

Introduction
Over the last several decades, many codes of research ethics have been written, and widely disseminated. Medical and professional societies have developed specialty-oriented guidelines and policies to ensure the ethical treatment of research participants and the appropriate conduct of investigators.1 Although sometimes falling short,2 Institutional Research Boards (IRBs) and biomedical ethics committees are tasked with assessment of proposed human subjects research so that investigators acknowledge research actions that might increase the risk of harm to research participants and strategize to minimize research-related risks. For the clinician investigator, the overarching message is that ethical research provides safeguards against research-related harm, respects the autonomy of the individual, and ensures that the burdens, risks, and potential benefits of research are fairly distributed.
This paper will discuss decisional capacity and vulnerabilities and how these characteristics must be recognized and addressed in the clinical research process. I will review the concept of informed consent, especially in light of decisional capacity and vulnerability, and also describe the limitations of the current informed consent processes from the standpoint of the clinician researcher. Finally, I will advance a theoretical model to stimulate further consideration of the effectiveness of the informed consent process under certain clinical circumstances.
Informed Consent for Research Participation
For the clinician scientist, the ethical principles that are the pillars of ethical research can sometimes be difficult to implement in the course of the planning and execution of a clinical trial. For example, as stated in the Belmont Report, respect for persons in the research process recognizes that individuals have the right to determine what happens to their person (i.e., autonomy), and persons with diminished autonomy (i.e., who lack capacity for self-determination) must be afforded special protections.3
Obtaining informed consent for research participation is one method that attempts to secure these ethical rights for potential research participants.4 Informed consent for research participation seeks to obtain an individual’s authorization of or refusal for enrollment into a clinical research trial.5 A well-conducted informed consent process should ensure subject autonomy and address potential vulnerability when potential research participants are asked to participate in a clinical trial.6 From a practical standpoint, this implies that the informed consent process is truly informed, the potential participant comprehends the study and its personal impact, and that person has decisional capacity to consent to research. Yet the process of informed consent as often practiced is geared toward meeting regulatory requirements and burdened by legal concerns and excessive details that may obscure the aspects of a study most directly related to the risks and benefits of subject participation.7 Informed consent is often considered to be a document and not a process, and may not involve the robust two-way communication that may be needed to ensure adequate contemplation and comprehension on the part of a research subject.8 Capacity determination is not always considered by investigators or described in research protocols, and diminished or fluctuating capacity of potential research participants is often not recognized by clinician investigators.9 Many aspects of the informed consent process have room for improvement.
Decisional Capacity For Research
Decisional capacity to consent to research is the ability of a potential research subject to understand and logically process the information that is necessary to make an informed decision regarding study participation.10 The capacity to make an informed consent decision requires both cognitive and emotional ability, and should be considered in the context of the specific proposed research.11 Research decisional capacity is not determined by a patient’s diagnosis or membership in a particular group, but by characteristics unique to that person. For example, a homeless patient with a mental illness may be easily managing his daily affairs; his diagnosis and presumed membership in a socially disadvantaged group may suggest impaired research decision making capacity, where none exists.12 Conversely, capacity is related to context; capacity to make daily decisions does not guarantee capacity to deal with more complex concepts, such as treatment or research decisions.13 Research decisional capacity is not static; capacity may fluctuate over time for several reasons, for example, as part of the natural aging process, as a consequence of emotional upheaval, or as the natural progression of a disease state.14
Decision-making capacity involves specific thought domains. Obviously the decision to participate in a research study requires participant understanding of the nature of the study. However, comprehension of study materials does not adequately assess the potential participant’s decision-making capacity, even though clinician scientists often use comprehension as a marker of decision-making capacity.15 When comprehension is confused with capacity, the assumption is that the decision to participate in research is valid; in fact, comprehension may simply represent recall of the presented information,16 and not the ability to recognize the personal consequences of research involvement on the participant’s life.17
Beyond comprehension, the capacity to make decisions requires the ability to conceptualize and compare the consequences of research involvement or non-involvement (reasoning), appreciation that the goals of research do not necessarily include direct personal benefits, appreciation of how involvement or non-involvement may directly impact the individual, and the ability to express a logical choice.18
Research decisional capacity therefore is required for valid informed consent for research participation. Because decision-making capacity is frequently overestimated by clinician scientists,19 formal capacity assessment of potential participants may be required by IRBs when there is any concern about diminished or fluctuating capacity.20
Capacity Assessment
Guidance on when to assess capacity, what tools to use, and how to document the findings is usually provided by IRBs.21 What is key for the investigator is to recognize when an assessment appears needed, to have methods available to provide a capacity assessment when indicated, and to have protocols in place to act on the findings. For example, if assessment indicates impaired research decisional capacity, will surrogate consent be sought or will that individual be excluded from the study? Figure 1 illustrates a method clinician investigators could use, once potential incapacity is suspected.
Figure 1.
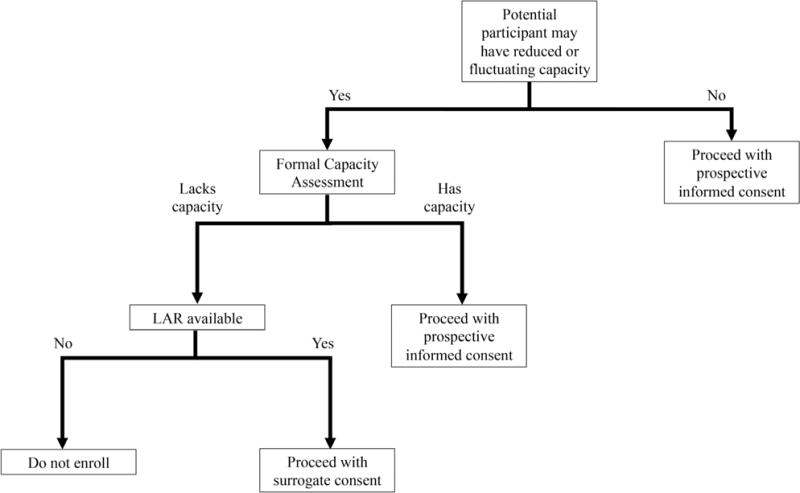
Determining the need for formal capacity assessment
The MacArthur Competence Assessment Tool for Clinical Research (MacCAT-CR) is frequently used to assess research decisional capacity. The MacCAT-CR is a structured interview that assesses four thought domains.22 Understanding (i.e., comprehension) is determined by questions related to the information that has been disclosed regarding the research study. Also assessed is the potential participant’s appreciation of how research involvement or non-involvement may affect their current situation. As a demonstration of reasoning, the MacCAT-CR assesses the ability of the potential participant to compare available alternatives to participation in terms of the consequences of research involvement or non-involvement. The tool is customized to the specific research protocol. Although responses are scored, no specific scale has been developed that indicates whether or not the potential participant does indeed have decision-making capacity.23 The lack of a standardized scoring system has proven confusing for some investigators, but this is intentional; the MacCAT-CR evaluates each domain separately and the assessor can assign different weights to each section, depending on the study context and the seriousness of the potential consequences of the participants’ choice regarding research involvement. Many authorities believe that as studies get riskier, a higher degree of decisional capacity should be demonstrated by the potential participant; thus, no specific level of capacity as determined by a specific MacCAT-CR score defines capacity in all circumstances.24 What is most important is the process the potential participant uses to make the decision regarding involvement in research (i.e., logical and sequential thinking).25
The University of California San Diego Brief Assessment of Capacity to Consent (UBACC) was developed to assist investigators in identifying potential research participants with questionable capacity to consent to a specific research study.26 The tool has a 10-item scale, including questions that focus on understanding and appreciation of the disclosed research information. The UBACC is best used as a screening tool; if the UBACC suggests possible diminished decision-making capacity, the investigator should consider performing a more comprehensive capacity assessment prior to enrolling the potential participant into the research study.27
Additional capacity assessment tools may be available for specific pathologies. If these tools are validated, their use is acceptable if justified to the IRB.
Vulnerability in Research
In addition to considering decisional capacity, clinical investigators also must consider the possibility that a potential research participant is not capable of self-protection the research process.28 This is one of several definitions of vulnerability in research. Potential research participants may be considered vulnerable by virtue of membership in a group, such as racial minorities,29 or personal characteristics, such as individuals in a subordinate position relative to the investigator.30 The research process itself may impose a degree of vulnerability on research subjects.31 This may occur when a study is poorly designed and therefore increases the risk of harm unbeknownst to subjects. Research-induced vulnerability may result from superficial consent discussions or overzealous presentations of potential direct benefit. Language that is above the educational level of the potential participant may make it difficult for potential participants to understand what is being asked of them, or embarrassed to ask questions. Vulnerability as related to research depends on the context of the study and characteristics of the potential participant. Vulnerability suggests that an individual has additional susceptibility to research-related harm or risk, or that additional safeguards are needed to protect the potential participant from research-related risk or harm.32
Vulnerability may also be caused by the current situation.33 Such situations may place a potential participant at a disadvantage for a number of reasons, such as because of a power differential between investigator and participant, fear of negative consequences if participation is declined, or stressful circumstances during which consent is requested. Similar to incapacity, situational vulnerability may be temporary; vulnerability at one point in time may no longer exist in another. For example, individuals who are in severe pain may be vulnerable while they are in severe pain. When the pain is great, the individual may be eager to participate in a research study because they believe that failure to enroll will deny them effective treatment. When they are no longer in severe pain, this misperception may no longer exist and the subject may be more critical of enrollment in the study.
Research-related vulnerability may be modifiable, and therefore from the standpoint of the clinical researcher, the assessment of vulnerability must include determination of whether investigator actions (or inactions) create research-related risks for the participant.
Examples of categories of potential vulnerabilities to research risk are shown in Table 1. Regardless of the cause, recognized or anticipated vulnerability requires that the investigator develop specific safeguards to minimize any potential harm provoked by research involvement.
Table 1.
Perceived or Real Vulnerability to Research Risks
| Group Membership | Research-Induced Vulnerability | Situational Vulnerability |
|---|---|---|
| Children Prisoners Pregnant women Members of the military Racial minorities Elders Refugees Minors |
Poorly designed study Inadequate or biased consent discussion Misrepresentation of research risks and benefit Presented material above the reading level of potential participant Pressure by investigator to quickly decide Material presented in a way potential participant cannot understand |
Stressful situation Sedated Non-English speaking Unable to read Fear of negative consequences Power differential Student or employee of investigator Patients of clinician-researcher Disenfranchised Economically disadvantaged |
Current Methods of Informed Consent
The current regulatory requirements clearly define two methods of obtaining informed consent for research participation. Prospective informed consent can be obtained directly from a potential participant when that individual is deemed to have the capacity to make decisions regarding their own research participation.34 An individual with diminished or fluctuating capacity may be represented by their legally authorized representative (LAR). In this circumstance, it is assumed that the LAR understands what the participant would likely choose if able to speak on their own behalf, and is acting in the best interest of the participant.35 The regulations also require identification of a participant’s membership in specific vulnerable groups, such as pregnant patients, prisoners, or children,36 and acknowledgment of other possible forms of vulnerability.
If prospective informed consent represents one end of a consent spectrum, at the other end of the spectrum are the regulations that allow an exception37 from or waiver38 of informed consent in emergency circumstances. Exception from or waiver of informed consent is only applicable in very narrow circumstances in which the devastating clinical status of the potential participants renders them incapable of providing meaningful prospective informed consent, and an LAR cannot be found within the proposed therapeutic window of the investigational agent or device.
The existing research regulations for informed consent for research participation address extremes of participant capacity and vulnerability. Prospective informed consent assumes that the potential subject has full capacity to provide a meaningful decision regarding their research participation; exception from and waiver of informed consent for emergency research requires total lack of capacity and imposition of vulnerability because of a devastating acute clinical condition.39 Other than surrogate consent, and waivers of some elements of informed consent,40 there are no specific research regulations or guidance for informed consent for clinical states between these extremes of participant capacity. As a result, investigators have attempted to retrofit study protocols to qualify for prospective informed consent, suggest methods to alter or waive the components of consent discussion, or to justify the use of an exception from informed consent even when not all criteria are met.41
A theoretical model that allows considerations of various degrees of research decisional capacity and vulnerability is illustrated in Figure 2. A spectrum of clinical conditions that may influence capacity and vulnerability is also shown. Although we do not have a “sliding scale” related to informed consent for research, models such as this suggest the use of alternative strategies to determine the potential participant’s values related to research participation. For example, some acutely ill but awake persons may not be able to fully appreciate an informed consent discussion, but may be able to express their emotional response to research involvement or be able to attend to a brief consent discussion; in this circumstance, assent or an abbreviated informed consent process42 may be possible. Valid informed refusal may also be possible in this circumstance; the ability to provide an informed refusal to participate in research is usually held to lower standards that those for informed consent.43 Sedation, severe pain, and other physiologically distressing conditions in an awake patient probably reduce decision-making capacity; with these conditions, assent may be possible, but even a brief consent discussion might be difficult. When patients are unstable but aware, any discussion about research involvement is unlikely to be meaningful, but this is an assumption and may not apply to all such patients. In these circumstances, the participant’s response to a simple disclosure of a research study or rejection of research in general may guide the assessment of the potential participant’s value system as it relates to research involvement.
Figure 2.
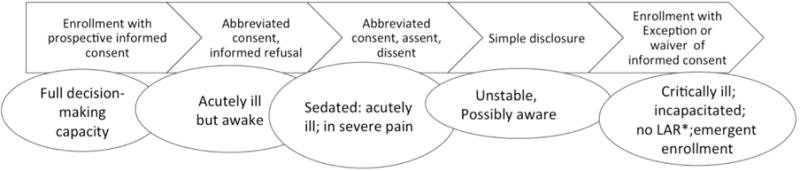
Proposed methods to assess attitudes toward research involvement among patients in various clinical states
Note that clinical conditions may allow overlap of suggested research discussions.
*LAR=legally authorized representative
Ascertaining the beliefs of a potential participant regarding their preferences about research participation may provide direction for LARs, investigators, and IRBs when a meaningful prospective informed consent process is not feasible. This approach moves ethical decision making to the bedside by asking LARs and investigators to consider real-time and individual patient factors that influence a participant’s ability to consent. It allows insights into the patient’s values related to research, and attempts to respect the opinion of the potential participant, even if they cannot engage in a detailed research discussion.
An approach such as this one may require expanding, revising, or reinterpreting current methods of obtaining consent for research involvement. Consideration of the options for informed consent suggested by this theoretical model or others like it may be controversial and will require thoughtful consideration of the basic concept of informed consent. Alternative ethical methods of informed consent may fill the gap created by the limitations- of the current regulations; they may also afford participants additional methods to express their values related to involvement in human subjects research.
Conclusions
The aim of clinical research is to provide new knowledge eventually applicable to improving the human condition. This requires the participation of human subjects, some of whom may be vulnerable or may lack decision-making capacity, who may have a clinical condition that renders them unable to speak on their own behalf, or may not have an available LAR. Existing regulations (or their current interpretation) for informed consent are difficult to apply to many clinical circumstances. Balancing the need for scientific advancement and the protection of human subjects is essential for the ethical conduct of research.
Acknowledgments
Preparation of this article and symposium was supported by the National Institutes of Health (NIH), National Human Genome Research Institute (NHGRI), and National Cancer Institute (NCI) grant R01HG008605 on “LawSeqSM: Building a Sound Legal Foundation for Translating Genomics into Clinical Application” (Susan M. Wolf, Ellen Wright Clayton, Frances Lawrenz, Principal Investigators)
Footnotes
Note
The author has no conflicts to declare.
References
- 1.Association of Clinical Research Professionals. Code of Ethics and Professional Conduct. 2016 available at < www.acrpnet.org/about/code-of-ethics> (last visited January 16, 2017); American College of Cardiology Foundation and the American Heart Association. The ACCF and AHA Codes of Conduct in Human Subjects Research. Journal of the American College of Cardiology. 2004;44(8):1724–1728. doi: 10.1016/j.jacc.2004.08.038. [DOI] [PubMed] [Google Scholar]; American College of Emergency Physicians. Policy Statement: Code of Ethics for Emergency Physicians. 2017 available at < www.ACEP.org/clinical-practice> (last visited January 16, 2017)
- 2.Lidz CW, et al. How Closely do Institutional Review Boards Follow the Common Rule? Academic Medicine. 2012;87(7):969–974. doi: 10.1097/ACM.0b013e3182575e2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Commission for the Protection of Human Subjects of Biomedical and Behavior Research. The Belmont Report. 1979 available at < https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/> (last visited January 16, 2017)
- 4.Grady C. Enduring and Emerging Challenges of Informed Consent. New England Journal of Medicine. 2015;372(9):855–862. doi: 10.1056/NEJMra1411250. [DOI] [PubMed] [Google Scholar]
- 5.Id.
- 6.Henderson G. Is Informed Consent Broken? American Journal of the Medical Sciences. 2011;342(4):267–272. doi: 10.1097/MAJ.0b013e31822a6c47. [DOI] [PubMed] [Google Scholar]
- 7.See Grady, supra note 4; Henderson, supra note 6.
- 8.See Henderson, supra note 6.
- 9.Kim S, et al. Do Clinicians Follow a Risk-Sensitive Model of Capacity-Determination? An Experimental Video Survey. Psychosomatics. 2006;47(4):325–329. doi: 10.1176/appi.psy.47.4.325. [DOI] [PubMed] [Google Scholar]; Jeste DV, et al. A New Brief Instrument for Assessing Decisional Capacity for Clinical Research. Archives of General Psychiatry. 2007;64(8):966–974. doi: 10.1001/archpsyc.64.8.966. [DOI] [PubMed] [Google Scholar]
- 10.Institutional Review Board, Office for Human Research Protection, Mayo Clinic. Informed Consent and Assessment of Capacity to Consent to Research. 2016 available at < http://mayocontent.mayo.edu/irb> (March 1, 2017)
- 11.Triebel KL, et al. Decision-making Capacity and Competency. In: Budd MA, et al., editors. Practical Psychology in Medical Rehabilitation (Cham, Switzerland: Spiner International Publishing. 2017. pp. 343–352. [Google Scholar]
- 12.Karlawish J, et al. Cognitive Impairment and PD Patients’ Capacity to Consent to Research. Neurology. 2013;81(9):801–807. doi: 10.1212/WNL.0b013e3182a05ba5. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also Triebel et al. supra note 11; Moran-Sanchez I, et al. Assessment of Capacity to Consent to Research Among Psychiatric Outpatients: Prevalence and Associated Factors. Psychiatric Quarterly. 2016;87(87):89–105. doi: 10.1007/s11126-015-9365-3. [DOI] [PubMed] [Google Scholar]
- 13.See Moran-Sanchez et al. supra note 12.
- 14.Bracken-Roche D, et al. The Vulnerability of Psychiatric Research Participants: Why This Research Ethics Concept Needs to be Revisited. Canadian Journal of Psychiatry. 2016;61(6):335–339. doi: 10.1177/0706743716633422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.See Kim et al. supra note 9; Jeste et al. supra note 9.
- 16.See Karlawish et al. supra note 12.
- 17.Appelbaum PS, Grisso T. MacArthur Competence Assessment Tool for Clinical Research (MacCAT-CR) Sarasota, FL: Professional Resources Press; 2001. p. 11. [Google Scholar]
- 18.Id.
- 19.See Kim et al. supra note 9; Grady, supra note 4.
- 20.University of California at San Diego Research Protections Program. Decision-making Capacity Guidelines. available at < http://irb.ucsd.edu> (last visited 1/16/17); University of Pittsburgh Human Subjects Protection Program. Decision-making Capacity. available at < http://irb.upitt.edu> (last visited January 16, 2017); University of Kentucky Office of Research Integrity. Impaired Consent Capacity Policy. available at < http://www.research.uky.edu/ori/ORIForms/62-Impaired-Consent-Capacity-Policy.pdf> (last visited January 16, 2017.; see also supra note 10
- 21.See University of Kentucky Office of Research Integrity, supra note 20.
- 22.See Appelbaum and Grisso, supra note 17.
- 23.See Moran-Sanchez et al. supra note 12. See also Appelbaum and Grisso, supra note 17.
- 24.Dunn LB, et al. Prevalence and Correlates of Adequate Performance on a Measure of Abilities Related to Decisional Capacity; Differences Among Three Standards for the MacCAT-CR in Patients with Schizophrenia. Schizophrenia Research. 2007;89(1–3):110–118. doi: 10.1016/j.schres.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Appelbaum PS. Assessment of Patients’ Competence to Consent to Treatment. New England Journal of Medicine. 2007;357(18):1834–1840. doi: 10.1056/NEJMcp074045. [DOI] [PubMed] [Google Scholar]
- 26.See Jeste et al. supra note 9.
- 27.See Appelbaum and Grisso, supra note 17.
- 28.Council for International Organizations of Medical Sciences (CIOMS) International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva, Switzerland: 2002. [PubMed] [Google Scholar]; International Conference on Harmonisation (ICH) Steering Committee. ICH Harmonized Tripartite Guideline. 1996. [Google Scholar]
- 29.See National Commission, supra note 3. See also CIOMS, supra note 28.
- 30.See ICH, supra note 28; CIOMS, supra note 28.
- 31.Rogers W, et al. Why Bioethics Needs a Concept of Vulnerability. International Journal of Feminist Approaches to Bioethics. 2012;5(2):11–22. [Google Scholar]; Aldridge J. Working with Vulnerable Groups in Social Research: Dilemmas by Default and Design. Qualitative Research. 2014;14(1):112–130. [Google Scholar]; Luna F. Elucidating the Concept of Vulnerability: Layers Not Labels. International Journal of Feminist Approaches to Bioethics. 2009;2(1):121–139. [Google Scholar]
- 32.See Aldridge, supra note 31.
- 33.See Luna, supra note 31; Bracken-Roche et al. supra note 14.
- 34.U.S. Department of Health and Human Services (DHHS) General Requirements for Informed Consent. 45 C.F.R. § 46.116, available at < https://www.hhs.gov/ohrp/regulations-and-policy/regulations/45-cfr-46/index.html#46.116> (last visited January 16, 2017)
- 35.See ICH, supra note 28; supra note 34.
- 36.University of Minnesota Human Research Protection Program. Policy-Research Involving Adults with Absent, Diminished, or Fluctuating Capacity to Consent to Participate in Research. 2017 available at < http://www.research.umn.edu/irb/toolkit.html> (last visited January 16, 2017)
- 37.U.S. Department of Health and Human Services (DHHS), 21 C.F.R. § 50.24 (1996); Ellis G, Lin M, Office for Protection from Research Risks, U.S. Department of Health and Human Services DHHS Informed Consent Requirements in Emergency Research. 1996 available at < https://www.hhs.gov/ohrp/regulations-and-policy/guidance/emergency-research-informed-consent-requirements> (last visited January 16, 2017)
- 38.U.S. Department of Health and Human Services (DHHS) Waiver of Informed Consent Requirements in Certain Emergency Research. 1996 45 C.F.R 46. [Google Scholar]
- 39.See ICH, supra note 28; see supra note 3.
- 40.U.S. Department of Health and Human Services (DHHS) IRB Waiver or Alteration of Informed Consent. 1996 45 C.F.R § 46.116d. [Google Scholar]
- 41.McKinney RE, et al. Use of Altered Informed Consent in Pragmatic Clinical Research. Clinical Trials. 2015;12(5):494–502. doi: 10.1177/1740774515597688. [DOI] [PMC free article] [PubMed] [Google Scholar]; Koenig B. Have We Asked Too Much of Consent? Hastings Center Report. 2014;44(4):33–34. doi: 10.1002/hast.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickert NW, et al. Confronting Ethical and Regulatory Challenges of Emergency Care Research with Conscious Patients. Annals of Emergency Medicine. 2016;67(4):538–545. doi: 10.1016/j.annemergmed.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biros MH, et al. Balancing Ethical Goals in Challenging Individual Participant Scenarios Occurring in a Trial Conducted with Exception from Informed Consent. Academic Emergency Medicine. 2015;22(3):340–346. doi: 10.1111/acem.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]