

Abstract
Although signaling of thrombin via its receptor protease-activated receptor 1 (Par1) is known to occur in atherothrombosis, its link to the actual pathogenesis of this condition is less clear. To better understand the role of thrombin–Par1 signaling in atherosclerosis, here we have studied their effects on cellular cholesterol efflux in mice. We found that by activating Par1 and cullin 3–mediated ubiquitination and degradation of ABC subfamily A member 1 (ABCA1), thrombin inhibits cholesterol efflux in both murine macrophages and smooth muscle cells. Moreover, disruption of the Par1 gene rescued ABCA1 from Western diet–induced ubiquitination and degradation and restored cholesterol efflux in apolipoprotein E–deficient (ApoE−/−) mice. Similarly, the Par1 deletion diminished diet-induced atherosclerotic lesions in the ApoE−/− mice. These observations for the first time indicate a role for thrombin–Par1 signaling in the pathogenesis of diet-induced atherosclerosis. We identify cullin 3 as a cullin-RING ubiquitin E3 ligase that mediates ABCA1 ubiquitination and degradation and thereby inhibits cholesterol efflux. Furthermore, compared with peripheral blood mononuclear cells (PBMCs) from ApoE−/− mice, the PBMCs from ApoE−/−:Par1−/− mice exhibited decreased trafficking to inflamed arteries of Western diet–fed ApoE−/− mice. This finding suggested that besides inhibiting cholesterol efflux, thrombin–Par1 signaling also plays a role in the recruitment of leukocytes during diet-induced atherogenesis. Based on these findings, we conclude that thrombin–Par1 signaling appears to contribute to the pathogenesis of atherosclerosis by impairing cholesterol efflux from cells and by recruiting leukocytes to arteries.
Keywords: ABC transporter, atherosclerosis, cholesterol, macrophage, signal transduction
Introduction
Atherosclerosis is the leading cause of death and disability in the Western world (1). Inflammation via triggering the expression or activation of molecules that affect the behavior of the vascular wall (namely endothelial cells (ECs)2 and smooth muscle cells (SMCs)) provokes the atherosclerotic lesion formation (2). The cardiovascular risk factors, such as hypercholesterolemia, that lead to accumulation of cholesteryl esters in macrophages and vascular cells may trigger inflammatory responses in the arterial wall and contribute to plaque formation and its calcification (3, 4). The earliest changes that precede the lesion formation of atherosclerosis occur in ECs, affecting their function (5). The dysfunctional endothelium via production of various cell adhesion molecules and chemoattractants may enhance the recruitment of monocytes to the intima, where they differentiate into macrophages (6). Macrophages with increased expression of scavenger receptors take up oxLDL and become foam cells, an early precipitous event in atherogenesis (7). Besides postintimal injury, several growth factors and cytokines released by different cell types, such as inflammatory cells, platelets, and ECs, could also lead to the phenotypic conversion of SMCs from the “contractile” state to the “synthetic” state, which then can migrate from media to intima, proliferate in the intima, and contribute to the pathogenesis of atherosclerosis (8). Similar to macrophages, SMCs express scavenger receptors and transform into foam cells (9, 10). Thus, both macrophages and SMCs appear to be critical players in the pathogenesis of atherosclerosis.
Besides its role in blood clotting, thrombin acts as a mitogen and chemotactic factor for many cell types, including fibroblasts and SMCs, and plays an important role in vascular wall remodeling (11, 12). Thrombin transmits its cellular effects via protease-activated receptors (Pars), namely Par1, Par3, and Par4, depending on the cell type (13). The presence of thrombin receptors in ECs, SMCs, and leukocytes suggests the potential involvement of thrombin in the pathophysiology of atherosclerosis (13). In fact, thrombin is produced at the sites of vascular injury by the interaction of tissue factor with circulating factor VII (14). It was also reported that the expression of factor VII and factor X is increased in macrophages within the atherosclerotic lesions in close proximity to tissue factor (15). Despite the presence of these coagulation factors within the atherosclerotic lesions, very little is known about the production of thrombin in the lesions and its role in the lesion progression (16). In this regard, the emerging evidence points out that thrombin, besides its hemostatic function, also plays a role in the modulation of inflammation and that it promotes the development of atherosclerosis (17, 18), although proof of its involvement in atherogenesis is lacking. Interestingly, a recent study showed that inhibition of thrombin attenuates high-fat diet–induced weight gain and nonalcoholic fatty liver disease (19). Besides its role in the regulation of vascular tone and permeability, thrombin and its receptors have also been reported to mediate endothelial dysfunction and intimal hyperplasia (20, 21), potential hallmarks in the development of atherosclerosis. The reported role of Pars in endothelial dysfunction and their increased expression in SMCs of synthetic phenotype (20–22) in combination with the role of thrombin in inflammation (23, 24) lead us to speculate a role for thrombin and its receptors in the pathogenesis of atherosclerosis, which is the focus of the present study.
Foam cell formation depends on the uptake and retention of lipoproteins, uptake of modified LDL, or decreased removal of cholesterol by macrophages and smooth muscle cells in the vessel wall (25, 26). Whereas scavenger receptors, such as SR-A1, SR-B1, and SR-B2 (CD36), mediate the uptake of modified LDL, the ABC transporters, namely ABCA1 and ABCG1, facilitate the removal of excess cholesterol from macrophages and smooth muscle cells (10, 27–30). Therefore, increased expression of scavenger receptors or decreased expression of ABCA1 or ABCG1 could influence pro-atherogenic effects. In fact, knockdown of ABCA1 and ABCG1 exacerbated the atherosclerotic lesion formation (31, 32). Despite the role of thrombin and its receptors in atherothrombosis (33) and the emerging evidence linking them to inflammation and atherogenesis (17, 18, 23, 24), there has been very little mechanistic insight into the role of the thrombin–Par1 axis in atherogenesis. Therefore, we asked the question of whether thrombin and its receptors have any influence on the modulation of cellular cholesterol levels and atherogenesis. Here, we report for the first time that thrombin via activation of its receptor Par1 promotes cullin 3–mediated ubiquitination and degradation of ABCA1 and thereby inhibits cholesterol efflux and enhances atherogenesis. We also found that when compared with PBMCs from ApoE−/− mice, the PBMCs from ApoE−/−:Par1−/− mice showed decreased trafficking to inflamed arteries of WD-fed ApoE−/− mice, which indicates that besides its role in the inhibition of cholesterol efflux, thrombin–Par1 signaling also mediates leukocyte recruitment during diet-induced atherogenesis.
Results
Thrombin inhibits cholesterol efflux both in macrophages and smooth muscle cells
To understand the role of thrombin in the pathogenesis of atherosclerosis, we studied its effects on cholesterol efflux. Thrombin inhibited cholesterol efflux in both mouse primary peritoneal macrophages and smooth muscle cells (Figs. 1A and 2A). The ABC transporters ABCA1 and ABCG1 play an important role in cholesterol efflux, and their disruption exacerbates atherogenesis (25–27). To find the possible mechanism of inhibition of cholesterol efflux by thrombin, we tested its effects on ABCA1 and ABCG1 levels. Thrombin, without having any effect on ABCG1 levels, substantially reduced ABCA1 levels both in macrophages and smooth muscle cells (Figs. 1B and 2B). To understand the mechanisms underlying thrombin-induced down-regulation of ABCA1 levels, we studied its effects on ABCA1 and ABCG1 mRNA levels. Thrombin had no effects on either ABCA1 or ABCG1 mRNA levels (Figs. 1C and 2C). To find whether thrombin-induced depletion of ABCA1 levels was due to its proteosomal degradation or autophagy, we next tested the effects of MG132 and chloroquine, potent proteosomal and autophagy inhibitors, respectively (34, 35). MG132, but not chloroquine, completely prevented ABCA1 from thrombin-induced degradation both in macrophages and smooth muscle cells (Figs. 1D and 2D). Consistent with these observations, MG132 also restored cholesterol efflux from inhibition by thrombin in these cells (Figs. 1E and 2E). In addition, we found that thrombin induces the ubiquitination of ABCA1 in a time-dependent manner (Figs. 1F and 2F), and its overexpression with a mild influence on basal cholesterol efflux rescued it from inhibition by thrombin (Figs. 1G and 2G). Furthermore, pulse–chase studies showed that following 1-h treatment with thrombin, its removal from the medium restored ABCA1 to its pre-thrombin treatment levels by 2 h (Figs. 1H and 2H).
Figure 1.

Thrombin inhibits cholesterol efflux via ABCA1 degradation in mouse primary peritoneal macrophages. A, peritoneal macrophages were labeled with [3H]cholesterol (1 μCi/ml) for 24 h, treated with and without thrombin (0.5 units/ml) for the indicated time periods, and subjected to a cholesterol efflux assay. B, equal amounts of protein from control and the indicated time periods of thrombin-treated macrophages were analyzed by Western blotting for ABCA1 and ABCG1 levels using their specific antibodies, and the ABCA1 blot was normalized to β-tubulin levels. C, equal amounts of RNA from control and the indicated time periods of thrombin-treated macrophages were analyzed by RT-PCR for ABCA1, ABCG1, and β-actin mRNA levels using their specific primers. D, macrophages were treated with and without thrombin in the presence and absence of MG132 (10 μm) or chloroquine (50 μm) for 1 h, cell extracts were prepared and analyzed by Western blotting for ABCA1 levels, and the blot was normalized to β-tubulin levels. E, all of the conditions were the same as in D except that cells were subjected to a cholesterol efflux assay. F, all of the conditions were the same as in B except that the cell extracts were immunoprecipitated with anti-ubiquitin antibodies, and the immunocomplexes were analyzed by Western blotting for ABCA1 levels. The same cell extracts were also analyzed by Western blotting for ABCA1 levels, and the blot was normalized to β-tubulin levels. G, macrophages were transfected with empty vector or pCMV-ABCA1, labeled with [3H]cholesterol (1 μCi/ml) for 24 h, treated with and without thrombin for 2 h, and subjected to cholesterol efflux assay. Cell extracts from a parallel set of experiment were analyzed by Western blotting for ABCA1 to show the overexpression of ABCA1, and the blot was normalized to β-tubulin. H, macrophages were treated with and without thrombin for the indicated time periods or for 1 h, followed by the indicated chase time period. Cell extracts were prepared and analyzed by Western blotting for ABCA1 levels, and the blot was normalized for CDK4 and β-tubulin levels. The bar graphs represent mean ± S.D. (error bars) values of three experiments. *, p < 0.05 versus control or vector; **, p < 0.05 versus thrombin or vector + thrombin.
Figure 2.

Thrombin inhibits cholesterol efflux via ABCA1 degradation in mouse aortic smooth muscle cells. A–H, all of the experimental conditions were the same as in Fig. 1, A–H, respectively, in their order except that instead of macrophages, aortic smooth muscle cells were used. *, p < 0.05 versus control or vector; **, p < 0.05 versus thrombin or vector + thrombin. Error bars, S.D.
Par1 mediates thrombin-induced ABCA1 ubiquitination and degradation
Pars, G protein–coupled receptors, mediate the cellular effects of thrombin (13, 20, 36). To identify the Par mediating thrombin-induced ABCA1 degradation, we used an siRNA approach. Depleting Par1 but not Par3 or Par4 levels by their siRNAs prevented thrombin-induced degradation of ABCA1 in macrophages as well as smooth muscle cells (Fig. 3, A and D). Depletion of Par1 also reversed the cholesterol efflux from inhibition by thrombin in these cells (Fig. 3, B and E). Based on these observations, we next tested the role of Par1 in thrombin-induced ABCA1 ubiquitination and degradation. As expected, depletion of Par1 levels significantly prevented ABCA1 ubiquitination and degradation in macrophages and smooth muscle cells (Fig. 3, C and F). To confirm the role of Par1 in thrombin-induced ABCA1 degradation, we isolated peritoneal macrophages and smooth muscle cells from WT and Par1−/− mice and tested the effect of thrombin on ABCA1 levels. Thrombin, while reducing the ABCA1 levels both in macrophages and smooth muscle cells of WT mice, had no effect on ABCA1 levels either in the macrophages or smooth muscle cells of Par1−/− mice (Fig. 4, A and C). In line with these observations, thrombin inhibited cholesterol efflux only in the macrophages and smooth muscle cells of WT mice and not Par1−/− mice (Fig. 4, B and D). These results show that the ABCA1 degradation and cholesterol efflux inhibition are mediated via Par1 both in macrophages and smooth muscle cells.
Figure 3.

Depletion of Par1 prevents thrombin-induced inhibition of cholesterol efflux and ABCA1 degradation in both macrophages and smooth muscle cells. A, macrophages were transfected with siControl, siPar1, siPar3, or siPar4 (100 nm), and 36 h later, cells were treated with and without thrombin for 1 h, cell extracts were prepared and analyzed by Western blotting for ABCA1 levels, and the blot was reprobed for Par1, Par3, Par4, or β-tubulin to show the effects of the siRNA on its target and off-target molecule levels. B, macrophages were transfected with siControl siPar1, siPar3, or siPar4 (100 nm), labeled with [3H]cholesterol (1 μCi/ml) for 24 h, and treated with and without thrombin for 2 h, and the cholesterol efflux was measured. C, macrophages were transfected with siControl or siPar1 (100 nm) and treated with and without thrombin for 1 h. Cell extracts were prepared and immunoprecipitated with anti-ubiquitin antibodies, and the immunocomplexes were analyzed by Western blotting for ABCA1 levels. The same cell extracts were also analyzed by Western blotting for Par1, ABCA1, and β-tubulin levels to show the effects of the siRNA on its target and off-target molecule levels. D–F, all of the experimental conditions were the same as in A–C, respectively, except that instead of macrophages, smooth muscle cells were used. The bar graphs represent mean ± S.D. (error bars) of three experiments. *, p < 0.05 versus siControl; **, p < 0.05 versus siControl + thrombin.
Figure 4.

Deletion of the Par1 gene blocks thrombin-induced inhibition of cholesterol efflux and ABCA1 degradation in macrophages and smooth muscle cells. A, peritoneal macrophages were isolated from WT and Par1−/− mice and treated with and without thrombin for 1 h. Cell extracts were prepared and analyzed by Western blotting for ABCA1 levels, and the blot was normalized to β-tubulin levels. B, peritoneal macrophages from WT and Par1−/− mice were labeled with [3H]cholesterol (1 μCi/ml) for 24 h and treated with and without thrombin for 2 h, and the cholesterol efflux was measured. C and D, all of the experimental conditions were the same as in A and B except that instead of macrophages, smooth muscle cells were used. The bar graphs represent mean ± S.D. (error bars) of three experiments. *, p < 0.05 versus control; **, p < 0.05 versus thrombin.
Disruption of the Par1 gene reduces diet-induced atherosclerosis in ApoE−/− mice
To obtain an additional line of evidence on the role of Par1 in thrombin-induced inhibition of cholesterol efflux and ABCA1 degradation, we generated ApoE−/−:Par1−/− mice and used them compared with ApoE−/− mice in these studies. Peritoneal macrophages and smooth muscle cells from ApoE−/− and ApoE−/−:Par1−/− mice fed with WD for 16 weeks were isolated and analyzed for cholesterol efflux and ABCA1 ubiquitination. We found decreased cholesterol efflux in macrophages and smooth muscle cells of ApoE−/− mice as compared with those from ApoE−/−:Par1−/− mice in response to thrombin (Fig. 5, A and C). Similarly, ABCA1 ubiquitination was found to be profoundly higher in macrophages and aorta of WD-fed ApoE−/− mice as compared with ApoE−/−:Par1−/− mice (Fig. 5, B and D). ABCA1 ubiquitination levels were found to be very low in macrophages as well as aorta of both ApoE−/− and ApoE−/−:Par1−/− mice maintained on CD as compared with WD-fed ApoE−/− mice (Fig. 5, B and D). To study the clinical relevance of these observations, both ApoE−/− and ApoE−/−:Par1−/− mice were fed with WD for 16 weeks starting from 8 weeks of age and analyzed for lipid profiles and atherosclerotic lesion formation. No major differences were observed in the body weight and plasma levels of total cholesterol, HDL, LDL, and triglycerides between ApoE−/− and ApoE−/−:Par1−/− mice in response to WD feeding (Table 1). Measurement of total atherosclerotic lesions by enface lipid staining revealed a significant increase in the atherosclerotic lesions in ApoE−/− mice that were fed with WD compared with ApoE−/− mice maintained on CD (Fig. 6A). On the other hand, the atherosclerotic lesions in WD-fed ApoE−/−:Par1−/− mice were found to be substantially reduced as compared with those of WD-fed ApoE−/− mice (Fig. 6A). The percentage of plaque area in the aortic roots of WD-fed ApoE−/− mice was found to be increased as compared with that of ApoE−/− mice that were kept on CD (Fig. 6B). Similar to atherosclerotic lesions, the percentage of plaque area in the aortic roots of WD-fed ApoE−/−:Par1−/− mice was found to be decreased as compared with that of WD-fed ApoE−/−mice (Fig. 6B). Consistent with these observations, immunofluorescence staining of the aortic root sections of WD-fed ApoE−/− mice showed increased co-localization of filipin with both Mac3 and SMCα-actin as compared with those of ApoE−/− mice kept on CD (Fig. 6C). The co-localization of filipin with both Mac3 and SMCα-actin was found to be significantly decreased in WD-fed ApoE−/−:Par1−/− mice as compared with WD-fed ApoE−/− mice (Fig. 6C). Consistent with these observations, immunofluorescence staining of the aortic root sections showed increased co-localization of Par1 with both Mac3 and SMCα-actin in WD-fed ApoE−/− mice as compared with ApoE−/− mice kept on CD (Fig. 6D). As expected, no immunostaining of Par1 was detected either in macrophages or smooth muscle cells of ApoE−/−:Par1−/− mice (Fig. 6D). In contrast, increased ABCA1 expression was observed both in macrophages and smooth muscle cells in the aortic root sections of WD-fed ApoE−/−:Par1−/− mice as compared with those of ApoE−/− mice (Fig. 6D). The ABCA1 expression levels in the aortic root sections of CD-fed ApoE−/− mice were found to be comparable with those in WD-fed ApoE−/−:Par1−/− mice (Fig. 6D).
Figure 5.
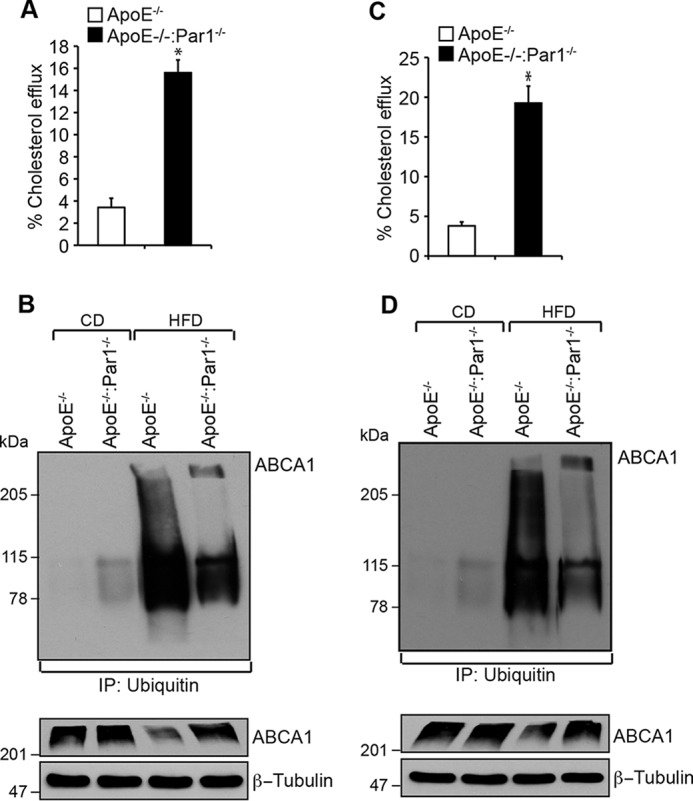
Par1 mediates the inhibition of cholesterol efflux and ABCA1 degradation in macrophages and smooth muscle cells. A, peritoneal macrophage were isolated from WD-fed ApoE−/− and ApoE−/−:Par1−/− mice, labeled with [3H]cholesterol (1 μCi/ml) for 24 h, and treated with and without thrombin for 2 h, and cholesterol efflux was measured. B, peritoneal macrophages from WD-fed ApoE−/− and ApoE−/−:Par1−/− mice were isolated, cell extracts were prepared and immunoprecipitated (IP) with anti-ubiquitin antibodies, and the immunocomplexes were analyzed by Western blotting for ABCA1 levels. The same cell extracts were also analyzed by Western blotting for ABCA1 levels, and the blot was normalized for β-tubulin. C and D, all of the experimental conditions were the same as in A and B except that instead of macrophages, smooth muscle cells or aorta were used. The bar graphs represent mean ± S.D. (error bars) of three experiments. *, p < 0.05 versus CD-fed ApoE−/− mice; **, p < 0.05 versus WD-fed ApoE−/− mice.
Table 1.
Plasma lipid profiles of ApoE−/− and ApoE−/−:Par1−/− mice fed with WD
| Genotype | Diet | Weight | Cholesterol | HDL | LDL | Triglycerides | n |
|---|---|---|---|---|---|---|---|
| g | mg/dl | mg/dl | mg/dl | mg/dl | |||
| ApoE−/− | WD | 32.8 ± 3 | 972 ± 183 | 652 ± 226 | 294 ± 124 | 217 ± 47 | 9 |
| ApoE−/−:Par1−/− | WD | 30.2 ± 2 | 875 ± 102 | 595 ± 143 | 229 ± 155 | 215 ± 143 | 7 |
Figure 6.

Genetic deletion of Par1 reduces atherosclerotic plaque progression. A, representative en face staining of aortas from ApoE−/− and ApoE−/−:Par1−/− mice fed with CD or WD for 16 weeks is shown, and the plaque area is presented as lesion percentage in the bar graph. B, representative Oil Red O staining of the aortic root sections of the mice described in A are shown, and the bar graph represents the quantification of the area positive for lipid staining. C and D, aortic root sections of the ApoE−/− and ApoE−/−:Par1−/− mice fed with CD or WD were stained for filipin, Par1, or ABCA1 in combination with Mac3 or SMCα-actin. Bar graphs represent mean ± S.D. (error bars) of seven animals. *, p < 0.05 versus WD-fed ApoE−/− mice. Scale bars, 100 μm (B), 200 μm (C), and 50 μm (D).
Cullin 3 mediates thrombin-induced ABCA1 ubiquitination and degradation
Previous studies have shown that cullin 3 plays a role in the regulation of lymphoid effector function, and mutations in cullin 3 were found to affect vascular smooth muscle cell function and cause hypertension (37–39). In addition, cullin 3 has been shown to regulate several cellular signaling events, including WNT signaling (40). However, nothing is known about their role in cholesterol transport. Therefore, to explore the potential mechanisms of ABCA1 ubiquitination and degradation, we have studied the role of cullin family of E3 ligases. Time course studies revealed that whereas cullin 1 and cullin 2 were found to be associated with ABCA1 constitutively, cullin 3 was observed to form a complex with ABCA1 in a time-dependent manner in response to thrombin in both macrophages and smooth muscle cells (Fig. 7, A and D). Based on these observations, we next tested the role of cullin 3 in thrombin-induced ABCA1 ubiquitination and degradation. Depletion of cullin 3 levels by its siRNA prevented thrombin-induced ABCA1 ubiquitination and degradation in macrophages and smooth muscle cells (Fig. 7, B and E). In accordance with these findings, siRNA-mediated knockdown of cullin 3 levels also rescued cholesterol efflux from inhibition by thrombin in both of these cells (Fig. 7, C and F). In line with these observations, increased immunostaining for cullin 3 was observed both in macrophages and smooth muscle cells of the aortic root sections of WD-fed ApoE−/− mice as compared with CD-fed ApoE−/− mice or WD-fed ApoE−/−:Par1−/− mice (Fig. 7G).
Figure 7.

Cullin 3 mediates ABCA1 ubiquitination and its degradation. A, peritoneal macrophages from WT mice were isolated and treated with and without thrombin for the indicated time periods; cell extracts were prepared and immunoprecipitated with anti-ABCA1 antibodies; and the immunocomplexes were analyzed by Western blotting for cullin 1, cullin 2, and cullin 3 using their specific antibodies. The blot was normalized for ABCA1 levels. B, peritoneal macrophages were transfected with cullin 3 siRNA (100 nm) and treated with and without thrombin for 1 h; cell extracts were prepared and immunoprecipitated with anti-ubiquitin antibodies; and the immunocomplexes were analyzed by Western blotting for ABCA1 levels. The same cell extracts were also analyzed by Western blotting for cullin 3, ABCA1, or β-tubulin levels to show the effect of the siRNA on its target and off-target molecules levels. C, peritoneal macrophages were transfected with cullin 3 siRNA (100 nm), labeled with [3H]cholesterol (1 μCi/ml) for 24 h, and treated with and without thrombin for 2 h, and the cholesterol efflux was measured. D–F, all of the experimental conditions were the same as in A–C, respectively, except that instead of macrophages, smooth muscle cells were used. G, aortic root sections of the ApoE−/− and ApoE−/−:Par1−/− mice fed with CD or WD were stained for cullin 3 in combination with Mac3 or SMCα-actin. The bar graphs represent mean ± S.D. (error bars) of three experiments. *, p < 0.05 versus control or siControl; **, p < 0.05 versus thrombin or siControl + thrombin. Scale bar in G, 50 μm.
PKCθ-mediated ABCA1 phosphorylation is required for its interaction with cullin 3 and degradation
In a parallel study, we found that Gα12, Pyk2, Gab1, and PKCθ-mediated CD36 expression downstream to Par1 is required for thrombin-induced foam cell formation.3 Based on these observations, we have examined the role of this signaling in thrombin-induced ABCA1 degradation and inhibition of cholesterol efflux. Depletion of Par1, Gα12, Pyk2, Gab1, or PKCθ using their specific siRNAs rescued ABCA1 levels and cholesterol efflux from thrombin-induced degradation and inhibition, respectively (Fig. 8, A and B). In addition, thrombin induced ABCA1 phosphorylation in a time-dependent manner, and this posttranslational modification correlated by its association with cullin 3 and its degradation (Fig. 8C). Excitingly, depletion of PKCθ levels by its siRNA inhibited thrombin-induced phosphorylation of ABCA1, its association with cullin 3, and its ubiquitination and degradation, restoring its steady-state levels (Fig. 8D). These results suggest that thrombin induces ABCA1 association with cullin 3 in a PKCθ-mediated phosphorylation-dependent manner, and these events require activation of Par1, Gα12, Pyk2, and Gab1 signaling. To confirm these observations in vivo, aortas were isolated from CD-fed ApoE−/− and WD-fed ApoE−/− and ApoE−/−:Par1−/− mice and analyzed for Gα12, Pyk2, Gab1, or PKCθ activation. Aortas from WD-fed ApoE−/− mice showed Gα12 activation, as measured by its dissociation from Par1, and increased phosphorylation of Pyk2, Gab1, and PKCθ as compared with CD-fed ApoE−/− or WD-fed ApoE−/−:Par1−/− mice (Fig. 8E). In addition, increased ABCA1 phosphorylation, its association with cullin 3, and its degradation were seen in WD-fed ApoE−/− mice as compared with CD-fed ApoE−/− or WD-fed ApoE−/−:Par1−/− mice (Fig. 8F). These results clearly show that Par1-dependent activation of Gα12, Pyk2, Gab1, and PKCθ via phosphorylation of ABCA1 and its association with cullin 3 leads to ABCA1 ubiquitination and degradation, resulting in inhibition of cholesterol efflux. In addition, prothrombin/thrombin levels were found to be increased by 50% in WD-fed ApoE−/− and ApoE−/−:Par1−/− mice as compared with CD-fed ApoE−/− mice (Fig. 8G).
Figure 8.

PKCθ-mediated phosphorylation of ABCA1 is required for its association with cullin 3 in its ubiquitination and degradation. A, Raw264.7 cells were transfected with the indicated siRNA, quiesced, and treated with and without thrombin for 2 h. Cell extracts were prepared and analyzed by Western blotting for ABCA1 levels using its specific antibodies, and the blots were reprobed for siRNA target and off-target molecules to show the efficacy and specificity of the indicated siRNA. B, all of the conditions were the same as in A except that after transfection, cells were labeled with [3H]cholesterol (1 μCi/ml) for 24 h, treated with and without thrombin for 2 h, and subjected to a cholesterol efflux assay. C, Raw264.7 cells were treated with and without thrombin for the indicated time periods, and cell extracts were prepared. Equal amounts of protein from each condition were immunoprecipitated (IP) with ABCA1 antibodies; the immunocomplexes were analyzed by Western blotting for pSer/Thr or cullin 3 antibodies; and the blot was reprobed for ABCA1. D, top, cells were transfected with siControl or siPKCθ, quiesced, and treated with and without thrombin for 2 h; cell extracts were prepared; equal amounts of protein from each condition were immunoprecipitated with ABCA1 antibodies; the immunocomplexes were analyzed by immunoblotting with pSer/Thr or cullin 3 antibodies; and the blot was reprobed for ABCA1. The same cell extracts were also analyzed by Western blotting for PKCθ and β-tubulin levels to show the efficacy of the siRNA on its target and off-target molecules. Bottom, equal amounts of proteins from same the cell extracts were also immunoprecipitated with anti-ubiquitin antibodies, and the immunocomplexes were analyzed by Western blotting for ABCA1 levels using its specific antibodies. E and F, aortas from CD-fed ApoE−/− or 12 weeks of WD-fed ApoE−/− and ApoE−/−:Par1−/− mice were isolated, tissue extracts were prepared, and equal amounts of proteins from each regimen were immunoprecipitated with Par1 or ABCA1 antibodies. The anti-Par1 and anti-ABCA1 immunocomplexes were analyzed by Western blotting for Gα12 levels and pSer/Thr or cullin 3 levels, respectively. The same tissue extracts were also analyzed by Western blotting for pPyk2, pGab1, and pPKCθ levels using their phospho-specific antibodies, and the blots were normalized for their total levels. G, whole blood was collected from CD-fed ApoE−/− or 12-week WD-fed ApoE−/− and ApoE−/−:Par1−/− mice, and plasma prothrombin/thrombin levels were measured using a kit from Abcam following the manufacturer's protocol. Error bars, S.D.
Par1 is required for leukocyte trafficking during diet-induced atherogenesis
Previously, we have reported that thrombin–Par1 signaling targeting Gα12, Pyk2, Gab1, p115 RhoGEF, Rac1, RhoA, and Pak1/2 plays a role in vascular smooth muscle cell and macrophage migration (41, 42). To find whether the thrombin–Par1 signaling was also involved in the trafficking of leukocytes during diet-induced atherogenesis, PBMCs were isolated from CD-fed ApoE−/− and ApoE−/−:Par1−/− mice and injected into WD-fed ApoE−/− mice, and 24 h later, the aortas were isolated and examined for the recruitment of leukocytes. As compared with PBMCs of ApoE−/− mice, the PBMCs from ApoE−/−:Par1−/− mice exhibited 3-fold less capacity of trafficking to the inflamed arteries of WD-fed ApoE−/− mice (Fig. 9). These findings indicate that Par1 plays a role in the recruitment of leukocytes during diet-induced atherogenesis.
Figure 9.

PBMCs from ApoE−/−:Par1−/− mice exhibit decreased trafficking. PBMCs isolated from ApoE−/− or ApoE−/−:Par1−/− mice were labeled with PKH67 cell tracker and injected into WD-fed ApoE−/− mice (1.0 × 105 cells/mice) via the tail vein. Twenty-four hours later, aortas were isolated, cleaned, fixed, opened, stained with DAPI, mounted on a slide with luminal side upward, and examined under a Zeiss inverted fluorescent microscope. PKH67-positive cells were counted and expressed as cells/field. Scale bar, 20 μm. Error bars, S.D.
Discussion
Thrombin's role in atherothrombosis has been well-studied (33, 43). Thrombin elicits its cellular effects via G protein–coupled receptors, namely Pars (13, 20, 36). The role of Pars in inflammation has also been demonstrated (36, 44). Many studies have shown that clotting factors, such as factor VII and factor X, were increased in the atherosclerotic lesions and that factor VII was generated in proximity to tissue factor (15, 16). These findings provide clues for the possible role of thrombin in atherosclerosis, at least via its reported roles in SMC migration and proliferation (11, 12, 21) and the fact that SMCs contribute to foam cell formation (9, 10). However, there is very little known about the role of thrombin and Pars in atherogenesis. Toward this end, the present findings provide the first evidence that thrombin inhibits cholesterol efflux. Decreased cholesterol efflux from macrophages and smooth muscle cells in the artery can lead to accumulation of cholesteryl esters, which can cause inflammation, thereby paving the way for atherogenesis (45). Indeed, a substantial body of evidence indicates that deletion of ABCA1 or ABCG1 alone or in combination impairs cholesterol efflux and exacerbates atherogenesis (30–32). In this context, our results show that thrombin-induced inhibition of cholesterol efflux was due to depletion of ABCA1. Furthermore, the observations that both cholesterol efflux inhibition and ABCA1 depletion were prevented by down-regulation of Par1 or disruption of its gene suggest that Par1 mediates the effects of thrombin on cholesterol efflux and ABCA1 levels in both macrophages and smooth muscle cells. Some reports showed that the ABC transporters, particularly ABCA1 and ABCG1, were vulnerable for ubiquitination and degradation affecting cholesterol efflux (46, 47). Although a few studies have shown a role for ubiquitination in the degradation of ABCA1 and ABCG1, the underlying mechanisms, particularly the role of E3 ubiquitin ligases in these effects, have not been explored. In this context, a recent study showed that HUWE1 and NEDD4-1, two HECT domain–containing ubiquitin E3 ligases, mediate ubiquitination and degradation of ABCG1 but not ABCA1 (48). In the present study, we demonstrate for the first time that Cullin-RING ubiquitin E3 ligases play a role in the ubiquitination and degradation of ABCA1. Specifically, our findings show that cullin 3 ubiquitinates and degrades ABCA1 in response to activation of thrombin–Par1 signaling both in macrophages and smooth muscle cells. Thus, these observations offer a mechanistic view that cullin 3 is activated by thrombin in a Par1-dependent manner in the ubiquitination and degradation of ABCA1 affecting cholesterol efflux.
The decreased cholesterol efflux by thrombin could also lead to the manifestation of atherosclerotic lesions. Indeed, disruption of Par1 was found to be atheroprotective in ApoE−/− mice, a murine model of experimental atherosclerosis (49). First, as compared with CD, feeding with WD for 16 weeks increased the expression of Par1 both in macrophages and smooth muscle cells of ApoE−/− mice. Second, as compared with CD, macrophage and smooth muscle cell accumulation of free cholesterol takes place in the atherosclerotic lesions of WD-fed ApoE−/− mice, and deletion of Par1 abrogates this effect. Third, ABCA1 levels were decreased in WD-fed ApoE−/− mice over CD, and deletion of Par1 rescued the ABCA1 from WD feeding–induced depletion. Decreased ABCA1 levels were also observed in cells in the plaque despite increased load of cholesterol (50, 51). In summary, the present findings for the first time demonstrate that thrombin–Par1 signaling via down-regulation of ABCA1 levels and inhibition of cholesterol efflux promotes atherogenesis. It appears that cullin 3, a cullin-RING ubiquitin E3 ligase, is a culprit in thrombin–Par1 signaling–mediated ABCA1 ubiquitination and degradation causing impaired cholesterol efflux. However, it is possible that in addition to inhibition of cholesterol efflux, thrombin–Par1 signaling could also be involved in the modulation of scavenger receptors and uptake of modified LDL particles in contributing to the pathogenesis of atherosclerosis, which needs to be explored.
In a parallel study, we found that Par1-, Gα12-, Pyk2-, and Gab1-dependent activation of PKCθ was involved in thrombin-induced CD36 expression and foam cell formation.3 Therefore, in exploring the mechanisms of cullin 3 association with ABCA1, we examined the role of Gα12-Pyk2-Gab1-PKCθ signaling and found that activation of this signaling is required for thrombin-induced ABCA1 degradation. It is interesting to note that thrombin induces the phosphorylation of ABCA1 in a PKCθ-dependent manner and that inhibition of its phosphorylation blocks ABCA1 association with cullin 3 and thereby prevents its ubiquitination and degradation. From these observations, it can be inferred that ABCA1 phosphorylation by PKCθ is essential for its association with cullin 3 in its ubiquitination and degradation. This conclusion can be further supported by the findings that prevention of ABCA1 degradation by down-regulation of Gα12, Pyk2, Gab1, or PKCθ rescued cholesterol efflux from inhibition by thrombin. These in vitro observations were also confirmed by in vivo findings, as WD-fed ApoE−/− mice showed increased Pyk2, Gab1, and PKCθ phosphorylation, which, in turn, correlated with increased ABCA1 phosphorylation and its association with cullin 3, leading to its degradation as compared with CD-fed ApoE−/− mice or WD-fed ApoE−/−:Par1−/− mice. Based on these findings, it may be stated that thrombin–Par1 signaling exerts a prominent role in the down-regulation of reverse cholesterol transporters, such as ABCA1, and impairs cholesterol removal from cells homing in the arterial wall, which over a period of time could precipitate into a fatty lesion. In addition, the reduced capacity of trafficking of PBMCs from ApoE−/−:Par1−/− mice as compared with those of ApoE−/− mice to inflamed arteries of WD-fed ApoE−/− mice indicates that besides affecting cholesterol efflux, thrombin–Par1 signaling might also be contributing to leukocyte recruitment during diet-induced atherogenesis. In fact, a large body of recent data shows that thrombin generation by dietary conditions is involved in the development of inflammation and atherosclerosis (23, 24). Furthermore, many studies have demonstrated the presence of various coagulation proteins in atherosclerotic plaques and linked them to promoting events such as inflammation and cell proliferation (16, 52). It was also shown that FII (prothrombin)−/WT:ApoE−/− mice with a diminished coagulation capacity exhibited reduced atherosclerotic lesions, decreased leukocyte infiltration, and altered collagen levels compared with control ApoE−/− mice (53). In addition, it was demonstrated that coagulation factors, such as tissue factor, factor Xa, and thrombin, play an important role in the progression of atherosclerotic plaques independent of their role in thrombus formation (54, 55). All of these observations provide clues for the role of endogenous thrombin generation in the development of atherosclerosis. In the present study, we found that WD induces prothrombin/thrombin levels both in ApoE−/− and ApoE−/−:Par1−/− mice. In addition, WD induced Par1 expression by severalfold as compared with CD in ApoE−/− mice. Thus, our findings provide the first mechanistic evidence for the role of the thrombin–Par1 axis in atherogenesis.
In summary, as outlined in Fig. 10, thrombin–Par1 signaling appears to contribute to the pathogenesis of atherosclerosis via impairing cholesterol efflux as well as recruitment of leukocytes to the artery.
Figure 10.
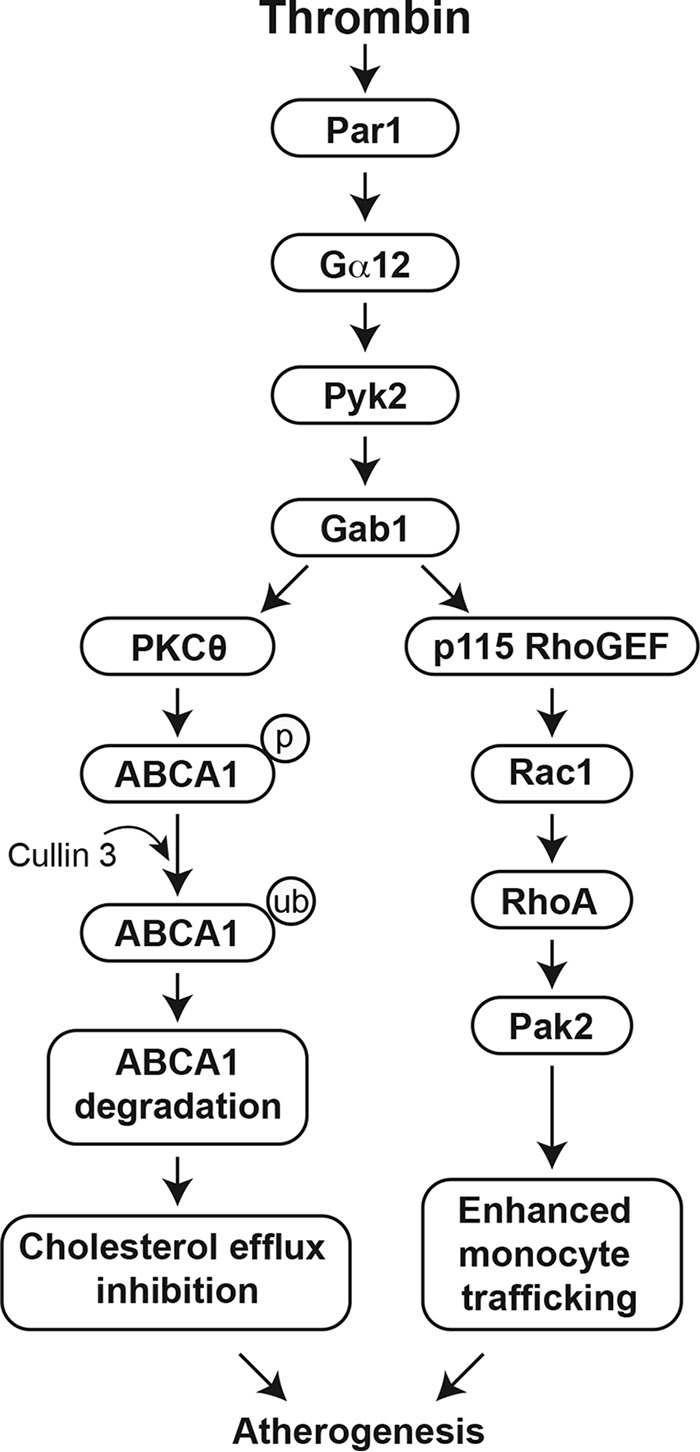
Schematic diagram outlining the possible mechanisms of a Par1 role in the inhibition of cholesterol efflux and enhanced leukocyte trafficking during diet-induced atherogenesis.
Materials and methods
Reagents
Chloroquine (C6628), filipin (F9765), PKH67 Green Fluorescent Cell Linker kit (PKH67GL), anti-SMCα-actin antibody (A2547), and thrombin (T8885) were purchased from Sigma-Aldrich. Collagenase II (LS004176) and elastase (LS006365) were obtained from Worthington. Anti-ABCA1 (ab18180), anti-ABCG1 (ab52617), anti-cullin 2 (ab166917), anti-cullin 3 (ab75851), anti-Pyk2 (ab32571), and anti-pSer/Thr (ab17464) antibodies and mouse prothrombin/thrombin Total ELISA Kit (ab157527) were purchased from Abcam (Cambridge, MA). Thrombin R (ATAP2/Par1) (SC-5605), anti-Par3 (SC-5598), anti-Par4 (SC-25466), anti-Mac3 (SC-19991), anti-cullin 1 (SC-17775), anti-cullin 3 (SC-166054), anti-CDK4 (SC-260), anti-Gab1 (SC-6292), anti-Gα12 (SC-409), and anti-β-tubulin (SC-9104) antibodies were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX). Anti-pGab1 (catalog no. 3233), anti-pPKCθ (catalog no. 9377), and anti-pPyk2 (catalog no. 3291) antibodies were bought from Cell Signaling Technology (Beverly, MA). Anti-ubiquitin antibody (P4D1) was purchased from Enzo Life Sciences (Farmingdale, NY). ApoA-I (BT-927) and HDL (BT-914) were bought from Biomedical Technologies (Stoughton, MA). MG132 (catalog no. 474790) was obtained from Calbiochem. pCMV6-ABCA1 (Myc-DDK tagged) overexpression plasmid was obtained from Origene (Rockville, MD). The Vectashield mounting medium (H-1000) was obtained from Vector Laboratories (Burlingame, CA). DH5α competent cells (18258-012), Lipofectamine 3000 transfection reagent (L3000-015), TRIzol (catalog no. 15596018), Hoechst 33342 (catalog no. 3570), and Prolong Gold antifade mounting medium (P36930) were purchased from Invitrogen. [3H]Cholesterol (specific activity 53 Ci/mmol) was obtained from PerkinElmer Life Sciences. Thioglycolate medium brewer-modified (catalog no. 21176) was purchased from BD Biosciences. GeneSilencer (TS00750) and GenePorter3000 (T203015) were from Gentalis (San Diego, CA). Mouse Gα12 siRNA (L-043467-00), mouse PKCθ siRNA (L-048426-00-0005), and control nontargeting siRNA (D-001810-10) were purchased from Dharmacon RNAi Technologies (Chicago, IL). Mouse cullin 3 (Silencer Select ID-188654), mouse Gab1 Silencer Select (ID-S66350), mouse Par1 (Silencer Select ID-S65790), mouse Par3 (Silencer Select S96752), mouse Par4 (Silencer Select ID-S65799), and mouse Pyk2 (Silencer Select ID-S72406) siRNAs were purchased from Ambion (Waltham, MA).
Cell culture
Mouse RAW264.7 cells, mouse primary peritoneal macrophages, and mouse aortic smooth muscle cells were cultured and maintained in DMEM or DMEM/F-12 containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. RAW264.7 cells were purchased from American Type Culture Collection (Manassas, VA). The cells were quiesced overnight in DMEM/F-12 medium without serum and used between four and 10 passages for the experiments.
Animals
ApoE−/− mice (stock number 002052, Jackson Laboratory, Bar Harbor, ME) were crossed with Par1−/− mice (stock number 00282, Jackson Laboratory) to obtain ApoE−/−:Par1−/− mice, and both of the strains were on C57BL/6 background. The F2 littermates were used in the study. Mice were bred and maintained according to the guidelines of the Institutional Animal Care and Use Facility of the University of Tennessee Health Science Center (Memphis, TN). Female and male C57BL/6, ApoE−/−, or ApoE−/−:Par1−/− mice fed with CD or WD (TD.88137, Envigo, Indianapolis, IN) for 16 weeks starting from 8 weeks of age were used. The institutional animal care and use committee of the University of Tennessee Health Science Center (Memphis, TN) approved all of the experiments involving animals.
Isolation of peritoneal macrophages
Mice were injected with 1 ml of 4% autoclaved thioglycolate intraperitoneally, and 4 days later, the animals were anesthetized with ketamine and xylazine, and the peritoneal lavage was collected in DMEM/F-12. Cells were plated at 3 × 105 cells/cm2 in DMEM containing 100 units/ml penicillin and 100 μg/ml streptomycin. After 3 h, the floating cells (mostly red blood cells) were removed by washing with cold PBS, and the adherent cells (macrophages) were used for the experiments.
Isolation of peripheral blood mononuclear cells (PBMCs)
ApoE−/− and ApoE−/−:Par1−/− mice were anesthetized with ketamine and xylazine. Blood was collected by cardiac puncture into BD Vacutainer K2 EDTA tubes and diluted with an equal volume of PBS. Blood was then overlaid on the Lymphoprep and centrifuged at 1500 rpm for 30 min at 4 °C. The PBMC layer was collected, washed with PBS, resuspended in PBS, and labeled with PKH67 green fluorescent cell tracker according to the manufacturer's instructions.
Isolation of aortic smooth muscle cells
Mouse aortic smooth muscle cells were isolated by collagenase II/elastase digestion; grown in DMEM/F-12 medium containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified CO2 incubator at 37 °C; and used between three and seven passages. The authenticity of SMCs was verified by SMα-actin staining.
Plasma lipid profiles
Blood was collected into BD Vacutainer Plus plasma tubes (catalog no. 367960, BD Biosciences) by cardiac puncture and centrifuged at 1,300 × g for 10 min at 4 °C to collect the plasma. Total plasma cholesterol, HDL, LDL, and triglyceride levels were measured by a Roche Diagnostics COBAS MIRA analyzer using the manufacturer's kits and protocol.
Cholesterol efflux assay
Peritoneal macrophages and smooth muscle cells were plated in 12-well plates at a density of 6 × 105 cells/well. Cells were incubated with [3H]cholesterol (1 μCi/ml) for 24 h followed by extensive washings with PBS. Cells were then equilibrated in serum-free DMEM containing 0.2% fatty acid-free BSA for 2 h. After equilibration, medium was replaced with fresh DMEM containing 0.2% fatty acid-free BSA and 10 μg/ml ApoA-I or HDL, and incubation was continued in the presence and absence of thrombin for 4 h at 37 °C. An aliquot of the efflux medium (100 μl) was removed for radioactivity determination. Cells were then rinsed with PBS and dried, and isopropyl alcohol was added for overnight extraction of cholesterol at room temperature. An aliquot of the extract (100 μl) was collected for radioactivity determination. Cholesterol efflux was expressed as a percentage of total cellular radioactivity released into the medium. Because both ApoA-I and HDL gave similar results in the initial experiments, we used ApoA-I throughout the rest of the study as cholesterol acceptor.
Transfections
Peritoneal macrophages were transfected with nontargeted control or Silencer Select siRNA at a final concentration of 100 nm using GeneSilencer reagent from Gentalis according to the manufacturer's instructions. In regard to plasmids, cells were transfected with plasmid DNAs at a final concentration of 2.5 μg/well in a 12-well culture plate or 5 μg/60-mm culture dish using GenePorter transfection reagent according to the manufacturer's instructions. After transfections, cells were recovered in complete medium overnight, growth-arrested for 12 h in serum-free medium, and used as required. Aortic smooth muscle cells were transfected with nontargeted control or Silencer Select siRNA at a final concentration of 100 nm using Lipofectamine 3000 transfection reagent according to the manufacturer's instructions. For plasmids, cells were transfected with plasmid DNAs at a final concentration of 2.5 μg/well in a 12-well culture plate or 5 μg/60-mm culture dish using Lipofectamine 3000 transfection reagent according to the manufacturer's instructions. After transfections, cells were recovered in complete medium overnight, growth-arrested for 24 h in serum-free medium, and used as required.
RT-PCR
Total cellular RNA was extracted from peritoneal macrophage cells and aortic smooth muscle cells using TRIzol reagent according to the manufacturer's protocol. Reverse transcription was performed with a high-capacity cDNA reverse transcription kit (Applied Biosystems). Complementary DNA was then used as a template for amplification using the following primers: mouse ABCA1 (NM_013454.3) forward (5′-CATTTCGAAGGAGACAAACATGTCA-3′) and reverse (5′-CATGGCTTTATTCGGAAAGTGGCAC-3′); mouse ABCG1 (NM_009593.2) forward (5′-AGAAGAAAGGATACAAGACCCTTTT-3′) and reverse (5′-CCCTTTCATGCCAGTCTCCCTGTAT-3′); mouse β-actin (NM_007393.5) forward (5′-AGCCATGTACGTAGCCATCC-3′) and reverse (5′-CTCTCAGCTGTGGTGGTGAA-3′). The amplification was performed using the Gene AMP PCR system 2400 (Applied Biosystems). The amplified PCR products were separated on 2% agarose gels and stained with ethidium bromide, and the pictures were captured using the Eastman Kodak Co. In-Vivo Imaging System (Rochester, NY).
Western blot analysis
After appropriate treatments, cell or tissue extracts were prepared and resolved by electrophoresis on 0.1% (w/v) SDS and 8 or 10% (w/v) polyacrylamide gels. The proteins were transferred electrophoretically onto a nitrocellulose membrane. After blocking in 5% (w/v) nonfat dry milk, the membrane was incubated with the appropriate primary antibody (1:1,000 dilution), followed by incubation with horseradish peroxidase–conjugated secondary antibody. The antigen-antibody complexes were detected with the enhanced chemiluminescence detection reagent kit (GE Healthcare).
Immunoprecipitation
After rinsing with cold PBS, cells were lysed in 250 μl of lysis buffer (PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/ml phenylmethylsulfonyl fluoride, 100 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm sodium orthovanadate) for 20 min on ice. The cell extracts were cleared by centrifugation at 12,000 rpm for 20 min at 4 °C. The cleared cell extracts containing an equal amount of protein from control and the indicated treatments were incubated with appropriate antibodies (1:100 dilution) overnight at 4 °C, followed by incubation with protein A/G-Sepharose beads for 2 h with gentle rocking. The beads were collected by centrifugation at 4,000 rpm for 1 min at 4 °C and washed four times with lysis buffer and once with PBS. The immunocomplexes were released by heating the beads in 40 μl of Laemmli sample buffer and analyzed by Western blotting for the indicated molecules using their specific antibodies.
Enface staining
Aortas were excised, cleared from fat, fixed in 4% paraformaldehyde, treated with 10% formalin, washed with 60% isopropyl alcohol, and stained with 0.5% Oil Red O. After staining, the aortas were rinsed with 60% isopropyl alcohol, and photographs were taken using a Nikon D7100 camera. The percentage of lesion surface area was measured using ImageJ (National Institutes of Health).
Aortic root sections
For immunohistochemistry and immunofluorescence staining, mice were perfused with 4% paraformaldehyde, and the hearts were collected. Sequential 10-μm aortic root sections were cut from the point of the appearance of the aortic valve leaflets with a Leica CM3050S cryostat machine (Leica Biosystems, Wetzlar, Germany) and used.
Oil Red O staining
After fixing with formalin, the aortic root sections were washed once with 60% isopropyl alcohol and stained with Oil Red O (0.5% in 60% isopropyl alcohol) for 15 min, followed by counterstaining with hematoxylin. The sections were observed under a Nikon Eclipse 50i microscope with ×4/NA 0.1, and the images were captured with a Nikon Digital Sight DS-L1 camera.
Immunofluorescence staining
The aortic root cross-sections were fixed with acetone/methanol (1:1) for 10 min; permeabilized in 0.2% Triton X-100 for 10 min; blocked with 5% goat serum in PBS containing 3% BSA for 1 h; incubated with mouse anti-human Par1, mouse anti-human ABCA1, or rabbit anti-human cullin 3 in combination with rat anti-human Mac3 or mouse anti-human SMCα-actin at 1:100 dilution, followed by incubation with Alexa Fluor 488–conjugated goat anti-rat, Alexa Fluor 488–conjugated goat anti-mouse, Alexa Fluor 568–conjugated goat anti-rabbit, or Alexa Fluor 568–conjugated goat anti-mouse secondary antibodies at 1:250 dilution; and counterstained with DAPI. To detect cholesterol content, the sections after incubation with the secondary antibodies were stained with filipin (0.05 mg/ml in PBS containing 3% BSA) for 2 h at room temperature. The sections were observed under a Zeiss inverted microscope (Zeiss Axiovision Observer.z1; magnification at ×10/NA 0.25), and the fluorescence images were captured with a Zeiss AxioCam MRm camera using the microscope operating software and image analysis software AxioVision version 4.7.2 (Carl Zeiss Imaging Solutions GmbH).
Leukocyte trafficking
ApoE−/− mice fed with WD for 3 months were anesthetized with ketamine and xylazine, and peripheral blood mononuclear cells isolated from ApoE−/− or ApoE−/−:Par1−/− mice were injected via tail vein (1.0 × 105 cells/mice). Twenty-four hours later, aortas were isolated, cleaned, fixed, opened, stained with DAPI, mounted on a slide with luminal side upward, and examined under a Zeiss inverted microscope (magnification at ×40/NA 0.16). The fluorescence images were captured with a Zeiss AxioCam MRm camera using the microscope operating software and image analysis software AxioVision version 4.7.2 (Carl Zeiss Imaging Solutions GmbH), and PKH67-positive cells were quantified.
Statistics
All of the experiments were repeated three times with similar results. Data are presented as the mean ± S.D. The treatment effects were analyzed by one-way analysis of variance or Student's t test, and the p values <0.05 were considered to be statistically significant. In the case of Western blotting and immunofluorescence analysis, one set of the representative data is presented.
Author contributions
S. R. performed cholesterol efflux, RT-PCR, Western blotting, isolation of mouse peritoneal macrophages and aortic smooth muscle cells, and Oil Red O staining; N. K. S. performed enface staining, Western blotting, and immunofluorescence; A. M. M. performed Western blot analysis; G. N. R. conceived the overall goal of the project, designed the experiments, interpreted the data, and wrote the manuscript.
This work was supported in part by National Institutes of Health Grant HL103575 (to G. N. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
S. Raghavan, N. K. Singh, S. Gali, A. M. Mani, and G. N. Rao, unpublished observations.
- EC
- endothelial cell
- SMC
- smooth muscle cell
- Par
- protease-activated receptor
- WD
- Western diet
- CD
- control diet
- HDL
- high-density lipoprotein
- LDL
- low-density lipoprotein
- DMEM
- Dulbecco's modified Eagle's medium
- PBMC
- peripheral blood mononuclear cell
- DAPI
- 4′,6-diamidino-2-phenylindole
- NA
- numerical aperture.
References
- 1. Maouche S., and Schunkert H. (2012) Strategies beyond genome-wide association studies for atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 170–181 10.1161/ATVBAHA.111.232652 [DOI] [PubMed] [Google Scholar]
- 2. Mach F., Schönbeck U., Sukhova G. K., Bourcier T., Bonnefoy J. Y., Pober J. S., and Libby P. (1997) Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 94, 1931–1936 10.1073/pnas.94.5.1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tall A. R., and Yvan-Charvet L. (2015) Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15, 104–116 10.1038/nri3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nègre-Salvayre A., Augé N., Camaré C., Bacchetti T., Ferretti G., and Salvayre R. (2017) Dual signaling evoked by oxidized LDLs in vascular cells. Free Radic. Biol. Med. 106, 118–133 10.1016/j.freeradbiomed.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 5. Tesauro M., Schinzari F., Iantorno M., Rizza S., Melina D., Lauro D., and Cardillo C. (2005) Ghrelin improves endothelial function in patients with metabolic syndrome. Circulation 112, 2986–2992 [DOI] [PubMed] [Google Scholar]
- 6. Gerszten R. E., Garcia-Zepeda E. A., Lim Y. C., Yoshida M., Ding H. A., Gimbrone M. A. Jr., Luster A. D., Luscinskas F. W., and Rosenzweig A. (1999) MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 398, 718–723 10.1038/19546 [DOI] [PubMed] [Google Scholar]
- 7. Rahaman S. O., Zhou G., and Silverstein R. L. (2011) Vav protein guanine nucleotide exchange factor regulates CD36 protein-mediated macrophage foam cell formation via calcium and dynamin-dependent processes. J. Biol. Chem. 286, 36011–36019 10.1074/jbc.M111.265082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alexander M. R., and Owens G. K. (2012) Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 74, 13–40 10.1146/annurev-physiol-012110-142315 [DOI] [PubMed] [Google Scholar]
- 9. Chellan B., Reardon C. A., Getz G. S., and Hofmann Bowman M. A. (2016) Enzymatically modified low-density lipoprotein promotes foam cell formation in smooth muscle cells via macropinocytosis and enhances receptor-mediated uptake of oxidized low-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 36, 1101–1113 10.1161/ATVBAHA.116.307306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ricciarelli R., Zingg J. M., and Azzi A. (2000) Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 102, 82–87 10.1161/01.CIR.102.1.82 [DOI] [PubMed] [Google Scholar]
- 11. Rauch B. H., Millette E., Kenagy R. D., Daum G., Fischer J. W., and Clowes A. W. (2005) Syndecan-4 is required for thrombin-induced migration and proliferation in human vascular smooth muscle cells. J. Biol. Chem. 280, 17507–17511 10.1074/jbc.M410848200 [DOI] [PubMed] [Google Scholar]
- 12. Wang D., Paria B. C., Zhang Q., Karpurapu M., Li Q., Gerthoffer W. T., Nakaoka Y., and Rao G. N. (2009) A role for Gab1/SHP2 in thrombin activation of PAK1: gene transfer of kinase-dead PAK1 inhibits injury-induced restenosis. Circ. Res. 104, 1066–1075 10.1161/CIRCRESAHA.109.196691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coughlin S. R. (2005) Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 3, 1800–1814 10.1111/j.1538-7836.2005.01377.x [DOI] [PubMed] [Google Scholar]
- 14. Mitropoulos K. A. (1994) Lipoprotein metabolism and thrombosis. Curr. Opin. Lipidol. 5, 227–235 10.1097/00041433-199405030-00011 [DOI] [PubMed] [Google Scholar]
- 15. Wilcox J. N., Noguchi S., Casanova J. R., and Rasmussen M. E. (2001) Extrahepatic synthesis of FVII in human atheroma and smooth muscle cells in vitro. Ann. N.Y. Acad. Sci. 947, 433–438 [DOI] [PubMed] [Google Scholar]
- 16. Borissoff J. I., Heeneman S., Kilinç E., Kassák P., Van Oerle R., Winckers K., Govers-Riemslag J. W., Hamulyák K., Hackeng T. M., Daemen M. J., ten Cate H., and Spronk H. M. (2010) Early atherosclerosis exhibits an enhanced procoagulant state. Circulation 122, 821–830 10.1161/CIRCULATIONAHA.109.907121 [DOI] [PubMed] [Google Scholar]
- 17. Borissoff J. I., Spronk H. M., and ten Cate H. (2011) The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 364, 1746–1760 10.1056/NEJMra1011670 [DOI] [PubMed] [Google Scholar]
- 18. Esmon C. T. (2014) Targeting factor Xa and thrombin: impact on coagulation and beyond. Thromb. Haemost. 111, 625–633 10.1160/TH13-09-0730 [DOI] [PubMed] [Google Scholar]
- 19. Kopec A. K., Joshi N., Towery K. L., Kassel K. M., Sullivan B. P., Flick M. J., and Luyendyk J. P. (2014) Thrombin inhibition with dabigatran protects against high-fat diet-induced fatty liver disease in mice. J. Pharmacol. Exp. Ther. 351, 288–297 10.1124/jpet.114.218545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leger A. J., Covic L., and Kuliopulos A. (2006) Protease-activated receptors in cardiovascular diseases. Circulation 114, 1070–1077 10.1161/CIRCULATIONAHA.105.574830 [DOI] [PubMed] [Google Scholar]
- 21. Austin K. M., Nguyen N., Javid G., Covic L., and Kuliopulos A. (2013) Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis. J. Biol. Chem. 288, 23105–23115 10.1074/jbc.M113.467019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyen K. T., Frye S. R., Eskin S. G., Patterson C., Runge M. S., and McIntire L. V. (2001) Cyclic strain increases protease-activated receptor-1 expression in vascular smooth muscle cells. Hypertension 38, 1038–1043 10.1161/hy1101.092840 [DOI] [PubMed] [Google Scholar]
- 23. Sanchez C., Poggi M., Morange P. E., Defoort C., Martin J. C., Tanguy S., Dutour A., Grino M., and Alessi M. C. (2012) Diet modulates endogenous thrombin generation, a biological estimate of thrombosis risk, independently of the metabolic status. Arterioscler. Thromb. Vasc. Biol. 32, 2394–2404 10.1161/ATVBAHA.112.250332 [DOI] [PubMed] [Google Scholar]
- 24. Kalz J., ten Cate H., and Spronk H. M. (2014) Thrombin generation and atherosclerosis. J. Thromb. Thrombolysis 37, 45–55 10.1007/s11239-013-1026-5 [DOI] [PubMed] [Google Scholar]
- 25. Sorci-Thomas M. G., and Thomas M. J. (2016) Microdomains, inflammation, and atherosclerosis. Circ. Res. 118, 679–691 10.1161/CIRCRESAHA.115.306246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McLaren J. E., Michael D. R., Ashlin T. G., and Ramji D. P. (2011) Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog. Lipid Res. 50, 331–347 10.1016/j.plipres.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 27. Zhou Y. F., Guetta E., Yu Z. X., Finkel T., and Epstein S. E. (1996) Human cytomegalovirus increases modified low density lipoprotein uptake and scavenger receptor mRNA expression in vascular smooth muscle cells. J. Clin. Invest. 98, 2129–2138 10.1172/JCI119019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crucet M., Wüst S. J., Spielmann P., Lüscher T. F., Wenger R. H., and Matter C. M. (2013) Hypoxia enhances lipid uptake in macrophages: role of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis 229, 110–117 10.1016/j.atherosclerosis.2013.04.034 [DOI] [PubMed] [Google Scholar]
- 29. Dubland J. A., and Francis G. A. (2016) So much cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr. Opin Lipidol. 27, 155–161 10.1097/MOL.0000000000000279 [DOI] [PubMed] [Google Scholar]
- 30. Yvan-Charvet L., Wang N., and Tall A. R. (2010) Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 30, 139–143 10.1161/ATVBAHA.108.179283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Westerterp M., Tsuchiya K., Tattersall I. W., Fotakis P., Bochem A. E., Molusky M. M., Ntonga V., Abramowicz S., Parks J. S., Welch C. L., Kitajewski J., Accili D., and Tall A. R. (2016) Deficiency of ATP-binding cassette transporters A1 and G1 in endothelial cells accelerates atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 36, 1328–1337 10.1161/ATVBAHA.115.306670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yvan-Charvet L., Welch C., Pagler T. A., Ranalletta M., Lamkanfi M., Han S., Ishibashi M., Li R., Wang N., and Tall A. R. (2008) Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 118, 1837–1847 10.1161/CIRCULATIONAHA.108.793869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jennings L. K. (2009) Role of platelets in atherothrombosis. Am. J. Cardiol. 103, 4A–10A 10.1016/j.amjcard.2008.11.017 [DOI] [PubMed] [Google Scholar]
- 34. Feng F. Y., Varambally S., Tomlins S. A., Chun P. Y., Lopez C. A., Li X., Davis M. A., Chinnaiyan A. M., Lawrence T. S., and Nyati M. K. (2007) Role of epidermal growth factor receptor degradation in gemcitabine-mediated cytotoxicity. Oncogene 26, 3431–3439 10.1038/sj.onc.1210129 [DOI] [PubMed] [Google Scholar]
- 35. Kimura T., Takabatake Y., Takahashi A., and Isaka Y. (2013) Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 73, 3–7 10.1158/0008-5472.CAN-12-2464 [DOI] [PubMed] [Google Scholar]
- 36. Coughlin S. R. (2000) Thrombin signalling and protease-activated receptors. Nature 407, 258–264 10.1038/35025229 [DOI] [PubMed] [Google Scholar]
- 37. Pelham C. J., Ketsawatsomkron P., Groh S., Grobe J. L., de Lange W. J., Ibeawuchi S. R., Keen H. L., Weatherford E. T., Faraci F. M., and Sigmund C. D. (2012) Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/Rho-kinase. Cell Metab. 16, 462–472 10.1016/j.cmet.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boyden L. M., Choi M., Choate K. A., Nelson-Williams C. J., Farhi A., Toka H. R., Tikhonova I. R., Bjornson R., Mane S. M., Colussi G., Lebel M., Gordon R. D., Semmekrot B. A., Poujol A., Välimäki M. J., et al. (2012) Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482, 98–102 10.1038/nature10814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mathew R., Seiler M. P., Scanlon S. T., Mao A. P., Constantinides M. G., Bertozzi-Villa C., Singer J. D., and Bendelac A. (2012) BTB-ZF factors recruit the E3 ligase cullin 3 to regulate lymphoid effector programs. Nature 491, 618–621 10.1038/nature11548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Angers S., Thorpe C. J., Biechele T. L., Goldenberg S. J., Zheng N., MacCoss M. J., and Moon R. T. (2006) The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-β-catenin pathway by targeting Dishevelled for degradation. Nat. Cell Biol. 8, 348–357 10.1038/ncb1381 [DOI] [PubMed] [Google Scholar]
- 41. Gadepalli R., Singh N. K., Kundumani-Sridharan V., Heckle M. R., and Rao G. N. (2012) Novel role of proline-rich nonreceptor tyrosine kinase 2 in vascular wall remodeling after balloon injury. Arterioscler. Thromb. Vasc. Biol. 32, 2652–2661 10.1161/ATVBAHA.112.253112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gadepalli R., Kotla S., Heckle M. R., Verma S. K., Singh N. K., and Rao G. N. (2013) Novel role for p21-activated kinase 2 in thrombin-induced monocyte migration. J. Biol. Chem. 288, 30815–30831 10.1074/jbc.M113.463414 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Martorell L., Martínez-González J., Rodríguez C., Gentile M., Calvayrac O., and Badimon L. (2008) Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb. Haemost. 99, 305–315 [DOI] [PubMed] [Google Scholar]
- 44. Samad F., and Ruf W. (2013) Inflammation, obesity, and thrombosis. Blood 122, 3415–3422 10.1182/blood-2013-05-427708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 10.1038/nature08938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ogura M., Ayaori M., Terao Y., Hisada T., Iizuka M., Takiguchi S., Uto-Kondo H., Yakushiji E., Nakaya K., Sasaki M., Komatsu T., Ozasa H., Ohsuzu F., and Ikewaki K. (2011) Proteasomal inhibition promotes ATP-binding cassette transporter A1 (ABCA1) and ABCG1 expression and cholesterol efflux from macrophages in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 31, 1980–1987 10.1161/ATVBAHA.111.228478 [DOI] [PubMed] [Google Scholar]
- 47. Aleidi S. M., Howe V., Sharpe L. J., Yang A., Rao G., Brown A. J., and Gelissen I. C. (2015) The E3 ubiquitin ligases, HUWE1 and NEDD4-1, are involved in the post-translational regulation of the ABCG1 and ABCG4 lipid transporters. J. Biol. Chem. 290, 24604–24613 10.1074/jbc.M115.675579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hsieh V., Kim M. J., Gelissen I. C., Brown A. J., Sandoval C., Hallab J. C., Kockx M., Traini M., Jessup W., and Kritharides L. (2014) Cellular cholesterol regulates ubiquitination and degradation of the cholesterol export proteins ABCA1 and ABCG1. J. Biol. Chem. 289, 7524–7536 10.1074/jbc.M113.515890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Plump A. S., Smith J. D., Hayek T., Aalto-Setälä K., Walsh A., Verstuyft J. G., Rubin E. M., and Breslow J. L. (1992) Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 71, 343–353 10.1016/0092-8674(92)90362-G [DOI] [PubMed] [Google Scholar]
- 50. Allahverdian S., Chehroudi A. C., McManus B. M., Abraham T., and Francis G. A. (2014) Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129, 1551–1559 10.1161/CIRCULATIONAHA.113.005015 [DOI] [PubMed] [Google Scholar]
- 51. Higashi Y., Sukhanov S., Shai S. Y., Danchuk S., Tang R., Snarski P., Li Z., Lobelle-Rich P., Wang M., Wang D., Yu H., Korthuis R., and Delafontaine P. (2016) Insulin-like growth factor-1 receptor deficiency in macrophages accelerates atherosclerosis and induces an unstable plaque phenotype in apolipoprotein E-deficient mice. Circulation 133, 2263–2278 10.1161/CIRCULATIONAHA.116.021805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Camera M., Brambilla M., Facchinetti L., Canzano P., Spirito R., Rossetti L., Saccu C., Di Minno M. N., and Tremoli E. (2012) Tissue factor and atherosclerosis: not only vessel wall-derived TF, but also platelet-associated TF. Thromb. Res. 129, 279–284 10.1016/j.thromres.2011.11.028 [DOI] [PubMed] [Google Scholar]
- 53. Borissoff J. I., Otten J. J., Heeneman S., Leenders P., van Oerle R., Soehnlein O., Loubele S. T., Hamulyák K., Hackeng T. M., Daemen M. J., Degen J. L., Weiler H., Esmon C. T., van Ryn J., Biessen E. A., Spronk H. M., and ten Cate H. (2013) Genetic and pharmacological modifications of thrombin formation in apolipoprotein e-deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil-dependent manner. PLoS One 8, e55784 10.1371/journal.pone.0055784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ten Cate H. (2012) Tissue factor-driven thrombin generation and inflammation in atherosclerosis. Thromb. Res. 129, S38–S40 10.1016/j.thromres.2012.02.028 [DOI] [PubMed] [Google Scholar]
- 55. Borissoff J. I., Joosen I. A., Versteylen M. O., Spronk H. M., ten Cate H., and Hofstra L. (2012) Accelerated in vivo thrombin formation independently predicts the presence and severity of CT angiographic coronary atherosclerosis. JACC Cardiovasc. Imaging 5, 1201–1210 10.1016/j.jcmg.2012.01.023 [DOI] [PubMed] [Google Scholar]