

Chronic obstructive pulmonary disease (COPD) is a debilitating respiratory disorder defined by progressive and largely irreversible airflow limitation (1). In an individual patient, various pathological conditions contribute to the clinical presentation of COPD, including: chronic bronchitis, characterized by persistent inflammation, remodelling and mucus hypersecretion in the airways; and emphysema, characterized by small airway/parenchymal destruction and alveolar airspace enlargement (1). Notably, exposure to cigarette smoke is a well-known risk factor for the development of COPD, with over 90% of COPD deaths occurring in individuals with a history of smoking (1,2). To gain mechanistic insight into how smoking promotes COPD, numerous studies have investigated the effects of tobacco smoke on biological and, in particular, immunological processes that may contribute to disease development and progression. Here, we present a broadly-encompassing framework within which to contextualize these effects (Figure 1). We propose that the effects of cigarette smoke on immune processes that drive COPD pathogenesis fall into three categories: (I) attenuation of cellular viability and barrier functions in the pulmonary environment; (II) mediation of local danger signalling; and (III) alteration of cellular responses to secondary inflammatory stimuli. Notably, many of these effects appear to represent the misappropriation of homeostatic immunological and biological processes into responses that promote continual tissue damage in the pulmonary environment. In this manner, the immune system is both the victim of cigarette smoke exposure with regards to host defense, and an aggressor in the pathogenesis of COPD.
Figure 1.
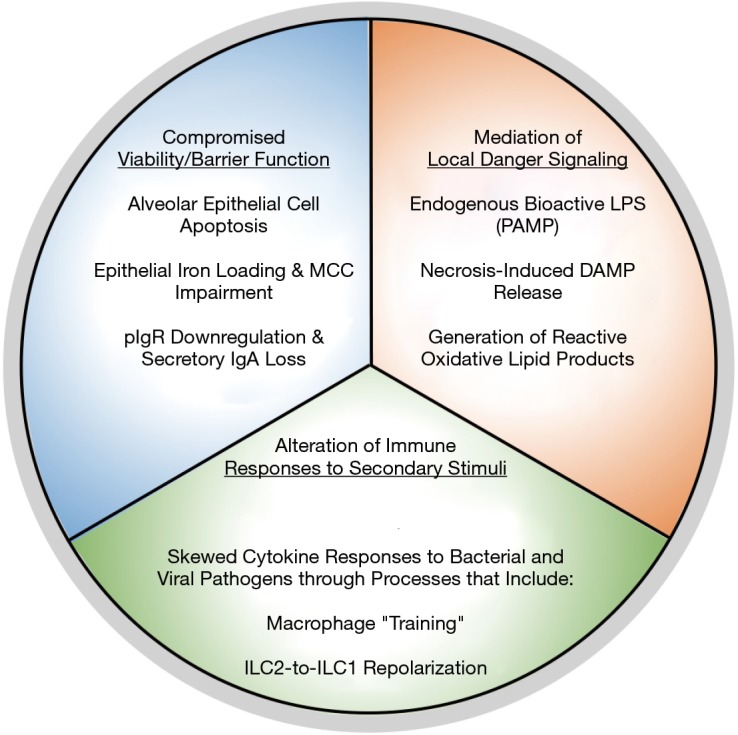
Categorization of interactions between cigarette smoke and the host immune system that have been implicated in facilitating COPD pathogenesis. Cigarette smoke has been demonstrated to: (I) compromise the viability and barrier function of cells in the pulmonary environment; (II) mediate local danger signaling, and; (III) alter the nature of immune responses to secondary inflammatory stimuli. The net product of these interactions results in the direct damage of pulmonary cells, as well as in an enhanced induction of inflammatory processes and recruitment of inflammatory cells, such as neutrophils and macrophages, into the lungs. Ultimately, these processes are thought to contribute to the development of lung pathologies, such as emphysema and chronic bronchitis, resulting in airflow limitation and the onset/progression of chronic obstructive pulmonary disease (COPD). MCC, mucociliary clearance; pIgR, poly-immunoglobulin receptor; IgA, immunoglobulin A; LPS, lipopolysaccharide; PAMP/DAMP, pathogen/danger-associated molecular pattern; ILC, innate lymphoid cell.
Attenuation of cellular viability and intrinsic barrier functions
Tobacco smoke is known to contain thousands of different chemical compounds, including: nicotine; toxic compounds, such as ammonia and carbon monoxide; reactive oxygen species; and carcinogens, such as benzo[a]pyrene and acrolein (3). Consequently, cigarette smoke has been described to have direct effects on the viability and intrinsic barrier function of resident pulmonary cells. For instance, cigarette smoke has been described to elicit apoptosis in alveolar epithelial cells (4). Importantly, high levels of apoptosis have been noted in the lungs of emphysematous COPD patients, suggesting that the smoke-induced programmed cell death may contribute to alveolar destruction (4). Notably, the apoptosis of cells exposed to toxic stimuli, such as cigarette smoke, likely represent evolutionarily-conserved processes aimed at maintaining tissue homeostasis. Specifically, if cells damaged by environmental stimuli are allowed to accumulate, they may undergo necrosis to cause damaging secondary inflammation. Apoptosis, in contrast, offers a controlled, non-inflammatory form of cell death. Moreover, given the mutagenic nature of cigarette smoke, DNA damage may lead to cellular transformation and the development of cancer. Thus, programmed cell death likely comprises a beneficial, homeostatic response to tobacco smoke exposure. However, in the context of chronic exposure these processes contribute to excessive epithelial cell death and, consequently, the loss of alveolar and small airway integrity, ultimately compromising lung function in smoking individuals.
In addition to compromising cell viability, cigarette smoke has been reported to impair intrinsic barrier functions within the respiratory tract. For instance, smoke exposure is known to reduce lung cilia length, and compromise pulmonary mucociliary clearance (5-7). In healthy individuals, this process normally facilitates the upward transit of foreign particles, such as inhaled pathogens and debris, out of the respiratory system and into the digestive tract, thereby precluding dangerous pulmonary inflammatory responses (8). Mechanistically, the detrimental effects of cigarette smoke on mucociliary clearance has been attributed to increased expression of iron-responsive element-binding protein (IRP) 2 and mitochondrial iron loading in ciliated epithelial cells within the lungs (6). Ultimately, this appears to promote mitochondrial dysfunction and epithelial cell death, thus impairing the mucociliary ‘escalator’ capacity of the respiratory epithelium (6). However, IRP-mediated mitochondrial iron metabolism is critical to various cellular processes, including the facilitation of protein function via the generation of iron-based enzyme co-factors, as well as proliferation (9). Thus, through the inappropriate activation of molecular pathways associated with cellular iron metabolism, cigarette smoke compromises mucociliary clearance, resulting in the accumulation of inflammatory pathogens and debris in the respiratory tract.
Moreover, cigarette smoke exposure may decrease the ability of host epithelial cells to maintain an intrinsic humoral barrier against inhaled microbes. For instance, in severe COPD, epithelial cells in the airways are observed to have reduced expression of the polymeric immunoglobulin receptor (pIgR) (10), which is responsible for the transport of secretory immunoglobulin (sIg)A from the lamina propria into the lumen (11). When localized within the mucous lining in the airway, sIgA normally serves to neutralize incoming microbes and prevent their contact with the apical epithelial surface, a process known as “immune exclusion” (12). However, data from both pIgR deficient mice (13) and the airways of COPD patients (14) have suggested that the absence of this receptor, and subsequent IgA loss, leads to bacterial invasion of the epithelium. Subsequently, this leads to the initiation of local, nuclear factor (NF)κB-mediated inflammatory processes, resulting in macrophage and neutrophil recruitment, and, over time, increased collagen deposition and airspace enlargement (13). Although the mechanisms that drive epithelial pIgR loss in severe COPD has yet to be conclusively determined, it has been suggested that the overexpression of transforming growth factor (TGF)-β1, a cytokine known to be produced in response to oxidative components within cigarette smoke (15), contributes to transcriptional downregulation of pIgR (10). Notably, TGF-β1 secretion has been implicated in facilitating pulmonary repair processes in response to cytotoxic damage, in part through the activation of lung fibroblasts (16). Thus, downregulation of pIgR in the lungs of patients with severe COPD, and the consequent reduction in IgA barrier integrity, may occur as a result of continual TGF-β1 secretion following chronic attempts to repair cigarette smoke-induced tissue damage (10). Overall, compromised cellular barrier function and viability in the context of the smoke-exposed lung seems to both compromise pulmonary pathogen resistance, and contribute to COPD pathogenesis.
Mediation of local danger signaling
In addition to directly affecting the viability and intrinsic function of resident lung cells, components of cigarette smoke are able to act as, create, or promote the release of danger signals. These signals, in turn, are able to induce inflammatory responses that promote collateral damage within the respiratory tract. For instance, cigarette smoke exposure has been shown to elicit Toll-like receptor (TLR)4 activation in both ex vivo human systems using peripheral blood-derived monocytes (17), as well as in in vivo murine models (18). Of note, this effect may be mediated through bioactive lipopolysaccharide (LPS) contained within combusted tobacco smoke (19), and/or in an LPS-independent manner (17). Downstream of TLR signaling, Myeloid differentiation primary-response protein (MyD)88/NFκB pathway activation is able to elicit the production of interleukin (IL)-8, chemokine C-X-C motif ligand (CXCL)-1, and other pro-inflammatory mediators that are capable of promoting the recruitment of monocytes and neutrophils into the lung (20). Of note, TLR4 activation by cigarette smoke resembles a normal immune response to common, LPS-expressing Gram-negative bacterial pathogens, such as Haemophilus (H.) influenzae, Moraxella catarrhalis, and Pseudomonas aeruginosa. Both monocyte-derived macrophages and neutrophils are useful in combatting such bacterial infections, given that they are professional phagocytes. However, when continually recruited in the context of chronic cigarette smoke exposure, these cells are thought to mediate collateral damage. Neutrophils and macrophages are able to produce proteolytic enzymes, including matrix metalloproteinases (MMPs), and neutrophil elastase (NE), that cleave integral extracellular matrix proteins such as collagen and elastin, thus facilitating alveolar destruction (20). In addition, macrophages are a source of TGF-β1 that can act on local fibroblasts to facilitate fibrotic remodelling of the airways, thereby contributing to bronchitis (20). Therefore, through the chronic activation of innate immune signaling pathways which normally facilitate the clearance of bacterial pathogens, cigarette smoke may drive inappropriate inflammatory processes that contribute to COPD-related pathologies, such as airspace enlargement and bronchitis.
In addition to containing components such as LPS, cigarette smoke may compound inflammatory processes by facilitating the release of endogenous danger signals. For instance, cigarette smoke is known to cause necrosis of epithelial cells and neutrophils, a form of inflammatory cell death that results in the release of danger-associated molecular patterns (DAMPs) (21,22). On one hand, the DAMPs released following necrosis are useful, in that they can recruit phagocytes to the site of cell death to mediate efferocytosis, the phagocytic clearance of cellular debris (23). However, chronic smoke exposure and necrotic DAMP release can contribute to the sustained recruitment of neutrophils and monocytes, which can in turn perpetuate lung tissue damage.
Finally, cigarette smoke can facilitate the generation of novel danger signals within the lung environment. For instance, components of tobacco smoke are known to facilitate lipid peroxidation, producing highly reactive oxidative products such as 4-hydroxy-2-nonenal (4-HNE) which accumulate in the lungs of COPD patients (24,25). Notably, oxidized lipids are readily consumed by alveolar macrophages, and stimulate, in mice, IL-1α secretion by these cells (26). This, in turn, is thought to elicit granulocyte-macrophage colony stimulating factor (GM-CSF) production by local alveolar epithelial cells, presumably to facilitate the recruitment of additional phagocytes to assist with damaged lipid clearance. Notably, IL-1α produced by alveolar macrophages in other contexts, such as following non-typeable H. influenzae (NTHi) infection, has been shown to facilitate neutrophilic influx into the lungs by stimulating the secretion of CXCL5 by alveolar epithelial cells (27). Thus, it appears that although IL-1α production in response to oxidized lipids within the smoke-exposed murine lung promotes homeostasis by increasing macrophage-mediated lipid clearance, it concurrently seems to facilitate collateral damage to lung tissue by facilitating the recruitment of proteolytic enzyme-producing macrophages and neutrophils into the pulmonary environment. Ultimately, whether mediated by the direct activation of pattern-recognition receptors and the release of pro-inflammatory cytokines, the generation of reactive intermediates, or through the release of DAMPs as a by-product of necrosis, cigarette smoke can facilitate the mediation of danger signaling within the lung to promote a pro-inflammatory environment prone to inducing tissue damage.
Alteration of cellular responses to secondary inflammatory stimuli
Finally, through chronic stimulation, cigarette smoke alters the nature of immune responses against secondary stimuli such as bacterial and viral pathogens. Specifically, smoke exposure fundamentally skews normal antimicrobial immune responses in a manner prone to inducing further damage upon pathogen exposure. Clinically, these aberrant immune responses to pathogens may explain the notably increased risk of lower respiratory tract infections observed in smokers (28), and may contribute to the presentation of acute exacerbations of COPD following microbial infection (29). For instance, cigarette smoke is well known to reduce the capacity of alveolar macrophages to perform phagocytosis (30), a process that is critically important to destroy pathogens that enter the lower airways, as well as facilitate the clearance of dead and dying cells. In addition, cigarette smoke skews the ability of these macrophages to produce specific pro-inflammatory cytokines in response to pathogen exposure. For instance, tobacco smoke has been observed to prime murine alveolar macrophages to produce excessive amounts of IL-1α and monocyte chemotactic protein (MCP)-1 in response to stimulation with NTHi (27,31). Concurrently, however, these cells are reduced in their ability to secrete tumour necrosis factor (TNF)-α. Of note, these findings are concurrent with observations of increased lung compliance and airspace enlargement in smoke-exposed, infected mice (27). This skewed expression of pro-inflammatory mediators following cigarette smoke may represent a form of “trained innate immunity”, or innate immune “memory”, wherein exposure to a stimuli (here, cigarette smoke) alters baseline cellular activity and/or responses to secondary stimuli for a prolonged period of time (32). On one hand, the inhibition of TNF-α secretion is reminiscent of endotoxin tolerance, a phenomenon that is thought to have evolved as a mechanism to autoregulate the potent inflammatory effects of endotoxin experienced in the context of sequential exposure to Gram-negative bacterial products (33). In contrast, the enhanced production of IL-1α and MCP-1 by smoke-exposed alveolar macrophages may constitute a trained response similar to that observed following vaccination with bacille Calmette-Guérin (BCG), an attenuated Mycobacterium tuberculosis strain, wherein human peripheral blood mononuclear cells were shown to produce greater levels of IL-1β and TNF-α in response to unrelated pathogens, such as Staphylococcus aureus and Candida albicans, after vaccination (34). Ultimately, while pathogen-induced innate memory or endotoxin tolerance may be beneficial in specific contexts, the supposed “trained innate immunity” observed in cigarette smoke-exposed alveolar macrophages can seemingly promote skewed antimicrobial inflammatory responses in response to pathogen exposure. This, in turn, may thereby promote collateral tissue damage and the development of COPD-associated pathologies.
Interestingly, inflammatory responses to viral stimuli seem to be additionally altered in the lung following cigarette smoke exposure. For instance, cigarette smoke increases IL-33 release by lung epithelial cells upon viral infection, and concurrently promotes increased expression of the IL-33 receptor ST2 on lung macrophages (35). Interestingly, IL-33-primed macrophages have been shown to produce heightened levels of TNF-α and IL-12 upon viral sensing, for example through TLR3 ligation (35). Given that IL-33 is thought to act as a DAMP following its release from lysed epithelial cells (36), this indirect pro-inflammatory effect of cigarette smoke on macrophages via IL-33 appears to co-opt a natural response to danger signals in the lung environment. Notably, concurrent with the effects of smoke on macrophage function is a change in the ability of innate lymphoid cells (ILCs) to respond to pulmonary pathogens (35,37). ILCs are a lymphoid-lineage group of cells that do not display antigen-specificity, whose effector functions largely entail specific cytokine release in various homeostatic and inflammatory contexts (38). In particular, smoke exposure has been shown to facilitate a shift within lung ILC2 cells, which typically produce IL-4 and other Th2-type cytokines, towards an ILC1-like phenotype (37). Subsequently, ILC1 cells were noted to have an augmented capacity to secrete cytokines such as IL-6, IL-12, TNF-α and interferon (IFN)-γ in response to influenza infection (37). Notably, infection with influenza virus independent of cigarette smoke exposure was also able to promote the accumulation of ILC1-like cells in the lung (37). These findings suggest the ILC2-to-ILC1 shift observed following cigarette smoke exposure represents an inappropriate activation of homeostatic, ILC-mediated antimicrobial mechanisms in the absence of infection, thereby predisposing the lung towards excessive inflammation upon virus exposure. Interestingly, in this study the proportion of circulating ILC1s was observed to be inversely correlated with forced expiratory volume in one second (FEV1% predicted) in COPD patients, and shown to increase in patients with frequent exacerbations (≥2 per year) (37). Consequently, it appears that cigarette smoke may contribute to COPD pathogenesis by directly facilitating ILC1 polarization in the lung, thereby enhancing inflammatory responses to inhaled viruses and promoting harmful acute disease exacerbations. Notably, if optimal antimicrobial immunity is characterized by immune responses of an appropriate measure that limit tissue damage, then the skewed phenotypic profiles of macrophages and ILCs observed in the context of cigarette smoke may indirectly represent a form compromised pulmonary host defense. Thus, these examples illustrate how host immunity is both victimized by tobacco smoke in terms of host defense, and concurrently seem to mediate COPD pathogenesis.
Conclusions
Chronic obstructive pulmonary disease (COPD) currently represents the third-leading cause of death worldwide (39). However, to date, only therapeutic interventions aimed at providing symptomatic relief, such as corticosteroids, bronchodilators, and phosphodiesterase-4 inhibitors, are available to treat patients with COPD (1). Troublingly, however, to date no treatments exist that were designed to interrupt the cellular or molecular mechanisms that drive disease progression. Ultimately, this lack of disease-modifying therapies stems from an incomplete understanding of the mechanisms that drive COPD pathogenesis. Thus, although the continual promotion of smoking cessation remains a priority, further mechanistic studies are essential to alleviating the burden of COPD on our health care systems.
It must be noted that, although approximately 90% of COPD-related deaths are attributable to prior or current smoking, the effects of cigarette smoke exposure cannot fully explain the pathogenesis of COPD, as only 25–40% of continuous smokers go on to develop the disease (2). A more comprehensive understanding of COPD pathogenesis must transcend this single variable, to include other risk factors such as genetics, development influences, pollution exposure, and concurrent atopy that may synergize to drive disease progression. However, given that cigarette smoke exposure is the leading risk factor for COPD development, understanding how it affects the immune system is critical to achieve an integrated mechanistic understanding of COPD pathogenesis, and ultimately developing disease-modifying interventions to halt disease progression. To this end, many studies have implicated the effects of cigarette smoke on various aspects of the host immune system as playing a role in driving the development of COPD-associated pathologies, such as bronchitis and emphysema. Using a simple framework, we suggest that the effects of cigarette smoke exposure on host immune processes may be classified as: directly damaging resident host cells in the pulmonary environment, and/or attenuating cellular barrier functions; serving as, creating, or promoting the release of danger signals to promote destructive inflammation; and altering the capability of immune cells to respond to secondary stimuli such as bacterial and viral pathogens. Through this framework, it becomes evident that many of the immunological and biological processes that are thought to contribute to COPD pathogenesis represent inappropriate utilization of homeostatic functions, such as programmed cell death and antimicrobial mechanisms. Furthermore, the examples provided illustrate that functions of the respiratory immune system are victimized by chronic tobacco smoke exposure to the detriment of host defense and, at the same time, misappropriated into antagonizing pulmonary homeostasis, promoting tissue destruction, and driving COPD pathogenesis.
Acknowledgements
The authors wish to thank Steven Cass and Danya Thayaparan for their editorial support.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2017. Available online: http://goldcopd.org.
- 2.Løkke A, Lange P, Scharling H, et al. Developing COPD: A 25 year follow up study of the general population. Thorax 2006;61:935-9. 10.1136/thx.2006.062802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perfetti TA, Rodgman A. The complexity of tobacco and tobacco smoke. Contrib to Tob Res 2011;24:215-32. [Google Scholar]
- 4.Morissette MC, Parent J, Milot J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J Chron Obstruct Pulmon Dis 2009;4:19-31. [PMC free article] [PubMed] [Google Scholar]
- 5.Leopold PL, O’Mahony MJ, Julie Lian X, et al. Smoking is Associated with Shortened Airway Cilia. PLoS One 2009;4:e8157. 10.1371/journal.pone.0008157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cloonan SM, Glass K, Laucho-Contreras ME, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med 2016;22:163-74. 10.1038/nm.4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lam HC, Cloonan SM, Bhashyam AR, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest 2013;123:5212-30. 10.1172/JCI69636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munkholm M, Mortensen J. Mucociliary clearance: Pathophysiological aspects. Clin Physiol Funct Imaging 2014;34:171-7. 10.1111/cpf.12085 [DOI] [PubMed] [Google Scholar]
- 9.Cloonan SM, Mumby S, Adcock IM, et al. The “Iron”-y of iron overload and iron deficiency in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2017;196:1103-12. 10.1164/rccm.201702-0311PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gohy ST, Detry BR, Lecocq M, et al. Polymeric immunoglobulin receptor down-regulation in chronic obstructive pulmonary disease: Persistence in the cultured epithelium and role of transforming growth factor-β. Am J Respir Crit Care Med 2014;190:509-21. 10.1164/rccm.201311-1971OC [DOI] [PubMed] [Google Scholar]
- 11.Macpherson AJ, McCoy KD, Johansen FE, et al. The immune geography of IgA induction and function. Mucosal Immunol 2008;1:11-22. 10.1038/mi.2007.6 [DOI] [PubMed] [Google Scholar]
- 12.Stokes CR, Soothill JF, Turner MW. Immune Exclusion is a function of IgA. Nature 1975;255:745-6. 10.1038/255745a0 [DOI] [PubMed] [Google Scholar]
- 13.Richmond BW, Brucker RM, Han W, et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat Commun 2016;7:11240. 10.1038/ncomms11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polosukhin VV, Richmond BW, Du RH, et al. Secretory IgA deficiency in individual small airways is associated with persistent inflammation and remodeling. Am J Respir Crit Care Med 2017;195:1010-21. 10.1164/rccm.201604-0759OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang RD, Wright JL, Churg A. Transforming growth factor-β1 drives airway remodeling in cigarette smoke-exposed tracheal explants. Am J Respir Cell Mol Biol 2005;33:387-93. 10.1165/rcmb.2005-0203OC [DOI] [PubMed] [Google Scholar]
- 16.Bartram U, Speer CP. The role of transforming growth factor β in lung development and disease. Chest 2004;125:754-65. 10.1378/chest.125.2.754 [DOI] [PubMed] [Google Scholar]
- 17.Karimi K, Sarir H, Mortaz E, et al. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir Res 2006;7:66. 10.1186/1465-9921-7-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doz E, Noulin N, Boichot E, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol 2008;180:1169-78. 10.4049/jimmunol.180.2.1169 [DOI] [PubMed] [Google Scholar]
- 19.Hasday JD, Bascom R, Costa JJ, et al. Bacterial endotoxin is an active component of cigarette smoke. Chest 1999;115:829-35. 10.1378/chest.115.3.829 [DOI] [PubMed] [Google Scholar]
- 20.Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev 2004;56:515-48. 10.1124/pr.56.4.2 [DOI] [PubMed] [Google Scholar]
- 21.Pouwels SD, Hesse L, Faiz A, et al. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am J Physiol Lung Cell Mol Physiol 2016;311:L881-92. 10.1152/ajplung.00135.2016 [DOI] [PubMed] [Google Scholar]
- 22.Heijink IH, Pouwels SD, Leijendekker C, et al. Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol 2015;52:554-62. 10.1165/rcmb.2013-0505OC [DOI] [PubMed] [Google Scholar]
- 23.Peter C, Wesselborg S, Herrmann M, et al. Dangerous attraction: Phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis. 2010;15:1007-28. 10.1007/s10495-010-0472-1 [DOI] [PubMed] [Google Scholar]
- 24.Aoshiba K, Koinuma M, Yokohori N, et al. Immunohistochemical evaluation of oxidative stress in murine lungs after cigarette smoke exposure. Inhal Toxicol. 2003;15:1029-38. 10.1080/08958370390226431 [DOI] [PubMed] [Google Scholar]
- 25.Rahman I, van Schadewijk A, Crowther AJL, et al. 4-Hydroxy-2-Nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;166:490-5. 10.1164/rccm.2110101 [DOI] [PubMed] [Google Scholar]
- 26.Morissette MC, Shen P, Thayaparan D, et al. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur Respir J 2015;46:1451-60. 10.1183/09031936.00216914 [DOI] [PubMed] [Google Scholar]
- 27.Nikota JK, Shen P, Morissette MC, et al. Cigarette Smoke Primes the Pulmonary Environment to IL-1 /CXCR-2-Dependent Nontypeable Haemophilus influenzae-Exacerbated Neutrophilia in Mice. J Immunol 2014;193:3134-45. 10.4049/jimmunol.1302412 [DOI] [PubMed] [Google Scholar]
- 28.Huttunen R, Heikkinen T, Syrjänen J. Smoking and the outcome of infection. J Intern Med 2011;269:258-69. 10.1111/j.1365-2796.2010.02332.x [DOI] [PubMed] [Google Scholar]
- 29.Sethi S. Infection as a comorbidity of COPD. Eur Respir J 2010;35:1209-15. 10.1183/09031936.00081409 [DOI] [PubMed] [Google Scholar]
- 30.Hodge S, Hodge G, Scicchitano R, et al. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol 2003;81:289-96. 10.1046/j.1440-1711.2003.t01-1-01170.x [DOI] [PubMed] [Google Scholar]
- 31.Gaschler GJ, Skrtic M, Zavitz CCJ, et al. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J Respir Crit Care Med 2009;179:666-75. 10.1164/rccm.200808-1306OC [DOI] [PubMed] [Google Scholar]
- 32.Netea MG, Joosten L, Latz E, et al. Trained immunity: A program of innate immune memory in health and disease. Science 2016;352:aaf1098. 10.1126/science.aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol 2009;30:475-87. 10.1016/j.it.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 34.Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci 2012;109:17537-42. 10.1073/pnas.1202870109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kearley J, Silver J, Sanden C, et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I Interleukin-33-dependent response to infection. Immunity 2015;42:566-79. 10.1016/j.immuni.2015.02.011 [DOI] [PubMed] [Google Scholar]
- 36.Chen GY, Nuñez G. Sterile inflammation: Sensing and reacting to damage. Nat Rev Immunol 2010;10:826-37. 10.1038/nri2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silver JS, Kearley J, Copenhaver A, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 2016;17:626-35. 10.1038/ni.3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Artis D, Spits H. The biology of innate lymphoid cells. Nature 2015;517:293-301. 10.1038/nature14189 [DOI] [PubMed] [Google Scholar]
- 39.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095-128. 10.1016/S0140-6736(12)61728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]