

Abstract
Hsp70 chaperones bind to various protein substrates for folding, trafficking, and degradation. Considerable structural information is available about how prokaryotic Hsp70 (DnaK) binds substrates, but less is known about mammalian Hsp70s, of which there are 13 isoforms encoded in the human genome. Here, we report the interaction between the human Hsp70 isoform heat shock cognate 71-kDa protein (Hsc70 or HSPA8) and peptides derived from the microtubule-associated protein Tau, which is linked to Alzheimer's disease. For structural studies, we used an Hsc70 construct (called BETA) comprising the substrate-binding domain but lacking the lid. Importantly, we found that truncating the lid does not significantly impair Hsc70's chaperone activity or allostery in vitro. Using NMR, we show that BETA is partially dynamically disordered in the absence of substrate and that binding of the Tau sequence GKVQIINKKG (with a KD = 500 nm) causes dramatic rigidification of BETA. NOE distance measurements revealed that Tau binds to the canonical substrate-binding cleft, similar to the binding observed with DnaK. To further develop BETA as a tool for studying Hsc70 interactions, we also measured BETA binding in NMR and fluorescent competition assays to peptides derived from huntingtin, insulin, a second Tau-recognition sequence, and a KFERQ-like sequence linked to chaperone-mediated autophagy. We found that the insulin C-peptide binds BETA with high affinity (KD < 100 nm), whereas the others do not (KD > 100 μm). Together, our findings reveal several similarities and differences in how prokaryotic and mammalian Hsp70 isoforms interact with different substrate peptides.
Keywords: chaperone, microtubule-associated protein (MAP), protein conformation, protein complex, nuclear magnetic resonance (NMR)
Introduction
Hsp70 chaperones assist in intracellular protein trafficking, protein refolding (1, 2), and protein degradation via the proteasome (3) and via autophagy (4). The human genome codes for 13 Hsp70 isoforms, one of which is the cytosolic and constitutively expressed Hsc70 (HSPA8) (5). In the last decade, Hsc70 has been linked to multiple protein misfolding disorders such as Alzheimer's, Parkinson's, and Huntington's diseases and type II diabetes (6). Like all Hsp70s, human Hsc70, modeled in Fig. 1, consists of a nucleotide-binding domain (NBD, residues 1–386)4 and a substrate-binding domain (SBD) that is further subdivided into a β-domain harboring the substrate-binding cleft (BETA, residues 395–508) and an α-helical lid (LID, residues 509–604). The SBD is followed by a dynamically unstructured region (residues 605–646) harboring the CHIP-binding site at its very C terminus (7).
Figure 1.
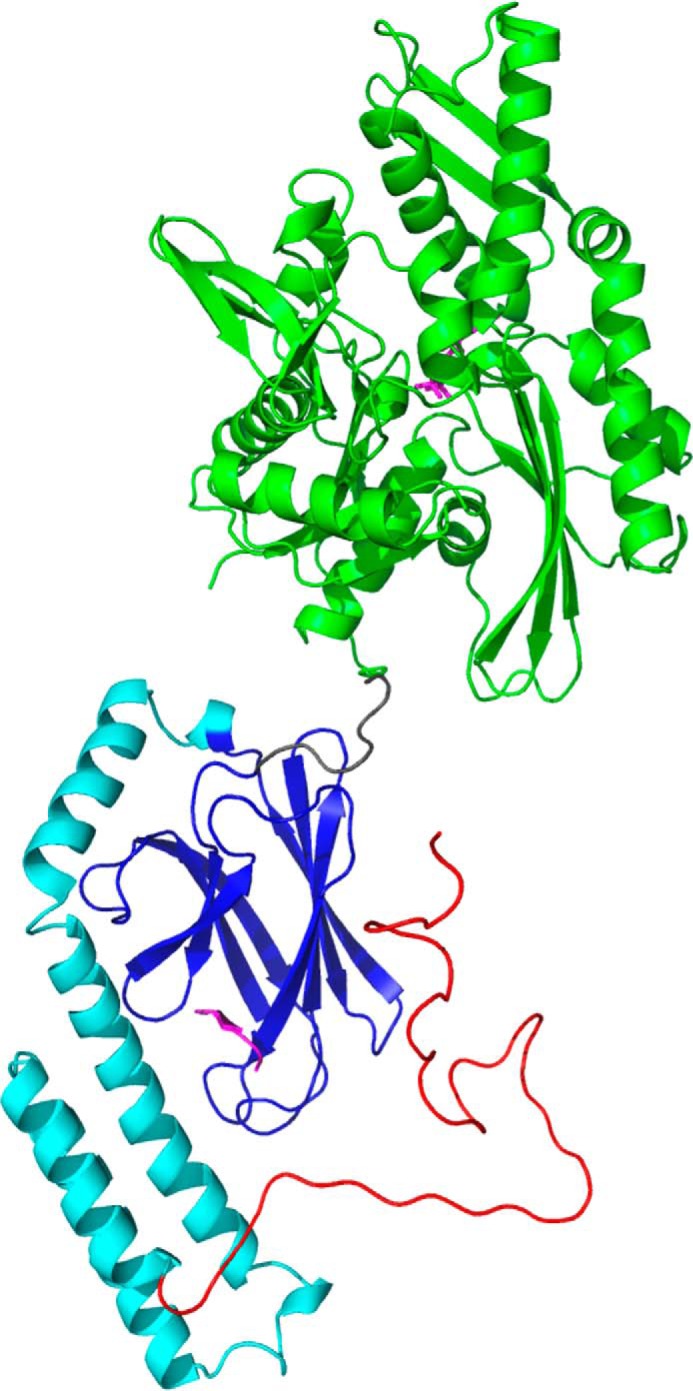
Overview of Hsc70. Shown is a model based on Hsc70 (HSPA8) NBD (PDB entry 3HSC; green) with ADP (magenta) and Hsp70 (HSPA1A) SBD (PDB entry 4PO2; BETA domain in blue, LID in cyan) with NRLLLTG (magenta) bound. The TAIL (red) has arbitrary conformation. The overall juxtaposition was taken after DnaK in ADP-NRLLLTG state (PDB entry 2KHO).
Much of the insight into the function of the Hsp70 chaperones has been obtained for DnaK, a homolog from Escherichia coli. DnaK, in the presence of ATP and the co-chaperones DnaJ and GrpE, hydrolyzes nucleotide and refolds denatured luciferase in vitro. During this cycle, DnaK in the ADP state binds tightly (KD < 0.1 μm) to exposed hydrophobic sequences in misfolded substrate proteins and helps these proteins unfold. Upon exchange of ADP for ATP, the affinity for the bound protein is reduced (KD = ∼1 μm), and the substrate is released to refold (8). The interactions of DnaK with peptides (9), nucleotides (8), and co-chaperones (10) has been extensively studied. However, far fewer quantitative details are known for the human proteins. What is clear is that the fundamental features of the biochemical cycle are intact. Hsc70, in the presence of ATP and co-chaperones, such as DnaJA2 and BAG2, also hydrolyzes nucleotide and refolds luciferase in vitro (11). One difference in humans is the large expansion of the number of co-chaperones, which seems to have diversified Hsp70's activities. Another difference is that the substrate proteins of human Hsp70s have not been categorized.
Much of our structural knowledge of this system is derived from crystallography and NMR studies of E. coli DnaK. Structures are available for the ADP- and ATP-bound states of the NBD (12, 13), as well as the apo- (14) and substrate-bound forms of the SBD (15, 16). There are also structures of near native NBD–SBD constructs in both the ADP-peptide (17) and ATP-apo (18, 19) states. In contrast, there is a paucity of human Hsp70 structures. Although a crystal structure for the human Hsc70 NBD (12) and a solution structure for the human substrate-bound SBD (20) were deposited a long time ago, it is not yet clear how nucleotide state or peptide substrate binding impact these structures. In addition, there are no equivalent structures of near native NBD–SBD Hsc70 constructs.
Nevertheless, most workers in the field assume that the structural results for DnaK can be safely extrapolated to the human chaperones. But is that true? Here we address one of these omissions by studying the binding of peptides to Hsc70 SBD.
As a model substrate system, we focused on the abundant neuronal microtubule-associated protein Tau because it one of the few proteins that is known to be a natural substrate for Hsc70 in cells (21). Hsc70's binding sites on Tau have been well characterized (22). Tau is an intrinsically unfolded protein (23) that plays a role in microtubule assembly. However, when Tau becomes hyperphosphorylated, it fails to bind microtubules and deposits in neurofibrillary tangles that correlate with Alzheimer's disease (24, 25). Hsc70 (HSPA8) and Hsp70 (HSPA1A) levels are elevated in cells containing neurofibrillary tangles, suggesting a relationship with Tau proteostasis (26). Significantly, Dickey, Gestwicki, and co-workers showed that Tau clearance is enhanced by compounds that lock Hsc70 in the ADP state (27, 28).
Hsc70 is involved in CHIP-mediated transport of substrates, including Tau, to the proteasome (29), as well as in degradation in the lysosome via chaperone-mediated autophagy (CMA) (30). Interestingly, the surfaces of Tau that are involved in these two degradation pathways are likely distinct: proteasomal degradation involves Hsc70 binding to specific sequences in Tau: 590KVQIINKK and 613VQIVYK (21), whereas interactions of Hsc70 with Tau during CMA require sequences with the polar KFERQ signature (31–33). Thus, one of the major questions in the Hsc70 field is how these various interactions take place and which ones occur in the classic SBD cleft. It is with Dr. Chad Dickey's research into the biomedical Tau–Hsc70 linkage in mind that we dedicate this manuscript to his memorial.
In this work, we use a truncated construct, BETA, to study the interactions between Hsc70 and Tau using fluorescence competition assays and NMR. The NMR data show that BETA contains regions of dynamic disorder in the apo-state. The addition of KVQIINKK, but not VQIVYK, results in significant rigidification. Nuclear Overhauser effect distance measurements show that KVQIINKK binds into the canonical substrate-binding cleft. These results suggested to us that BETA might be a useful construct for broadly studying Hsc70–peptide interactions. Accordingly, we studied a number of additional peptides, including the prototypical sequence, NRLLLTG (15), the CMA KFERQ peptide, a peptide derived of huntingtin (34), and the C-peptide of insulin (35). The results further elaborate the diverse interactions of Hsc70 and suggest that BETA is a useful tool for rapidly studying other substrates of human Hsp70s.
Results
The main focus of the current study is the interaction of the human Tau-derived peptide 590KVQIINKK, which we will refer to as TAU1, with human Hsc70. Fig. 1 shows a modeled structure of full-length Hsc70, showing its NBD, SBD-BETA, SBD-LID, and Tail for reference. Because the full-length 70-kDa protein is too large for detailed NMR studies, we set out to design a truncated form (<30 kDa) that would bind to TAU1 and be representative of the WT protein. This was not an easy endeavor. We found that many Hsc70 SBD constructs tend to bind to themselves, either intramolecularly (20) or intermolecularly. Even Hsc70 385–604, composed of just the β-domain and the helical LID, bound the TAU1 peptide with too low of an affinity (KD = ∼200 μm as estimated by NMR, not shown). We reasoned that the LID domain might be interfering with entry into the cleft. Therefore, we decided to further truncate the domain down to the BETA basket, with exact sequence MSNA395D–G508-G3H6 (Fig. 1, blue). The N-terminal MSNA is a cloning artifact that does not seem to impact structure or function. We describe below that this domain, which we refer to as BETA, binds TAU1 with high affinity. Furthermore, it permits detailed studies into how human Hsc70 binds to other “substrate” peptides.
Fig. 2 shows 15N-1H TROSY HSQC spectra of BETA in the absence and presence of TAU1. Significantly, the addition of TAU1 causes the emergence of many new signals (red). Because we do not observe the loss of any resonances that were present in the apo spectrum upon adding TAU1, we conclude that the appearing resonances are broadened beyond detection in the apo state. In other words, resonances that were not visible in the spectrum of apo BETA were broadened beyond detection because of dynamical disorder on the microsecond time scale. To understand whether the interaction of TAU1 with Hsc70 was retained in full-length Tau, we purified a splice isoform that is expressed in adult brains, termed 4R0N Tau. 4R0N Tau also causes appearances of new resonances in BETA, suggesting that similar contacts might be made with the full-length protein (Fig. 2, insets).
Figure 2.

15N-1H TROSY HSQC spectra of Hsc70 BETA. The boxes show enlargements. Blue, 35 μm BETA. Red, 35 μm BETA + 200 μm TAU1 (GKVQIINKKG). Black, 35 μm BETA + 57 μm WT 4R0N TAU.
To further characterize the interaction, we collected 13C-1H methyl HSQC NMR data at low concentrations. Fig. 3A shows the appearance of new methyl resonances upon binding of (unlabeled) TAU1, with the most prominent being those of Ile440 13Cδ1-1H3 (9, 0.25 ppm) and 13Cγ2-1H3 (17.5, 0.45 ppm). The addition of more TAU1 only changes intensities of the appearing cross-peaks and not chemical shifts, indicating slow off-rates and high affinity (KD ≪ 10 μm; see also Table 1). The addition of 4R0N Tau causes appearances of new resonances just as the TAU1 peptide (Fig. 3C), suggesting a basically similar interaction. Another Tau-derived sequence that has been proposed to bind Hsc70, 613VQIVFK, caused hardly any change and thus binds with weak affinity if at all (Fig. 3D and Table 1).
Figure 3.

Methyl region of the 13C-1H HSQC spectrum of BETA (MSNA395d-G508G3H6). A, blue, 11 μm BETA; red, with 11 μm TAU1, GKVQIINKKG. B, blue, 35 μm BETA; red, with TAU1, 300 μm GKVQIINKKG. C, blue, 11 μm BETA; red, with 19 μm WT 4R0N TAU. D, blue, 11 μm BETA; red, with 12 μm TAU2, VQIVYK. The conditions were as follows: 25 mm Tris, pH 7.20, 50 mm NaCl, 1 mm EDTA, 2 mm DTT, 0.02% azide, 5% D2O, with SIGMAFAST proteinase inhibitors.
Table 1.
Binding of substrates to Hsc70 and its truncations by fluorescence polarization
TAU1, GKVQIINKKG; insulin C-peptide (C-peptide); TAU2, VQIVYK; huntingtin, MATLEKLMKAFESLKSF. The Fam-HLA tracer was at 20 nm, wt-Hsc70 at 5 μm, NBD-BETA and BETA both at 1 μm. The inhibitory constant KI1 was computed from numerical integration of kinetic equations (see “Experimental procedures”); KI2 was computed from Equation 3 in Ref. 54.
| Wild type |
NBD-BETA (1–508) |
BETA (395–508) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 | KI1 | KI2 | EC50 | KI1 | KI2 | EC50 | KI1 | KI2 | |
| μm | μm | μm | |||||||
| 4R0N TAU | 1.1 ± 0.4 | <1.0 | 4.2 | 0.5 ± 0.3 | 0.2 | 0.75 | 0.8 ± 0.3 | 0.2 | 0.75 |
| TAU1 | 6.3 ± 2.0 | <1.0 | 5.9 | 2.6 ± 1.5 | 0.3 | 1.3 | 3.1 ± 1.6 | 0.5 | 1.6 |
| NRLLLTG | 15 ± 11 | 4.0 | 14 | 9.4 ± 3.9 | 2.0 | 4.7 | 15 ± 4.7 | 2.0 | 7.8 |
| C-peptide | 4.4 ± 5.4 | <1.0 | 4.1 | 0.8 ± 0.4 | <0.1 | 0.4 | 1.0 ± 0.2 | <0.1 | 0.5 |
| TAU2 | >200 | >50 | >50 | ||||||
| huntingtin | >200 | >50 | >50 | ||||||
| KFERQ | >200 | >200 | >200 | ||||||
We chose to work with the Hsc70 BETA construct because a DnaK NBD–SBD construct, also without LID, has been shown to be functional in multiple chaperone assays, establishing the isolated DnaK BETA construct as a valid domain for structural studies (14) (36). However, we needed to re-explore this issue for the human protein. To do this, we created Hsc70 1–508, which includes the NBD and the BETA domains but lacks the LID and Tail. We used a fluorescence polarization assay to measure binding of FAM–HLA to Hsc70 1–508 and full-length, human Hsc70. We found that FAM–HLA bound tightly to Hsc70 1–508 (KD = ∼0.1 μm in the presence of ADP), whereas binding is nucleotide-dependent (at least 10-fold weaker binding with ATP) (Fig. 4A), establishing that nucleotide–substrate allostery is intact in this construct. Interestingly, we also found that FAM–HLA binding to Hsc70 1–508 is ∼10–30 times stronger than to full-length Hsc70. Indeed, it was difficult to reach concentrations high enough to measure binding of FAM–HLA to wt Hsc70 because of solubility limits. Next, we compared the ability of Hsc70 1–508 and full-length Hsc70 to be stimulated by J protein in steady-state ATPase assays. We found that Hsc70 1–508 was readily stimulated by a representative J protein, DnaJA2, and that this stimulation was almost identical to full-length protein (Fig. 4B). One of the most complex activities of Hsc70 is its ability to refold denatured firefly luciferase. This activity requires multiple rounds of substrate binding and release coordinated with ATP hydrolysis and J protein interactions. To investigate whether Hsc70 1–508 was capable of this function, we tested it in a luciferase refolding assay in combination with ATP and DnaJA2. Although Hsc70 1–508 was not as efficient as wt Hsc70, it had significant refolding activity (Fig. 4C). The reduction in maximum refolding efficiency as compared with wt Hsc70 is not too surprising, because it is known that the efficiency depends on the kinetics of substrate binding and release, which likely are perturbed without the LID (37). From this experiment, we were also interested in testing whether Hsc70 1–508 might coordinate with DnaJA2. We noticed that the refolding activity of Hsc70 1–508 shows a maximum at approximately the same concentration of DnaJA2 (∼0.5 μm), as seen with wt Hsc70, suggesting that it can indeed coordinate with the J protein. At higher concentrations of DnaJA2 (0.5 μm), the apparent refolding activity decreases for both proteins, likely because of accumulation of DnaJA2 on unfolded luciferase (11). Finally, it is known that substrate binding to prokaryotic DnaK enhances ATP hydrolysis but that this allostery is lost in human Hsc70 (11). Consistent with this idea, we found that the ATPase activity of wt Hsc70 and Hsc70 1–508 are not/hardly affected by addition of NR peptide, whereas DnaK is strongly stimulated (Fig. 4D). Together, we conclude that Hsc70 1–508 mimics native Hsc70 in these assays and is largely functional as a chaperone. This result gave us confidence that Hsc70–BETA would be suitable for studying the structural consequences of Hsc70–substrate interactions.
Figure 4.

Functional assays reveal that Hsc70(1–508) (NBD–BETA) behaves similarly to wt-Hsc70. A, HLA tracer binding in the ADP and ATP states, as measured by fluorescence polarization. Dark blue, NBD–BETA ADP; light blue, NBD–BETA ATP; black, wt-Hsc70 ADP; gray, wt-Hsc70 ATP. Note that the wt-Hsc70 curves could not reach saturation because of limitations on protein solubility. B, DJA2 stimulation of ATP turnover, as measured by malachite green assays. Blue, NBD–BETA; black, wt-Hsc70. C, refolding of denatured luciferase. Blue, NBD–BETA; black, wt-Hsc70; gray, no Hsc70 control. Note that DJA2 will first stimulate and then inhibit ATP turnover, showing a maximal stoichiometry. D, the ATPase activity of neither wt-Hsc70 nor NBD–BETA can be stimulated by peptide substrates. Red, DnaK by NR; black, wt-Hsc70 by TAU1; blue, NBD–BETA by TAU1.
As a further test of this idea, we determined the affinities of 4R0N Tau, TAU1, and other peptides to different Hsc70 constructs using a fluorescence competition assay with a FAM–HLA peptide (Fig. 5). To cross-validate this assay by NMR, we show in Fig. S1 of the supporting data that FAM–HLA causes appearances of many resonances (Fig. S1A) at similar frequencies as TAU1 does (Fig. S1B), so it is reasonable to assume that they compete for the same site. The competition assays in Fig. 5 show that the series of peptides compete for the NBD–BETA and BETA constructs with similar potency, whereas the competition with wt Hsc70 consistently required approximately three times higher concentrations. However, the affinity of wt Hsc70 for tracer was also weaker, so to better compare the values we converted the EC50 values to inhibitory constants (see “Experimental procedures”). By this analysis, we found that the binding affinities of the peptides for all three constructs are indistinguishable (Table 1). Interestingly, it appears that 4R0N Tau binds somewhat tighter than TAU1, perhaps because of the contribution from secondary interactions. As a control, we also ensured that none of the peptides interacts with the tracer (Fig. S2). Together, these results are consistent with the idea that BETA is a suitable model for studying substrate interactions.
Figure 5.
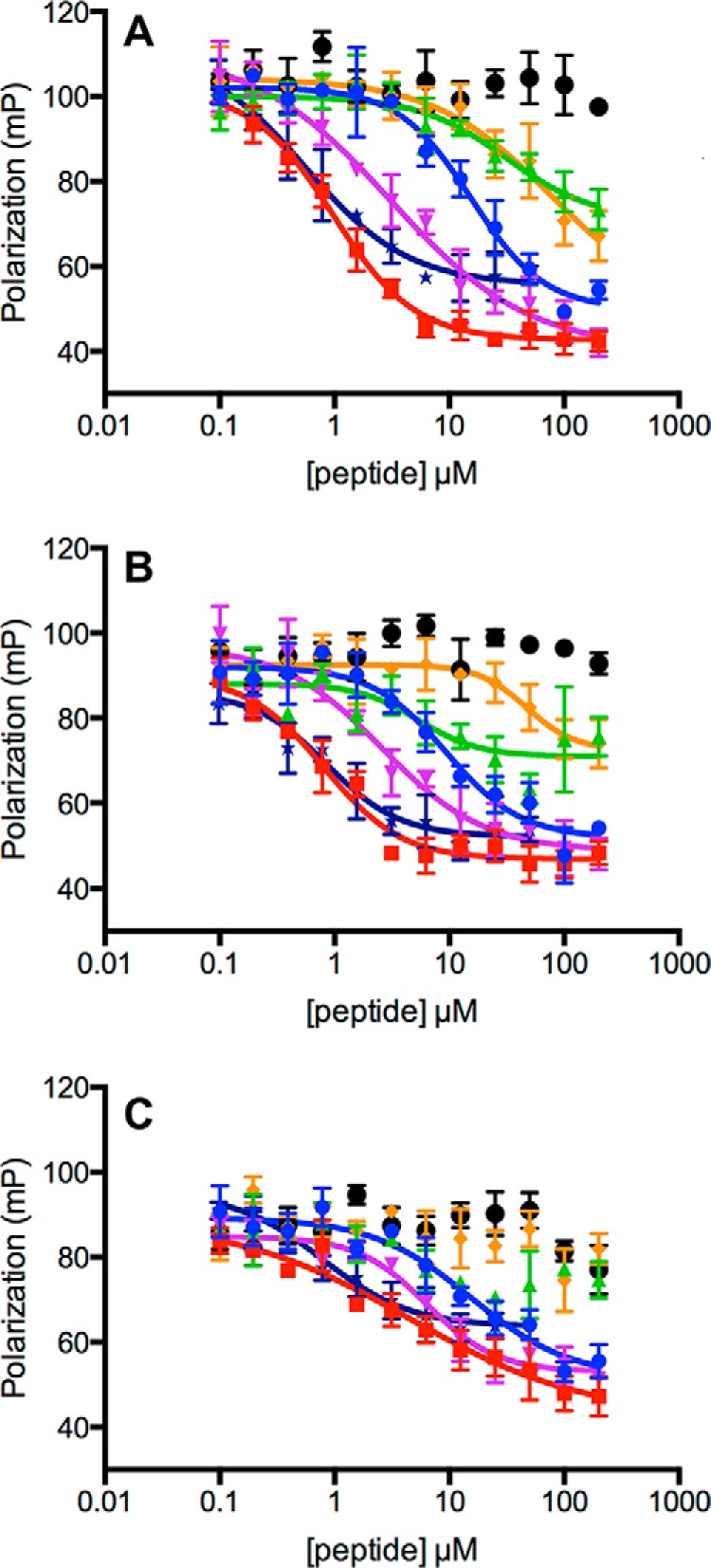
Competition of various peptides with FAM–HLA for binding to different Hsc70 constructs in the presence of 1 mm ADP. A, Hsc70–BETA at 1 μm. B, Hsc70 NBD–BETA (1–508) at 1 μm. C, wt-Hsc70 at 5 μm. Red, insulin C-peptide; purple, TAU1; blue, NRLLLTG; orange, TAU2; green, huntingtin; black, KFERQ; dark blue, 4R0N TAU. Also see Table 1.
The other major paralog of the Hsp70 family in the cytosol is Hsp72 (HSPA1A), also called inducible Hsp70. It is known that Hsc70 binds differently than Hsp72 to 0N4R Tau (22), so we wondered whether relative affinity for TAU1 or TAU2 might partly explain this difference. Using the FAM–HLA tracer, we measured the relative affinity of 0N4R Tau and the peptides for wt Hsp72 and the equivalent Hsp72 BETA construct (residues 395–508). We found that most of the peptides, except for the C-peptide of insulin, bind equivalently or weaker to Hsp72 than Hsc70 (Fig. S3 and Table S1). This was especially prominent for TAU1, which bound >10-fold less well to Hsp72 BETA than Hsc70 BETA.
As a next step, we investigated whether the aforementioned dynamical changes in the SBD also occur upon binding of TAU1 to full-length Hsc70. However, the 70 kDa full-length Hsc70 does not give rise to good NMR spectra, because of dominance of the resonances of the unstructured tail (38), so we used a construct that includes NBD, BETA, and LID, without the TAIL. This construct was enabled for substrate binding by including a mutation (L543Y) as suggested by the Lab of Dr. Gierasch (39). This mutation, in context of DnaK or Hsp72 (HSPA1A), restores the peptide-binding affinity that is lost upon removal of the TAIL. Satisfyingly, we found that TAU1 peptide binding causes the appearance of resonances in the TROSY HSQC NMR spectrum of Hsc70(1–604) (Fig. 6, red spots) and that the majority of those occur at the same positions as they do in the isolated BETA domain (black circles).
Figure 6.

15N-1H TROSY HSQC spectra of near wt Hsc70(1–604). Blue, 45 μm Hsc70(1–604) L542Y in the ADP state. Red, 45 μm Hsc70(1–604) L542Y in the ADP state +50 μm TAU1 (GKVQIINKKG). Black, 45 μm BETA + 200 μm TAU1 (GKVQIINKKG).
To explore where the interaction of TAU1 takes place, we carried out de novo resonance assignments for the backbone and most methyl-bearing side chains of BETA complexed to TAU1 and plotted the TAU1-induced NMR spectral changes on the NMR structure available for BETA domain (40). From this analysis, many of the broadened resonances are found in and around the canonical binding cleft (Fig. 7). However, the extent of the spectral perturbation is too large to confidently map the binding of TAU1 to the cleft itself.
Figure 7.

The TAU1-induced NMR spectral changes plotted on the Hsc70 BETA domain (PDB entry 7HSC). Red, amides and methyls for which the BETA apo NMR signal intensity is < 50% of that of BETA with TAU1. Blue, resonances that were less perturbed. Gray, no information.
Subsequently, we compared the binding of peptide of TAU1 to BETA with the binding of NRLLLTG (NR) to BETA. Comparing TAU1 and NR binding is interesting because crystal and solution structures are available for NR bound to DnaK SBD (15, 16) and human Hsp70 (HSPA1A) SBD (41), showing the peptide to be deeply inserted into the hydrophobic cleft. Fig. 8A shows that NR, just like TAU1, causes the appearance of many resonances, so it is reasonable to assume that both NR and TAU1 bind into the same cleft. However, as the overlay shows (Fig. 8B), the spectra of NR- and TAU1-bound BETA are not identical, especially for the peaks that appear upon binding, indicating structural differences between the complexes at the binding sites.
Figure 8.

Comparing the 15N-1H TROSY-HSQC spectra of BETA with NRLLLTG and with GKVQIINKKG. A, 35 μm BETA in blue superposed on 35 μm BETA + 200 μm NRLLLTG (green). B, 35 μm BETA + 200 μm NRLLLTG (green) superposed on 35 μm BETA + 200 μm GKVQIINKKG (red).
Thus, we cannot rely on the HSPA1A and DnaK structures with NR for the details of the binding of TAU1 to Hsc70. We thus wondered what the details of the TAU1 binding are, such as which residues of TAU1 are buried into the pocket and what the direction of binding is. To address this question, we set out to identify NOEs between assigned resonances of TAU1 and resonances of BETA. However, to obtain reliable assignments and NOEs for TAU1 resonances, one needs 13C,15N-labeled TAU. Hence we decided to design and assign a construct that contains the TAU1 sequence in cis: (Hsc70 (385–533)-QPGGGKVQIINKKLD). Fig. S4 shows that virtually all cross-peaks of the 1H-15N NMR spectrum of the 113-residue BETA domain bound to TAU1 in trans overlay with a subset of the cross-peaks of the 153-residue BETA–LID–cis–TAU1 construct, providing confidence that the two preparations yield the same complex.
Short of carrying out a de novo NMR structure determination, we identified 153 nonsequential NOEs in a set of 3D NOESY spectra (examples in Figs. S5 and S6) of the current construct that are compatible with the structure of PDB entry 7HSC (Fig. S7). 11 additional NOEs define the interaction between the TAU1 sequence and the core of the BETA domain (see Fig. S7 and the listing under “Experimental procedures”). We carried out a molecular dynamics structure refinement based on a constrained structure of PDB entry 7HSC structure with the TAU1 peptide positioned by the identified NOEs (see “Experimental procedures” and Figs. 9, A and B). From this analysis, we find that the second Ile of TAU1 is buried in the pocket.
Figure 9.

A, molecular replacement structure for Hsc70 BETA domain with the TAU peptide bound. Color coding is as in Fig. 6. The sequence GKVQIINKKG is shown in yellow. The sphere is the CD methyl group of the second Ile of the in cis sequence GKVQIINKKG. B, details of the molecular replacement structure for Hsc70 BETA domain with the in cis TAU1 sequence bound. On Hsc70, hydrophobic residues are in green, amphipathic residues (Thr, Tyr) in peat green, positive residues in blue, negative residues in red, and polar residues in gray. Residues VII of the in cis sequence GKVQIINKKG are labeled as Val539, Ile541, and Ile542 (magenta). Other residues of TAU1 are in yellow.
With the Hsc70 BETA construct in-hand, we also investigated the binding of other peptides. Fig. S8 shows that the peptide GAGSLQPLALEGSLQKR, derived from the insulin C-peptide, interacts strongly with Hsc70. According to the competition assay, the affinity is submicromolar, not only for BETA but also for wt-Hsc70 and wt-HSPA1A (Table 1 and Table S1). Conversely, the autophagy recognition peptide KFERQ and a Huntingtin-derived peptide (MATLEKLMKAFESLKSF) do not compete with tracer binding for wt-Hsc70 or HSPA1A (Table 1 and Table S1 and Fig. S8). The significance of these findings will be discussed below. More broadly, this work thus also establishes the BETA construct as a useful tool for studying the interaction of Hsc70 with its substrates or drug candidates.
Discussion
In this study, we find structural evidence that TAU1 (KVQIINKK) binds to the substrate-binding cleft of Hsc70, with the same N → C polarity as NRLLLTG in DnaK. The NOE data provide evidence that the Cδ1H3 group of the second TAU1 Ile residue is buried deeply in the binding pocket. This is similar to the binding of the Cδ1H3 of the second leucine in NRLLLTG in the case of DnaK but different from the binding of NRLLLTG with Hsp72 (HSPA1A), where the Cδ1H3 of the third leucine is inserted into the pocket (41). Although this discovery may not be earth-shattering, it adds to our knowledge of Hsc70–substrate interactions in a number of ways.
First, the Hsc70 SBD-binding cleft shows unexpected substrate specificity. For example, the TAU1 sequence binds tightly (KD ≈ 0.4 μm; Table 1), whereas the TAU2 sequence (SVQIVYK) does not (EC50 > 50 μm). Furthermore, we found that the consensus model peptide, NRLLLTG, does not bind very tightly to Hsc70 BETA (KD ≈ 2 μm; Table 1). Current methods for predicting the substrates of Hsp70 are based on peptide array data obtained with prokaryotic DnaK, so it may be worth revisiting these ideas.
Second, we found that an Hsc70 construct lacking the LID domain still has many of the activities of the wt protein in vitro, including protein-refolding ability. Similar findings were made for a LID-less DnaK construct (14). The BETA domain binds the different peptides equally well as wt-Hsc70 and the NBD–BETA construct in the ADP state (Table 1); hence the absence of the LID does not cause the BETA domain to move to a lower-affinity “ATP state.” Indeed, the LID-less NBD–SBD construct can still have low or high affinity for substrate depending on the nucleotide state of the NBD (Fig. 4A). Hence, the dramatic difference in position of the LID in the structures of wt-DnaK in ADP (17) and ATP (19) states does not seem to be a hallmark of the allosteric state. So, what then is the function of the LID? Slepenkov and Witt (36) have shown that the DnaK LID decreases the kinetics of both substrate binding and release by at least 1 order of magnitude. Others have found that the DnaK tail may aid in recruiting it to substrate (42). For Hsc70, the LID extended tail contains the C-terminal EEVD motif, which is critical for binding to CHIP (29). In addition, recent NMR and mutagenesis studies indicate that the Hsc70 LID binds to phosphoserine lipids in the lysosome and endosome outer envelopes, processes that are important in CMA (43). Thus, the LID seems to play more important roles in protein–protein and protein–lipid interactions than in substrate binding.
Third, we find that many NMR resonances of the binding loops around the substrate cleft of the Hsc70 BETA domain are absent in the apo state, likely because of dynamical disorder on the NMR chemical shift time scale (milli/microseconds). It seems likely that these motions allow the cleft to structurally adept itself different hydrophobic sequences. This finding appears to be corroborated in other systems. For example, dynamical disorder was also detected in the isolated apo-BETA domain of DnaK E. coli by NMR (14). In the crystal structures of DnaK in the ATP state, in which the LID is disengaged from the SBD and in which no substrate is present, the crystallographic B-factors in the binding loops (40–60 Å2) are significantly higher than for other regions of the protein (∼20 Å2). However, from those studies, one cannot distinguish whether the absence of a docked LID or the absence of a bound substrate causes the disorder. Here, we show that the dynamic disorder is due to the lack of substrate, because it is quenched upon substrate binding even without LID present. The amount of disorder (i.e. the number of missing resonances) is more substantial in Hsc70 than in DnaK BETA (14) and but the extent of the disorder is smaller in Hsc than in DnaK. For example, the NH NMR cross-peaks of DnaK residues Thr417 and Ile418, more than 20 Å removed from the substrate binding cleft, are broadened in the apo state of DnaK, whereas they are present in the Hsc70 apo state. In DnaK, these resonances reappear when the peptide NR is bound. As was then hypothesized (14, 44), these DnaK residues may be part of the interface to the NBD and may allosterically communicate the cleft's status to the NBD. Indeed, the crystal structures of DnaK in the ATP state (18, 19) show Thr417 and Ile418 at the center of the SBD–NBD interface, lending much credence to these earlier conjectures. The allosteric mechanism connecting cleft and interface to the NBD in DnaK has been further explored by the Gierasch lab, showing substrate-induced conformational changes forming a “path” from SBD cleft to NBD interface (45). However, according to the current structural/dynamical data on Hsc70 BETA domain, there is no clear conformational/dynamic path from the cleft area to the presumed interface with the NBD (at the right-hand side in Fig. 7). The NH resonances of the corresponding residues Thr419 and Ile420 are visible in the apo NMR data, although Ile420 is weak, and becomes somewhat stronger when TAU1 is bound. This behavior is interesting in the context of a surprising asymmetry in the allostery of wt Hsc70. Specifically, we found that substrate binding to Hsc70 has little effect on ATP hydrolysis (Fig. 4D), whereas the effect is large for DnaK. Thus, whereas NBD → SBD allosterics are fully functional in Hsc70 (Fig. 4A), there seems to be little SBD → NBD communication. Of course, the basic thermodynamic cycle should still exist; because Hsc70 with ADP binds TAU1 tighter than Hsc70 with ATP, TAU1 must drive the NBD toward the ADP conformation by a mechanism that remains unclear. Moreover, it remains unclear why the human system has lost substrate-induced activation of ATP hydrolysis, but the current BETA construct might be one structural tool for studying this question.
Fourth, the Hsc70 BETA construct allowed us to study a variety of substrate interactions. Hsc70 also plays a key role in CMA and recognizes its autophagy clients through their exposed KFERQ sequences (46). This sequence does not conform to the typical hydrophobic sequence that one expects for a cleft-binding peptide, such as TAU1 or NR. Therefore, we were not surprised to find that the KFERQ peptide does not bind to the canonical substrate binding cleft (Table 1 and Fig. S8). We were also interested in the binding of the Huntingtin-derived peptide (MATLEKLMKAFESLKSF). Hsc70 is known to bind directly to Huntingtin and to suppress Huntington's phenotypes in many animal models. It has been suggested that the mentioned sequence, which flanks the poly-Q tract, constitutes an Hsc70-binding site (47). Our results (Table 1 and Fig. S8) show that it does not bind to the Hsc70 substrate binding cleft, suggesting either that the flank region is not involved or that another noncanonical interaction with Hsc70 is responsible. Lastly, we found that the insulin C-peptide, GAGSLQPLALEGSLQKR, binds tightly (KD < 100 nm; Table 1) to the Hsc70 BETA cleft (Table 1 and Fig. S8). We expect that the ER Hsp70 chaperone Bip also binds to this peptide where such interaction would have functional significance.
Experimental procedures
Protein expression and purification
A Pet21 plasmid coding for Hsc70 BETA (residues SNA395D–G508GGGHHHHHH) was purchased from VectorBuilder (Cyagen). It was transfected into and expressed in E. coli BL21 cells in M9 medium containing 1 g/liter 15NH4Cl and 2 g/liter glucose 13C6 (Cambridge Isotopes Laboratories). The homogenized cell contents were loaded on nickel–nitrilotriacetic acid (Qiagen) and extensively washed with Tris buffer. When the wash eluate was clear, the bound BETA domain was unfolded/refolded from 4 m guanidine HCl while on the column to remove bound peptides/proteins. After elution with imidazole, the protein was exchanged into 100 mm (NH4)2CO3 (buffered at pH 7 with 1 mm Tris) and lyophilized, because it was in our hands not possible to concentrate the construct beyond 20 μm using ultra-filtration. The freeze-dried material was resuspended into 1 ml of H2O to yield a 200 μm sample (by Bradford and NMR).
The Pet21 plasmid coding for Hsc70 BETA-TAU CIS construct, MSNA395D–Q533 534QPGGGKVQIINKKLDGGGHHHHHH, was also purchased from VectorBuilder. The protein was expressed in E. coli BL21 cells in M9 medium containing 1 g/liter 15NH4Cl and 2 g/liter Glucose 13C6. The protein was purified over nickel–nitrilotriacetic acid without refolding and concentrated using Centricon (Millipore) ultrafiltration. The sample was dialyzed into 25 mm Tris, 25 mm KCl, 1 mm EDTA, 1 mm DTT, 0.02% NaN3 in 90% H2O/D2O, pH 7.2. The peptides mentioned in this work were purchased from Peptide 2.0 (Chantilly, VA).
NMR experimentation
NMR backbone assignments were independently determined for BETA (with unlabeled KVQIINKK bound) and BETA–cis–TAU, from HNCO, HNCACO, HNCA, HNCOCA, HNCACB, and HNCOCACB TROSY experiments on Bruker Avance2 spectrometers operating at 16 and 14 Tesla, respectively. The semiautomatic assignment program EZ-ASSIG 4.1 was used for both (48). Additional assignments were made from intraresidue and sequential NOEs identified in (1H-TM-15N-1H) 3D NOESY-TROSY spectra. The methyl groups were assigned for BETA using 3D H(C)CH COSY, 3D (H)CCH COSY, and TOCSY spectra. The assignments were transferred to the methyl spectrum of BETA–cis–TAU. The additional methyls in BETA–cis–TAU were assigned from intraresidue and sequential NOEs with the backbone NH using 3D 1H-TM-13C-1H and 13C-TM-13C-1H NOESY HSQC-Watergate spectra. Most NMR experiments were obtained using nonuniform-sampling schemes with 15% point density, using the Hyberts–Wagner protocols (49). The backbone (HN, N, CA, CB, and CO) assignments are virtually contiguous and 85% complete except for the region Lys531–Gly537, for which no single resonance could be identified, and for the N- and C-terminal cloning artifacts. The methyl group assignments (Ala, Thr, Met, Ile, Leu, and Val) are 80% complete (see Fig. S6). The assignments are deposited at the Biological Magnetic Resonance Bank (accession no. 27395).
The bulk of the NOEs for the structure verification/determination were assigned by hand in Sparky displays (50) from 1H-TM-15N-1H 3D NOESY-TROSY (Fig. S5) and 13C-TM-15N-1H 3D HMQC-NOESY-TROSY spectra (Fig. S6), together with the 3D 1H-TM-13C-1H NOESY-HSQC and 13C-TM-13C-1H HMQC-NOESY-HSQC-Watergate spectra. The obtained NOEs were compared with NOEs computed from the NMR structure for the self-bound Hsc70 SBD (385–543) (PDB entry 7HSC) and from the crystal structure of Hsp70 SBD with NRLLLTG bound (PDB entry 4PO2). The NOE computation was a complete relaxation matrix numerical integration based on cross-relaxation rates obtained from distances in the structure using formal relaxation equations assuming no local dynamics and an experimentally determined rotational correlation time (9 ns) (51).
Modeling
The “molecular replacement” structure for BETA–cis–TAU presented here was obtained starting from the Hsc70 SBD (385–543) coordinates (PDB entry 7HSC) (20), omitting residues 534–543 and replacing them with QPGGGKVQIINKKLD in extended conformation pointing away from the protein core. This conformation was minimized using AMBER 16 (52), using the generalized Born-solvent replacement method with all CA restrained to their current positions (53). The sequence QPGGGKVQIINKKLD was pulled into the substrate-binding cleft during a 50-ps molecular dynamics run by the NOEs: T429HN-I541HN, T429HN-I542HN, T429HN-I542MD, T429HN-I541HA, T430HN-I541HA, T430HN-I542MD, I541HN-T429MG, I542HN-T429MG, I542HN-I541MD, V438MG-I542MD, and I440MD-I542MD, whereas residues 395–508 remained restrained to their CA positions. The conformation was equilibrated, cooled down, and minimized.
Fluorescence polarization assay
Experiments were performed as described previously (11). Solutions of Hsc70 were prepared at the concentrations specified with saturating amounts of ATP or ADP (1 mm final) in fluorescence polarization assay buffer (100 mm Tris, 20 mm KCl, 6 mm MgCl2, 0.01% Triton X-100, pH 7.4). The solutions were added at a total volume of 16 μl to the wells of a low-volume, black, 384-well, round-bottom plate (catalog no. 4511, Corning). Then 4 μl of the fluorescent peptide probe 5Fam-RENLRIALRY (Fam-HLA) was added to a final concentration of 20 nm, and the plate was allowed to incubate, in the dark, at room temperature for 1 h. At the end of the incubation period, fluorescence polarization was read at an excitation wavelength of 485 nm and an emission wavelength of 530 nm in a Molecular Devices Spectramax M5 plate reader. The results were fit to a 1:1 variable slope model in GraphPad PRISM 7.0.
Converting EC50 to KI
We solved the kinetic equations for the scheme
by numerical integration. T is the tracer, and I is the inhibitor. For NBD–BETA and BETA, the following parameters were used: KT = kTf/kTb = 200 nm (see Fig. 4A), whereas KI = kIf/kIb was varied from 10 nm to 50 μm for different calculations. Both forward rates were arbitrarily set to 106 m−1 s−1. The integration starting conditions were: [P](t = 0) = 1 μm, [T](t = 0) = 20 nm, [PT](t = 0) = 0, [PI](t = 0) = 0, with [I](t = 0) varying from 0 to 200 μm. The total integration time was set to 3/slowest kinetic rate, allowing the equilibrium concentrations of all five reaction species to be obtained. For wt-Hsc70, the following parameters were used: KT = 3 μm (see Fig. 4A) and [P](t = 0) = 5 μm. For each run with different KI, the value of [I] for which [PT] was reduced to 50% of its value without I was compared with the experimental EC50 value.
ATP hydrolysis
A version of this assay was performed as previously described (11). Briefly, solutions of Hsc70 and DJA2 were prepared in malachite green buffer (100 mm Tris, 20 mm KCl, 6 mm MgCl2, 0.01% Triton X-100, pH 7.4) and added to the wells of a clear, 96-well plate. Hsc70 was held at a constant concentration of 1 μm, whereas DJA2 was prepared and added in a dilution series. A solution of ATP was added to a final concentration of 1 mm in a total assay volume of 25 μl/well. The plate was covered, briefly centrifuged, and then allowed to incubate at 37 °C for 1 h. During incubation, the Malachite Green reagent was prepared, as a 2:2:1:1 mixture of water, malachite green (0.081% w/v), polyvinyl alcohol (2.3% w/v), and ammonium heptamolybdate tetrahydrate (5.7% w/v in 6 m HCl). At the end of the incubation period, 80 μl of malachite green reagent, followed by 10 μl of saturated sodium citrate, was added to each well, and absorbance was read at 620 nm in a Molecular Devices Spectramax M5 plate reader. A phosphate standard was added to the plate to convert absorbance values to pmol ATP hydrolyzed/min. The results were fit to a Michaelis–Menten with variable intercept model in GraphPad PRISM 7.0.
Luciferase refolding
Experiments were performed as previously described, with some modifications (11). Denatured luciferase was prepared by incubating native firefly luciferase (Promega) with 8 m guanidine HCl for 1 h at room temperature. A solution of Hsc70 and denatured luciferase (final concentrations, 1 μm and 100 nm, respectively) was prepared in refolding buffer (23 mm HEPES, 120 mm KAc, 1.2 mm MgAc, 15 mm DTT, 61 mm creatine phosphate, 35 units/ml creatine kinase, 5 ng/μl BSA, pH 7.4) and added to the wells of a white 96-well plate. DJA2 was also added to the wells in a dilution series. A solution of ATP was added to each well at a final concentration of 1 mm, in a total reaction volume of 25 μl/well. The plate was covered, briefly centrifuged, and then allowed to incubate at 37 °C for 30 min. Although the plate was incubating, the Steady-Glo reagent (Promega) was prepared, as described previously (11), of which 25 μl was added to each well following the 1-h incubation, and luminescence was read immediately in a Molecular Devices Spectramax M5 plate reader. To improve clarity, the results were fit to a spline.
Author contributions
I. R. T., A. A., T. W., B. A. N., A. B., and E. R. P. Z. data curation; I. R. T. and E. R. P. Z. formal analysis; I. R. T. and E. R. P. Z. investigation; I. R. T., J. E. G., and E. R. P. Z. writing-review and editing; J. E. G. and E. R. P. Z. conceptualization; J. E. G. and E. R. P. Z. resources; J. E. G. and E. R. P. Z. supervision; J. E. G. and E. R. P. Z. funding acquisition; J. E. G. and E. R. P. Z. validation; J. E. G. and E. R. P. Z. methodology; J. E. G. and E. R. P. Z. writing-original draft; J. E. G. and E. R. P. Z. project administration; E. R. P. Z. software; E. R. P. Z. visualization.
Supplementary Material
Acknowledgments
E. R. P. Z. thanks Drs. Scott Robson and Sven Hyberts from Prof. G. Wagner's lab at Harvard Medical for guidance in implementing nonuniform-sampling sampling and processing for the 3D NMR spectra. E. R. P. Z. also thanks Dr. Patrick O'Brien, Dr. Alexander Ninfa, and Michael Baldwin (all at Michigan) for invaluable advice and encouragement.
This work was supported by National Institutes of Health Grant R01NS059690 (to J. E. G. and E. R. P. Z.) and the Protein Folding and Disease Program of the University of Michigan (to E. R. P. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Dedicated to the memory of Chad A. Dickey.
This article contains Table S1 and Figs. S1–S9.
- NBD
- nucleotide-binding domain
- SBD
- substrate-binding domain
- BETA
- β-domain harboring the substrate-binding cleft
- LID
- α-helical lid
- CMA
- chaperone-mediated autophagy
- HSQC
- heteronuclear single quantum coherence
- PDB
- Protein Data Bank
- CHIP
- C terminus of Hsp-interacting protein
- TROSY
- transverse relaxation optimized spectroscopy.
References
- 1. Schröder H., Langer T., Hartl F. U., and Bukau B. (1993) Dnak, Dnaj and Grpe form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 12, 4137–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chiappori F., Merelli I., Milanesi L., Colombo G., and Morra G. (2016) An atomistic view of Hsp70 allosteric crosstalk: from the nucleotide to the substrate binding domain and back. Sci. Rep. 6, 23474 10.1038/srep23474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Höhfeld J., and Patterson C. (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96 10.1038/35050618 [DOI] [PubMed] [Google Scholar]
- 4. Kaushik S., and Cuervo A. M. (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kampinga H., Hageman J., Vos M., Kubota H., Tanguay R., Bruford E., Cheetham M., Chen B., and Hightower L. (2009) Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chap. 14, 105–111 10.1007/s12192-008-0068-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evans C. G., Chang L., and Gestwicki J. E. (2010) Heat shock protein 70 (hsp70) as an emerging drug target. J. Med. Chem. 53, 4585–4602 10.1021/jm100054f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang J., Ballinger C. A., Wu Y., Dai Q., Cyr D. M., Höhfeld J., and Patterson C. (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 276, 42938–42944 10.1074/jbc.M101968200 [DOI] [PubMed] [Google Scholar]
- 8. McCarty J. S., Buchberger A., Reinstein J., and Bukau B. (1995) The role of ATP in the functional cycle of the DnaK chaperone system. J. Mol. Biol. 249, 126–137 10.1006/jmbi.1995.0284 [DOI] [PubMed] [Google Scholar]
- 9. Rüdiger S., Germeroth L., Schneider-Mergener J., and Bukau B. (1997) Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16, 1501–1507 10.1093/emboj/16.7.1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gamer J., Multhaup G., Tomoyasu T., McCarty J. S., Rüdiger S., Schönfeld H. J., Schirra C., Bujard H., and Bukau B. (1996) A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 15, 607–617 [PMC free article] [PubMed] [Google Scholar]
- 11. Rauch J. N., and Gestwicki J. E. (2014) Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J. Biol. Chem. 289, 1402–1414 10.1074/jbc.M113.521997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flaherty K. M., DeLuca-Flaherty C., and McKay D. B. (1990) 3-Dimensional structure of the ATPase fragment of a 70k heat-shock cognate protein. Nature 346, 623–628 10.1038/346623a0 [DOI] [PubMed] [Google Scholar]
- 13. Bhattacharya A., Kurochkin A. V., Yip G. N., Zhang Y., Bertelsen E. B., and Zuiderweg E. R. (2009) Allostery in Hsp70 chaperones is transduced by subdomain rotations. J. Mol. Biol. 388, 475–490 10.1016/j.jmb.2009.01.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pellecchia M., Montgomery D. L., Stevens S. Y., Vander Kooi C. W., Feng H. P., Gierasch L. M., and Zuiderweg E. R. (2000) Structural insights into substrate binding by the molecular chaperone DnaK. Nat. Struct. Biol. 7, 298–303 10.1038/74062 [DOI] [PubMed] [Google Scholar]
- 15. Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. E., and Hendrickson W. A. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614 10.1126/science.272.5268.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stevens S. Y., Cai S., Pellecchia M., and Zuiderweg E. R. (2003) The solution structure of the bacterial HSP70 chaperone protein domain DnaK(393–507) in complex with the peptide NRLLLTG. Protein Sci. 12, 2588–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bertelsen E. B., Chang L., Gestwicki J. E., and Zuiderweg E. R. (2009) Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. U.S.A. 106, 8471–8476 10.1073/pnas.0903503106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qi R., Sarbeng E. B., Liu Q., Le K. Q., Xu X., Xu H., Yang J., Wong J. L., Vorvis C., Hendrickson W. A., Zhou L., and Liu Q. (2013) Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct Mol. Biol. 20, 900–907 10.1038/nsmb.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kityk R., Kopp J., Sinning I., and Mayer M. P. (2012) Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol. Cell 48, 863–874 10.1016/j.molcel.2012.09.023 [DOI] [PubMed] [Google Scholar]
- 20. Morshauser R. C., Hu W., Wang H., Pang Y., Flynn G. C., and Zuiderweg E. R. (1999) High-resolution solution structure of the 18 kDa substrate-binding domain of the mammalian chaperone protein Hsc70. J. Mol. Biol. 289, 1387–1403 10.1006/jmbi.1999.2776 [DOI] [PubMed] [Google Scholar]
- 21. Sarkar M., Kuret J., and Lee G. (2008) Two motifs within the Tau microtubule-binding domain mediate its association with the hsc70 molecular chaperone. J. Neurosci. Res. 86, 2763–2773 10.1002/jnr.21721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jinwal U. K., Akoury E., Abisambra J. F., O'Leary J. C. 3rd, Thompson A. D., Blair L. J., Jin Y., Bacon J., Nordhues B. A., Cockman M., Zhang J., Li P., Zhang B., Borysov S., Uversky V. N., Biernat J., et al. (2013) Imbalance of Hsp70 family variants fosters Tau accumulation. FASEB J. 27, 1450–1459 10.1096/fj.12-220889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cleveland D. W., Hwo S. Y., and Kirschner M. W. (1977) Physical and chemical properties of purified Tau factor and the role of Tau in microtubule assembly. J. Mol. Biol. 116, 227–247 10.1016/0022-2836(77)90214-5 [DOI] [PubMed] [Google Scholar]
- 24. Trojanowski J. Q., Schmidt M. L., Shin R. W., Bramblett G. T., Rao D., and Lee V. M. (1993) Altered Tau and neurofilament proteins in neuro-degenerative diseases: diagnostic implications for Alzheimer's disease and Lewy body dementias. Brain Pathol. 3, 45–54 10.1111/j.1750-3639.1993.tb00725.x [DOI] [PubMed] [Google Scholar]
- 25. Lei P., Ayton S., Finkelstein D. I., Adlard P. A., Masters C. L., and Bush A. I. (2010) Tau protein: relevance to Parkinson's disease. Int. J. Biochem. Cell Biol. 42, 1775–1778 10.1016/j.biocel.2010.07.016 [DOI] [PubMed] [Google Scholar]
- 26. Cuervo A. M., Stefanis L., Fredenburg R., Lansbury P. T., and Sulzer D. (2004) Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- 27. Young Z. T., Rauch J. N., Assimon V. A., Jinwal U. K., Ahn M., Li X., Dunyak B. M., Ahmad A., Carlson G. A., Srinivasan S. R., Zuiderweg E. R., Dickey C. A., and Gestwicki J. E. (2016) Stabilizing the Hsp70–Tau complex promotes turnover in models of tauopathy. Cell Chem. Biol. 23, 992–1001 10.1016/j.chembiol.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rousaki A., Miyata Y., Jinwal U. K., Dickey C. A., Gestwicki J. E., and Zuiderweg E. R. (2011) Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J. Mol. Biol. 411, 614–632 10.1016/j.jmb.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDonough H., and Patterson C. (2003) CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 8, 303–308 10.1379/1466-1268(2003)008%3C0303:CALBTC%3E2.0.CO%3B2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cuervo A. M. (2004) Autophagy: many paths to the same end. Mol. Cell Biochem. 263, 55–72 10.1023/B:MCBI.0000041848.57020.57 [DOI] [PubMed] [Google Scholar]
- 31. Wang Y., Martinez-Vicente M., Krüger U., Kaushik S., Wong E., Mandelkow E. M., Cuervo A. M., and Mandelkow E. (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170 10.1093/hmg/ddp367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Caballero B., Wang Y., Diaz A., Tasset I., Juste Y. R., Stiller B., Mandelkow E. M., Mandelkow E., and Cuervo A. M. (2018) Interplay of pathogenic forms of human Tau with different autophagic pathways. Aging Cell 17, 12692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cuervo A. M., Dice J. F., and Knecht E. (1997) A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 272, 5606–5615 10.1074/jbc.272.9.5606 [DOI] [PubMed] [Google Scholar]
- 34. Wyttenbach A., Swartz J., Kita H., Thykjaer T., Carmichael J., Bradley J., Brown R., Maxwell M., Schapira A., Orntoft T. F., Kato K., and Rubinsztein D. C. (2001) Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum. Mol. Genet. 10, 1829–1845 10.1093/hmg/10.17.1829 [DOI] [PubMed] [Google Scholar]
- 35. Ko A. S., Smyth D. G., Marktussen J., and Sundby F. (1971) The amino acid sequence of the C-peptide of human proinsulin. Eur. J. Biochem. 20, 190–199 10.1111/j.1432-1033.1971.tb01378.x [DOI] [PubMed] [Google Scholar]
- 36. Slepenkov S. V., and Witt S. N. (2002) Kinetic analysis of interdomain coupling in a lidless variant of the molecular chaperone DnaK: DnaK's lid inhibits transition to the low affinity state. Biochemistry 41, 12224–12235 10.1021/bi0263208 [DOI] [PubMed] [Google Scholar]
- 37. Buczynski G., Slepenkov S. V., Sehorn M. G., and Witt S. N. (2001) Characterization of a lidless form of the molecular chaperone DnaK: deletion of the lid increases peptide on- and off-rate constants. J. Biol. Chem. 276, 27231–27236 10.1074/jbc.M100237200 [DOI] [PubMed] [Google Scholar]
- 38. Smith M. C., Scaglione K. M., Assimon V. A., Patury S., Thompson A. D., Dickey C. A., Southworth D. R., Paulson H. L., Gestwicki J. E., and Zuiderweg E. R. (2013) The E3 ubiquitin ligase CHIP and the molecular chaperone Hsc70 form a dynamic, tethered complex. Biochemistry 52, 5354–5364 10.1021/bi4009209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Swain J. F., Schulz E. G., and Gierasch L. M. (2006) Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J. Biol. Chem. 281, 1605–1611 10.1074/jbc.M509356200 [DOI] [PubMed] [Google Scholar]
- 40. Morshauser R. C., and Zuiderweg E. R. (1999) High resolution four-dimensional HMQC-NOESY-HSQC spectroscopy. J. Magn. Reson. 139, 232–239 10.1006/jmre.1999.1802 [DOI] [PubMed] [Google Scholar]
- 41. Zhang P., Leu J. I., Murphy M. E., George D. L., and Marmorstein R. (2014) Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PLoS One 9, e103518 10.1371/journal.pone.0103518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smock R. G., Blackburn M. E., and Gierasch L. M. (2011) Conserved, disordered C terminus of DnaK enhances cellular survival upon stress and DnaK in vitro chaperone activity. J. Biol. Chem. 286, 31821–31829 10.1074/jbc.M111.265835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morozova K., Clement C. C., Kaushik S., Stiller B., Arias E., Ahmad A., Rauch J. N., Chatterjee V., Melis C., Scharf B., Gestwicki J. E., Cuervo A. M., Zuiderweg E. R., and Santambrogio L. (2016) Structural and biological interaction of hsc-70 protein with phosphatidylserine in endosomal microautophagy. J. Biol. Chem. 291, 18096–18106 10.1074/jbc.M116.736744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Montgomery D. L., Morimoto R. I., and Gierasch L. M. (1999) Mutations in the substrate binding domain of the Escherichia coli 70 kDa molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling. J. Mol. Biol. 286, 915–932 10.1006/jmbi.1998.2514 [DOI] [PubMed] [Google Scholar]
- 45. Zhuravleva A., and Gierasch L. M. (2015) Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc. Natl. Acad. Sci. U.S.A. 112, E2865–E2873 10.1073/pnas.1506692112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cuervo A. M., and Dice J. F. (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 10.1126/science.273.5274.501 [DOI] [PubMed] [Google Scholar]
- 47. Monsellier E., Redeker V., Ruiz-Arlandis G., Bousset L., and Melki R. (2015) Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576 10.1074/jbc.M114.603332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zuiderweg E. R., Bagai I., Rossi P., and Bertelsen E. B. (2013) EZ-ASSIGN, a program for exhaustive NMR chemical shift assignments of large proteins from complete or incomplete triple-resonance data. J. Biomol. NMR 57, 179–191 10.1007/s10858-013-9778-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hyberts S. G., Arthanari H., Robson S. A., and Wagner G. (2014) Perspectives in magnetic resonance: NMR in the post-FFT era. J. Magn. Reson 241, 60–73 10.1016/j.jmr.2013.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goddard T. D., and Kneller D. G. (2000) SPARKY 3, University of California, San Francisco [Google Scholar]
- 51. Zuiderweg E. R. (2016) Backbone and Methyl resonance assignments of the 42 kDa human Hsc70 nucleotide binding domain. J. Biomol. NMR Assignments, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr, Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mongan J., Case D. A., and McCammon J. A. (2004) Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 25, 2038–2048 10.1002/jcc.20139 [DOI] [PubMed] [Google Scholar]
- 54. Cheng Y., and Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 10.1016/0006-2952(73)90196-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.