding also demonstrates the double-edged sword of using a ligand such as AXT050 for targeting. Too little surface functionalization and the NPs circulate for a long time but without sufficient active targeting to the tissue of interest, and too much active targeting and the NPs are cleared to off-target sites too quickly (Figure 6D). The relatively high amount of the injected dose that reaches the tumor is quite promising when compared to the median 0.9 % targeted NP accumulation of an injected dose to the tumor as has been reported by Wilhelm et al. by analyzing the literature from the past decade.41 Faster clearance by the reticulo-endothelial system of the 100% NPs that has the least PEG shielding could explain both its short half-life and low tumor accumulation. Non-specific accumulation of the NPs occurred in liver, kidneys, and spleen, which are the expected routes of clearance, but minimally in lungs and heart, organs that are desirable to avoid for safety considerations. It is also important to note that no acute systemic toxicity was observed in the animals injected with any formulation of NPs at the doses given and the NPs were chosen so that they were composed of biodegradable and bioeliminable materials.
DISCUSSION
The half-life of PLGA-PEG NPs and factors affecting clearance time in vivo has been widely studied, with half-life increasing significantly, often several orders of magnitude, upon PEGylation of PLGA NP.42 This is thought to decrease recruiting of opsonins on the surface of NPs that cue macrophages for clearance. Other means of elimination include off-target endocytosis, renal clearance, hepatic clearance, splenic filtration, and degradation of the polymer prior to arriving on target. While PEGylation allows longer circulation and potential passive accumulation in leaky vasculature, it does not enable specific binding to tumors or their vasculature.
Biomimetic peptide-PEG-PLGA NPs are a promising method of drug delivery addressing difficulties in both targeting and controlled release. Surface properties of NPs are incredibly important for facilitating successful transport from the site of injection to the tumor site,43 thus targeting ligands used to increase uptake in the tumor must be optimized to prevent clearance prior to reaching the tumor site. Many targeting ligands have been used to target biomarkers to tumors and tumor vasculature, including integrin-targeted cyclic-RGD peptide, folate receptor-targeted folic acid, and prostate-specific membrane antigen-targeted small molecules.44–46 However, since previously reported ligands generally only function as targeting agents, cancer treatments with these particles rely on the delivery of additional therapeutic components such as chemotherapies like paclitaxel, doxorubicin, and docetaxel, that are physically encapsulated at low loading content within these particles. In the current study, we wanted to utilize and evaluate a novel biomimetic molecule on NP surfaces that could function as both a targeting agent and a multi-modal therapeutic agent. By chemically conjugating the peptide to PEG-PLGA polymer, rather than relying on just physical encapsulation, we can increase the loading of the peptide in nanoparticles, enable multivalent display from the surface of the particle to the surface of the cell, and consequently induce a greater therapeutic effect.
The slow off-rate constant and nanomolar KD between the short AXT050 peptide and its cellular target suggests a strong interaction between the AXT050 coated nanoparticles and the surfaces of cancer cells and angiogenic endothelial cells where αvβ3 is overexpressed. Multivalency and avidity between AXT050 presented from the NP surface to integrin αvβ3 clustered on the cellular surface could further enhance the strength of the interaction and duration of effect.47 This could also explain the shorter half-life and reduced tumor accumulation of 100% NP compared to 10% NP, since greater surface exposure of the peptide could result in strong binding of the NPs with integrin receptors at sites with low expression of integrin αvβ3 in addition to the tumor microenvironment where there is high integrin αvβ3.
In vitro targeting assays clearly showed affinity of the targeted NPs for integrin αvβ3, an important upregulated target in tumor neovasculature. In addition, the therapeutic potential of the NPs was also validated through in vitro biological activity assays against human triple-negative breast cancer (anti-tumorigenic) and MEC (anti-angiogenic) cells. We observed activity of the functionalized particles against tumor cells and endothelial cells in short-term adhesion assays through their surface interactions despite the spatial restrictions that this places on the peptide.
In addition, the surface peptide has further activity over time dependent on the hydrolytic breakdown of the particle, similar to release of encapsulated drug. We also demonstrated a strategy for creating stable nanoparticles with a higher loading of therapeutic compound by attaching our therapeutic agent to the surface of our particles in addition to encapsulating it within the particles. We demonstrated that enhanced persistence time in the blood circulation is not dramatically affected when using modestly coated AXT050 targeted particles (10%), but heavily targeted particles (100%) are quickly cleared, resulting in little chance for NPs to effectively accumulate in the tumor site. We hypothesize that this is due to the tradeoff between the targeting properties of the biomimetic peptide and the corresponding loss of stealth properties.
While this work has demonstrated that AXT050 peptide can be to be an effective targeting agent for nanomedicine by increasing the accumulation of NPs to human triple-negative breast cancer tumors significantly while also reducing liver accumulation, clearance and remaining accumulation in the liver and spleen still remain as issues. The non-specific accumulation in the liver and spleen is mostly due to clearance and elimination via the mononuclear phagocyte system (MPS). It is important to note that the peptide, unlike a traditional chemotherapy, is designed to only to affect cells with upregulated integrin αvβ3, in particular tumor cells and their neovasculature, and will therefore trigger a minimal effect on the specific function of these off-target clearance cells. Nonetheless, the potential dose-responsive systemic side effects of such a NP system should be further investigated in multiple preclinical animal models to ensure the safety of this approach.
CONCLUSIONS
We have found that AXT050, a multi-modal peptide with anti-tumorigenic and anti-angiogenic properties, can function as both a targeting ligand and a therapeutic bioactive agent useful for NP-mediated delivery to human triple-negative breast cancer cells. We found that these AXT050-targeted nanoparticles were able to target integrin αVβ3 on the surface of cells in culture, which was confirmed through a series of binding experiments. The AXT050 NPs exhibit in vitro activity in adhesion and proliferation assays against human triple-negative breast cancer MB-MDA-231 cells and MEC endothelial cells, both through surface presentation of peptide and controlled release of the peptide from the nanoparticle. We also determined a biphasic response with peptide surface coating density and tumor targeting in vivo. Optimal 10% PLGA-PEG-AXT050/90% PEG-PLGA nanoparticles were found to exhibit an extended 103 min half-life and accumulate 14% of the peptide dose in human triple-negative breast cancer tumors in an orthotopic xenograft mouse model. As the components of this NP system are each biodegradable and bioeliminable, and as the AXT050 peptide NPs have the potential to target and disrupt both cancer cells and endothelial cells, this technology may be promising for applications in cancer nanomedicine.
Supplementary Material
Acknowledgments
The authors acknowledge and thank Usha Rai for technical assistance with the animal studies.
The authors thank the NIH for funding support (R21CA152473, R01EB022148, U43CA179506). JK thanks Samsung for fellowship support.
ABBREVIATIONS
- PLGA
Poly(lactic-co-glycolic acid)
- PLGA-PEG
Poly(lactic-co-glycolic acid)-block-polyethylene glycol
- NP
nanoparticle
- MEC
microvascular endothelial cell
- EPR
enhanced permeability and retention
- TAMRA
5-carboxytetramethyl-rhodamine
- HSA
human serum albumin
- HPLC
high performance liquid chromatography
Footnotes
SUPPLEMENTARY MATERIALS
Supplementary materials available online contain supplemental figures and the full statistical results from Figure 5. The supplemental figures include biodistribution per organ area for non-targeted NPs, conjugation efficiency of PLGA-PEG-NHS to AXT050 peptide, the anti-adhesion effect of AXT050 in free peptide, intact NP and degraded NP forms, and the effects of AXT050 peptide and peptide-PEG molecules on in vitro adhesion of MECs.
POTENTIAL CONFLICTS OF INTEREST JJG, ASP, NP, RS, and EB report affiliation and equity with the biotechnology company AsclepiX Therapeutics. JJG and ASP are co-founders of AsclepiX Therapeutics and serve as the CTO and CSO respectively. NP, RS, and EB are current/former employees of AsclepiX Therapeutics. Potential conflicts of interest are managed by the Johns Hopkins University Committee on Outside Interest.
References
- 1.Corti A, Pastorino F, Curnis F, Arap W, Ponzoni M, Pasqualini R. Targeted drug delivery and penetration into solid tumors. Med Res Rev. 2012;32(5):1078–91. doi: 10.1002/med.20238. [DOI] [PubMed] [Google Scholar]
- 2.Khawar IA, Kim JH, Kuh HJ. Improving drug delivery to solid tumors: priming the tumor microenvironment. J Control Release. 2015;201:78–89. doi: 10.1016/j.jconrel.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Sriraman SK, Aryasomayajula B, Torchilin VP. Barriers to drug delivery in solid tumors. Tissue Barriers. 2014;2:e29528. doi: 10.4161/tisb.29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–22. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 5.Graf N, Bielenberg DR, Kolishetti N, Muus C, Banyard J, Farokhzad OC, Lippard SJ. alpha(V)beta(3) integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt(IV) prodrug. ACS Nano. 2012;6(5):4530–9. doi: 10.1021/nn301148e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alibolandi M, Ramezani M, Sadeghi F, Abnous K, Hadizadeh F. Epithelial cell adhesion molecule aptamer conjugated PEG-PLGA nanopolymersomes for targeted delivery of doxorubicin to human breast adenocarcinoma cell line in vitro. Int J Pharm. 2015;479(1):241–51. doi: 10.1016/j.ijpharm.2014.12.035. [DOI] [PubMed] [Google Scholar]
- 7.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105(45):17356–61. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–76. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu H, Chen H, Zeng X, Wang Z, Zhang X, Wu Y, Gao Y, Zhang J, Liu K, Liu R, et al. Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance. Biomaterials. 2014;35(7):2391–400. doi: 10.1016/j.biomaterials.2013.11.086. [DOI] [PubMed] [Google Scholar]
- 10.Danhier F, Lecouturier N, Vroman B, Jerome C, Marchand-Brynaert J, Feron O, Preat V. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133(1):11–7. doi: 10.1016/j.jconrel.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 11.Liang C, Yang Y, Ling Y, Huang Y, Li T, Li X. Improved therapeutic effect of folate-decorated PLGA-PEG nanoparticles for endometrial carcinoma. Bioorg Med Chem. 2011;19(13):4057–66. doi: 10.1016/j.bmc.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chopra S, Lim JM, Karnik R, Farokhzad O. Role of Electrostatic Interactions in Protein Loading in PLGA-PEG Nanoparticles. 2014 40th Annual Northeast Bioengineering Conference (Nebec) 2014 [Google Scholar]
- 14.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73(8):2412–7. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danhier F, Le Breton A, Preat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9(11):2961–73. doi: 10.1021/mp3002733. [DOI] [PubMed] [Google Scholar]
- 16.Xie J, Shen Z, Li KC, Danthi N. Tumor angiogenic endothelial cell targeting by a novel integrin-targeted nanoparticle. Int J Nanomedicine. 2007;2(3):479–85. [PMC free article] [PubMed] [Google Scholar]
- 17.Fasehee H, Dinarvand R, Ghavamzadeh A, Esfandyari-Manesh M, Moradian H, Faghihi S, Ghaffari SH. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: in vitro and in vivo investigations. J Nanobiotechnology. 2016;14:32. doi: 10.1186/s12951-016-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Chui WK, Ho PC. Design of a multifunctional PLGA nanoparticulate drug delivery system: evaluation of its physicochemical properties and anticancer activity to malignant cancer cells. Pharm Res. 2009;26(5):1162–71. doi: 10.1007/s11095-009-9837-y. [DOI] [PubMed] [Google Scholar]
- 19.Arosio D, Casagrande C. Advancement in integrin facilitated drug delivery. Adv Drug Deliv Rev. 2016;97:111–43. doi: 10.1016/j.addr.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci U S A. 2008;105(7):2586–91. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moghimi SM, Hunter AC, Andresen TL. Factors controlling nanoparticle pharmacokinetics: an integrated analysis and perspective. Annu Rev Pharmacol Toxicol. 2012;52:481–503. doi: 10.1146/annurev-pharmtox-010611-134623. [DOI] [PubMed] [Google Scholar]
- 23.Xia Q, Li H, Xiao K. Factors Affecting the Pharmacokinetics, Biodistribution and Toxicity of Gold Nanoparticles in Drug Delivery. Curr Drug Metab. 2016;17(9):849–861. doi: 10.2174/1389200217666160629114941. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. Self-assembled lipid–polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–94. doi: 10.1016/j.jconrel.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Mirando AC, Popel AS, Green JJ. Gene delivery nanoparticles to modulate angiogenesis. Adv Drug Deliv Rev. 2016 doi: 10.1016/j.addr.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karagiannis ED, Popel AS. A systematic methodology for proteome-wide identification of peptides inhibiting the proliferation and migration of endothelial cells. Proc Natl Acad Sci U S A. 2008;105(37):13775–80. doi: 10.1073/pnas.0803241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosca EV, Koskimaki JE, Pandey NB, Tamiz AP, Popel AS. Structure-activity relationship study of collagen-derived anti-angiogenic biomimetic peptides. Chem Biol Drug Des. 2012;80(1):27–37. doi: 10.1111/j.1747-0285.2012.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosca EV, Penet MF, Mori N, Koskimaki JE, Lee E, Pandey NB, Bhujwalla ZM, Popel AS. A biomimetic collagen derived peptide exhibits anti-angiogenic activity in triple negative breast cancer. PLoS One. 2014;9(11):e111901. doi: 10.1371/journal.pone.0111901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee E, Lee SJ, Koskimaki JE, Han Z, Pandey NB, Popel AS. Inhibition of breast cancer growth and metastasis by a biomimetic peptide. Sci Rep. 2014;4:7139. doi: 10.1038/srep07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, Green JJ. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small. 2015;11(13):1519–25. doi: 10.1002/smll.201402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhise NS, Shmueli RB, Gonzalez J, Green JJ. A novel assay for quantifying the number of plasmids encapsulated by polymer nanoparticles. Small. 2012;8(3):367–73. doi: 10.1002/smll.201101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haubner R, Wester HJ, Reuning U, Senekowitsch-Schmidtke R, Diefenbach B, Kessler H, Stocklin G, Schwaiger M. Radiolabeled alpha(v)beta3 integrin antagonists: a new class of tracers for tumor targeting. J Nucl Med. 1999;40(6):1061–71. [PubMed] [Google Scholar]
- 36.Decuzzi P, Pasqualini R, Arap W, Ferrari M. Intravascular delivery of particulate systems: does geometry really matter? Pharm Res. 2009;26(1):235–43. doi: 10.1007/s11095-008-9697-x. [DOI] [PubMed] [Google Scholar]
- 37.Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Xiang D, Shigdar S, Yang W, Li Q, Lin J, Liu K, Duan W. Epithelial cell adhesion molecule aptamer functionalized PLGA-lecithin-curcumin-PEG nanoparticles for targeted drug delivery to human colorectal adenocarcinoma cells. Int J Nanomedicine. 2014;9:1083–96. doi: 10.2147/IJN.S59779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosca EV, Koskimaki JE, Pandey NB, Wolff AC, Popel AS. Development of a biomimetic peptide derived from collagen IV with anti-angiogenic activity in breast cancer. Cancer Biol Ther. 2011;12(9):808–17. doi: 10.4161/cbt.12.9.17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mosqueira VC, Legrand P, Gulik A, Bourdon O, Gref R, Labarre D, Barratt G. Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG-modified nanocapsules. Biomaterials. 2001;22(22):2967–79. doi: 10.1016/s0142-9612(01)00043-6. [DOI] [PubMed] [Google Scholar]
- 41.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials. 2016;1(5) [Google Scholar]
- 42.Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Livaniou E, Evangelatos G, Ithakissios DS. Effect of copolymer composition on the physicochemical characteristics, in vitro stability, and biodistribution of PLGA-mPEG nanoparticles. Int J Pharm. 2003;259(1–2):115–27. doi: 10.1016/s0378-5173(03)00224-2. [DOI] [PubMed] [Google Scholar]
- 43.Verma A, Stellacci F. Effect of surface properties on nanoparticle-cell interactions. Small. 2010;6(1):12–21. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 44.Danhier F, Vroman B, Lecouturier N, Crokart N, Pourcelle V, Freichels H, Jerome C, Marchand-Brynaert J, Feron O, Preat V. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with paclitaxel. J Control Release. 2009;140(2):166–73. doi: 10.1016/j.jconrel.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 45.Esmaeili F, Ghahremani MH, Ostad SN, Atyabi F, Seyedabadi M, Malekshahi MR, Amini M, Dinarvand R. Folate-receptor-targeted delivery of docetaxel nanoparticles prepared by PLGA-PEG-folate conjugate. J Drug Target. 2008;16(5):415–23. doi: 10.1080/10611860802088630. [DOI] [PubMed] [Google Scholar]
- 46.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–20. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tassa C, Duffner JL, Lewis TA, Weissleder R, Schreiber SL, Koehler AN, Shaw SY. Binding affinity and kinetic analysis of targeted small molecule-modified nanoparticles. Bioconjug Chem. 2010;21(1):14–9. doi: 10.1021/bc900438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.