

Abstract
Transcellular propagation of protein aggregate “seeds” has been proposed to mediate the progression of neurodegenerative diseases in tauopathies and α-synucleinopathies. We previously reported that tau and α-synuclein aggregates bind heparan sulfate proteoglycans (HSPGs) on the cell surface, promoting cellular uptake and intracellular seeding. However, the specificity and binding mode of these protein aggregates to HSPGs remain unknown. Here, we measured direct interaction with modified heparins to determine the size and sulfation requirements for tau, α-synuclein, and β-amyloid (Aβ) aggregate binding to glycosaminoglycans (GAGs). Varying the GAG length and sulfation patterns, we next conducted competition studies with heparin derivatives in cell-based assays. Tau aggregates required a precise GAG architecture with defined sulfate moieties in the N- and 6-O-positions, whereas the binding of α-synuclein and Aβ aggregates was less stringent. To determine the genes required for aggregate uptake, we used CRISPR/Cas9 to individually knock out the major genes of the HSPG synthesis pathway in HEK293T cells. Knockouts of the extension enzymes exostosin 1 (EXT1), exostosin 2 (EXT2), and exostosin-like 3 (EXTL3), as well as N-sulfotransferase (NDST1) or 6-O-sulfotransferase (HS6ST2) significantly reduced tau uptake, consistent with our biochemical findings, and knockouts of EXT1, EXT2, EXTL3, or NDST1, but not HS6ST2 reduced α-synuclein uptake. In summary, tau aggregates display specific interactions with HSPGs that depend on GAG length and sulfate moiety position, whereas α-synuclein and Aβ aggregates exhibit more flexible interactions with HSPGs. These principles may inform the development of mechanism-based therapies to block transcellular propagation of amyloid protein–based pathologies.
Keywords: Tau protein (Tau), Alzheimer disease, heparan sulfate, α-synuclein, amyloid-β (Aβ), neurodegeneration, neurodegenerative disease, CRISPR/Cas, proteoglycan, HSPG, tauopathies
Introduction
Tau and α-synuclein aggregates bind heparan sulfate proteoglycans (HSPGs)3 on the cell surface, which mediate uptake and intracellular seeding (1). However, the specificity of this interaction is unknown. Heparan sulfate (HS) is a linear glycosaminoglycan (GAG) composed of disaccharide repeats of uronic acid (iduronic or glucuronic acid) and glucosamine with sulfate moieties at discrete N-, C2-, C3-, and C6-residues (Fig. 1) and is synthesized in all known eukaryotic cells (2, 3). Heparin is a linear GAG that is elaborated and secreted by mast cells. It is chemically similar to HS and frequently used as a model compound for HS. Heparin differs from HS by shorter average chain lengths, higher contents of iduronic acid, and a higher average number of sulfate moieties per disaccharide unit, which leads to higher charge density per residue (2). HS chains are covalently attached to core proteins, forming HSPGs. Many HSPGs are secreted into the extracellular matrix or are membrane-associated either via a transmembrane domain or a glycosylphosphatidylinositol anchor (2, 3). The structure of the HS chains is determined by the activity of synthetic enzymes, availability of precursor molecules, and flux through the Golgi apparatus. The structural diversity of HSPGs among different cell and tissue types accounts for their interaction with a myriad of proteins. The so-called heparan sulfate interactome is composed of proteins involved in cell attachment, migration, invasion and differentiation, morphogenesis, organogenesis, blood coagulation, lipid metabolism, inflammation, and injury response (2, 4). Some HS-binding proteins bind HS carbohydrates with great specificity. The interaction between HSPGs and their binding proteins can depend on modification, domain, charge, and sugar conformation (2, 5, 6). Some interactions are based on defined chemistry. For example, the binding of a protein may require spatially separated patches of sulfated residues on the GAGs. Purely charge-based associations are more flexible, as they are based on electrostatic interactions between the positively charged residues on the binding protein and the negatively charged sulfate residues on the HS chain (2). We do not know whether the interactions of tau and α-synuclein with HSPGs are specific to sulfation positioning or are purely based on charge density.
Figure 1.
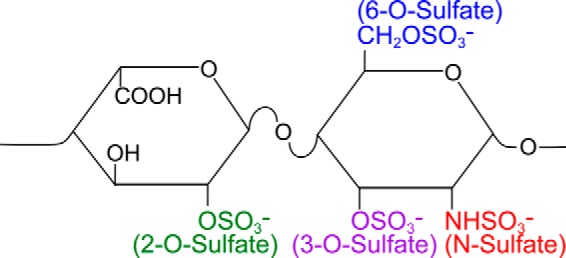
Basic disaccharide unit of heparan sulfate and heparin. The basic disaccharide unit of heparan sulfate and heparin is composed of uronic acid (glucuronic acid or iduronic acid, left) and glucosamine (right). Iduronic acid results from epimerization of glucuronic acid. Heparin differs from HS by shorter average chain lengths, higher contents of iduronic acid, and a higher charge density per residue. Uronic acid residues are sulfated at the 2-O-position, whereas glucosamine residues are sulfated at the N-, 3-O-, and 6-O-positions (2, 3).
We previously observed that exogenously applied tau and α-synuclein aggregates bind to HSPGs on the cell surface to trigger cellular uptake and induce intracellular seeding (1). Thus, the HSPG–aggregate interaction may represent a critical step for the spread of pathology in neurodegenerative diseases. Other laboratories have similarly observed that Aβ and prion protein bind to HSPGs on the cell surface to trigger their internalization (7–11). It is not clear which HSPG properties drive the interaction with aggregates, and if the required GAG composition for binding differs among amyloid proteins.
We have analyzed the GAG-binding requirements for recombinant tau, α-synuclein, and Aβ-amyloid fibrils. We used a small heparin mimetic library to determine the critical size and sulfation requirements for HS to bind to aggregates in biochemical and cell-based assays and CRISPR/Cas9 knockout to test specific components of the HSPG synthesis pathway. Binding of tau fibrils to HSPGs depends on relatively specific modifications, whereas binding of α-synuclein and Aβ fibrils seemed to be more complex and variable between different experimental approaches.
Results
Carbohydrate microarrays identify unique binding patterns for amyloid proteins
Carbohydrate microarrays allow rapid analysis of interactions between proteins and GAGs (12). We used this approach to investigate the structural components of heparin that are required for binding to tau, α-synuclein, Aβ, and huntingtin fibrils (Fig. 2). We employed a library of modified heparin polysaccharides (Neoparin, Alameda, CA) to investigate sulfation requirements for binding: heparin (Hep), N-desulfated heparin (De-N), 6-O-desulfated heparin (De-6-O), 2-O-desulfated heparin (De-2-O), O-desulfated heparin (De-O), fully desulfated heparin (De-S, in which >90% of all sulfates are removed) and oversulfated heparin (Over-S, which contains >3.5 sulfates per disaccharide unit).
Figure 2.

Carbohydrate microarray analyses identify unique amyloid–GAG interactions. Biotin-labeled tau, α-synuclein, and Aβ fibrils at 1 μm (monomer equivalent) were applied to a glass surface coated with heparin derivatives at 0.5, 1, 5, 10, and 15 μm (left to right): heparin (Hep), oversulfated heparin (Over-S), fully desulfated heparin (De-S), O-desulfated heparin (De-O), N-desulfated heparin (De-N), 2-O-desulfated heparin (De-2-O), and 6-O-desulfated heparin (De-6-O). We applied an anti-biotin IgG antibody conjugated to Cy5 and quantified relative fibril binding by measuring fluorescence. Each protein was analyzed in triplicate, and the data represent an average value for 10 spots at a given carbohydrate concentration. The average values were standardized to the highest concentration of heparin (15 μm) in each data set. Note distinct binding patterns for N-desulfated heparin, 2-O-desulfated heparin, and 6-O-desulfated heparin. Error bars show S.E.
We applied nanoliter volumes of heparins at concentrations from 0.5 to 15 μm to glass microarray surfaces coated with poly-l-lysine. We then applied biotinylated full-length tau, α-synuclein, Aβ42, and huntingtin exon 1 (HttExon1Q50) fibrils to the microarray, and we visualized the bound proteins with an anti-biotin antibody tagged with Cy5. Tau, α-synuclein, and Aβ aggregates bound heparin in a concentration-dependent manner (Fig. 2). Huntingtin fibrils exhibited no binding (data not shown) and were not analyzed further. None of the fibrils bound desulfated heparin, suggesting that sulfation is a critical component of the aggregate–GAG interaction (Fig. 2). Our results agreed with previous reports that tau, α-synuclein, and Aβ, but not Htt, are heparin-binding proteins (1, 7, 13, 14).
The different seeds exhibited unique sulfation requirements for binding. Tau efficiently bound heparin and 2-O-desulfated heparin, whereas 6-O-desulfation, N-desulfation, or O-desulfation abolished its binding (Fig. 2). α-Synuclein and Aβ fibrils each required O-sulfation for heparin binding. However, the removal of N-, 6-O-, or 2-O-sulfation did not significantly inhibit their binding, whereas desulfated heparin abolished their binding (Fig. 2). Thus, for α-synuclein and Aβ fibrils, no single sulfate moiety but overall sulfation was required to mediate binding to the GAG microarray.
Inhibition of amyloid uptake requires specific heparin sulfation
We and others have previously observed that heparin inhibits the cellular binding and uptake of tau, α-synuclein, and Aβ (1, 7). Heparin directly interacts with HS binding domains and prevents aggregate binding to HSPGs on the cell surface. Thus, we tested whether the structural determinants of the aggregate–GAG interaction observed in the microarray assay would translate to aggregate internalization and propagation in cells.
We labeled tau, α-synuclein, and Aβ fibrils with a fluorescent Alexa Fluor 647 dye and applied them to C17.2 cells in culture for 4 h (tau and α-synuclein) or 20 h (Aβ), optimal times determined empirically. We measured aggregate uptake with flow cytometry by quantifying the median fluorescence intensity (MFI) per cell. Heparin decreased seed internalization dose-dependently for all three fibril types (Fig. 3).
Figure 3.
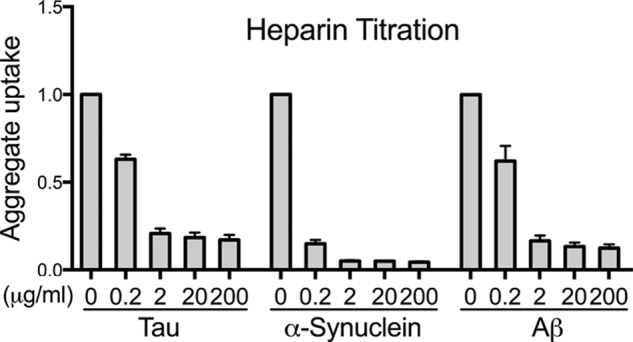
Heparin blocks aggregate uptake of Tau, α-synuclein, and Aβ. Tau, α-synuclein, or Aβ seeds labeled with Alexa Fluor 647 fluorescent dye were applied to C17.2 cells with increasing doses of heparin (0.2, 2, 20, and 200 μg/ml). We quantified aggregate uptake based on the MFI per cell by flow cytometry. Heparin dose-dependently decreased cellular uptake in all cases. Each condition was recorded in triplicate, and values represent the average of three separate experiments. Data reflect uptake relative to the untreated samples. Error bars show S.D.
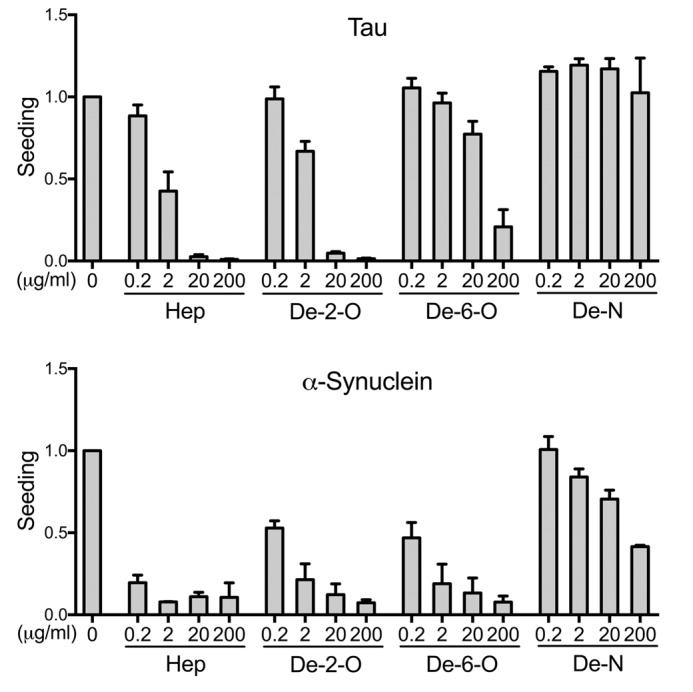
We next tested the desulfated heparins as inhibitors of aggregate internalization (Fig. 4). Tau aggregate uptake was strongly inhibited by 2-O-desulfated heparin, similar to standard heparin. Thus, removal of 2-O-sulfates did not disrupt tau uptake inhibition. The inhibitory potency of 6-O-desulfated heparin was reduced compared with 2-O-desulfated heparin and standard heparin, whereas N-desulfated heparin had virtually no inhibitory effect (Fig. 4). In summary, as for heparin binding in vitro, competitors for tau uptake in cells required 6-O- and N-sulfation, whereas 2-O-sulfation was dispensable.
Figure 4.

Heparin-mediated inhibition of aggregate uptake requires specific sulfation patterns. Normal heparin and 2-O-, 6-O-, and N-desulfated forms were tested as inhibitors of cellular uptake for tau, α-synuclein, and Aβ aggregates in C17.2 cells. N- and 6-O-desulfated heparins weakly inhibited and 2-O-desulfated heparins strongly inhibited tau uptake. For α-synuclein and Aβ, removal of N-sulfation and to a lesser degree 6-O-sulfation and 2-O-sulfation reduced inhibitor efficacy. Each condition was recorded in triplicate, and values represent the average of three separate experiments. Data reflect uptake relative to the untreated group. Error bars show S.D.
The structural requirements differed for the inhibition of α-synuclein and Aβ (Fig. 4). Compared with standard heparin, removal of N-sulfation strongly reduced the inhibitory activity of the compounds on aggregate uptake. The removal of 6-O- or 2-O-sulfation from heparin also reduced its inhibitory effect on α-synuclein and Aβ uptake compared with unmodified heparin. Thus, N-sulfation was critical, but other sulfates also contributed to aggregate binding. This result corroborated the microarray data in that all sulfate moieties seemed to contribute to aggregate binding to GAGs. However, the relevance of N-sulfation was different between these two experimental approaches, with no alteration of binding by the removal of N-sulfation in the microarray setting and a critical loss of uptake inhibition in the uptake assay.
Inhibition of aggregate uptake requires a critical chain length
To determine the role of polysaccharide chain length, we tested the inhibitory activity of fractionated heparins in the cell uptake assay. Heparin fragments composed of 4, 8, 12, and 16 sugar molecules (referred to as 4-, 8-, 12-, and 16-mer, respectively) were incubated with labeled aggregates, applied to C17.2 cells, and tested for inhibition of uptake (Fig. 5). For tau, shorter heparin fragments (4- and 8-mer) had no activity, whereas the longer fragments (12- and 16-mer) modestly inhibited uptake (Fig. 5). No compound had a potency similar to full-length heparin (∼50-mer), even when taking into account the number of individual saccharide units. We concluded that larger heparin chains were necessary to inhibit tau uptake.
Figure 5.

Inhibition of aggregate uptake requires a critical heparin size. The 4-, 8-, 12-, and 16-mer heparins were evaluated as inhibitors of tau, α-synuclein, and Aβ aggregate internalization in C17.2 cells. Potency increased with the GAG chain length. Each condition was recorded in triplicate, and values represent the average of three separate experiments. Data reflect uptake relative to the untreated group. Error bars show S.D.
Aβ fibrils exhibited greater sensitivity to shorter polysaccharides, and 12- and 16-mer inhibited uptake. As for tau, the uptake inhibition of Aβ increased with the heparin chain length. α-Synuclein aggregates were also dose-dependently inhibited by all fractionated heparins, with greater inhibitory activity of the 12- and 16-mer compared with the shorter heparins (Fig. 5). Thus, depending on their target, heparins required critical and distinct chain lengths to function as uptake inhibitors. We concluded that tau, α-synuclein, and Aβ aggregates each have specific structural determinants for GAG binding, including sulfation pattern and size.
Structural requirements for inhibition of seeding
Amyloid aggregates could gain entry to cells by multiple mechanisms, some of which could lead to seeding activity, and others not. Thus, we tested heparins in an established seeding assay that consists of a monoclonal “biosensor” cell line that stably expresses tau repeat domain (RD) harboring the disease-associated mutation P301S (Fig. S1), fused to yellow or cyan fluorescent proteins (RD-CFP/YFP) (15, 16). Upon binding to the cell surface, tau aggregates trigger their own internalization and induce intracellular aggregation of RD-CFP/YFP, enabling fluorescence resonance energy transfer (FRET). We used flow cytometry to quantify the number of cells exhibiting FRET. An α-synuclein biosensor that expresses full-length α-synuclein with the disease-associated mutation A53T tagged to either CFP or YFP (syn-CFP/YFP) functioned similarly (16). We did not test for Aβ seeding due to the lack of a functional biosensor cell line. We incubated tau or α-synuclein fibrils overnight with heparins, prior to direct exposure of the biosensor cells and incubation for 48 h. To improve yield (due to low seeding efficiency) we re-exposed the α-synuclein biosensor cell line to aggregate–heparin complexes after passaging for an additional 48 h prior to flow cytometry. Simultaneous application of heparin with tau and α-synuclein fibrils to the biosensor cell lines reduced seeding dose-dependently (Fig. 6).
Figure 6.
Sulfation pattern specifies inhibition of seeding. 2-O-, 6-O-, and N-desulfated heparins were evaluated for inhibition of tau seeding in the P301S FRET biosensor cell line and α-synuclein seeding in the A53T FRET biosensor cell line. We used flow cytometry to quantify seeding based on the percentage of FRET-positive cells. Inhibition of tau seeding required N-sulfation and, to a lesser extent, 6-O-sulfation. By contrast, α-synuclein primarily required N-sulfation and 6-O- and 2-O-sulfation to a lesser degree. In every experiment, each condition was tested in triplicate. Values represent the average of three separate experiments for tau, and two separate experiments for α-synuclein. The data reflect seeding relative to the untreated group. Error bars show S.D.
We next used the desulfated heparins as competitors in the seeding assay (Fig. 6). 2-O-Desulfated heparin blocked tau seeding almost as well as heparin, whereas 6-O-desulfated heparin lost most activity, and N-desulfated heparin lost all activity against tau seeding. These observations were consistent with our prior finding that N-sulfation and 6-O-sulfation are important for cellular tau binding and internalization, whereas 2-O-sulfation is not. For α-synuclein, N-desulfated heparin lost most of its inhibitory activity. 6-O-Desulfated heparin and 2-O-desulfated heparin also had reduced inhibitory activity compared with standard heparin. This indicated that concordant with results from our uptake studies, N-sulfation and to a lesser degree 6-O-sulfation and 2-O-sulfation contributed to the inhibition of α-synuclein seeding.
We next tested fractionated heparins (4-, 8-, 12-, and 16-mer). We observed no appreciable inhibitory activity against tau and only slight inhibitory activity against α-synuclein with shorter heparin fragments (4- and 8-mer), and weak activity with longer fragments (12- and 16-mer) for both fibril types. The results were qualitatively similar to the uptake assay, indicating that the inhibitory activity of the heparins against seeding increased with the saccharide chain length (Fig. 7).
Figure 7.

GAG size specifies inhibition of seeding. The potency of different heparins against tau seeding in the P301S FRET biosensor cell line and α-synuclein seeding in the A53T FRET biosensor cell line depended on the chain lengths of the heparins. The 4-, 8-, 12-, and 16-mer heparins were evaluated as inhibitors of tau and α-synuclein seeding. The inhibitory potency of the heparin fragments increased with GAG chain length. In every experiment, each condition was tested in triplicate. Values represent the average of three separate experiments for tau, and two separate experiments for α-synuclein. Data reflect seeding relative to the untreated group. Error bars show S.D.
HSPG synthetic genes required for uptake of aggregates
The HSPG synthesis pathway is a complex hierarchical cascade taking place in the Golgi apparatus, involving ∼30 enzymes. After initial formation of a linkage region, extension enzymes (EXT1 and EXT2) catalyze the addition of alternating units of glucuronic acid and GlcNAc. The dual activity enzyme N-deacetylase/N-sulfotransferase then catalyzes N-deacetylation and N-sulfation of some GlcNAc residues. Next, a variable number of glucuronic acid residues are epimerized to iduronic acid (2, 3). Finally, sulfate groups are attached to C3 and C6 of glucosamine and C2 of iduronic acid and some glucuronic acid residues (Fig. 1) (2, 3). The final structure of the HS chain in terms of length, sugar composition, and sulfation pattern varies substantially between different cell and tissue types. We tested specific genes in the synthesis pathway to elucidate the structural determinants of cell-surface HSPGs and to further test the requirement of specific sulfation patterns for aggregate internalization.
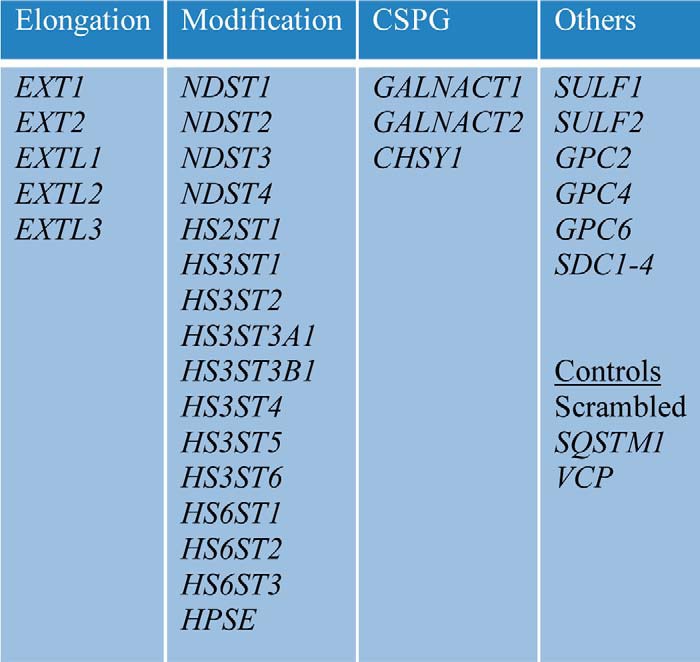
We used CRISPR/Cas9 to individually knock out all the genes in the HSPG synthesis pathway and screened for inhibition of aggregate uptake in HEK293T cells (Table 1). We selected 5–6 separate human gRNAs for each gene and cloned them into the lentiCRISPRv2 vector (17). Plasmids containing the gRNAs for each gene were pooled and used for lentivirus production. Cells were exposed to lentivirus and cultured for at least 10 days under puromycin selection prior to phenotypic screening. As a positive control, we included the gRNAs to target the valosin-containing peptide (VCP) gene, because the knockout of this gene is lethal in mammalian cells. In addition, we transduced the available gRNAs individually to identify the functional gRNAs for selected genes. We then used a single gRNA to produce polyclonal knockout cell lines and confirmed the presence of indels in the predicted DNA regions by Tracking of Indels by Decomposition (TIDE) (Fig. S3) (18). We tested several commercially available antibodies to verify gene knockout, but due to the low endogenous expression levels of the genes in the HSPG pathway (The Human Protein Atlas version 18, www.proteinatlas.org (19, 20) and Fig. S4), we were not able to identify antibodies that reliably detected endogenous protein levels (data not shown).
Table 1.
Genes included in the genetic screen
Major genes involved in the synthesis of HSPGs were screened for effects on tau and α-synuclein uptake. We also tested genes coding for proteoglycans core proteins (glypicans, syndecans) and SULF1/2, which diminish HSPG sulfation via arylsulfatase and endoglucosamine-6-sulfatase activities. Genes involved in the synthesis of the chondroitin proteoglycan (CSPG) synthesis pathway, as well as SQSTM1, were targeted as negative controls. To confirm gene knockout we targeted VCP, whose knockout is lethal after 5 days.
The knockout of five genes strongly inhibited tau uptake: EXT1, EXT2, EXTL3, NDST1, and HS6ST2. Four of these five genes (EXT1, EXT2, EXTL3, and NDST1) were also required for α-synuclein uptake (Fig. 8, Table 2, and Fig. S2). We had previously observed that EXT1 is required for cellular uptake of tau aggregates (1). EXT1 is a glycosyltransferase that polymerizes heparan sulfate chains, and knockout of the EXT1 gene reduces HSPG expression without affecting other proteoglycan subtypes (chondroitin and dermatan sulfate proteoglycans) (21). EXT1 and EXT2 are co-polymerases, and both are required for proper HS chain elongation in vivo (22). EXTL3 likewise is a glycosyltransferase involved in the initiation and the elongation of the HS chain, and reduced levels create longer HS with fewer side chains (22).
Figure 8.

HSPG genes critical for the internalization of tau and α-synuclein aggregates. Genes implicated in HSPG synthesis were individually targeted in HEK293T cells using CRISPR/Cas9 to create polyclonal knockout lines. The cell lines were then tested for internalization of fluorescently labeled tau and α-synuclein aggregates by measuring MFI per cell with flow cytometry. The cells were treated in parallel with fluorescently labeled transferrin to control for nonspecific reduction of clathrin-mediated uptake. Knockout of four genes (EXT1, EXT2, EXTL3, and NDST1) reduced both tau and α-synuclein uptake, whereas the knockout of HS6ST2 only reduced tau uptake. None of the gene knockouts reduced transferrin uptake. Data were collected in duplicate on 2 different days for each cell line and each aggregate type and normalized to uptake from control cells treated with scrambled gRNA (Sc). For transferrin uptake, the data from all 4 experimental days was combined. Red columns indicate the gene knockouts with the strongest effects. Error bars show S.D. For statistical analyses, we combined the averages for each experimental day. ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; one-way ANOVA, Dunnett.
Table 2.
Genes required for tau and α-synuclein uptake
The knockout of the listed genes reduced aggregate uptake by >50% and was statistically significant with p < 0.001 for tau and p < 0.0001 for α-synuclein in the genetic candidate screen (Fig. 8); One-way Anova, Dunnett.
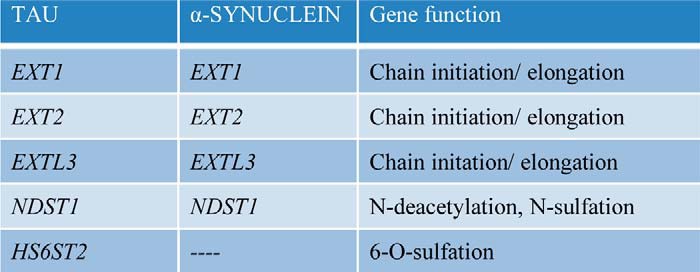
Both NDST1 and HS6ST2 knockouts substantially reduced tau aggregate internalization (Fig. 8 and Fig. S2A). NDST1 places N-sulfate groups on glucosamine residues, whereas HS6ST2 attaches sulfate groups to 6-O-glucosamine residues. NDST1 is a dual activity enzyme with N-acetylase/N-sulfotransferase activity and is located upstream of other sulfotransferases. N-Sulfation is therefore a key regulatory step for subsequent modifications (3), and the knockout of this enzyme is likely to induce changes of other sulfate moieties as well. However, in conjunction with our previous data that demonstrated a loss of uptake inhibition by removal of N-sulfates and 6-O-sulfates, we concluded that N- and 6-O-sulfate moieties are critical for HSPG-mediated uptake of tau aggregates. On the contrary, knockout of genes coding for 2-O- and 3-O-sulfotransferases had no effect (e.g. HS2ST1 and HS3ST3A1) on tau uptake. Importantly, gRNAs targeting genes for CSPG synthesis enzymes (CSGALNACT and CHSY1) did not affect tau uptake (Fig. 8).
For α-synuclein, only knockout of NDST1 strongly reduced aggregate uptake (Fig. 8 and Fig. S2B). Knockout of other sulfotransferases mildly reduced uptake (e.g. HS2ST1), increased uptake (e.g. HS6ST2, albeit not significantly), or had no effect (Fig. 8). Consistent with the cell uptake and seeding data, enzymes responsible for N-sulfation were important for α-synuclein uptake, whereas those responsible for sulfation at other positions did not play a critical role. No gene knockouts reduced the internalization of labeled transferrin, which is controlled by clathrin-mediated endocytosis, indicating that our observed effects were likely specific to macropinocytosis (Fig. 8).
To confirm specific effects of knockout for the main genes of interest (EXT1, NDST1, and HS6ST2), we produced knockout cell lines in HEK293T cells and the 2 FRET biosensors by transiently transfecting the gRNA/Cas9 plasmids (so that they would eventually be lost) and culturing them for at least 10 days. We then generated rescue plasmids with the HA-tagged cDNA followed by internal ribosomal entry site (IRES) and an mCherry-coding sequence. We used the cDNA plasmids to produce lentivirus and treated the knockout cell lines for at least 3 days to drive cDNA expression prior to uptake and seeding experiments (Fig. 9). We performed a Western blot using an anti-HA antibody to detect plasmid overexpression (Fig. S5). The bands for EXT1-HA (expected mass = 87.7 kDa) and NDST1-HA (expected mass = 102.4 kDa) were located at the correct locations (lanes 3 and 5, respectively, in Fig. S5). For HS6ST2-HA (expected mass = 70.2 kDa), several bands were visible between ∼60 and 100 kDa, possibly due to protein aggregation or changes of protein properties due to the HA tag. We only detected HA tag in cells overexpressing the rescue constructs.
Figure 9.

Rescue of tau and α-synuclein uptake and seeding after gene knockout. We generated knockout (KO) cell lines using HEK293T cells for uptake, the P301S FRET biosensor cell line for tau seeding, and the A53T FRET biosensor cell line for α-synuclein seeding for the main genes of interest (EXT1, NDST1, and HS6ST2) and tested them with and without genetic rescue (R) by lentiviral cDNA. For tau, knockout of all three genes was rescued by overexpression for both uptake and seeding. For α-synuclein, knockout of EXT1 and NDST1 was rescued for uptake and seeding. However, HS6ST2 knockout increased α-synuclein uptake and did not change α-synuclein seeding, whereas HS6ST2 overexpression decreased α-synuclein uptake and increased seeding. Data were collected from at least three different experiments in triplicate for tau uptake and seeding and for α-synuclein uptake, and from two different experiments for α-synuclein seeding. The uptake or seeding in knockout cell lines was normalized to the uptake/seeding detected in scrambled control cells (Sc). Error bars show S.D. For statistical analyses, we combined the averages for each experiment. n.s., not significant, *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; one-way ANOVA, Holm-Šídák testing.
Next, we used the knockout and rescue cell lines for uptake and seeding experiments (Fig. 9). We gated for mCherry-expressing cells and quantified uptake and seeding within this population. Knockout of all three genes was partly or fully reversible for tau uptake and seeding when rescued with cDNA lentivirus (Fig. 9). Similarly, knockout of EXT1 and NDST1 was partly or fully reversible for α-synuclein uptake and seeding. HS6ST2 knockout increased α-synuclein uptake, similar to our previous data (Fig. 8, albeit not significant here), and did not change α-synuclein seeding. HS6ST2 overexpression decreased α-synuclein uptake, yet paradoxically increased seeding. We concluded that HS6ST2 modulation has complex effects on α-synuclein uptake and seeding possibly related to dysregulation/compensation by multiple pathways.
In summary, targeted genetic knockout of the HSPG biosynthesis pathway indicated that both tau and α-synuclein internalization were mediated by HSPGs and dependent on specific sulfotransferases. In concordance with microarray and heparin competition experiments, tau uptake seemed to depend strongly on N- and 6-sulfation via NDST1 and HS6ST2. By contrast, α-synuclein uptake depended strongly on N-sulfation by NDST1 and to a lesser degree on 2-O-sulfation by HS2ST1.
Discussion
Using a combination of biochemical and cell-based assays, we have investigated the HSPG structure–function relationships that govern the uptake of tau, α-synuclein, and Aβ aggregates. The aggregate–GAG interaction, especially for tau, is determined by the pattern of sulfate moieties and the GAG length. We corroborated our findings with a genetic candidate screen of the HSPG synthesis pathway and identified specific sulfotransferases that play a role in tau and α-synuclein uptake. Our data indicate that the interaction between HSPGs and aggregates may be a regulated and specific biological process.
Distinct sulfation requirements for aggregate binding
We originally employed a library of modified heparin polysaccharides spotted on a microarray to determine that the binding of tau aggregates depended on 6-O- and N-sulfation, whereas α-synuclein and Aβ aggregate binding depended on overall sulfation. We tested predictions from these studies in cell-based uptake and seeding assays, using heparins as competitive inhibitors of aggregate binding to the cell surface. All data consistently indicated a requirement of N- and 6-O-sulfation for tau aggregate binding, uptake, and seeding.
In contrast, for α-synuclein and Aβ fibrils, the removal of N-, 6-O-, or 2-O-sulfation did not significantly inhibit their binding to the microarray, indicating that no single sulfate moiety but overall sulfation was required to mediate binding in this experimental approach. In the uptake experiments, α-synuclein and Aβ aggregate uptake strongly required N-sulfation, whereas 2-O-sulfation and 6-O-sulfation were moderately required. We observed the same binding pattern for α-synuclein seeding. Thus, although N-sulfation seemed to be more relevant in the cell-based assays, the microarray data and the cell data congruently showed that all three sulfate moieties play a role for α-synuclein and Aβ internalization. Interestingly, a recent report by Ihse et al. (23) found that the general degree of sulfation drove cellular α-synuclein uptake rather than specific modifications within the GAG chain. By contrast, another study demonstrated that baculovirus uptake exhibits specificity similar to tau, requiring N- and 6-O-sulfation, but not 2-O-sulfation, for binding and entry into mammalian cells (24).
Zhao et al. (25) recently described a requirement of 6-O-sulfation for tau binding to GAGs in vitro, and Rauch et al. (26) identified a requirement for 6-O-sulfation for tau internalization in induced pluripotent stem cell-derived neurons and mouse brain slice culture. Differences in the observed importance of N-sulfation could derive from individual protein preparations, cell types, and assays. Given the variations in fibril structure that can occur during formation in vitro, it is possible that this could also play a role, as we have previously observed distinct patterns of neuronal vulnerability to different tau strains, which are composed of distinct fibrillar structures (27, 28).
GAG chain length is critical
GAG chain length appears critical to mediate aggregate binding and uptake of all amyloids studied, although we did observe differences. Short heparin fragments (4- and 8-mer) exhibited no inhibitory activity for tau, whereas the longer fragments (12- and 16-mer) had only low inhibitory activity. By contrast, Aβ and α-synuclein aggregates displayed some degree of dose-dependent inhibition for all fractionated heparins, with increasing inhibitory activity for longer fragments. We conclude that chain length requirements for binding will vary among amyloid proteins, although all three protein aggregates appear to bind longer chains with greater avidity.
Specific sulfotransferases required for uptake
After screening all HSPG candidate genes, we observed that knockout of only five genes (EXT1, EXT2, EXTL3, NDST1, and HS6ST2) strongly reduced tau aggregate uptake. We had previously validated EXT1 as a modifier of tau uptake (1). EXT2, EXTL3, NDST1, and HS6ST2 have not previously been recognized to play a role in tau aggregate uptake. EXT2 and EXTL3 have similar functions to EXT1, whereas NDST1 mediates N-deacetylation/N-sulfation, and HS6ST2 mediates 6-O-sulfation. Knockout of HS2ST1, the sole sulfotransferase for 2-O-sulfation, did not alter tau uptake. Taken together, the genetic data corroborate the microarray and pharmacological data that tau uptake is mediated by N- and 6-O-sulfation of HSPGs.
For α-synuclein, the knockout of EXT1, EXT1, EXTL3, and NDST1, but not HS6ST2, clearly reduced uptake. Knockout of HS2ST1 mildly reduced α-synuclein uptake, and knockout of HS6ST2 slightly increased uptake and did not change α-synuclein seeding. The overexpression of HS6ST2 decreased α-synuclein uptake and paradoxically increased α-synuclein seeding. Thus, the genetic data suggest that N-sulfation, and to a lesser degree 2-O-sulfation, is important for uptake. The genetic data partly corroborate the pharmacological data in that N-sulfation is more relevant for α-synuclein uptake and seeding than the other moieties. However, no alteration of binding was observed upon removal of single sulfate moieties in the microarray setting. We conclude that binding of α-synuclein to HSPGs is more complex than for tau and shows some variability between different experimental approaches.
No other N- and/or 6-O-sulfotransferases were required for α-synuclein and tau uptake. Some of the paralogs (e.g. NDST3, NDST4, and HS6ST3) have low or nondetectable expression levels in HEK293 cells (The Human Protein Atlas version 18 (19, 20)). It is possible that each paralog possesses unique substrate specificities and is not compensated by other members of the HSPG family (e.g. NDST2 and HS6ST1). In addition, in the mammalian brain, the HSPG composition varies substantially between different brain areas, cell types, and developmental stages, and little is known so far on the relevance of specific HS sulfation patterns (29). Thus, in future studies, it will be informative to determine which paralogs drive N- and 6-O-sulfation in different cell and tissue types. We also recognize that future work must evaluate the requirements for the genes identified in this study in the context of post-mitotic central nervous system neurons.
Conclusion
Our results indicate considerable specificity for tau aggregate interaction with HSPGs. This relationship is not simply based on charge density but on the position of sulfate moieties as well. Thus, further studies to investigate the structural parameters governing aggregate uptake in vivo could be fruitful. A large body of literature indicates that sulfated GAGs inhibit the infectivity of the prion protein in vitro and in vivo (8, 30–32), and our data suggest that these concepts might also be applied to guide drug treatment for other amyloid disorders, such as tauopathies and α-synucleinopathies.
Materials and methods
Protein preparation, fibrillization, and labeling
Recombinant full-length WT tau and α-synuclein were purified and fibrillized as described previously (1). Huntingtin protein (Exon1 Gln-50) was prepared by solid-phase synthesis by the Keck Biotechnology Resource Laboratory at Yale University and was labeled and fibrillized as described previously (1). Synthetic Aβ42 was purchased from Anaspec unlabeled or with a biotin tag on the N-terminal amino acid. Biotin-Aβ42 was reconstituted in 1% NH4OH, lyophilized, and stored at −80 °C. Unlabeled Aβ42 was reconstituted in 1% NH4OH, diluted in PBS, and stored at −80 °C. Fibrillization of Aβ42 was achieved by bringing the protein concentration to 100 μm (biotin-Aβ42) or 110 μm (unlabeled Aβ42) in 10 mm HCl and incubating for 24 h at 37 °C. α-Synuclein and Aβ42 fibrils were dialyzed into PBS overnight using dialysis cassettes (ThermoFisher Scientific). For uptake assays, fibrils were incubated with Alexa Fluor 647 succinimidyl ester dye (Invitrogen) for 1 h at room temperature (molar ratios monomer/dye ∼1:12.5 for tau and ∼1:4 for α-synuclein and Aβ42), quenched with 100 mm glycine for 1 h at room temperature, and dialyzed overnight into PBS using dialysis cassettes (ThermoFisher Scientific) to remove excess dye. The labeled fibrils were stored at 4 °C until use.
Heparin polysaccharide suppliers
The first experiment with microarrays was performed with heparins from Neoparin (Alameda, CA), which went out of business upon completion of the first part of our studies. Therefore, the heparins for the following studies were purchased from AMSbio (Cambridge, MA). The glycochemistry of these molecules is complicated, and the quality of the products depends on the origin of the raw material used, the synthesis methods, and the final purity of the preparations. We noticed differences in inhibitory activity when performing uptake assays with the same heparin from two different companies. However, we still derived qualitatively similar results, e.g. that certain defined moieties such as N-sulfation and 6-O-sulfation are crucial for tau uptake. Also, not all the heparins used in the microarrays were available from AMSbio, which limited the number of heparins that could be tested in the second part of this study.
Carbohydrate microarrays
As described previously (12), solutions of the polysaccharides (10 μl per well in a 384-well plate) were spotted onto poly-l-lysine–coated slides by using a Microgrid II arrayer (Biorobotics; Cambridge, UK) at room temperature and 50% humidity. We used solution concentrations between 0.5 and 15 μm, and the arrayer printed 1 nl of each concentration 10 times. The arrays were incubated in a 70% humidity chamber overnight, followed by storage in a low-humidity, dust-free desiccator. For protein binding studies, the slides were blocked with 3% bovine serum albumin (BSA) in PBS (5 ml) with gentle rocking at 37 °C for 1 h. Recombinant tau and α-synuclein fibrils were biotinylated with EZ-LinkTM Sulfo-NHS-Biotin biotinylation kit (ThermoFisher Scientific) according to the manufacturer's instructions, lyophilized, and stored at −80 °C until use. Biotin-Aβ42 was purchased from Anaspec, fibrillized as described above, lyophilized, and stored at −80 °C until use. The biotinylated fibrils were reconstituted in 1% BSA in PBS, and 50-μl quantities at a concentration of 1–2 μm were added to the slides, followed by incubation in a humidity chamber for 1 h. Next, the slides were washed five times for 3 min each in PBS (5 ml) while gently rocking. After the washes, the slides were incubated with an anti-biotin IgG antibody conjugated to Cy5 in the dark with gentle rocking for 1 h (1:5000, Molecular Probes), washed with PBS followed by H2O, and then dried under a gentle stream of N2. All incubations and washes were carried out at room temperature. The microarrays were analyzed with a GenePix 5000a scanner at 635 nm for Cy5, and fluorescence quantification was performed using GenePix 6.0 software with correction for local background. Each protein was analyzed in triplicate, and the data represent an average value for 10 spots at a given carbohydrate concentration. The average values were standardized to the fluorescence recorded for the highest concentration of heparin (15 μm) in each data set. Statistical significance was determined using ANOVA with post hoc Tukey's HSD for individual comparisons; p values <0.05 were considered statistically significant. A similar experiment was performed with biotinylated monomeric tau, α-synuclein, Aβ, and huntingtin (data not shown), but no binding could be detected (data not shown).
Uptake assay for heparin experiments
C17.2 cells were plated at 4000 cells per well in a 96-well plate. Fluorescently labeled aggregates (50 nm monomer equivalent) were sonicated for 30 s at an amplitude of 65 (corresponding to ∼80 watts; QSonica) and preincubated overnight at 4 °C in media containing the heparins at four different concentrations (0.2–200 μg/ml). The next morning, the aggregate–heparin complexes were applied to cells for 4 h (tau and α-synuclein) or 20 h (Aβ) for internalization in media vehicles of 150 μl per well by media exchange. Incubation times were optimized empirically. Cells were harvested with 0.25% trypsin for 5 min and resuspended in flow cytometry buffer (HBSS plus 1% FBS and 1 mm EDTA) before flow cytometry. Cells were counted with the LSRFortessa SORP (BD Biosciences). We determined the MFI per cell to quantify cellular aggregate internalization. Each experiment was conducted three independent times with technical triplicates per condition, and a minimum of 2000 single cells was analyzed per replicate. Conditions with single cell counts below 2000 were excluded from analysis. We determined the average MFI of the replicates for each condition and standardized to aggregate uptake without inhibitor treatment within each experiment. The standardized averages of each condition were then combined for the graphs shown in this paper. Data analysis was performed using FlowJo version 10 software (Treestar Inc.) and GraphPad Prism version 7 for Mac OS X.
Seeding assays for heparin experiments
Tau
We previously used HEK293T cells to generate a stable monoclonal P301S FRET biosensor cell line by overexpressing tau RD with the disease-associated mutation P301S (Fig. S1), and tagged at the C terminus with either CFP or YFP to detect tau seeding (15, 16). The seeding assay was conducted as described except that biosensor cells were plated at a density of 15,000 cells/well in a 96-well plate. Recombinant tau fibrils were sonicated for 30 s at an amplitude of 65 (corresponding to ∼80 watts, QSonica) prior to use. Fibrils (100 nm monomer equivalent) were preincubated overnight at 4 °C in media containing the heparins at four different concentrations (0.2–200 μg/ml). At 20–30% confluency, the seed–heparin complexes were applied to the cells in media volumes of 50 μl per well, and cells were incubated for an additional 48 h. We did not use Lipofectamine to drive internalization of seeds because we sought to monitor HSPG-mediated uptake and the inhibitory activity of heparin blocking this process. Cells were harvested with 0.05% trypsin and post-fixed in 2% paraformaldehyde for 10 min and then resuspended in flow cytometry buffer (HBSS plus 1% FBS and 1 mm EDTA). The LSRFortessa SORP (BD Biosciences) was used to perform FRET flow cytometry. We quantified FRET as described previously (15, 16) with the following modification; we identified single cells that were YFP- and CFP-positive and subsequently quantified FRET-positive cells within this population. For each data set, three independent experiments with three technical replicates were performed. For each experiment, a minimum of ∼10,000 single cells per replicate was analyzed. Wells with YFP/CFP-positive cell counts below 5000 cells were excluded from the analysis. Data analysis was performed using FlowJo version 10 software (Treestar Inc.) and GraphPad Prism version 7 for Mac OS X.
α-Synuclein
The monoclonal A53T FRET biosensor cell line was previously established in our laboratory to detect α-synuclein seeding, using HEK293T cells stably expressing full-length α-synuclein with the disease-associated mutation A53T, and tagged either with CFP or YFP (15, 16). The seeding assay was performed similarly to the tau seeding assay, except that biosensor cells were plated at a density of 10,000 cells/well in a 96-well plate. Recombinant α-synuclein fibrils (200 nm monomer equivalent) were sonicated for 30 s at an amplitude of 65 (corresponding to ∼80 watts, QSonica) prior to use. The fibrils were preincubated overnight at 4 °C in media containing the heparins at four different concentrations (0.2–200 μg/ml). At 20% confluency, the fibril–heparin complexes were applied to the cells in media vehicles of 50 μl per well, and cells were incubated for 48 h. Cells were then split 1:5 into a new 96-well plate and incubated overnight. The next day, a second treatment with α-synuclein fibril–heparin complexes was performed as above. After an additional 48 h, cells were harvested for flow cytometry. For the data shown, we performed two independent experiments, with three technical replicates of each condition in each case. Data analysis was performed as described above for tau seeding.
Candidate screen using CRISPR/Cas9 knockout and lentiviral transduction
5–6 human gRNA sequences per gene were selected from the GeCKO version 2 libraries (17). For all gRNA sequences not beginning with guanine, a single guanine nucleotide was added at the 5′-end of the sequence to enhance U6 promoter activity. DNA oligonucleotides were synthesized by IDT DNA and cloned into the lentiCRISPRv2 vector (17) for lentivirus production. The plasmids for 5–6 gRNAs for each gene were pooled together. Lentivirus was created as described previously (33): HEK293T cells were plated at a concentration of 1 × 106 cells/well in a 6-well plate. 18 h later, cells were transiently co-transfected with PSP helper plasmid (1200 ng), VSV-G (400 ng), and gRNA plasmids (400 ng) using 7.5 μl of TransIT-293 (Mirus). 72 h later, the conditioned medium was harvested and centrifuged at 1000 rpm for 5 min to remove dead cells and debris. Lentivirus was concentrated 50× using lenti-X concentrator (Clontech) with the concentrated pellet being resuspended in PBS with 25 mm HEPES (pH 7.4). For transduction, a 1:25 to 1:40 dilution of virus suspension was added to HEK293T cells at a cell confluency of 20% in a 96-well plate. 20 h post-transduction, infected cells were treated with 2 μg/ml puromycin (Life Technologies, Inc.) and cultured for an additional 2 days, followed by passaging 1:5 and a second round of virus and puromycin application. The cells were kept in culture for at least 10 days after the first lentiviral transduction before using them for uptake and seeding experiments. After identification of the genes of interest, we made knockout cell lines using the available gRNAs individually to identify the functional gRNAs. We identified 2–3 gRNAs per gene and used these for further knockout experiments.
Confirmation of gene editing by TIDE (18)
We used a single functional gRNA for each gene to produce knockout cell lines for analysis by TIDE as established by Brinkman et al. (18) to confirm the presence of indels in predicted DNA regions (Table S1 and Fig. S3). We extracted genomic DNA using a protocol by Mendell and co-workers (34) with the following modifications: cell pellets from knockout cell lines and scrambled control cells (106 cells per sample) were frozen overnight at −80 °C, thawed the next day, and resuspended in 500 μl of tissue lysis buffer (500 μl of 10× STE buffer (10 mm EDTA (pH 8.0), 100 mm Tris-HCl (pH 8.0), 1 m NaCl), 100 μl of 0.5 m EDTA, 50 μl of 20 mg/ml proteinase K solution (ThermoFisher Scientific), 200 μl of 10% SDS, 4.15 ml H2O) and incubated for 5 h with shaking at 550 rpm and 55 °C. Tubes were allowed to cool down before adding 1 μl of 10 mg/ml RNase A (ThermoFisher Scientific), followed by incubation at 37 °C for 1 h while shaking at 550 rpm. Extractions were performed in Phase Lock Gels Light (VWR) using 500 μl of pH 7.9-buffered phenol (ThermoFisher Scientific), followed by phenol/chloroform/isoamyl alcohol (ThermoFisher Scientific), followed by chloroform (Sigma). Each extraction step was followed by centrifugation at 16,000 × g for 5 min. The upper aqueous phase was then transferred into a new 2-ml tube, mixed with 1.3 μl of glycoblue, followed by 1 ml of 100% ethanol. The DNA was precipitated at −80 °C for 1 h, followed by centrifugation at 15,000 × g for 15 min at 4 °C. Pellets were washed with 1 ml of 75% ethanol, centrifuged again (15,000 × g, 15 min, 4 °C), dried for a few minutes at room temperature, and solubilized in 50 μl of water at room temperature for 30 min. DNA concentration was determined with a spectrophotometer (DeNovix DS-11 FX+). PCR primers were designed according to the developer's instructions for TIDE (18). PCRs were performed using 100–500 ng of genomic DNA with 2× Taq-Pro Red Complete Polymerase (Denville Scientific, Inc.) following the manufacturer's instructions. PCR conditions were 3 min at 95 °C (1×), followed by 15 s at 95 °C, 15 s at 55 °C, and 1 min at 72 °C (30×) and 10 min at 72 °C (1×). The PCR product was run on a 1% agarose gel to verify the product size and gel-extracted using the QIAQuick gel extraction kit (Qiagen). Purified PCR samples (12 μl, ∼4–8 ng/μl) were Sanger sequenced at the sequencing core facility at UT Southwestern Medical Center, Dallas. TIDE analysis was performed according to the software instructions (Table S1 and Fig. S3). The data were derived with a forward primer for HS6ST2 and reverse primers for EXT1 and NDST1. Because Brinkman et al. (18) found that the results of TIDE analysis are nearly identical for both DNA strands, we did not sequence the opposite strands. R2 represents the goodness of fit and should be close to 0.9 for reliable data (18). R2 was slightly lower for EXT1 and HS6ST2, but the aberrant sequence signal was clearly visible in each graph (Fig. S3).
Uptake assay for knockout screen
The uptake assay for the knockout screen was conducted as described above with the following modifications. Knockout cell lines were plated at 20,000 cells per well in a 96-well plate. The next day, aggregates labeled with Alexa Fluor 647 were sonicated for 30 s at an amplitude of 65 (corresponding to ∼80 watt, QSonica), mixed with transferrin labeled with Alexa Fluor 488 (Life Technologies, Inc.), and applied to the cells in a vehicle of 50 μl of media for 4 h. Final concentrations of tau and α-synuclein in the media were 25 nm (monomer equivalent) and of transferrin 25 μg/ml. Cells were harvested with 0.25% trypsin for 5 min and resuspended in flow cytometry buffer (HBSS plus 1% FBS and 1 mm EDTA) before flow cytometry. Cells were counted with the LSRFortessa SORP (BD Biosciences). Data were collected from the same cell lines in duplicate on two different days. For each replicate, ∼10,000 single cells were analyzed. Uptake was normalized to the uptake detected in scrambled control cells. For the analysis, we combined the averages for each experimental day to determine the multiplicity adjusted p values using one-way ANOVA and Dunnett testing. Data analysis was performed using FlowJo version 10 software (Treestar Inc.) and GraphPad Prism version 7 for Mac OS X.
Making knockout cell lines for rescue experiments
Two to three functional gRNA plasmids identified as described above were pooled and used for transfection. 30,000 HEK293T cells or FRET biosensors cells (for tau and α-synuclein seeding) were plated per well in a 96-well plate. The next day, 100 ng of DNA was combined with OptiMEM (Gibco, Life Technologies, Inc.) to a final volume of 10 μl per well. Similarly, 0.5 μl of Lipofectamine-2000 per well (Invitrogen, Life Technologies, Inc.) was combined with OptiMEM (Gibco, Life Technologies, Inc.) to a final volume of 10 μl per well in a master mix solution. Both solutions were mixed after 5 min of incubation and incubated for an additional 20 min at room temperature, followed by dropwise addition to the cells at about 70% of cell confluency. We applied puromycin once at 2 μg/ml 24 h after transfection. Cells were cultured to confluency, passaged, and transfected in a similar way two more times. Cells were maintained in culture at least 10 days after the last transfection and prior to uptake and seeding experiments.
Cloning for rescue experiments
We used our previously described lentiviral FM5-YFP plasmid (27), replaced the human ubiquitin C promoter with a human CMV promoter, replaced the YFP sequence with an HA-tag coding sequence, and added an IRES element followed by an mCherry-coding sequence downstream of the insert. The resulting FM5/HA IRES mCherry vector bicistronically expresses an HA-tagged insert and mCherry protein from the same promoter. For the rescue experiments, EXT1 (pCMV3-Ext1-HA; Sino Biological), NDST1 (pUC75-NDST1; Genscript), and HS6ST2 (pANT7-cGST-HS6ST2; DNASU Plasmid Depository, Arizona State University (35)) cDNA was subcloned into the FM5/HA IRES mCherry vector using Gibson assembly Master Mix (New England Biolabs) according to the manufacturer's instructions. All genes were sequenced before use.
Rescue experiments
We used the cDNA IRES mCherry constructs to produce lentivirus and concentrated it 50× as described above. We applied the lentivirus at a 1:600 dilution to the knockout cell lines for 3 days prior to uptake and seeding experiments. We also used higher and lower lentivirus concentrations (dilutions between 1:160 and 1:2400), which led to toxicity for higher concentrations and reduced the percentage of mCherry-positive cells at lower concentrations, without significantly improving the rescue effects. We performed uptake and seeding experiments as described, with the following modifications. For uptake experiments, cells were treated with 100 nm (monomer equivalent) Alexa Fluor 647-labeled recombinant fibrils (tau or α-synuclein). For tau seeding experiments, cells were treated 2 h after plating at a final concentration of 100 nm (monomer equivalent) recombinant tau fibrils for 72 h. For α-synuclein seeding experiments, α-synuclein fibrils were sonicated for a total of 30 min (alternating 30 s on and 90 s off to avoid heating of the water bath) at an amplitude of 50 (QSonica) prior to use. Freshly sonicated α-synuclein fibrils were applied to the cells 2 h after plating at a final concentration of 800 nm and incubated for 72 h. Flow cytometry data were collected from the same cell lines in triplicate on at least 2 different days. For data acquisition and analysis, we gated for mCherry-expressing cells and quantified uptake and seeding within this cell population. The quantification of uptake (median fluorescence intensity of Alexa Fluor 647) and seeding (percentage of FRET positive cells) was then performed as described above. For each replicate, ∼5000 single cells for uptake experiments and 10,000 cells for seeding experiments were analyzed. Uptake and seeding were normalized to the uptake/seeding detected in control cells treated with scrambled gRNA. For the analysis, we combined the averages for each experimental day to determine the multiplicity adjusted p values using one-way ANOVA and Holm-Šídák testing. Data analysis was performed using FlowJo version 10 software (Treestar Inc.) and GraphPad Prism version 7 for Mac OS X.
Western blotting to confirm overexpression of rescue plasmids
The HEK293T knockout cell lines and the scrambled control cell line were grown to confluency in a 6-well cell culture dish (1 well per cell line). Cells were pelleted at 1000 rpm, washed with PBS, and frozen at −80 °C. Pellets were thawed on ice, resuspended in 100 μl of lysate buffer (0.05% Triton X-100, protease inhibitor, PBS), incubated for 10 min at 4 °C while rotating, and centrifuged for 5 min at 1000 × g. The supernatant was preserved and quantified for total protein content by the Bradford assay. Samples were boiled for 5 min with SDS-PAGE sample buffer, and 10 μg of total protein were loaded into a 4–12% polyacrylamide gel (NuPAGE). Using electrophoresis, samples were run for ∼60 min and transferred to a PVDF membrane (semi-wet transfer method). After blocking in 5% nonfat dry milk, membranes were incubated with primary antibody (1:4000 anti-HA tag antibody-ChIP Grade (ab9110, Abcam)) at room temperature for 2 h. Following an incubation for 2 h at room temperature with secondary antibody (1:4000; anti-Rb HRP-labeled; Jackson Immunotherapy), membranes were imaged by the ECL Prime Western blotting detection system (ThermoFisher Scientific) using a digital imager (Syngene).
Author contributions
B. E. S., B. B. H., V. A. M., J. V.-A., L. C. H.-W., and M. I. D. conceptualization; B. E. S., B. B. H., G. M. M., and L. C. H.-W. data curation; B. E. S., B. B. H., G. M. M., V. A. M., J. V.-A., W. L. P., L. C. H.-W., and M. I. D. formal analysis; B. E. S., B. B. H., G. M. M., V. A. M., J. V.-A., and W. L. P. investigation; B. E. S., B. B. H., G. M. M., V. A. M., and J. V.-A. methodology; B. E. S. writing-original draft; B. E. S., B. B. H., J. V.-A., L. C. H.-W., and M. I. D. writing-review and editing; L. C. H.-W. and M. I. D. supervision; L. C. H.-W. and M. I. D. project administration; M. I. D. resources; M. I. D. funding acquisition.
Supplementary Material
Acknowledgments
We thank Allison Ruchinskas and Omar Kashmer for recombinant protein preparation. We thank Dana Dodd and Sushobhna Batra for helpful comments on the manuscript.
This work was supported by RWTH University Aachen, Faculty of Medicine, Germany, through the Rotation Program for Junior Researchers (to B. E. S.), the Carl and Florence E. King Foundation (to B. E. S.), National Institutes of Health Grant 1F31NS079039 (to B. B. H.), Medical Scientist Training Program Washington University St. Louis Grant 5T32GM07200 (to B. B. H.), the University of Texas Southwestern Medical Student Summer Research Fellowship (to W. L. P.), the Rainwater Charitable Foundation (to M. I. D.), and the Cure Alzheimer's Fund (to M. I. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S5 and Table S1.
- HSPG
- heparan sulfate proteoglycan
- HS
- heparan sulfate
- GAG
- glycosaminoglycan
- Aβ
- β-amyloid
- YFP
- yellow fluorescent protein
- CFP
- cyan fluorescent protein
- RD
- repeat domain
- ANOVA
- analysis of variance
- CMV
- cytomegalovirus
- HBSS
- Hanks' buffered saline solution
- IRES
- internal ribosomal entry site
- TIDE
- tracking of indels by decomposition
- gDNA
- genomic DNA
- MFI
- median fluorescence intensity.
References
- 1. Holmes B. B., DeVos S. L., Kfoury N., Li M., Jacks R., Yanamandra K., Ouidja M. O., Brodsky F. M., Marasa J., Bagchi D. P., Kotzbauer P. T., Miller T. M., Papy-Garcia D., and Diamond M. I. (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147 10.1073/pnas.1301440110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 10.1146/annurev-biochem-060713-035314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lindahl U., and Kjellén L. (2013) Pathophysiology of heparan sulphate: many diseases, few drugs. J. Intern. Med. 273, 555–571 10.1111/joim.12061 [DOI] [PubMed] [Google Scholar]
- 4. Bishop J. R., Schuksz M., and Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 10.1038/nature05817 [DOI] [PubMed] [Google Scholar]
- 5. Wang Z., Hsieh P. H., Xu Y., Thieker D., Chai E. J., Xie S., Cooley B., Woods R. J., Chi L., and Liu J. (2017) Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate. J. Am. Chem. Soc. 2017 10.1021/jacs.7b01923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsieh P. H., Thieker D. F., Guerrini M., Woods R. J., and Liu J. (2016) Uncovering the relationship between sulphation patterns and conformation of iduronic acid in heparan sulphate. Sci. Rep. 6, 29602 10.1038/srep29602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kanekiyo T., Zhang J., Liu Q., Liu C. C., Zhang L., and Bu G. (2011) Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. J. Neurosci. 31, 1644–1651 10.1523/JNEUROSCI.5491-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Horonchik L., Tzaban S., Ben-Zaken O., Yedidia Y., Rouvinski A., Papy-Garcia D., Barritault D., Vlodavsky I., and Taraboulos A. (2005) Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 280, 17062–17067 10.1074/jbc.M500122200 [DOI] [PubMed] [Google Scholar]
- 9. Magalhães A. C., Baron G. S., Lee K. S., Steele-Mortimer O., Dorward D., Prado M. A., and Caughey B. (2005) Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 25, 5207–5216 10.1523/JNEUROSCI.0653-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ben-Zaken O., Tzaban S., Tal Y., Horonchik L., Esko J. D., Vlodavsky I., and Taraboulos A. (2003) Cellular heparan sulfate participates in the metabolism of prions. J. Biol. Chem. 278, 40041–40049 10.1074/jbc.M301152200 [DOI] [PubMed] [Google Scholar]
- 11. Caughey B., and Raymond G. J. (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 67, 643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shipp E. L., and Hsieh-Wilson L. C. (2007) Profiling the sulfation specificities of glycosaminoglycan interactions with growth factors and chemotactic proteins using microarrays. Chem. Biol. 14, 195–208 10.1016/j.chembiol.2006.12.009 [DOI] [PubMed] [Google Scholar]
- 13. Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., and Crowther R. A. (1996) Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553 10.1038/383550a0 [DOI] [PubMed] [Google Scholar]
- 14. Castillo G. M., Ngo C., Cummings J., Wight T. N., and Snow A. D. (1997) Perlecan binds to the β-amyloid proteins (Aβ) of Alzheimer's disease, accelerates Aβ fibril formation, and maintains Aβ fibril stability. J. Neurochem. 69, 2452–2465 [DOI] [PubMed] [Google Scholar]
- 15. Furman J. L., Holmes B. B., and Diamond M. I. (2015) Sensitive detection of proteopathic seeding activity with FRET flow cytometry. J. Vis. Exp. 2015, e53205 10.3791/53205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holmes B. B., Furman J. L., Mahan T. E., Yamasaki T. R., Mirbaha H., Eades W. C., Belaygorod L., Cairns N. J., Holtzman D. M., and Diamond M. I. (2014) Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, E4376–E4385 10.1073/pnas.1411649111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brinkman E. K., Chen T., Amendola M., and van Steensel B. (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thul P. J., Akesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., Alm T., Asplund A., Bjork L., Breckels L. M., Backstrom A., Danielsson F., Fagerberg L., Fall J., Gatto L., et al. (2017) A subcellular map of the human proteome. Science 356, eaal3321 10.1126/science.aal3321 [DOI] [PubMed] [Google Scholar]
- 20. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 21. Lidholt K., Weinke J. L., Kiser C. S., Lugemwa F. N., Bame K. J., Cheifetz S., Massagué J., Lindahl U., and Esko J. D. (1992) A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 89, 2267–2271 10.1073/pnas.89.6.2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Busse-Wicher M., Wicher K. B., and Kusche-Gullberg M. (2014) The exostosin family: proteins with many functions. Matrix Biol. 35, 25–33 10.1016/j.matbio.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 23. Ihse E., Yamakado H., van Wijk X. M., Lawrence R., Esko J. D., and Masliah E. (2017) Cellular internalization of α-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 7, 9008 10.1038/s41598-017-08720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makkonen K. E., Turkki P., Laakkonen J. P., Ylä-Herttuala S., Marjomäki V., and Airenne K. J. (2013) 6-O- and N-sulfated syndecan-1 promotes baculovirus binding and entry into mammalian cells. J. Virol. 87, 11148–11159 10.1128/JVI.01919-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao J., Huvent I., Lippens G., Eliezer D., Zhang A., Li Q., Tessier P., Linhardt R. J., Zhang F., and Wang C. (2017) Glycan determinants of heparin-tau interaction. Biophys. J. 112, 921–932 10.1016/j.bpj.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rauch J. N., Chen J. J., Sorum A. W., Miller G. M., Sharf T., See S. K., Hsieh-Wilson L. C., Kampmann M., and Kosik K. S. (2018) Tau internalization is regulated by 6-O sulfation on heparan sulfate proteoglycans (HSPGs). Sci. Rep. 8, 6382 10.1038/s41598-018-24904-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanders D. W., Kaufman S. K., DeVos S. L., Sharma A. M., Mirbaha H., Li A., Barker S. J., Foley A. C., Thorpe J. R., Serpell L. C., Miller T. M., Grinberg L. T., Seeley W. W., and Diamond M. I. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288 10.1016/j.neuron.2014.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaufman S. K., Sanders D. W., Thomas T. L., Ruchinskas A. J., Vaquer-Alicea J., Sharma A. M., Miller T. M., and Diamond M. I. (2016) Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 92, 796–812 10.1016/j.neuron.2016.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farhy Tselnicker I., Boisvert M. M., and Allen N. J. (2014) The role of neuronal versus astrocyte-derived heparan sulfate proteoglycans in brain development and injury. Biochem. Soc. Trans. 42, 1263–1269 10.1042/BST20140166 [DOI] [PubMed] [Google Scholar]
- 30. Tsuboi Y., Doh-Ura K., and Yamada T. (2009) Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology 29, 632–636 10.1111/j.1440-1789.2009.01058.x [DOI] [PubMed] [Google Scholar]
- 31. Doh-ura K., Ishikawa K., Murakami-Kubo I., Sasaki K., Mohri S., Race R., and Iwaki T. (2004) Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J. Virol. 78, 4999–5006 10.1128/JVI.78.10.4999-5006.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diringer H., and Ehlers B. (1991) Chemoprophylaxis of scrapie in mice. J. Gen. Virol. 72, 457–460 10.1099/0022-1317-72-2-457 [DOI] [PubMed] [Google Scholar]
- 33. Araki T., Sasaki Y., and Milbrandt J. (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013 10.1126/science.1098014 [DOI] [PubMed] [Google Scholar]
- 34. Golden R. J., Chen B., Li T., Braun J., Manjunath H., Chen X., Wu J., Schmid V., Chang T. C., Kopp F., Ramirez-Martinez A., Tagliabracci V. S., Chen Z. J., Xie Y., and Mendell J. T. (2017) An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202 10.1038/nature21025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seiler C. Y., Park J. G., Sharma A., Hunter P., Surapaneni P., Sedillo C., Field J., Algar R., Price A., Steel J., Throop A., Fiacco M., and LaBaer J. (2014) DNASU plasmid and PSI:Biology-materials repositories: resources to accelerate biological research. Nucleic Acids Res. 42, D1253–D1260 10.1093/nar/gkt1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.