

Abstract
Therapeutic cancer vaccines require adjuvants leading to robust type I interferon and proinflammatory cytokine responses in the tumor microenvironment to induce an anti-tumor response. Cyclic dinucleotides (CDNs), a potent Stimulator of Interferon Receptor (STING) agonist, are currently in phase I trials. However, their efficacy may be limited to micromolar concentrations due to the cytosolic residence of STING in the ER membrane. Here we utilized biodegradable, poly(beta-amino ester) (PBAE) nanoparticles to deliver CDNs to the cytosol leading to robust immune response at > 100-fold lower extracellular CDN concentrations in vitro. The leading CDN PBAE nanoparticle formulation induced a log-fold improvement in potency in treating established B16 melanoma tumors in vivo when combined with PD-1 blocking antibody in comparison to free CDN without nanoparticles. This nanoparticle-mediated cytosolic delivery method for STING agonists synergizes with checkpoint inhibitors and has strong potential for enhanced cancer immunotherapy.
Keywords: STING agonist, PBAE nanoparticle formulation, cancer immunotherapy
Graphical abstract
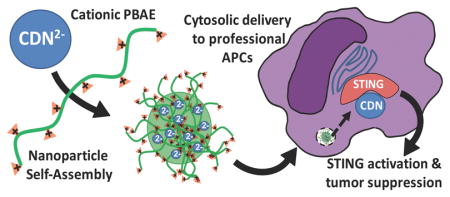
Background
The innate arm of the immune system recognizes pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs) to initiate effective immunological responses, and activation of the innate arm of the immune system can be critically important in priming an effective adaptive response. Cyclic dinucleotides (CDNs) have been demonstrated to be highly effective PAMP molecules capable of robust activation of interferon regulatory factor 3 (IRF3).1 Recently much attention has been focused on STING which localizes to the endoplasmic reticulum, a potent inducer of type I IFNs in response to their binding of CDNs.2 CDNs are small molecule secondary messengers used by bacteria and produced as endogenous products of cytosolic DNA sensing cyclic GMP-AMP synthase (cGAS) that are recognized by STING. Both the canonical 2,3 linked bacterial cyclic di-GMP and cyclic di-AMP, as well as the non-canonical eukaryotic 2,5 linked cGAMP have been shown to directly bind STING and subsequently initiate TBK1-IRF3 and NFkB dependent type I IFN proinflammatory immune responses.3 With the recent clinical success of immune checkpoint inhibitors4–6 in multiple cancer patients and the overall positive correlations between proinflammatory tumor microenvironment and clinical responses to PD-1 blocking antibodies7–10, intratumoral stimulation of STING may potentially synergize with immune checkpoint inhibitors. While CDNs are currently undergoing clinical trials for safety, there are concerns that simple intratumoral CDN injection is a suboptimal means to stimulate the cytosolic STING signaling pathway.
Since CDN molecules are anionic, we investigated whether cationic polymers that have traditionally been used to deliver DNA and siRNA may enable improved cytosolic delivery of CDNs as well. Nanoparticles have been engineered for the intracellular delivery of DNA particles to facilitate efficient transfection in relevant cells while minimizing toxicity. Poly (beta-amino esters) (PBAEs), a class of synthetic, cationic polymers, have been found to be effective as non-viral gene delivery agents for a wide variety of cell types both in vitro and in vivo.11–14 Importantly, PBAEs are relatively easy to synthesize with structural diversity,15 are effective at binding nucleic acids,16 and hydrolytically degradable under physiological conditions, which greatly reduces their cytotoxicity despite being cationic in nature.12,13,17–19 Moreover, they can have specific cellular uptake to target cell types20–22 and can also enable effective endosomal escape to the cytoplasm, likely due to their buffering capacity.19 With these properties we hypothesized that PBAE nanoparticles would improve the cytosolic delivery of anionic CDN molecules, enabling anti-tumor responses at lower CDN doses. We investigated whether PBAEs could enhance CDN delivery to immune cells in vitro, the physical and biological properties of the PBAE/CDN nanoparticles, and their anti-tumor efficacy in vivo.
Methods
Chemicals
All chemicals were of analytical grade and obtained from Sigma Aldrich Chemicals (St Louis, MO, USA). RPMI 1640 medium (Gibco BRL, Grand Island, NY, USA), fluorochrome conjugated antibodies (BD Biosciences, San Jose, CA, USA), MTT Aqueous One cell viability kit (Promega, Madison, WI, USA). Cyclic dinucleotides were provided by Aduro Biotech (Berkeley, CA, USA). QUANTI-blue and THP1-ISG cells were purchased from Invivogen (San Diego, CA, USA). Monocyte isolation kit (Miltenyi Biotec, Auburn, CA, USA) was used to isolate monocytes from peripheral blood mononuclear cells (PBMC)s and Mouse anti-PD-1 antibody (clone G4) was generated in-house using mouse hybridoma cells. Monomers for PBAE synthesis were purchased as specified in the Supplementary Information Table 1.
Polymer Synthesis
Poly(beta-amino ester)s (PBAEs) were synthesized in a two-step Michael addition reaction following protocols previously published.18 The naming scheme for PBAEs used follows that established by Tzeng et al.23 with the first digit denoted by base monomer carbon number, the second digit denoted by sidechain monomer carbon number, and the third digit to describe the polymer’s endcapping group. The PBAE chosen for CDN delivery was synthesized at a molar ratio of 1.1:1 from monomers 1,4-butanediol diacrylate (B4) and 4-amino-1-butanol (S4) and then endcapped with a 0.2 M solution of 1-(3-aminopropyl)-4-methylpiperazine (E7) in anhydrous tetrahydrofuran. PBAEs were then precipitated twice in a 10x volume of anhydrous diethyl ether, isolated via centrifugation at 3,000 rcf and stored under vacuum for two days to remove excess diethyl ether. PBAEs were dissolved in anhydrous DMSO at a concentration of 100 μg/μL and stored at −20°C in individual use aliquots. PBAE molecular weight was determined via gel permeation chromatography (GPC; Waters, Milford, MA) using methods previous published.15,24,25 For 1H NMR, approximately 5 mg of diethyl ether precipitated polymer was dissolved in CDCl3 and NMR spectrum was acquired using a Bruker 500 MHz NMR.
Nanoparticle characterization
CDNs and polymer were diluted in sodium acetate buffer (25 mM, pH 5.0) to the same concentrations as used for the in vitro immunostimulatory treatment. Diluted solutions of CDN and polymer were then mixed in a 1:1 volume ratio and allowed to incubate for 10 minutes to formulate nanoparticles. A Nanosight NS500 (Malvern Instruments, UK) was used to determine number-averaged nanoparticle hydrodynamic diameter. A Zetasizer Nano ZS (Malvern Instruments, UK) was used to determine intensity Z-average hydrodynamic diameter and zeta potential of particles diluted six-fold in 150 mM PBS (pH 7.4). For nanoparticle tracking analysis, nanoparticles were diluted 500 fold in 150 mM PBS (pH 7.4) to give 20–80 particles per frame. Camera and analysis settings were maintained for all comparisons between individually prepared samples. All data points presented are for mean ± standard error of the mean of value of individually prepared samples assessed in triplicate. For transmission electron microscopy (TEM), nanoparticles were formed at a 500 w/w ratio between RR-CDG and PBAE 447 at the same concentration as for in vitro immuno-stimulatory treatments. Ten microliters were then added to a corona plasma treated carbon film 400 square mesh TEM grid and allowed to dry for one hour. TEM images were then acquired using a Philips CM120. Nanoparticles without CDN were prepared in an identical manner, constituting polymer only.
Mice and Cell Culture
Our study was performed in compliance with approved Johns Hopkins Hospital (JHH) Animal Review Board protocol. Female 6–8 weeks old C57BL/6 mice were obtained from The Jackson Laboratory and maintained according to animal care facilities of JHH. THP1-Blue™ ISG (interferon-stimulated genes, Invivogen), THP1 human monocytes were grown at concentrations between 0.2–1.0*106 cells/mL in suspension in vertical T25/75 flasks. THP1cells, B16-F1 melanoma cells, and human monocytes were all cultured in RPM-1640 medium supplemented with 10% FBS and penicillin (100 U/ mL) and streptomycin (100 mg/mL) and maintained in a humidified incubator at 37°C in a 5% CO2 atmosphere. RAW 264.7 murine macrophages were likewise cultured in DMEM medium supplemented with 10% FBS, penicillin (100 U/ mL) and streptomycin (100 mg/mL).
Immunostimulatory treatment
Lipopolysaccharide (LPS) was diluted in fresh media from frozen aliquots of 0.1 μg/μL to the concentration required for the specified well dose. CDNs were stored at −20°C in aliquots at 10 μg/μL and were diluted in 25 mM NaAc, pH 5.0, to the required concentration for the specific well dose to be delivered in 20 μL for all treatments. PBAEs were stored at 100 μg/μL in DMSO at −20°C until used. Nanoparticles were formed by diluting PBAEs in 25 mM NaAc buffer, pH 5.0, which was then mixed in a 1:1 v/v ratio with 25 mM NaAc buffer containing CDNs at the concentration required for the doses specified. The nanoparticle solution was incubated for 10 minutes to allow for particle formation after which for freshly prepared particles, 20 μL were added to each well. For lyophilized particles, after particle formation endotoxin free sucrose from a stock of 600 μg/μL was added to each sample to a concentration of 30 μg/μL. Samples were then frozen at −80°C and lyophilized. Lyophilized samples were stored at −20°C with desiccant and resuspended in ultrapure water before being added to wells for treatment. After 3–5 hours of incubation, treatment media was removed from all wells, cells were washed twice and resuspended in 200 μL fresh media.
Quanti-Blue and Cell Titer Assays
THP1-Blue™ ISG cells were specifically designed to monitor the interferon (IFN) signaling pathway in a physiologically relevant cell line. They derive from the human THP-1 monocyte cell line by stable integration of an IFN regulatory factor (IRF)- inducible SEAP reporter construct. THP1-Blue™ ISG cells express a secreted embryonic alkaline phosphatase (SEAP) reporter gene under the control of an ISG54 minimal promoter in conjunction with five IFN stimulated response elements. As a result, THP1-Blue™ ISG cells allow the monitoring of IRF activation by determining the activity of SEAP. The levels of IRF-induced SEAP in the cell culture supernatant are readily assessed with QUANTI-Blue™, a SEAP detection reagent (Invivogen). The Quanti-Blue assay was used to measure THP1-Blue cell SEAP activity as a means of assessing IRF3 activation. For the assay, Quanti-Blue detection medium was prepared according to manufacturer’s instruction. Following 18h treatment, 20 μL of supernatants were added to 180 μL of Quanti-Blue detection medium in a new 96 well round bottom plate. The Quanti-Blue assay plate was incubated at 37°C for 1–4h and then the absorbance was read at 630 nm. Mean absorbance of a blank well of Quanti-Blue detection media with fresh RPMI media was subtracted to determine raw Quanti-Blue assay values.
Cell viability assay
Cell viability was assessed using an MTT assay one day following treatment. For the assay, MTT reagent was diluted 10-fold into complete medium and incubated with cells in 96 well plates for 2 h. Absorbance was then analyzed at 490 nm using a multi-plate reader (Synergy 2, Biotek), blank-subtracted and normalized to the untreated cell values to give relative cell viability.
Cellular uptake experiments
Cells were plated at a density of 150,000 cells/mL in complete medium with 100 μL of media per well in 96 well plates. PBAE particles labeled with Cy5 were formed with RR-CDG at a 500 w/w ratio, then were added to cells at a dose of 20 μg polymer/well and incubated for 1h at 37°C after which cells were washed with PBS twice and analyzed via flow cytometry. A BD Accuri C6 (BD Biosciences) flow cytometer with two lasers (488 and 633 nm) with four channels corresponding to green, yellow, red, and far-red fluorescence (FL1 at 530 ± 15 nm, FL2 at 565 ± 10nm, FL3 a t610 ± 10 nm and FL4 at 675 ± 12.5 nm, respectively) was used for all flow cytometry experiments in combination with a HyperCyt autosampler (IntelliCyt). Cell counts per well and nanoparticle concentrations in media were equal between cell types. Statistical significance was assessed between the uptake levels of the treated cells by one-way ANOVA with multiple comparisons corrected for by Tukey’s method.
For confocal microscopy experiments, PBMCs transfected in suspension in 96 well round bottom plates as described above were stained with Hoechst 33342 (H3570; Thermo Fisher) to label nuclei. Cells were resuspended in live cell imaging solution and transferred to 8 well Nunc Lab-Tek chambered borosilicate coverglass well plates (155411; Thermo Fisher) at 37,500 cells/well for imaging. Images were acquired using a Zeiss LSM 780 microscope with Zen software and 63 oil immersion lens. Specific laser channels used were 405 nm diode, 488 nm argon, 561 nm solid-state, and 639 nm diode lasers.
Tumor growth and Measurement
For in vivo therapeutic experiments, C57BL/6 mice were injected with B16-F1 melanoma cells at a concentration of 2*105 cells/100 μL in the flank. The mice were grouped (n=7) according to the experimental plan. Post days 3, 6, 9 and 12 post-tumor inoculation, mice were treated intratumorally with HBSS, CDN in HBSS, resuspended empty PBAE nanoparticles or resuspended PBAE + CDN nanoparticles via 50 μL intratumoral injection. For anti-PD-1 groups, 100 μg was injected twice weekly on days 3, 7, 10, 14 and 17 after mice were inoculated with tumors.
Statistical analysis
Figures present mean ± standard error of the mean calculated from three replicates unless otherwise noted. All the above experiments were repeated at least twice whereas for experiments with 96 well plates used either three or four well replicates. Statistical significance was designated as follows: ****p<.0001; ***p<.001; **p<.01; *p<.05. Statistical analysis was assessed using Graphpad Prism software (La Jolla, CA) at a global alpha value of 0.05.
Results
CDNs are potent activators of IRF3 at micromolar doses
Recent publications have shown CDNs to be potent activators of the adaptive immune system via activation of type I IFN and proinflammatory cytokine response leading to potent efficacy in multiple models of established cancer.1 As a result, we selected the CDNs shown in Figure 1A to act as adjuvant molecules for studies in vitro. ML-RR-CDA is a modified version of cyclic-di-AMP formulated for increased in vivo stability and human STING activation, while RR-CDG was a phosphodiesterase resistant version of cyclic-di-GMP (Figure 1A).1 Since the anti-tumor efficacy of CDN is partly mediated through activity in antigen-presenting cells (APCs),26 we optimized PBAE formulated CDN nanoparticles in the human monocyte cell line, THP1-Blue. The THP1-Blue human monocyte cell line used for in vitro screening expresses the reporter gene secreted embryonic alkaline phosphatase (SEAP) driven by the IRF3 promoter activation that is activated by ligand binding to the cytosolic protein Stimulator of interferon gene (STING). THP1-Blue cells were shown to respond strongly to extracellular concentrations of 10 μM CDN following a four hour incubation time, while showing minimal response at or below the level of 1000 nM concentrations of CDN (Figure 1B/C).
Figure 1. CDNs are potent immune activators at μM concentrations.

(A) STING activating CDNs ML-RR-CDA and RR-CDG modified for increased stability were used in all studies. (B) ML-RR-CDA was a potent activator of immune response down to 10 μM extracellular concentrations, while (C) RR-CDG was effective down to 10 μM concentrations when assessed using the THP1-Blue cell line with Quanti-Blue assay to detect IRF3 promoter activity.
PBAE nanoparticles improve the efficiency of CDN delivery to human monocytes in vitro
An initial array of biodegradable PBAE structures were assessed for their ability to increase STING activation in the human monocyte cell line (THP1-Blue) at low extracellular doses of CDN (Supplementary Figure 1). These structures were selected for having previously been shown to be effective for delivery of plasmid DNA or RNA molecules, which are notably larger and more electronegative molecules than CDNs.13,27 While both traditional hydrophobic PBAE and the disulfide backbone polymer tested were capable of improving the efficiency of CDN delivery to human monocytes and STING activation at low overall CDN doses, they had differential activity. For example, polymers PBAE 446 and PBAE R647 generally had less than half the activity of the lead polymer PBAE 447. On the other hand, polymer 537 demonstrated significant cytotoxicity, even at a relatively low dose of 31.25 nM, whereas the other PBAEs evaluated did not. Overall, PBAE 447 (Figure 2A), a structure containing 4 carbons between acrylate groups and 4 carbons between its amine group and its hydroxyl group, was selected for its balance of being most effective at IRF3 activation while maintaining high cell viability among the structures evaluated.
Figure 2. PBAE 447 synthesis and characterization.

(A) PBAEs were synthesized from small molecule monomers B4 and S4 in a Michael addition reaction to yield the base polymer B4S4. The acrylate terminated polymer was then end-capped with small molecule E7 to yield PBAE 447. (B) PBAE nanoparticles assessed using NTA had a hydrodynamic diameter distribution with a mean and SEM of 110 ± 41 nm. (C) TEM of PBAE+CDN nanoparticles at a w/w ratio of 500 showed dried diameters mean and SEM of 77 ± 5 nm. Scale bar 100 nm. (D) Nanoparticle diameter assessed by dynamic light scattering (DLS) Z-average diameter, nanoparticle tracking analysis (NTA) and transmission electron microscopy (TEM) showed varying nanoparticle diameters. Error bars show mean + SEM of three independently prepared samples (DLS and NTA) or 20 nanoparticles (TEM). (E) Zeta potential mean and SEM of 9.1 ± 1.3 mV for nanoparticles formed at a 500 w/w ratio with CDN in 150 mM PBS. PBAE nanoparticles with CDN had a statistically higher positive zeta potential (*P<0.05).
PBAE 447 polymer and PBAE 447/CDN nanoparticle physical characterization
PBAEs were synthesized via Michael addition reaction as our lab has described15 and the synthesis scheme is shown in Figure 2A. PBAE 447, selected as optimal for CDN delivery, was measured to have MN 10,700 g/mol, MW 38,200 g/mol and a polydispersity of 3.58 via gel permeation chromatography against polystyrene standards. Analysis via 1H NMR of the synthesized acrylate and endcapped polymers showed that endcapping was effective (as demonstrated by the disappearance of the acrylate peaks); the resulting endcapped polymers had an MN of 7 kDa (Supplementary Figure 2). Nanoparticles formed from PBAE 447 fabricated at a w/w ratio of 500:1 with CDN were characterized using Dynamic Light Scattering (DLS), Nanoparticle Tracking Analysis (NTA), and transmission electron microscopy (TEM) (Figure 2), demonstrating that they had a diameter of approximately 100 nm and slightly positive zeta-potential of approximately +10 mV. The discrepancy between hydrodynamic diameter reported by DLS and NTA has been noted previously for polyplex nanoparticles25 where larger polyplex nanoparticles are over-represented in hydrodynamic measurements calculated by an intensity-averaged measurement (DLS) compared to a number-averaged measurement (NTA). In addition, the nanoparticles were observed to have smaller diameters in dry conditions (TEM) than in hydrated conditions, when hydrodynamic diameters were calculated. Notably, unlike some polyplex nanoparticles such as polyethlyenimine, this particular PBAE structure forms nanoparticles via the hydrophobic effect even in the absence of any anionic species for complexation. Although the observed means appeared slightly higher between the diameters of PBAE nanoparticles with the small CDNs compared to those without CDNs as assessed by DLS, NTA, and TEM, no statistically significant differences were found between these measurements. The zeta potential of PBAE nanoparticles with the small CDNs was found to be slightly statistically significantly higher than those without them, presumably because the CDNs would electrostatically attract additional cationic PBAE polymer, which could coat the CDNs and increase cationic surface charge.
PBAE 447/CDN nanoparticle biological optimization
Following selection of PBAE 447 as the lead structure, we optimized the dose of PBAE added per well (Figure 3A and 3B), resulting in a optimized does of 20 μg for PBAE 447. PBAE 447 was highly effective for delivery of ML-RR-CDA, giving equivalent IRF3 activation to free CDN at a 100-fold lower dose of dinucleotide (Figure 3C and 3D). ML-RR-CDA was also shown to be a significantly more potent activator than RR-CDG in the THP1-Blue cells at lower doses (Figure 3E and 3F) and was selected for additional studies.
Figure 3. Optimization of PBAE 447 delivery of CDN in vitro.

The dose per well of PBAE polymer 447 doses was screened for (A) efficacy of CDN delivery and (B) cell viability. A dose of 20 μg/well was selected for future in vitro experiments to balance efficacy and cytotoxicity. (C) PBAE 447 was able to improve IRF-3 activation at over 100-fold lower extracellular doses compared to free ML-RR-CDA, with minimal effect on (D) cell viability. CDN molecules, ML-RR-CDA and RR-CDG were then tested directly for STING (E) activation and (F) cell viability in vitro, showing significantly higher activation for ML-RR-CDA over RR-CDG in THP1-Blue human monocytes at lower concentrations of CDN when tested with Holm-Sidak corrected multiple T-tests between groups. (****P<0.0001, **P<0.01, *P<0.05). Error bars show mean +/− SEM of three wells.
PBAE nanoparticles are highly effective for immune cell uptake
Currently, ML-RR-CDA are being developed for clinical purposes as an intratumoral injectable adjuvant. We hypothesized that PBAE-CDN nanoparticles would enhance the potency of CDNs by selective, efficient cellular uptake into immune cells in the tumor microenvironment. As a result we first examined the efficiency of nanoparticle uptake into monocytes and macrophages as compared to tumor cells in vitro. PBAE 447/CDN nanoparticles fluorescently labeled with Cy5 were shown to be taken up very effectively by THP1 human monocytes and RAW 264.7 murine macrophages, showing highly significant uptake over B16-F1 tumor cells cultured and treated under the same conditions (Figure 4A). Nanoparticle uptake into primary human myeloid cells was also noted, with over 90% of human donor monocytes showing nanoparticle uptake with no significant differences between the degree of uptake between separate donor samples (Figure 4B). Additionally, fluorescently labeled 447/CDN nanoparticles were clearly internalized following one hour incubation with human donor monocytes as evaluated by confocal microscopy (Figure 4C).
Figure 4. PBAE/CDN nanoparticles show selective and effective uptake by monocyte and macrophage populations.

(A) PBAE 447+RR-CDG nanoparticles were internalized by THP-1 human monocytes and RAW 264.7 murine macrophages more effectively than B16-F1 murine melanoma cells. Statistics performed as One-way ANOVA with Dunnett corrected multiple comparisons to B16-F1 uptake (****P<0.0001). (B) PBMCs from different donors were shown to take up CDN nanoparticles effectively, with no significant differences between donors. Error bars show mean +/− SEM of the geometric mean uptake of four wells. (C) Confocal microscopy shows RR-CDG+447-Cy5 labeled nanoparticles (green) effectively internalized by PBMCs. Nuclei stained with Hoechst (blue).
PBAE+CDN nanoparticles maintain efficacy post-lyophilization and can be stably stored for at least 9 months
To facilitate long-term storage of the hydrolysable nanoparticles, lyophilization studies were performed following a protocol that we developed for plasmid DNA nanoparticles28 to determine if similar stability could be achieved for CDN nanoparticles PBAE+CDN nanoparticles were mixed with sucrose to give a final concentration of 30 mg/mL sucrose, then frozen at −80°C and lyophilized. The particles were then rehydrated with water and tested to compare their relative efficacy to fresh nanoparticles. Lyophilized and stored nanoparticles that showed no detectable changes in efficacy or cell viability compared to the freshly prepared nanoparticles (Figure 5A and 5B). Impressively, the lyophilized nanoparticles remained highly effective for IRF3 activation in THP1-Blue cells following 9 months of storage at −20°C in containers with desiccant (Figure 5C).
Figure 5. PBAE/CDN nanoparticles are stable following lyophilization and storage.

Lyophilization had no effect on the (A) efficacy or (B) cell viability of PBAE+CDN nanoparticles when pre-mixed with sucrose as a cryoprotectant. Results analyzed by Holm-Sidak corrected multiple t-tests. (C) Lyophilized PBAE+CDN nanoparticles formulated at a dose of 20 μg polymer and 62.5 nM CDN remained highly effective at IRF3 activation for at least 9 months following lyophilization when stored at −20°C.
Intratumoral injections of CDN are highly effective at reducing tumor growth in vivo
Checkpoint inhibition with anti-PD-1 antibodies has become the standard of care for many cancers, but there remains a need to improve upon checkpoint inhibition alone.29 One attractive approach is combination immunotherapy.30 To investigate whether PBAE nanoparticle CDN formulations might offer value in the setting of combination immunotherapy, we examined whether co-administration of PBAE+CDN nanoparticles with PD-1 antibody would offer a survival benefit vs combination immunotherapy with free CDN alone in a melanoma treatment model in mice. B16-F1 murine melanoma tumors were established in C57BL/6 mice with subcutaneous flank injections of 200,000 cells. Starting on day three, intratumoral injections were performed every three days following the protocol outlined in Figure 6A, with the final injections on day 12. Administration of 2 μg PBAE+CDN nanoparticles with anti-PD-1 antibody resulted in significantly reduced tumor growth compared to combination immunotherapy with unencapsulated CDN 2 μg dose or empty nanoparticles (Figure 6B, P<0.0001 for the comparison). Additionally, when combined with PD-1 blockade, there was no statistically significant difference between treatment with 2 μg PBAE+CDN nanoparticles with anti-PD-1 antibody vs CDN 20 μg high dose therapy (Figure 6B) although the high bolus dose of 20 μg CDN was more apparently effective at completely eliminating tumors.
Figure 6. PBAE+CDN NP therapy reduces tumor growth in vivo in the presence of anti-PD-1.

(A) Tumor growth rate study of B16-F1 melanoma tumors injected subcutaneously in C57BL/6 mice. Intratumoral injections were started on day three when tumors were palpable in all animals. (B) Twice weekly intraperitoneal administration of anti-PD-1 with the nanoparticle formulation treatment (NP + CDN 2 μg) resulted in statistically reduced tumor growth compared to CDN 2 μg without nanoparticles when compared with Holm-Sidak corrected multiple t-tests. All error bars show mean ± SD of five animals.
Discussion
CDNs are highly potent adjuvants that can impact clinical cancer immunotherapy, leading to robust activation of the IRF3 transcription factor through STING, which is capable of triggering tumor regression when activated in the right context.1,26,31 With promising pre-clinical results reported to date, CDNs are currently under investigation in the FDA Phase I clinical trials for advanced metastatic solid tumors and lymphomas (NCT02675439) by Aduro Biotech working with Novartis to determine the tolerable dosing window.31 A clinical trial of intratumoral injected CDN as a combination therapy with anti-PD-1 is likewise underway in 2017 (NCT03172936). With this rapid advance the clinic, however, there are concerns regarding their narrow therapeutic window.7,10 This can be partly attributed to poor uptake and intracellular delivery of the CDNs into the cytosol of the immune cells in the tumor microenvironment and the need to use high doses to achieve efficacy.
Taking this into consideration, we hypothesized that CDN therapy could be improved at lower overall doses using a nanomedicine formulation that was biodegradable, enabled enhanced cytosolic delivery, and targeted professional APCs. The ER bound STING receptor in APCs does not have access to exogenous CDNs until they either reach the cytosol or are internalized to the endosomal space of activated cells.31,32 Because CDN molecules are both low molecular weight and water soluble due to their anionic charge, they are prone to rapid diffusion from the site of the tumor and lack efficient targeting to antigen presenting cells at the tumor site; this could potentially lead to off-target effects, resulting in overstimulation or autoimmunity side-effects that have been problematic for some patients in the case of other immunotherapies.33
Here we showed that delivery of CDNs via nanomedicine formulation with cationic and biodegradable PBAEs improves their ability to activate IRF3 in vitro at >100 fold lower extracellular concentrations. The polymeric PBAE/CDN nanoparticles were shown to have a slightly positive surface charge and a particle diameter of approximately 100 nm. Self-assembled PBAE nanoparticles have previously been used for delivery of plasmid DNA, minicircle DNA, and siRNA but have not been previously evaluated for delivery of smaller molecules, including cyclic dinucleotides to immune cells in the tumor microenvironment.11,14,17,34
While the small size of CDN molecules impedes the ability to measure their direct cellular uptake as they cannot be fluorescently labeled without affecting their chemical properties, their net negative charge (2−) should enable cationic PBAEs to effectively encapsulate them. We showed that fluorescently labeled PBAE nanoparticles were efficiently internalized to the THP1 human monocyte cell line as well as three human donor monocyte samples, with greater uptake than tumor cells under the same conditions. This degree of selective uptake may be attributable to the endcap utilized for the polymer tested, which has been shown to be able to convey both cell type specificity and endosomal uptake mechanism specificity.18,35,36
STINGVAX formulations incorporating CDN previously were shown to greatly reduce tumor growth and resulted in complete remission of some tumors for CDN doses of 20 μg, but had limited efficacy at lower CDN doses.1 Here we showed that a nanoparticle formulation allows for an order of magnitude reduction in the necessary dose to eliminate established poorly immunogenic B16-F1 when administered as a combination therapy with the checkpoint inhibitor anti-PD-1 antibody. This strategy may have implications for pursuing clinical approval of these drug molecules in the future if identification of effective dosing regimens proves to be challenging in current clinical trials. Moving towards this goal, we demonstrated the application of lyophilization and long-term storage over 9 months without loss of efficacy for this nanomedicine formulation. In addition to facilitating the translational potential of this therapy, this nanoparticle formulation consisted of only two components, unlike some previous STING based therapies incorporating CDN, attenuated tumor cells, and cytokines that could face significant manufacturing and regulatory hurdles and expense. The manufacturing of these nanoparticles, via mixing of two charged components, is amenable to continuous manufacture by a device like the microfluidic device as we have recently described for assembly of PBAE/DNA nanoparticles.28 Thus, biodegradable STING agonist nanoparticles composed of PBAE 447/CDN are promising for enhanced cancer immunotherapy.
Supplementary Material
Acknowledgments
Funding support:The authors thank the NIH for support of this research (NIH R01EB016721, R01EB022148, and R01CA195503; Wilmer Core Grant (5P30EY001765); NIH S10 OD016374; NIH R01CA178613). The authors thank the Bloomberg-Kimmel Institute for Cancer Immunotherapy for support and thank the Johns Hopkins University for support from a Discovery Award. DRW thanks the NSF for a Graduate Research Fellowship (DGE-0707427). YJK thanks the Baker Lab for support.
The authors thank Dr. Kristen Kozielski for the synthesis of polymer PBAE R647 used in the screening study.
Abbreviations
- (CDN)s
Cyclic dinucleotides
- STING
Stimulator of Interferon Receptor
- PBAEs
poly(beta-amino ester)s
- IFN
interferon
- SEAP
secreted embryonic alkaline phosphatase
- cGAS
cyclic GMP-AMP synthase
- PAMPs
pathogen-associated molecular patterns
- IRF3
interferon regulatory factor 3
- PBMCs
peripheral blood mononuclear cells
- B4
1,4-butanediol diacrylate
- S4
4-amino-1-butanol
- E7
1-(3-aminopropyl)-4-methylpiperazine
- TEM
transmission electron microscopy
- ISG
interferon-stimulated genes
- LPS
Lipopolysaccharide
- NaAc
sodium acetate
- SEAP
secreted embryonic alkaline phosphatase
- NTA
Nanoparticle Tracking Analysis
References
- 1.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Science translational medicine. 2015;7:283ra52–283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubensky TW, Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Therapeutic advances in vaccines. 2013;1:131–143. doi: 10.1177/2051013613501988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. New England Journal of Medicine. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brahmer JR, Pardoll DM. Immune checkpoint inhibitors: making immunotherapy a reality for the treatment of lung cancer. Cancer immunology research. 2013;1:85–91. doi: 10.1158/2326-6066.CIR-13-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. New England Journal of Medicine. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. The lancet oncology. 2016;17:956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 9.Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;2012:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, et al. Overall survival and long-term safety of nivolumab (anti–programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non–small-cell lung cancer. Journal of clinical oncology. 2015;33:2004–2012. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynn DM, Langer R. Degradable Poly(β-amino esters): Synthesis, Characterization, and Self-Assembly with Plasmid DNA. Journal of the American Chemical Society. 2000;122:10761–10768. [Google Scholar]
- 12.Tzeng SY, Guerrero-Cázares H, Martinez EE, Sunshine JC, Quiñones-Hinojosa A, Green JJ. Non-viral gene delivery nanoparticles based on poly(β-amino esters) for treatment of glioblastoma. Biomaterials. 2011;32:5402–10. doi: 10.1016/j.biomaterials.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mangraviti A, Tzeng SY, Kozielski KL, Wang Y, Jin Y, Gullotti D, et al. Polymeric Nanoparticles for Nonviral Gene Therapy Extend Brain Tumor Survival in Vivo. ACS Nano. 2015;9:1236–1249. doi: 10.1021/nn504905q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang F, Cho S-W, Son SM, Bogatyrev SR, Singh D, Green JJ, et al. Genetic engineering of human stem cells for enhanced angiogenesis using biodegradable polymeric nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3317–22. doi: 10.1073/pnas.0905432106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunshine JC, Akanda MI, Li D, Kozielski KL, Green JJ. Effects of base polymer hydrophobicity and end-group modification on polymeric gene delivery. Biomacromolecules. 2011;12:3592–600. doi: 10.1021/bm200807s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop CJ, Ketola TM, Tzeng SY, Sunshine JC, Urtti A, Lemmetyinen H, et al. The effect and role of carbon atoms in poly(beta-amino ester)s for DNA binding and gene delivery. J Am Chem Soc. 2013;135:6951–7. doi: 10.1021/ja4002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozielski KL, Tzeng SY, Green JJ. A bioreducible linear poly(β-amino ester) for siRNA delivery. Chemical communications (Cambridge, England) 2013;49:5319–21. doi: 10.1039/c3cc40718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sunshine JC, Peng DY, Green JJ. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Molecular pharmaceutics. 2012;9:3375–83. doi: 10.1021/mp3004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sunshine JC, Peng DY, Green JJ. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol Pharm. 2012;9:3375–83. doi: 10.1021/mp3004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shmueli RB, Sunshine JC, Xu Z, Duh EJ, Green JJ. Gene delivery nanoparticles specific for human microvasculature and macrovasculature. Nanomedicine. 2012;8:1200–7. doi: 10.1016/j.nano.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamboni CG, Kozielski KL, Vaughan HJ, Nakata MM, Kim J, Higgins LJ, et al. Polymeric nanoparticles as cancer-specific DNA delivery vectors to human hepatocellular carcinoma. J Control Release. 2017;263:18–28. doi: 10.1016/j.jconrel.2017.03.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bishop CJ, Majewski RL, Guiriba TR, Wilson DR, Bhise NS, Quinones-Hinojosa A, et al. Quantification of cellular and nuclear uptake rates of polymeric gene delivery nanoparticles and DNA plasmids via flow cytometry. Acta Biomater. 2016;37:120–30. doi: 10.1016/j.actbio.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzeng SY, Green JJ. Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for siRNA and DNA delivery to human brain cancer. Advanced healthcare materials. 2013;2:468–80. doi: 10.1002/adhm.201200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tzeng SY, Hung BP, Grayson WL, Green JJ. Cystamine-terminated poly(beta-amino ester)s for siRNA delivery to human mesenchymal stem cells and enhancement of osteogenic differentiation. Biomaterials. 2012;33:8142–51. doi: 10.1016/j.biomaterials.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhise NS, Gray RS, Sunshine JC, Htet S, Ewald AJ, Green JJ. The relationship between terminal functionalization and molecular weight of a gene delivery polymer and transfection efficacy in mammary epithelial 2-D cultures and 3-D organotypic cultures. Biomaterials. 2010;31:8088–96. doi: 10.1016/j.biomaterials.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell reports. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozielski KL, Tzeng SY, De Mendoza BAH, Green JJ. Bioreducible cationic polymer-based nanoparticles for efficient and environmentally triggered cytoplasmic siRNA delivery to primary human brain cancer cells. ACS nano. 2014;8:3232–41. doi: 10.1021/nn500704t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson DR, Mosenia A, Suprenant MP, Upadhya R, Routkevitch D, Meyer RA, et al. Continuous microfluidic assembly of biodegradable poly(beta-amino ester)/DNA nanoparticles for enhanced gene delivery. J Biomed Mater Res A. 2017;105:1813–1825. doi: 10.1002/jbm.a.36033. [DOI] [PubMed] [Google Scholar]
- 29.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 30.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer. 2017;5:16. doi: 10.1186/s40425-017-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corrales L, McWhirter SM, Dubensky TW, Gajewski TF. The host STING pathway at the interface of cancer and immunity. The Journal of clinical investigation. 2016;126:2404–2411. doi: 10.1172/JCI86892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gangadhar TC, Vonderheide RH. Mitigating the toxic effects of anticancer immunotherapy. Nature Reviews Clinical Oncology. 2014;11:91–99. doi: 10.1038/nrclinonc.2013.245. [DOI] [PubMed] [Google Scholar]
- 34.Little SR, Lynn DM, Ge Q, Anderson DG, Puram SV, Chen J, et al. Poly-beta amino ester-containing microparticles enhance the activity of nonviral genetic vaccines. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9534–9. doi: 10.1073/pnas.0403549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunshine J, Green JJ, Mahon KP, Yang F, Eltoukhy Aa, Nguyen DN, et al. Small-Molecule End-Groups of Linear Polymer Determine Cell-type Gene-Delivery Efficacy. Advanced Materials. 2009;21:4947–4951. doi: 10.1002/adma.200901718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J, Sunshine JC, Green JJ. Differential polymer structure tunes mechanism of cellular uptake and transfection routes of poly(β-amino ester) polyplexes in human breast cancer cells. Bioconjugate chemistry. 2014;25:43–51. doi: 10.1021/bc4002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.