

Abstract
Innate immunity is critical in the early containment of influenza A virus (IAV) infection, and surfactant protein D (SP-D) plays a crucial role in the pulmonary defense against IAV. In pigs, which are important intermediate hosts during the generation of pandemic IAVs, SP-D uses its unique carbohydrate recognition domain (CRD) to interact with IAV. An N-linked CRD glycosylation provides interactions with the sialic acid–binding site of IAV, and a tripeptide loop at the lectin-binding site facilitates enhanced interactions with IAV glycans. Here, to investigate both mechanisms of IAV neutralization in greater detail, we produced an N-glycosylated neck-CRD fragment of porcine SP-D (RpNCRD) in HEK293 cells. X-ray crystallography disclosed that the N-glycan did not alter the CRD backbone structure, including the lectin site conformation, but revealed a potential second nonlectin-binding site for glycans. IAV hemagglutination inhibition, IAV aggregation, and neutralization of IAV infection studies showed that RpNCRD, unlike the human analogue RhNCRD, exhibits potent neutralizing activity against pandemic A/Aichi/68 (H3N2), enabled by both porcine-specific structural features of its CRD. MS analysis revealed an N-glycan site-occupancy of >98% at Asn-303 of RpNCRD with complex-type, heterogeneously branched and predominantly α(2,3)-sialylated oligosaccharides. Glycan-binding array data characterized both RpNCRD and RhNCRD as mannose-type lectins. RpNCRD also bound LewisY structures, whereas RhNCRD bound polylactosamine-containing glycans. The presence of the N-glycan in the CRD increases the glycan-binding specificity of RpNCRD. These insights increase our understanding of porcine-specific innate defense against pandemic IAV and may inform the design of recombinant SP-D–based antiviral drugs.
Keywords: host–pathogen interaction, innate immunity, influenza virus, collectin, N-linked glycosylation, sialic acid, antiviral agent, structural biology, drug design, viral immunology, porcine immunology, pulmonary defense, recombinant protein, surfactant protein D
Introduction
Influenza is a highly contagious respiratory disease, caused by influenza A viruses (IAVs),2 that affects a wide range of natural hosts, including humans, pigs, and birds. On average, influenza infects 5–10% of the human population annually (1). Pigs are known to be highly susceptible to infection by both avian and human IAVs, and co-infection can generate novel pandemic viruses as illustrated by the most recent pandemic of 2009 (2, 3). Understanding the molecular mechanisms that underlie the role of swine as important intermediary hosts has become an increasingly relevant question in influenza virus research. Infection, maintenance, and adaptation of IAVs in host organisms are mainly determined by the (early) innate immune response that is considered fundamental for early containment of infection. Several soluble innate immune inhibitors contribute to immediate protection against respiratory infections (4). Lung collectin surfactant protein D (SP-D), in particular, expresses strong antiviral activity against IAVs as demonstrated by in vitro and in vivo studies (5, 6). This collagenous C-type lectin is constitutively expressed in the airways where it can interact directly with inhaled IAVs, resulting in viral aggregation and blocking of IAV attachment to epithelial cells, thereby protecting against IAV infection. In addition, SP-D acts as an opsonin and modulates the phagocytic activity of alveolar macrophages and neutrophils that contributes to more efficient clearance of IAV particles (7).
SP-D is a cruciform-shaped four-armed dodecameric glycoprotein (8), and each collagenous trimeric arm clusters three Ca2+-dependent mannose-type C-type lectin domains (carbohydrate recognition domains, CRDs). These domains facilitate multivalent, high-affinity interactions with patterns of high-mannose asparagine-linked glycans present on the hemagglutinin (HA) of IAV (9, 10). HA is the spike protein responsible for viral attachment through its sialic acid (SA) receptor–binding site to SA-rich glycoproteins on host cells, and therefore is an important virulence factor of IAV. In contrast to Ca2+-dependent viral glycan–targeting β-inhibitors like SP-D, another class of so-called “γ-inhibitors” acts in a Ca2+-independent manner via presentation of SA ligands to the HA receptor–binding site. This interaction interferes with the receptor-mediated attachment of the virus to SA moieties expressed on the surface of host epithelial cells. These so-called γ-inhibitors include the sialylated collectins surfactant protein A (SP-A) and lung glycoprotein-340. These collectins, in contrast to SP-D, are capable of neutralizing poorly glycosylated IAVs like A/Puerto Rico/8/34(H1N1) (PR-8) and pandemic IAVs, strains that typically lack N-linked glycans on the HA head (11–13).
Interestingly, previous studies showed that full-length porcine SP-D (pSP-D) expresses substantially stronger anti-IAV activity as compared with SP-Ds from other animal species, including human SP-D (hSP-D) (12). This is mainly due to two unique structural features present in the CRD of pSP-D. First, similar to SP-A, pSP-D has a highly sialylated N-linked glycan on its CRD at Asn-303 (Fig. 1) that generates a dual mechanism of interaction with IAV, turning pSP-D into a γ-inhibitor as well as a β-inhibitor against IAV (14). Second, sequence alignment of the CRDs of all characterized SP-Ds revealed the presence of three extra amino acids near the carbohydrate-binding site of the CRD in pSP-D, located between residues Gly-326 and Gly-327 of the hSP-D sequence (Gly-326A–Ser-326B–Ser-326C or “GSS-loop”; Fig. 1) (15). X-ray crystallographic analysis, docking studies with octamannose, and molecular dynamics simulations were performed with a recombinant trimeric fragment containing only the neck and CRD domain (NCRD) of pSP-D (RpNCRD), lacking the N-linked glycan (RpNCRD–dNG). These studies provided insight into the three-dimensional structure of the CRD from pSP-D complexed with or without d-mannose (16). The combined data suggested that the 3-amino acid insertion alters the lectin site conformation, shaping a flexible extended loop that potentially facilitates recognition of distal portions of highly-branched mannose-rich oligosaccharides like those expressed on the IAV surface. Previous in vitro studies with IAV indeed showed that pSP-D displays profound inhibitory activity against IAV due to both distinct structural features outlined above (16).
Figure 1.

Sequence alignment of neck-CRD region of SP-Ds and rSP-A. Shown is the CRD of pSP-D aligned with SP-Ds from other animal species and rat SP-A. Amino acid sequence alignment of the CRDs of porcine SP-D (pSP-D, GenBankTM accession code AF132496) with human SP-D (hSP-D, X65018), bovine SP-D (bSP-D, X75911), mouse SP-D (mSP-D, L40156), rat SP-D (rSP-D, M81231), and rat SP-A (rSP-A, U43092) is shown. Residue numbering according to hSP-D sequence and dashes indicate spaces that are inserted to maximize the identity across the alignment. The amino acid residues for pSP-D are shown in bold and also include the residues that comprise the neck region (underlined, residues 203–235). Dots indicate residues in common with the porcine sequence. The consensus sequence of all SP-Ds (conSP-D) is given in italics, and the secondary structural regions are indicated as gray arrows (β-sheets) and dark gray bars (α-helices) below the conSP-D sequence, deduced from the crystal structure of rat SP-A. The N-glycosylation sites are indicated by boxed triplet sequences. The indicated N303Q substitution at position 303 results in the deglycosylated RpNCRD–dNG. The three amino acid insertions found in the CRD of pSP-D are highlighted in black. SP-D groove regions (boxed in gray), cysteine bridge (dotted line), and key residues necessary for coordination of Ca2+ ions and hydroxyl groups of oligosaccharides (asterisks) are indicated.
As the next challenge, we aimed to investigate the native, N-glycosylated neck-CRD fragment of pSP-D (RpNCRD) to dissect the dual mechanism of interaction between pSP-D and IAV in more detail. Given the susceptibility of pigs for IAV co-infection and reassortment that can cause pandemics in humans, we also investigated the antiviral potential of RpNCRD against pandemic IAV. These experiments were also performed to assess the potential of RpNCRD-based novel antivirals to reduce disease severity in humans during future pandemic outbreaks of IAV. To this end, we used the HEK293 expression system to generate a fully functional mammalian N-glycosylated RpNCRD, studied its molecular structure by X-ray crystallography, and compared it with that of RpNCRD–dNG and RhNCRD. IAV neutralization studies revealed that RpNCRD, in contrast to RhNCRD, exhibited potent neutralizing activity against pandemic IAV (A/Aichi/68(H3N2); Aichi68), mediated by the CRD as well as the N-linked glycan present on RpNCRD. Given the contribution of the N-linked glycan on RpNCRD to its viral neutralization activity, we also characterized the location, site-occupancy, and composition of the N-glycan in RpNCRD. Furthermore, glycan-binding array studies were performed to characterize the CRD-binding characteristics of recombinant full-length pSP-D (RpSP-D) and the human analogue RhSP-D. The binding studies were also carried out with RpSP-D–dNG to study the impact of N-glycosylation of the CRD on the carbohydrate-binding properties of RpSP-D.
Results
Crystallographic analysis of RpNCRD
The structure of RpNCRD is very similar to that previously determined for the nonglycosylated pig SP-D, RpNCRD–dNG (16), and both are similar to RhNCRD with the exception of the GSS-insertion sequence in the long loop (Fig. 2). Superimposition of RpNCRD and RpNCRD–dNG results in a root-mean-square-deviation (r.m.s.d.) of 0.25 and 0.24 Å, respectively, calculated using 155 and 154 α-carbons. No major rearrangements in the trimeric structure are noted, which is evident from the similarity of the unit cell dimensions of the crystals. Therefore, the impact of N-glycosylation on the global structure of RpNCRD is minimal. The initial crystals were grown in the presence of glycerol, which can act as a ligand for SP-D at the high concentrations used in the growth solutions. Therefore, an attempt was made to remove the glycerol by soaking crystals in solutions lacking glycerol before data collection (Table 1), with the expectation of observing an unliganded lectin site. Surprisingly, the resulting maps show clear electron density for the first seven sugars of a biantennary complex–type glycan (Fig. 3B) from a neighboring molecule in the crystal (Fig. 3A). On each antenna, there is density present for a single mannose (MAN503A and MAN503B) and one GlcNAc (NAG504A and NAG504B). There is neither density for galactose or SAs nor other antennae, which could be present in the structure but disordered. The glycan makes several interactions with the CRD through the sugars of the antennae (Fig. 3C). MAN503A is bound in a similar way as the mannose in the previously published complex of RpNCRD–dNG (16), making canonical lectin site Ca2+ coordination interactions with the vicinal equatorial diol formed by its O3 and O4 atoms, as well as hydrogen bonds to the side chains of Asn-323 and Asn-341. MAN503A also forms, through its O6 atom. There are additional interactions between the CRD and the glycan in the structure. NAG504A, the next sugar in the chain after MAN503A, forms a hydrogen bond to the side chain of Asn-325 through its N-acetyl carbonyl group. NAG504B, the terminal sugar on the other antenna, forms a hydrogen bond to the side chain amino group of Lys-343 through its O6 atom. Finally, MAN503B makes several interactions with the CRD, where the O3 and O4 atoms make three hydrogen bonds with the side chains of Glu-333 and Arg-349.
Figure 2.
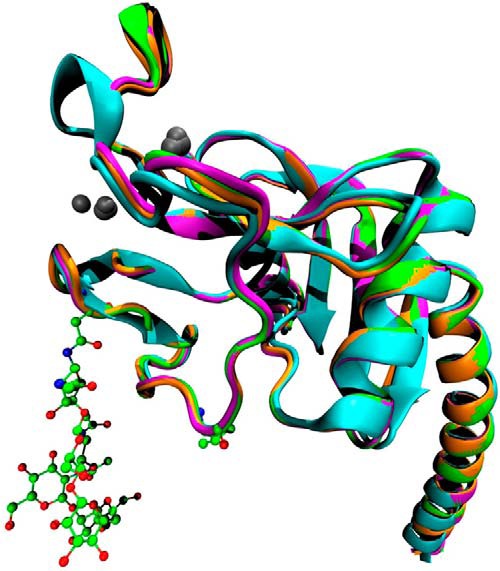
Tertiary protein structure of RpNCRD. Shown is a ribbon diagram of the superimposition of the RhNCRD (cyan), RpNCRD–dNG (magenta), RpNCRD (green), and RpNCRD complexed with glycerol (orange). Asn-303 and Val-313 are shown in ball-and-stick to indicate the locations of the N-glycosylation sites from pSP-D (with glycans shown) and rat SP-A (52), respectively. Ca2+ ions are represented as gray spheres.
Table 1.
Data collection and refinement statistics of RpNCRD crystal structure
The number in parentheses is the value for reflections in the outermost shell. The average B factor for protein atoms is calculated with the isotropic contribution to the B factor only, ignoring the contribution of the TLS terms.
| Parameter | Value |
|
|---|---|---|
| Glycerol complex | Glycerol soaked out | |
| Data collection | ||
| Space group | P63 | P63 |
| Unit cell dimensions | a = b = 66.798 Å | a = b = 66.015 Å |
| c = 64.974 Å | c = 64.658 Å | |
| Resolution | 15–1.9 Å (1.97–1.90 Å) | 15–1.9 Å (1.97–1.90 Å) |
| No. of reflections | 12,304 | 12,220 |
| Data cutoff | I ≤ −3σ | I ≤ −3σ |
| I/σ(I) | 23.6 (3.2) | 25.1 (4.5) |
| Completeness | 94.0% (67.4%) | 96.0% (82.2%) |
| Redundancy | 4.7 (2.8) | 6.5 (4.5) |
| Rmerge | 0.058 (0.356) | 0.063 (0.376) |
| Refinement | ||
| Rwork | 0.201 | 0.223 |
| Rfree | 0.247 | 0.250 |
| No. of protein atoms | 1216 | 1213 |
| No. of waters | 93 | 86 |
| No. of other atoms | 72 | 107 |
| Average B factors | ||
| Protein | 46.3 | 45.6 |
| Waters | 48.1 | 50.0 |
| Sugars/glycerol/calcium | 63.1 | 66.5 |
| r.m.s.d. from ideal values | ||
| Bond lengths (Å) | 0.014 | 0.013 |
| Bond angles (°) | 1.085 | 0.805 |
| Chiral volume (Å3) | 0.069 | 0.051 |
| Ramachandran plot | ||
| Favored | 98.71% | 98.06% |
| Allowed | 1.29% | 1.94% |
| Outliers | 0.00% | 0.00% |
Figure 3.

Electron density of the RpNCRD structure. A, electron density map calculated using Fourier coefficients 2Fobs − Fcalc of the final refined model showing the glycan structure in the backsoaked RpNCRD crystal structure. The map shows clear density for seven saccharide residues linked to Asn-303 from a neighboring molecule in the crystal. Sugar residue Man503B is bound in the lectin site, where it makes typical coordinating interactions with the lectin site Ca2+ through the 3- and 4-hydroxyl groups of the mannose ring. B, schematic diagram of the N-glycan with GlcNAc residues (NAG) shown as blue squares and α-d-mannose residues (MAN) and β-d-mannose residues (BMA) as green circles. C, interaction map is shown between the glycan and residues on the RpNCRD within 5 Å of the glycan. Both Asn-303 and the glycans belong to a neighboring molecule in the crystal from the other indicated residues.
Comparison of the RpNCRD structure (i.e. derived from crystals in which glycerol had been soaked out prior to data collection) with the RpNCRD–glycerol complex structure shows that the presence of the glycerol molecule in the lectin site (glycerol is present in the crystallization medium at a concentration of about 2 m) displaces one glycan antenna (MAN503A and NAG504A) from the lectin site by binding to the lectin site Ca2+. These sugars are not seen in the resulting electron density maps and are most likely disordered. However, the other antenna is observed in the back-soaked structure where it makes similar interactions with residues Lys-343, Gln-333, and Arg-349. The conservation of these interactions suggests that these residues may bind these same sugars in complexes of SP-D with biological substrates.
Neutralization of pandemic IAV by RpNCRD, RpNCRD–dNG, and RhNCRD
To determine the contribution of the N-linked glycan present in the CRD of pSP-D to interactions with pandemic IAV, we first determined differences in viral aggregation of Aichi68 by RpNCRD as compared with RpNCRD–dNG (Fig. 4A). Although incubation of virus in buffer alone did not result in any aggregate formation, RpNCRD was able to aggregate Aichi68, and loss of the N-glycan resulted in a significant drop in viral aggregation (RpNCRD–dNG). These differences in binding and aggregation of Aichi68 were underlined by assessing the HA potency of the obtained aggregates by adding erythrocytes to aliquots of these mixtures (Fig. 4B). Absence of aggregation results in free viral particles that agglutinate RBCs, resulting in high viral HA titers by such samples (control, highest hemagglutination). In contrast, samples obtained after viral aggregation in the presence of increasing amounts of RpNCRDs (2.0–4.0–8.0 μg/ml) showed a dose-dependent decrease in HA for both preparations. Importantly, the RpNCRD samples from the aggregation assay were significantly less potent in blunting HA activity as compared with RpNCRD–dNG, indicating that the N-glycan contributed to a more potent interaction between the NCRD of pSP-D and Aichi68. RhNCRD, in contrast to RpNCRD or RpNCRD–dNG, is not able to aggregate Aichi68, in agreement with what had been previously published (17).
Figure 4.

Aggregation of Aichi68 by RpNCRDs followed by HA titration. A, aggregation of Aichi68 viral particles caused by 4.0 μg/ml RpNCRDs (virus only as negative control). Results shown are means ± S.E. of six experiments and indicate changes in light transmission through stirred suspensions of influenza A virus particles (0–12 min). B, aliquots (10 μl) from samples obtained after completion of Aichi68 aggregation (12 min) in the presence of 0, 2.0, 4.0, or 8.0 μg/ml RpNCRD or RpNCRD–dNG were serially diluted, and hemagglutination was measured 2 h after addition of RBCs. Result is expressed as number of wells with increasing dilutions of aggregation sample that still showed viral HA. Statistical analysis was performed with the unpaired Student's t test; *, p < 0.05; **, p < 0.005 compared with RpNCRD.
More evidence for involvement of the N-glycan in RpNCRD in binding pandemic IAV and for the differences in antiviral activity between RhNCRD and RpNCRD–dNG (both lacking the N-glycan thus revealing differences in species-specific lectin–mediated interactions only) was obtained by HAI and neutralization of infectivity (Fig. 5, A and B, respectively). The HAI assay showed that whereas RhNCRD was very weak in inhibiting Aichi68, RpNCRD was significantly stronger in HAI activity as compared with both other preparations (RpNCRD > RpNCRD–dNG > RhNCRD). The same order of neutralizing potency was illustrated by neutralization of Aichi68 infectivity of MDCK cells. A dose-dependent decrease in infectivity was demonstrated for all three preparations, whereas virus neutralization by RpNCRD was significantly stronger compared with RhNCRD (at all concentrations tested) and with RpNCRD–dNG (10 μg/ml).
Figure 5.

HA inhibition and neutralization of infectivity of Aichi68 by RpNCRDs and RhNCRD. A, SP-D NCRD-mediated inhibition of HA by Aichi68. Serially diluted NCRDs (highest concentration tested was 10 μg/ml) were mixed with 4 hemagglutinating units of virus, incubated for 10 min at room temperature, followed by addition of RBCs and incubated for 2 h at room temperature. Results shown are minimal concentrations of NCRDs required to fully inhibit HA by Aichi68 and calculated from four different experiments. B, focus assay showing inhibition of Aichi68 infection of MDCK cells by SP-D NCRDs. Aichi68 virus was pre-incubated for 30 min at 37 °C with increasing amounts (0–40 μg/ml) of NCRDs and subsequently applied to monolayers of MDCK cells. After 7 h at 37 °C, the cells were stained for infectious foci using anti-nucleoprotein antibodies and fluorescence detection, and results were expressed as a percentage of control foci obtained in the presence of virus only (n = 4). Statistical analysis was performed with the unpaired Student's t test; *, p < 0.05; **, p < 0.005 compared with RpNCRD.
Characterization of the N-glycan in RpNCRD
The presence of an N-linked glycan at Asn-303 of RpNCRD was determined by analysis of glycopeptide tandem mass spectra (Fig. 6). Isolated precursor ion 1063.1666(4+) could be matched to the intact glycopeptide mass of SMTDIKTEGNF–Hex6HexNAc5dHex1NeuAc3. Upon collisional dissociation, the precursor generated low mass oxonium ions corresponding to protonated mono-, di-, and trisaccharide ions. These ions confirmed the presence of glycosylation on the selected precursor ion. Precursor identity was confirmed by peptide backbone ions, naked intact peptide ion, and peptide + saccharide ions (so-called stub glycopeptide ions), as described previously (18).
Figure 6.

Tandem mass spectrum (product-ion spectrum) for chymotryptic pSP-D precursor m/z 1063.1666(4+) corresponding to glycopeptide SMTDIKTEGNF-Hex6HexNAc5dHex1NeuAc3. Analysis was performed on a Q-TOF mass spectrometer using collisional dissociation. Inset shows the same spectrum zoomed in to better visualize the low abundance spectral peaks in the 500–2900 m/z range. Symbolic representation of glycans is as specified by Consortium for Functional Glycomics (http://www.functionalglycomics.org/static/consortium/Nomenclature.shtml).3 Glycan topology shown is speculative based on inferred glycan compositions. Peptide backbone product ions are labeled using standard mass spectrometric notation (53, 54). Such product ions containing the N-glycan core HexNAc unit are indicated. Product ions resulting from losses of monosaccharides from the precursor ion are given as Pep + monosaccharide composition. Glycan oxonium ions (55) are visualized according to the Symbol Nomenclature for Glycans (39). Abbreviations used are as follows: Hex, hexoses; HexNac, N-acetylhexosamines; dHex, deoxyhexoses; NeuAc, N-acetylneuraminic acid; NeuGc, N-glycolylneuraminc acid.
For site-occupancy analysis, peak areas for extracted ion chromatograms of labeled and unlabeled peptides, identified using database searches, were integrated and compared with calculate the site occupancy (Table 2). One of the considerations with identification of the site of N-glycosylation was that some of the asparagine residues may spontaneously undergo deamidation to form aspartic acid or iso-aspartic acid. If present, this reaction reduces the mass difference between the unglycosylated and the deglycosylated peptides (18O) to 2 Da instead of 3 Da. The rate of deamidation increases with pH and is highest for sequence motifs of “NG.” Considering the possibility for deamidation, chromatograms were extracted for masses corresponding to both deamidated and nondeamidated forms of the peptides (unlabeled) containing the sequon. It was noticed that most of the unlabeled/unglycosylated sequon-containing peptides had undergone deamidation. To confirm this, a deamidation control was analyzed using LC-MS under the same conditions, except the mass spectra were acquired in the linear ion trap. This control sample was a chymotryptic digest of RpNCRD, without any N-glycan release by PNGase F treatment. The sample showed that almost all of the unglycosylated sequon-containing peptides were present in the deamidated form (results not shown). The abundances of such unglycosylated sequon-containing peptides were low compared with the glycosylated forms. Quantification of the extracted ion chromatograms was performed for the identified labeled and unlabeled peptides with two and three miscleavages, respectively, with peptide sequences “SMTDIKTEGNFTYPTGEPL” and “LSMTDIKTEGNFTYPTGEPL” (Table 2). The site occupancy values were corrected for the effective H218O concentration in the PNGase F reaction. Effective H218O was only 85% due to the presence of buffer and enzyme in the reaction. The average site occupancy was determined as 98.75 ± 0.55% from these data. Representative MALDI-TOF mass spectra showing incorporation of either 16O or 18O at the site of glycosylation upon PNGase F treatment, are provided as supporting information for N-deglycosylated RpNCRD (Fig. S1A) and for both derived peptide fragments indicated above (Fig. S1, B and C).
Table 2.
Summary of RpNCRD N-glycosylation site-occupancy analysis
The N-glycosylation site on RpNCRD was labeled by enzymatic deglycosylation in the presence of H218O followed by LC-MS/MS. The ratio of abundances of labeled to unlabeled peptides was used to calculate exact site-occupancy, as described in the text.
| Technical replicate | 18O-Labeled peptide SMTDIKTEGNFTYPTGEPL (precursor 1052) | 18O-Labeled peptide LSMTDIKTEGNFTYPTGEPL (precursor 1109) | Average site occupancya | Average site occupancyb | S.D. |
|---|---|---|---|---|---|
| % | % | % | % | % | |
| 1 | 83.44 | 84.10 | 83.94 | 98.75 | 0.55 |
| 2 | 83.45 | 84.30 | |||
| 3 | 83.28 | 84.77 | |||
| 4 | 83.68 | 84.49 |
a Ratio of labeled/glycosylated peptide is shown.
b Ratio of labeled/glycosylated peptide, corrected for effective H218O in the reaction, is shown.
N-Glycan profiling was performed using hydrophilic interaction LC–MS (HILIC-MS) on the released N-glycan from RpNCRD obtained after PNGase F treatment. The resulting glycan compositions and relative abundances are depicted in Fig. 7A. Only those glycans are shown that had a relative abundance greater than 1% of total abundance from all identified glycans. Interpretation of the data points toward a heterogeneous distribution of typical mammalian complex N-glycans consisting of bi-, tri-, and tetra-antennary structures that are largely fucosylated with variable sialylation with N-acetylneuraminic acid (NeuAc) residues. The complete N-glycan profile is provided as part of the supporting information (see Fig. S2A). Neuraminidase treatment of the N-glycan, followed by HILIC-MS analysis, enabled us to determine the degree of sialylation of the N-glycan. The efficiency of NeuAc cleavage by this method was verified by including a transferrin-derived N-glycan control and resulted in 95% reduction of the NeuAcs present (results not shown). The relative abundance of the number of NeuAc residues present on the N-glycan is reported in Fig. 7B (average %). Nonsialylated glycans accounted for ∼50% of the relative abundance from all glycans assigned in the data. The remaining signal was divided into sialylated glycans carrying one, two, or three NeuAc residues. The complete N-glycan profile obtained after neuraminidase treatment is included in Fig. S2B.
Figure 7.

Glycan profiling/composition analysis on released N-glycans from RpNCRD. A, profile analysis of N-glycan derived from RpNCRD showing glycan structures with relative abundances greater than 1% of total identified. Percent relative abundances are average of three replicates; error bars represent standard deviation. Glycan symbolic representation is as defined by the Consortium for Functional Glycomics (http://www.functionalglycomics.org/static/consortium/Nomenclature.shtml).3 Glycan topologies shown are speculative. The x axis shows numeric codes that refer to the monosaccharides identified in the N-glycan. Bar colors correspond with number of NeuAcs present on the N-glycan as depicted in B. B, relative abundances of N-glycan compositions summarized by number of SAs (NeuAc) present in the N-glycan. C, composite relative abundances of SP-D N-glycans identified after chemical derivatization of NeuAc for discrimination between α(2,3)-linked and α(2,6)-linked NeuAc-containing glycans. Abbreviations used are as follows: Hex, hexoses; HexNac, N-acetylhexosamines; dHex, deoxyhexoses; NeuAc, N-acetylneuraminic acid; NeuGc, N-glycolylneuraminc acid.
To determine the linkage of NeuAc residues present on the N-glycan of RpNCRD, glycan profiling using HILIC-MS was performed after chemical derivatization as described under the “Experimental procedures” and Fig. S2. The results for SA linkage have been summarized in Fig. 7C. Ions were assigned to the different groups shown in the figure based on lowest parts/million error matches against derivatized glycan compositions. Ions accounting for ∼12% of the total relative abundance from matched ions could not be matched unambiguously (had more than one match). As shown in Fig. 7C, ∼35% of the abundance of sialylated N-glycans released from RpNCRD was contributed by glycans containing α(2–3)-linked SAs, whereas a smaller fraction was found to be contributed by those containing α(2–6)-linked SAs or both α(2–3) and α(2–6)-linked SAs (<13%). A profile of all the derivatized glycans that matched unambiguously with % relative abundances has been presented in Fig. S2C.
Glycan array binding analysis of RpSP-D, RpSP-D–dNG, and RhSP-D
To identify putative differences in glycan-binding potential of RpSP-D in presence or absence of the N-linked glycan in its CRD, and to compare their binding characteristics with those of SP-D from humans (RhSP-D), detailed analysis of their sugar-binding specificity was assessed using a mammalian glycan array (version 5.0) that consists of 611 natural and synthetic mammalian glycans, provided and screened by the Protein–Glycan Interaction Core (formerly Core H) of the Consortium for Functional Glycomics (http://www.functionalglycomics.org3 (19)). For all SP-Ds tested, highly specific carbohydrate-binding profiles were obtained (Fig. 8, A–C), and those carbohydrates that bound with the highest affinity and accuracy are shown as cartoons in each panel; full names of these carbohydrates are listed in Table S1.
Figure 8.

Binding specificity of recombinant SP-Ds assessed by glycan array screening. RpSP-D (A), RpSP-D–dNG (B), and RhSP-D (C) were incubated with the CFG glycan array 5.0 (611 different mammalian carbohydrate structures with their numbers indicated on the x axis), and relative binding of SP-D to specific oligosaccharides was detected with rabbit anti-pSP-D (A and B) or rabbit anti-hSP-D antibodies (C), followed by Alexa488-labeled conjugate. Each panel displays only structures of oligosaccharides of peaks with intensities >10% of highest peak while having % coefficient of variation values <20%. An overview of all best binders for all three SP-D's (in order of decreasing glycan binding affinity) is provided in Table 1. Glycan symbolic representation is used according to the Symbol Nomenclature for Glycans (39).
SP-D belongs to the mannose-type C-type lectins, and this is reflected in the strongest affinity of all three SP-D preparations tested for mannose-containing oligosaccharides. Especially RpSP-D shows high specificity for mannose-type glycans (top six binders all contain terminal mannose residues, see Table S1), in addition to minor but remarkably distinct peaks for LewisY structures (peaks 70, 71, and 458) that are absent in the profile of RhSP-D. The glycan array–binding profile of RhSP-D (Fig. 8C) was also determined previously by Zimmer et al. (CFG)3 using an older version of the glycan array, containing fewer glycan structures (CFG request 1179, cbp_1159, array version 3.0 containing 320 glycans). The results of RhSP-D binding properties analyzed here showed a similar predominantly mannose-type binding profile as compared with the previously established binding profile on hSP-D by Zimmer (CFG).3 However, our data also revealed an hSP-D binding preference for polylactosamine-rich tri-antennary glycans (Fig. 8, 572–573 and 586 and 588), which were not present on version 3.0 of the CFG glycan array. Interestingly, the binding profile of RpSP-D became less specific after removal of the N-linked glycosylation present in its CRD (RpSP-D–dNG, Fig. 8B). Whereas the major mannose-binding properties (Fig. 8, 315–317 and 212) and LewisY binding (70–72) remain conserved, RpSP-D–dNG shows a somewhat broader glycan-binding profile compared with the parental form containing the N-glycan, as illustrated by the appearance of several additional peaks. In particular the increase in binding to polylactosamine-rich tri-antennary glycans (Fig. 8, 72–573 and 586 and 588) is striking, because this binding property is one of the major binding properties of RhSP-D which, similar to RpSP-D-dNG, does not carry an N-glycan in its CRD.
Discussion
Pigs are susceptible to infection with avian, human, and swine influenza viruses, and co-infection may result in genetic reassortment between these viruses. This may lead to the emergence of novel IAVs and initiate pandemics within the human population, as occurred most recently in 2009, leading to the H1N1 pandemic. The innate immune response is considered fundamental in controlling viral spread at early stages of infection. Previous in vitro neutralization studies with pSP-D have revealed that this secreted immune protein exhibits distinct antiviral activity against IAVs as compared with SP-D from other animal species (12, 20), and therefore, it is considered to play an important role in early defense against IAV infections in pigs. Previously, as a first step to investigate the distinct mechanisms of interactions between pSP-D and IAV in more detail, we studied the 3D structure of a recombinant trimeric fragment of pSP-D that, because heterogeneous glycans typically hinder crystallization, lacked the N-glycosylation in its CRD (RpNCRD–dNG) (16). Comparison of that crystal structure with the previously solved structure of RhNCRD (21) revealed the presence of a flexible 326-GSS–loop that, based upon molecular dynamics studies, potentially enables interactions with distal portions of the viral mannose-containing glycans expressed on the HA of IAV. Moreover, a second unique mode of interaction with IAV had been described for pSP-D, and this involved the sialylated N-linked glycan present in the CRD of pSP-D (22) that provides interactions with the SA-binding domain of HA. Both the unique GSS-loop structure and the N-glycosylation in the CRD are considered important for the distinct activity of pSP-D, and it is not known whether these structural features might affect each other or act synergistically. To investigate the combined mechanisms of viral neutralization in more detail, we managed to crystallize RpNCRD that was subjected to structural analysis and compared with the structure of RpNCRD–dNG. To our knowledge, the impact of N-glycosylation on the 3D structure and carbohydrate-binding characteristics of (C-type) lectins has not been described before.
Crystal structure of RpNCRD shows a bound complex N-glycan and reveals a potential second site of glycan interaction
In many crystal structures of glycoproteins, the N-glycans, in part due to their heterogeneous structure, are disordered and therefore unable to be modeled in the electron density maps. In fact, in most cases disordered N-glycans hinder crystallization of a glycoprotein. Surprisingly, in the case of RpNCRD, the N-glycan structure is ordered and visible in the maps and can be categorized as a complex-type oligosaccharide. The ordered glycan structure is likely the result of binding of the N-linked glycan from Asn-303 of one molecule to the lectin site of a neighboring molecule in the crystal. Thus, the crystal is a network of linked RpNCRD trimers. The structure is reminiscent of the cross-linked trimers observed for mannose-binding lectin (MBL), also called mannose-binding protein or mannan-binding protein, complexed with Man9 (23), where different arms of the oligomannose are bound in lectin sites of neighboring molecules in the crystal. However, in the case of MBL, each end of the Man9 is bound by a lectin site through one of the nonreducing end mannoses. In the case of RpNCRD, one end of the sugar chain is covalently attached to Asn-303 of one SP-D monomer, whereasthe other end is bound by a lectin site through a nonterminal mannose sugar.
In the crystal structure, a complex glycan is shown bound by RpNCRD, indicating that binding of the collectin to viral glycoproteins may not be limited to high-mannose oligosaccharides. Given the open nature of the lectin site, this result is not too surprising because the biantennary glycan has several sites that meet the requirement of a vicinal, equatorial diol for binding to the lectin site Ca2+. These sites in complex glycans may be shielded from pSP-D by the antennae. However, previous studies have shown that RhNCRD can bind to the core hexose residues in lipopolysaccharides (24), so binding core saccharides in complex glycans may also be possible. The glycan array data show that pSP-D is able to bind complex glycans, consistent with the crystal structure, but oligomannose structures are the preferred substrate. In addition, pSP-D may not bind to higher-order complex–type glycans because the additional antennae of tri-, tetra-, and penta-antennary glycans may have substitutions that would abolish the target vicinal equatorial diols recognized by the lectin site. The glycan modeled here is a biantennary glycan, and complex glycans can contain up to five arms. In higher-order glycans, additional arms would be added at either the O4 or O6 atoms of MAN503A or MAN503B. Additions to the O6 atom of these residues would not be predicted to affect the binding; however, substitution at O4 would completely inhibit potential binding to the lectin site, because O4 makes coordination interactions with the lectin site Ca2+ and must be unsubstituted. Thus, the potential exists in the pSP-D lectin site to bind these complex glycans, but the binding determinants are fairly strict and become less likely to be satisfied as the complexity of the glycan increases.
pSP-D has a more sterically restricted lectin site compared with that of hSP-D due to the presence of three residues Gly-326A–Ser-326B–Ser-326C inserted in the long loop. Previous computational studies suggested a role for these residues in binding to branching sugars on oligomannose (16); however, in the current structure, no interactions are observed between the insertion sequence and the biantennary glycan. Instead, MAN503B forms hydrogen bonds to the protein through residues Gln-333 and Arg-349.
The crystal structure was solved both in the presence and absence of glycerol. The glycerol displaces the sugar bound in the lectin site; however, the sugars on the opposite antennae of the glycan are not displaced. In both cases, MAN503B is making hydrogen bonds to the side chains of protein residues Gln-333 and Arg-349. These observations suggest that this pair of residues could create a secondary binding site for sugars in other RpNCRD–glycan complexes. Typically, collectin–glycan structures show the majority of interaction through the lectin site Ca2+, with little or no interaction between other protein residues and the glycan, so observation of a secondary site is noteworthy. Both Gln-333 and Arg-349 are conserved in human SP-D; however, no crystal structure of WT human SP-D to date has shown interactions between these residues and ligands. In the R343V mutant of human SP-D, the altered binding mode created by the mutation allows both Gln-333 and Arg-349 to make hydrogen bonds to the nonlectin site bound mannose in the 1,2-dimannose complex (25), and molecular dynamics have suggested that Gln-333 may be the more important interaction (26). Arg-343 (Lys-343 in pSP-D) is located between the lectin site and Gln-333 and Arg-349, and the mutation opens up space to access this site. Thus, Gln-333 and Arg-349 could be participating in binding sugars in SP-D complexes with its substrates, but this does not explain the observed affinity difference between pig and human SP-Ds.
RpNCRD neutralizes pandemic IAV mediated by the unique lectin site and the N-linked glycan
Lectin-mediated interactions play a key role in the recognition of IAV by SP-D, and the molecular basis for this carbohydrate-dependent mechanism of interaction was previously described in detail via X-ray crystal structure analyses and modeling studies on complexes of HA and trimeric NCRD fragments of SP-D (26–28). IAV strains that lack glycosylation on the HA are less susceptible or even resistant to neutralization by SP-D, and this is generally observed for human pandemic IAV strains as well as H5N1 strains, all having only few glycan attachments on their HAs (18, 29–31). Importantly, comprehensive studies with recombinant full-length porcine SP-D (RpSP-D) performed on a broad panel of 30 IAV strains of varying subtype and host origin demonstrated its distinct antiviral properties against a much broader range of IAVs, including pandemic IAVs like A/California/09 (H1N1), as compared with RhSP-D (20, 32). Because SP-D interacts with IAV via its CRD, the mechanism of this distinct antiviral activity was further investigated with recombinant NCRD fragments from pSP-D and hSP-D. These studies showed that unlike NCRDs from other SP-D species, RpNCRD was able to inhibit infectivity of seasonal IAV in vitro (16).
As a next step, we investigated in more detail the distinct activity of RpNCRD against Aichi68, a pandemic IAV with only one predicted N-glycan at the top of the HA head and shown to be partially resistant for neutralization by hSP-D and highly resistant to RhNCRD (17, 26). As demonstrated by viral aggregation, HA inhibition, and inhibition of infectivity, we show that RpNCRD exhibits distinct antiviral activity against Aichi68 (Figs. 4 and 5). RpNCRD–dNG also showed a relatively strong protective effect, although the activity was significantly reduced as compared with RpNCRD. These data not only show that RpNCRD, unlike RhNCRD, is able to interact with high affinity to pandemic IAV, but also demonstrate that this interaction is mainly due to the unique carbohydrate-binding domain of pSP-D, further enhanced by interactions mediated by the N-linked glycan. Because we could not observe any differences in the CRD backbone structure between RpNCRD and RpNCRD–dNG, we conclude that the N-glycan enhances interactions of pSP-D with IAV directly and not indirectly by changing the lectin-binding properties of the CRD due to the presence of the N-linked glycan. In addition to increasing the binding affinity for virus particles, we hypothesized that the N-glycan in pSP-D might also play a role in inducing viral aggregation via a mechanism that involves Ca2+-induced “self-recognition” similar to that of the N-glycan–dependent aggregation of lipid vesicles, as described for the other CRD-glycosylated lung collectin SP-A (33). However, several experimental approaches (turbidimetric analysis of aggregate formation, isothermal titration calorimetry, and biotinylation) could not show evidence for such a mechanism for RpNCRD (data not shown). Of note, the relative contribution of the N-linked glycan to the MDCK infection inhibition activity of RpNCRD against pandemic Aichi68 is not substantially different from that against seasonal Phil82 (16), despite the differences in viral glycosylation between both IAV strains. This observation points toward a dual-mode interaction between pSP-D and IAV in which N-linked glycan-mediated interactions are not significantly affected by its lectin-mediated interactions with HA glycans of IAV. Reversely, glycan array binding studies revealed that, to some extent, the N-linked glycan in the CRD of RpNCRD broadens the specificity of carbohydrate binding of the lectin site, but it is difficult to state whether this results in changes of the lectin-mediated binding affinity for IAV.
RpNCRD is fully N-glycosylated with a heterogeneous complex oligosaccharide that is partially sialylated with α(2,3)-linked NeuAcs
Glycoprotein populations may exist with the absence of a glycan present at the putative glycosylation site, and N-linked glycosylation sequons are not always completely occupied. Because the N-glycosylated and nonglycosylated forms of RpNCRD interact differently with IAV, it is of importance to determine what fraction of the RpNCRD populations lacks an N-linked glycan at site Asn-303. Using MS, we first identified the exact site of glycosylation by direct analysis of intact glycopeptides. Results confirmed the presence of glycosylation at Asn-303, in line with previously published data obtained by N-terminal sequencing of tryptic peptides from bronchoalveolar lavage (BAL)-derived pSP-D (22). Although CHO cells are the most commonly used mammalian expression cells, we decided to use the HEK293E expression system to generate human-type post-translational modification of RpNCRD, important given its potential as a future antiviral drug to treat infections in humans. RpSP-D production by HEK293 cells resulted in a nearly full N-glycan site-occupancy (>98%) by a typical mammalian complex N-glycan, almost fully fucosylated but with heterogeneity in antenna number (2–4) and sialylation (0–3 NeuAc residues per N-glycan). Previously, profiling by fluorophore-assisted carbohydrate electrophoresis (FACE) analysis of the N-glycan present in the CRD of pSP-D derived from porcine BAL also indicated a complex N-glycan with strong heterogeneity in size (22). The only other detailed reports on glycan structures present on collectins describe the structure of the N-linked glycan that is conserved in the collagen domain of hSP-D (34) and the N-linked glycan present in the CRD of SP-A (35, 36), both complex-type glycans. The bi-antennary nonsialylated N-glycan in the collagen was shown not to be involved in interactions with IAV, and it most likely fulfills a role in assembly and stabilization of the collagen domain of SP-D. The N-linked glycan in SP-A, however, is also located in the CRD, similar to pSP-D, and was also shown to be important for interactions with IAV. Despite the major differences in protein source (alveolar proteinosis patient-derived BAL versus HEK293 cells) and differences in location of the N-linked glycan in their respective CRDs (denoted in Figs. 1 and 2), the structural characteristics of the N-glycan present on human SP-A and RpNCRD are remarkably similar. Both are structurally highly heterogeneous, with di- and tri-antennaries as dominant structures and smaller amounts of tetra-antennaries, a high degree of core and/or outer arm fucosylation, and partial terminal sialylation. The similarity in their CRD–N-glycan structures suggests that trimming and glycan maturation in the Golgi is mainly controlled by CRD-specific sequence motifs and/or nascent secondary structures. Of note, hSP-D lacks the CRD-glycan, and no significant differences were found in synthesis, assembly, and secretion between RpNCRD and RpNCRD–dNG (16) or between RpSP-D and its N-glycan deletion mutant RpSP–D-dNG (37). These findings illustrate that CRD glycosylation of collectins is not essential for proper folding, trafficking, or solubility of SP-D, but it is mainly of physiological importance because of its ability to either modulate protein function, change antigenicity, and/or improve protein stability by lowering susceptibility to proteases.
Previously, binding studies with conjugated lectins on BAL-derived full-length pSP-D showed full terminal sialylation with almost exclusively α(2,6)-linked SAs (14, 22). In contrast, only half of the N-glycans on RpNCRD were sialylated and contained predominantly α(2,3)-linked SAs (NeuAcs), with a smaller amount of α(2,6)-linked SAs or ambiguous linkages. Previous studies with BAL-derived pSP-D showed that these SAs are important for the N-glycan–mediated interactions between pSP-D and IAVs, and it was also demonstrated that the type of SA linkage present on pSP-D can be important for inhibition of IAV strains that have a corresponding receptor-binding specificity (14). Therefore, care should be taken with translation of results with regard to the SA-mediated interactions by RpNCRD for the SA-mediated antiviral activity of pSP-D in vivo, especially in the case of IAV strains that are poorly or not glycosylated, and lectin-mediated interactions between pSP-D and IAV are compromised. Because RpNCRD is only partially sialylated (50%), improvement of SA content could further enhance the potency of this antiviral protein, important with regard to development of RpNCRD-inspired novel therapeutic drugs in humans (e.g. RhNCRD equipped with activity-enhancing porcine-specific structural elements). Co-transfection of HEK293 cells with SA linkage–specific sialyltransferases might result in higher terminal sialic acid occupancy and enable manipulation of the SA-linkage profile preferentially generating N-glycans that have a balanced mixture of α(2,3)-linked and α(2,6)-linked SAs. This would potentially maximize N-glycan–mediated interactions with an even wider variety of IAV strains, regardless of the SA-linkage specificity of their HA receptor-binding sites, adding to the versatility and potency of RhNCRD-based antivirals.
RpSP-D also binds LewisY, and its N-glycan contributes to sugar-binding specificity
SP-D belongs to the C-type lectin superfamily, and the presence of the characteristic “EPN” motif in the long loop of the CRD (Fig. 1), conserved in all SP-Ds, including human and porcine, defines SP-D as a “mannose-type” lectin. However, saccharide affinity chromatography studies have demonstrated that species-specific differences in ligand recognition occur, primarily due to variations in residues present near the lectin-binding site (38). So far, differences in glycan-binding properties between the CRDs from pSP-D and hSP-D have only been studied by IC50 measurements using a variety of mono- and disaccharides and, with the exception of N-acetylmannosamine (higher IC50 for pSP-D), did not reveal any major differences between their carbohydrate-binding profiles (12). However, glycan array screening of a library that contains a broad range of natural and synthetic mammalian glycans provides essential information on lectin binding selectivity toward more complex, multibranched oligosaccharide ligands, which are of special interest given the extended loop on pSP-D. Although the crystal structure of RpNCRD does not reveal direct involvement of the GSS-loop in generating additional interactions with the bi-antennary complex N-glycan as visualized in the structure, it cannot be excluded that this might be different for other types of (high-mannose) N-glycans under more physiological conditions. The degree of multimerization may affect sugar-binding affinities, but RpSP-D, RpSP-D–dNG, and RhSP-D were all purified and analyzed by the same procedures, and only dodecameric fractions of SP-D were used for screening to allow comparison of binding profiles. The obtained glycan-binding profiles of RpSP-D (Fig. 8A) and RhSP-D (Fig. 8C) looked remarkably similar, with preference for bi- and tri-antennary mannose-rich glycans, in line with strong SP-D–mediated neutralization of IAVs that contain high-mannose glycans on their HA (10). As noted previously, the crystal structure analysis of RpNCRD showed possible interactions with complex type N-glycans, as reflected by the appearance of smaller peaks in the glycan array chromatograms for RpSP-D and also for RhSP-D. Furthermore, all of the high-mannose glycans on the array that were bound strongest by RpSP-D and RhSP-D contained predominantly terminal α1–2–linked mannose, similar to what was shown previously for RhNCRD by Zimmer (56) (results CFG request 1179) and Crouch et al. (25). Based upon the similarities in glycan array–binding profiles, we conclude that the observed differences in lectin-mediated binding of IAVs between pSP-D and hSP-D are due to differences in affinity caused by the GSS-loop and perhaps other amino acid substitutions in the vicinity of the lectin-binding site, while not having a major impact on selectivity of binding (both SP-Ds bind preferentially mannose-type oligosaccharides). Interestingly, removal of the N-linked glycan in the CRD of pSP-D resulted in a less specific glycan-binding profile, although the major peaks were identical for RpSP-D and RpSP-D–dNG. A possible explanation for the differences observed is that the presence of the (heterogeneous) N-glycan structure could generate either steric hindrance or charge repulsion, e.g. between the sialylated N-glycan in the CRD and SAs present on glycan structures of the glycan array, and this could explain the more stringent binding properties and slightly enhanced specificity of RpSP-D as compared with RpSP-D–dNG.
Although RpSP-D and RhSP-D expressed a shared preference for high-mannose oligosaccharides, the glycan-binding array profile did show some notable differences in glycan-binding selectivity between both SP-Ds. RpSP-D also bound LewisY structures, whereas RhSP-D showed additional preference for polylactosamine-rich di- and tri-antennary glycan structures. LewisY antigens, structurally related to the blood group antigens, are commonly found in O-linked glycans on glycoproteins and some glycosphingolipids. In mammalian species, these are expressed in only a few cell types, e.g. certain epithelial cells and at low levels in gastric tissue; enhanced expression of LeY is often associated with tumor growth, metastasis, and proliferation. Interestingly, fucosylated antigens may also be important for host–microbe interactions. Lipopolysaccharide from Helicobacter pylori expresses LeY antigens, identical to those occurring in the human gastric mucosa and during infection; this molecular mimicry may result in antibody production against gastric glycoprotein targets and cause autoimmune inflammation (40). It is speculated that enhanced recognition of LeY-expressing pathogens by pSP-D could contribute to more efficient clearance of bacterial infections in pigs and by masking bacterial LeY-epitopes could also help to prevent the production of autoreactive antibodies. From an innate immune perspective, the distinct recognition of polylactosamine structures by RhNCRD is also intriguing. These fundamental glycan structures present on glycoproteins and glycolipids may act as ligands for extracellular lectins such as galectin (41), and this might also be the case for SP-D in human mucosal tissues. This type of recognition might play a role in modulating the threshold of the immune response at the cellular level.
In summary, this study provides detailed high-resolution structural insights into the porcine-specific SP-D–mediated innate immune defense against IAVs and how distinct lectin-mediated binding, combined with unique interactions mediated by the sialylated lectin–glycan of pSP-D, results in strong inhibition of a broader range of IAVs than other SP-Ds, including human. In general, pigs show few clinical symptoms upon infection by IAV, and restriction of IAV infection by distinct porcine-specific innate defenses might delay antibody-mediated adaptive immune responses in pigs. It is speculated that the profound pSP-D–mediated IAV neutralization facilitates co-infection by different IAV strains and therefore, in part, promotes reassortment in this host species. The use of HEK293-expressed SP-Ds made it possible to shed light on the mechanisms that underlie the interactions between IAV and pSP-D; however, care should be taken with interpretation of data with respect to N-glycan–mediated interactions because RpNCRD shows a smaller SA content and different SA-linkage pattern as compared with natural pSP-D. This difference might, to some extent, affect the activity and selectivity of RpSP-D toward IAV strains as compared with that of endogenous pSP-D in the lungs of pigs. From the perspective of developing novel SP-D–based antivirals, however, full structural and functional characterization of HEK293-expressed RpNCRD as described in this study is an important step forward toward the design and development of highly effective, sialoglycosylated RhSP-D–based therapeutics and/or prophylactics against a broad range of IAVs and possibly other respiratory pathogens.
Experimental procedures
Recombinant expression of porcine and human SP-Ds
Constructs for RpNCRD and RpNCRD–dNG were generated as described previously (16), covering the NCRD region starting at Gly-223 (numbers according to van Eijk et al. (15)) and ending 142 nucleotides downstream from the stop codon. Recombinant collectins were produced using HEK293E cells, stably expressing Epstein-Barr virus nuclear antigen 1 (EBNA1). HEK293E cells were transiently transfected as described previously (42). The pSP-D sequence-containing pUPE105.03 expression plasmids were purified using an endotoxin-free plasmid maxi prep kit. Expression media containing RpNCRD and RpNCRD–dNG, which has the N303Q substitution (by site-directed mutagenesis, as described (16)) that results in a deleted N-glycosylation site in its CRD (Fig. 1), were harvested 6 days post-transfection. Both RpNCRD preparations contained an extra N-terminal Gly–Ser sequence derived from the BamHI-cloning site. After removal of HEK293 cells by centrifugation, large scale expression media were concentrated 10-fold and diafiltrated into 5 mm Hepes, 0.9% NaCl, 5 mm EDTA, pH 7.4, using the Quixstand Hollow Fiber System (GE Healthcare) using a cartridge with a 10-kDa cutoff. The RhSP-D–derived NCRD fragment (RhNCRD) was produced in Escherichia coli as described previously (43). The full-length recombinant collectins RpSP-D, RpSP-D–dNG, and RhSP-D, used for glycan array–binding studies, were constructed and expressed in HEK293 cells as published (37).
Purification of recombinant porcine and human SP-Ds
RpNCRD, RpNCRD–dNG, and full-lengths RpSP-D, RpSP-D–dNG, and RhSP-D were isolated from HEK293E cell-free diafiltrated medium as described previously (16), with the exception of Tris-HCl, which was replaced by 5 mm Hepes, pH 7.4, in all buffers. Instead of a single overnight incubation at 4 °C, the medium was subjected to a batchwise overnight incubation in the presence of 5 mm CaCl2 and mannan-agarose (5-ml bed volume/liter of medium). Mannan-bound RSP-Ds were eluted with 5 mm Hepes, 0.9% NaCl, 5 mm EDTA, pH 7.4, filtered through a 0.2-μm filter, and concentrated to <5 ml via ultrafiltration with Amicon Ultracel centrifugal filters (10-kDa cutoff; Millipore, Amsterdam, The Netherlands). Gel-filtration chromatography was performed using an ÄKTApurifier10 system (GE Healthcare) that was equipped with a Hiload 16/60 Superdex200 PREP grade column, using 5 mm Hepes, pH 7.4, 0.9% NaCl as elution buffer. Fractions from distinct peaks were pooled and concentrated by 10-kDa Amicon Ultracel centrifugation and analyzed for protein content. RhNCRD was purified by maltosyl-agarose chromatography followed by gel-filtration chromatography and characterized as described previously (43). Endotoxin levels were determined with the ToxinSensor LAL assay kit (Genscript, Piscataway, NJ), and endotoxin values of all SP-D preparations were below 50 pg/μg protein.
Biochemical characterization of RpNCRDs
SDS-PAGE analysis, Western blot analysis, and N-glycanase (Prozyme, Hayward, CA) treatment to determine the presence of the N-linked oligosaccharide on RpNCRD was performed as described in detail for the characterization of natural pSP-D (22); RpNCRD–dNG (16) was included for comparison analysis. Digestion of RpNCRD with PNGase F resulted in a mobility shift toward the position of RpNCRD–dNG, indicating that the difference in apparent molecular mass between RpNCRD and RpNCRD–dNG was due to N-glycosylation of the CRD of RpNCRD. Western blot analysis followed by immunostaining using a previously characterized rabbit anti-porcine SP-D IgG antibody showed similar immunoreactivity for both RpNCRD and RpNCRD–dNG. Chemical cross-linking of SP-D by amidation of primary amine groups and subsequent analysis by 15% SDS-PAGE, as described previously (37), showed a major band of ∼65 kDa for RpNCRD and ∼55 kDa for RpNCRD–dNG, consistent with the presence of trimers in both NCRD preparations (results not shown). RhNCRD was purified and characterized as described previously (43). This preparation was previously shown to consist almost exclusively of trimers and bound mannosyl- or maltosyl-agarose in a Ca2+-dependent manner. Biochemical analysis of full-length collectins RpSP-D, RpSP-D–dNG, and RhSP-D was performed in a similar fashion as published previously (37).
Crystallization of RpNCRD
Purified RpNCRD (in 0.9% (w/v) sodium chloride and 5 mm EDTA) was concentrated to ∼16 mg/ml by ultrafiltration (Amicon Ultracel centrifugal filters, 30-kDa cutoff; Millipore). Mannose and CaCl2 were then added to final concentrations of 20 and 15 mm, respectively. RpNCRD was then crystallized in hanging drops by mixing 5 μl of protein with 5 μl of reservoir solution (0.1 m BisTris, pH 6.5, 0.2 m MgCl2, 15% (v/v) glycerol, and 20–22% w/v PEG 3350) on a siliconized coverslip, inverting the coverslip, and sealing over a well containing 0.5 ml of reservoir solution on a VDX plate. Crystals grew within a few days after equilibration at 17 °C. For data collection, crystals were either frozen directly in the cold nitrogen stream (glycerol complex), or the glycerol was soaked out by soaking sequentially in reservoir solution with 15% and then 30% PEG400 added for about 10 min each prior to freezing. X-ray diffraction data were collected on a RAXIS-IV image plate system with a Rigaku RU-300 rotating anode as the X-ray source. Image data were integrated, merged, and scaled using the programs DENZO and Scalepack (Table 1) (44). The crystals had the same space group as the published RpNCRD–dNG structure in complex with mannose (P63 space group) (16). Therefore, the crystal structures were solved by using the published coordinates, less the waters and ligands, with the diffraction data as inputs for the AutoBuild program in Phenix (45). The input model was excluded from the final building steps to reduce phase bias. At this point, the models were refined using iterative cycles of manual rebuilding in Coot (46) and refinement with Phenix. Throughout the refinement, metal restraints were used for the Ca2+-coordinating residues as determined by the ReadySet program in Phenix, except for the sugar–lectin site interactions between crystallographically related monomers, for which restraints were determined manually. For the final refinement step, TLS refinement was used with two zones defined for the neck region (residues 205–235) and the CRD (residues 236–355). The final structures show good geometry and agreement with the experimental data (Table 1).
Virus preparations
The A/Aichi/68(H3N2) strain (Aichi68) was obtained from American Type Culture Collection (ATCC, Manassas, VA) and was grown in the chorioallantoic fluid of 10-day-old chicken eggs and purified on a discontinuous sucrose gradient as described previously (14). The virus was dialyzed against PBS to remove sucrose, aliquoted, and stored at −80 °C until needed. After thawing, the viral stocks contained ∼5 × 108 infectious focus–forming units/ml.
Viral aggregation and hemagglutination inhibition by aggregates
Aggregation of IAV particles (Aichi68) was assessed for RpNCRD and RpNCRD–dNG and was carried out as described previously (47). For both collectin preparations, 2.0, 4.0, or 8.0 μg/ml was mixed with a suspension of virus (Aichi68) in a final volume of 1 ml. During stirring, the light transmission was monitored for 12 min using an SLM/Aminco 8000C spectrofluorimeter (excitation and emission wavelengths were 350 nm). Viral aggregation resulted in an increase in light transmission, and results were expressed as a percentage of control light transmission at t = 0. As a negative control, virus without addition of NCRDs was included. Experiments were performed five times, and statistical analysis was performed with the Student's t test.
To determine the inhibition of HA by Aichi68 due to SP-D–induced aggregation of the virus, 10 μl of the resulting sample after aggregation was added to a round-bottom 96-well containing 90 μl of PBS++ (PBS with 1 mm CaCl2 and 0.5 mm MgCl2) and serially diluted followed by addition of 50 μl of a type O human erythrocyte suspension (in PBS++) to each well, and plates were incubated for 2 h at room temperature. Results were expressed as number of wells with increasing dilutions of aggregation sample that still showed viral HA. Experiments were performed four times, and statistical analysis was performed with the Student's t test.
Hemagglutination inhibition assay
The hemagglutination titer of Aichi68 was determined by titration of virus samples in PBS++ with thoroughly washed human type O, Rh(−) red blood cells as described (48). HAI was measured by serially diluting NCRD preparations (25 μl) in round-bottom 96-well plates using PBS++ as a diluent. After adding 25 μl of IAV (4 hemagglutination units) in each well, the IAV/NCRD mixture was incubated for 10 min at room temperature. Thereafter, 50 μl of a type O human erythrocyte suspension (in PBS++) was added to each well, and plates were incubated for 2 h at room temperature. The minimal concentration of NCRDs required to fully inhibit the hemagglutination activity of the viral suspension was detected by the formation of erythrocyte pellets in wells having the lowest amounts of NCRDs. The highest concentration tested was 10 μg/ml. Experiments were performed in duplicate and carried out four times. Statistical analysis was performed with the Student's t test.
Neutralization of infectivity
Confluent monolayers of Madin-Darby canine kidney (MDCK) cells were prepared in 96-well plates and infected with IAV (Aichi) for 45 min at 37 °C, which was preincubated for 30 min at 37 °C in the absence (control) or presence of increasing amounts of NCRDs. After washing with serum-free and glucose-free Dulbecco's modified Eagle's medium, the monolayers were incubated for 7 h at 37 °C, washed again, fixed, and FITC-labeled for IAV nucleoprotein as described (49). Fluorescent foci, which appeared to be single infected cells in general, were counted. Results were expressed as a percentage of control foci present after infection with collectin-treated virus as compared with untreated virus. All data points were performed in duplicate, and experiments were performed four times. Statistical analysis was performed with the Student's t test.
Structural analysis of N-linked glycan
Glycosylation site identification/confirmation
The amino acid sequence of RpNCRD indicates the presence of one N-linked glycosylation sequon, Asn-303–Phe-304–Thr-305 (Fig. 1). To confirm the presence and location of the N-linked glycosylation on RpNCRD, glycopeptide analysis was performed on chymotryptic RpNCRD, as described previously (18). RpNCRD samples were subjected to reduction (DTT), alkylation (iodoacetamide), and chymotryptic digestion in the presence of 2,2,2-trifluoroethanol as denaturant. Reactions were stopped by heating at 95 °C for 5 min and the addition of TFA to lower the pH. The samples were then dried down in a centrifugal evaporator. Chymotryptic glycopeptides were analyzed by C18 reversed-phase LC-MS/MS with online HILIC enrichment on an Agilent 6550 Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA) with a chip-cube ionization source coupled to an Agilent 1260 LC system, capable of delivering nanoliter/min flow rates. Data-dependent MS/MS was performed using quadrupole mass selection and collisional dissociation, on glycopeptides eluting from the chromatograph. Glycopeptide data were analyzed manually, and the presence of glycosylation at the N-linked glycosylation sequon was confirmed.
Glycan site-occupancy determination
Glycan site-occupancy was analyzed by enzymatic deglycosylation of chymotryptic SP-D peptides/glycopeptides in the presence of H218O. The enzymatic release of N-linked glycans using PNGase F leads to conversion of the occupied Asn residue to an Asp residue. When the deglycosylation is performed in the presence of H218O, the heavier oxygen (18O) gets incorporated into the glycosylation site leading to an ∼3-unit mass shift that can be easily detected in the mass spectrometer. Chymotryptic digests were deglycosylated with PNGase F (Sigma) in the presence of H216O or H218O, as described previously (50). Deglycosylated samples were subsequently desalted using PierceTM C18 spin columns (ThermoFisher Scientific) and analyzed using MALDI-MS on a Bruker UltraflextremeTM MALDI-TOF/TOF mass spectrometer (Bruker Daltonics, Billerica, MA), using 2,5-dihydroxybenzoic acid as matrix.
The labeled (N-deglycosylated) RpNCRD-derived peptide samples were then subjected to LC-MS/MS using a Nano-AcquityTM chromatograph and a Symmetry® 5-μm C18 trapping column (180 μm × 20 mm) and BEHTM 30 C18 analytical column (100 μm × 100 mm, Waters), coupled to a LTQ-OrbitrapTM XL mass spectrometer (ThermoFisher Scientific, San Jose, CA) via an Advion® NanomateTM, nano-ESI robot. Specific details on sample preparation, elution, and settings for MS/MS analysis are provided in Fig. S1. As a control for spontaneous deamidation, a chymotryptic digest was analyzed without any N-deglycosylation. Precursor ion abundances for the peptides verified with MS/MS were used for quantification. Peak areas for extracted ion chromatograms of labeled and unlabeled peptides were integrated in Thermo Xcalibur data analysis software and compared with calculate site occupancy. The experiment was carried out using four technical replicates.
N-Glycan profiling
RpNCRD was subjected to reduction and alkylation followed by tryptic digestion. PNGase F release was performed as described above in the presence of aprotinin to inhibit residual tryptic activity during deglycosylation. Released N-glycans were fractionated from peptides using Pierce C18 spin columns and then desalted using PD minitrap G-10 desalting columns (GE Healthcare). N-Glycan profiling was performed by hydrophilic interaction LC-MS (HILIC-MS) on the released N-glycan from RpNCRD obtained after PNGase F treatment. Full details on equipment, sample preparation, and elution procedures and settings for HILIC-MS are provided in Fig. S2. Data were analyzed manually using Agilent Mass Hunter Qualitative Analysis software (Version B.05.00), and glycan compositions were assigned to m/z values using an in-house excel-based tool, using a 20 ppm mass error tolerance. The extracted ion chromatograms for monoisotopic m/z values were integrated to get peak areas and relative ion abundances for each glycan composition. Three analytical replicates were analyzed, and average relative ion abundances were plotted only for the glycans that were present in all three replicates. The adducted ion forms and charge states were combined for each glycan composition.
For analysis of the degree of sialylation of the N-glycan released from RpNCRD, the N-glycan was subjected to neuraminidase treatment (New England Biolabs) using buffers and recommendations supplied by the manufacturer. Transferrin N-glycans were included as control to verify the efficiency of desialylation. After desalting, the samples were dried, resuspended, and analyzed using HILIC LC-MS as described above.
For analysis of SA linkages, a chemical derivatization method was used as described by Reiding et al. (51). Briefly, released N-glycans from RpNCRD were dissolved in 20 μl of 0.25 m 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC) and 0.25 m 1-hydroxybenzotriazole (HOBt) in ethanol, at 37 °C for 1 h. Subsequently, 20 μl of acetonitrile was added to the samples and incubated at −20 °C for 15 min. Glycan standards with known SA linkage (NeuAc) were purchased from V-Labs (Covington, LA) and used as controls for the reaction. EDC and HOBt were purchased from Sigma. The samples were dried in a centrifugal evaporator and analyzed using HILIC LC-MS as described above.
Data were analyzed manually using Agilent Mass Hunter Qualitative Analysis software (Version B.05.00), and glycan compositions were assigned to m/z values using an in-house excel–based tool, using a 20 ppm mass error tolerance. The extracted ion chromatograms for monoisotopic m/z values were manually integrated to get peak areas and relative ion abundances for each glycan composition. Three analytical replicates were analyzed, and average relative ion abundances were plotted only for the glycans that could be identified in all three replicates. The abundances from adducted ion forms and charge states were summed for each glycan composition.
Glycan array analysis
Glycan-array screening was performed at Core H of the Consortium for Functional Glycomics (CFG), Emory University School of Medicine, Atlanta, GA. The glycan array is a microarray containing a library of natural and synthetic glycans with amino linkers printed onto N-hydroxysuccinimide–activated glass microscope slides (SCHOTT Nexterion) to form a covalent amide linkage. Printed array version 5.0 containing glycan structures with CFG numbers 1611 was used. The procedure for testing the glycan array as well as all glycan structures used and their corresponding CFG numbers are available at the website of the CFG (http://www.functionalglycomics.org/fg/).3
Glycan array screening was performed with full-length SP-Ds instead of the truncated NCRDs to enable detection of glycan array-bound SP-D by previously characterized polyclonal antisera against pSP-D or hSP-D (22). These antisera are known to interact primarily with epitopes present in the NCRD part of the SP-D molecule, and this interaction requires the use of multimeric SP-D to facilitate interactions with glycans on the array and, simultaneously, interactions with the primary antibodies of the same domain via another trimeric arm of SP-D. The samples were diluted in binding buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm CaCl2, 2 mm MgCl2, 1% BSA, 0.05% Tween 20) and applied onto the surface of a single slide, covered with a microscope coverslip, and incubated in a humidified chamber for 60 min. Washing was carried out by successive rinses in 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm CaCl2, 2 mm MgCl2, 0.05% Tween 20 (four times), and 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm CaCl2, 2 mm MgCl2 (four times) after each step. Binding of primary antibody was detected by incubation with Alexa488-labeled secondary antibody followed by the same washing protocol and, after the last step, with H2O and immediately subjected to imaging. Fluorescence intensities were detected by using a ScanArray 5000 (PerkinElmer Life Sciences) confocal scanner. Image analyses were carried out using IMAGENE image analysis software (BioDiscovery, El Segundo, CA). No background subtractions were performed. Data were plotted by using Microsoft EXCEL software.
Author contributions
M. v. E., J. Z., I. V. D., and B. A. S. conceptualization; M. v. E., M. J. R., K. K., N. L., M. R. W., and T. R. C. data curation; M. v. E. and I. V. D. validation; M. v. E., M. J. R., K. K., and T. R. C. investigation; M. v. E., M. J. R., K. K., N. L., J. Z., M. R. W., and M. H. methodology; M. v. E. writing-original draft; M. v. E. project administration; M. J. R., K. K., M. R. W., K. L. H., and B. A. S. formal analysis; M. J. R., K. L. H., B. A. S., and H. P. H. writing-review and editing; J. Z., K. L. H., I. V. D., B. A. S., and H. P. H. supervision; I. V. D. and M. H. resources.
Supplementary Material
Acknowledgments
We acknowledge the Protein–Glycan Interaction Core (Emory University School of Medicine, Atlanta, GA) of the Consortium for Functional Glycomics (supported by National Institutes of Health Grant GM62116 from NIGMS) for performing the glycan array analysis.
This work was supported, in whole or in part, by Technology Foundation STW Open Technology Program Grant 10388 (to M. V. E. and H. P. H.) and National Institutes of Health Grants P41GM104603, S10OD010724, and PO1AI083222 (to J. Z., K. K., and B. S.), and RO1 HL069031 (to K. H. and M. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S2, Table S1, supporting Methods, and supporting Ref. 1.
The atomic coordinates and structure factors (codes 6BBD and 6BBE) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- IAV
- influenza A virus
- Aichi68
- IAV strain A/Aichi/68(H3N2)
- CRD
- carbohydrate recognition domain
- GSS-loop
- pSP-D–specific insertion of Gly-Ser-Ser adjacent to residue Gly-326 of the SP-D consensus sequence
- HAI
- hemagglutination inhibition
- hSP-D
- human SP-D
- MDCK
- Madin-Darby canine kidney
- NCRD
- neck region plus CRD from SP-D
- NeuAc
- N-acetylneuramic acid
- PNGase F
- peptide-N-glycosidase F
- pSP-D
- porcine SP-D
- r.m.s.d.
- root mean square deviation
- RhNCRD
- recombinant human SP-D–derived trimeric neck-CRD
- RhSP-D
- recombinant hSP-D
- RpNCRD
- recombinant porcine SP-D–derived trimeric neck-CRD
- RpNCRD–dNG
- RpNCRD with deleted N-glycosylation site
- RpSP-D
- recombinant pSP-D
- SA
- sialic acid
- SP-A
- surfactant protein A
- SP-D
- surfactant protein D
- EDC
- 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide
- HOBt
- 1-hydroxybenzotriazole
- HA
- hemagglutinin
- MBL
- mannose-binding lectin
- BAL
- bronchoalveolar lavage
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HILIC
- hydrophilic interaction LC
- CFG
- Consortium for Functional Glycomics
- RBC
- red blood cell.
References
- 1. World Health Organization (2016) Influenza (seasonal); fact sheet No. 211. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Neumann G., Noda T., and Kawaoka Y. (2009) Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459, 931–939 10.1038/nature08157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garten R. J., Davis C. T., Russell C. A., Shu B., Lindstrom S., Balish A., Sessions W. M., Xu X., Skepner E., Deyde V., Okomo-Adhiambo M., Gubareva L., Barnes J., Smith C. B., Emery S. L., et al. (2009) Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325, 197–201 10.1126/science.1176225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tripathi S., White M. R., and Hartshorn K. L. (2015) The amazing innate immune response to influenza A virus infection. Innate Immun. 21, 73–98 10.1177/1753425913508992 [DOI] [PubMed] [Google Scholar]
- 5. Ng W. C., Tate M. D., Brooks A. G., and Reading P. C. (2012) Soluble host defense lectins in innate immunity to influenza virus, soluble host defense lectins in innate immunity to influenza virus. J. Biomed. Biotechnol. 2012, 732191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khatri K., Klein J. A., White M. R., Grant O. C., Leymarie N., Woods R. J., Hartshorn K. L., and Zaia J. (2016) Integrated omics and computational glycobiology reveal structural basis for influenza A virus glycan microheterogeneity and host interactions. Mol. Cell. Proteomics 15, 1895–1912 10.1074/mcp.M116.058016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hartshorn K. L. (2010) Role of surfactant protein A and D (SP-A and SP-D) in human antiviral host defense. Front. Biosci. 2, 527–546 [DOI] [PubMed] [Google Scholar]
- 8. Crouch E., Persson A., Chang D., and Heuser J. (1994) Molecular structure of pulmonary surfactant protein D (SP-D). J. Biol. Chem. 269, 17311–17319 [PubMed] [Google Scholar]
- 9. Hartshorn K., Chang D., Rust K., White M., Heuser J., and Crouch E. (1996) Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am. J. Physiol. 271, L753–L762 [DOI] [PubMed] [Google Scholar]
- 10. Hartshorn K. L., White M. R., Voelker D. R., Coburn J., Zaner K., and Crouch E. C. (2000) Mechanism of binding of surfactant protein D to influenza A viruses: importance of binding to haemagglutinin to antiviral activity. Biochem. J. 351, 449–458 [PMC free article] [PubMed] [Google Scholar]
- 11. Hartshorn K. L., White M. R., Shepherd V., Reid K., Jensenius J. C., and Crouch E. C. (1997) Mechanisms of anti-influenza activity of surfactant proteins A and D: comparison with serum collectins. Am. J. Physiol. 273, L1156–L1166 [DOI] [PubMed] [Google Scholar]
- 12. van Eijk M., White M. R., Crouch E. C., Batenburg J. J., Vaandrager A. B., Van Golde L. M., Haagsman H. P., and Hartshorn K. L. (2003) Porcine pulmonary collectins show distinct interactions with influenza A viruses; role of the N-linked oligosaccharides in the carbohydrate recognition domain. J. Immunol. 171, 1431–1440 10.4049/jimmunol.171.3.1431 [DOI] [PubMed] [Google Scholar]
- 13. Hartshorn K. L., White M. R., Mogues T., Ligtenberg T., Crouch E., and Holmskov U. (2003) Lung and salivary scavenger receptor glycoprotein-340 contribute to the host defense against influenza A viruses. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L1066–L1076 10.1152/ajplung.00057.2003 [DOI] [PubMed] [Google Scholar]
- 14. van Eijk M., White M. R., Batenburg J. J., Vaandrager A. B., van Golde L. M., Haagsman H. P., and Hartshorn K. L. (2004) Interactions of influenza A virus with sialic acids present on porcine surfactant protein D. Am. J. Respir. Cell Mol. Biol. 30, 871–879 10.1165/rcmb.2003-0355OC [DOI] [PubMed] [Google Scholar]
- 15. van Eijk M., Haagsman H. P., Skinner T., Archibald A., Reid K. B., and Lawson P. R. (2000) Porcine lung surfactant protein D: complementary DNA cloning, chromosomal localization, and tissue distribution. J. Immunol. 164, 1442–1450 10.4049/jimmunol.164.3.1442 [DOI] [PubMed] [Google Scholar]
- 16. van Eijk M., Rynkiewicz M. J., White M. R., Hartshorn K. L., Zou X., Schulten K., Luo D., Crouch E. C., Cafarella T. R., Head J. F., Haagsman H. P., and Seaton B. A. (2012) A unique sugar-binding site mediates the distinct anti-influenza A activity of pig surfactant protein D. J. Biol. Chem. 287, 26666–26677 10.1074/jbc.M112.368571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nikolaidis N. M., White M. R., Allen K., Tripathi S., Qi L., McDonald B., Taubenberger J., Seaton B. A., McCormack F. X., Crouch E. C., and Hartshorn K. L. (2014) Mutations flanking the carbohydrate binding site of surfactant protein D confer antiviral activity for pandemic influenza A viruses. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L1036–L1044 10.1152/ajplung.00035.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khatri K., Staples G. O., Leymarie N., Leon D. R., Turiák L., Huang Y., Yip S., Hu H., Heckendorf C. F., and Zaia J. (2014) Confident assignment of site-specific glycosylation in complex glycoproteins in a single step. J. Proteome Res. 13, 4347–4355 10.1021/pr500506z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blixt O., Head S., Mondala T., Scanlan C., Huflejt M. E., Alvarez R., Bryan M. C., Fazio F., Calarese D., Stevens J., Razi N., Stevens D. J., Skehel J. J., van Die I., Burton D. R., et al. (2004) Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 17033–17038 10.1073/pnas.0407902101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hillaire M. L., van Eijk M., Vogelzang-van Trierum S. E., Fouchier R. A., Osterhaus A. D., Haagsman H. P., and Rimmelzwaan G. F. (2014) Recombinant porcine surfactant protein D inhibits influenza A virus replication ex vivo. Virus Res. 181, 22–26 10.1016/j.virusres.2013.12.032 [DOI] [PubMed] [Google Scholar]
- 21. Shrive A. K., Tharia H. A., Strong P., Kishore U., Burns I., Rizkallah P. J., Reid K. B., and Greenhough T. J. (2003) High-resolution structural insights into ligand binding and immune cell recognition by human lung surfactant protein D. J. Mol. Biol. 331, 509–523 10.1016/S0022-2836(03)00761-7 [DOI] [PubMed] [Google Scholar]
- 22. van Eijk M., van de Lest C. H., Batenburg J. J., Vaandrager A. B., Meschi J., Hartshorn K. L., van Golde L. M., and Haagsman H. P. (2002) Porcine surfactant protein D is N-glycosylated in its carbohydrate recognition domain and is assembled into differently charged oligomers. Am. J. Respir. Cell Mol. Biol. 26, 739–747 10.1165/ajrcmb.26.6.4520 [DOI] [PubMed] [Google Scholar]
- 23. Weis W. I., Drickamer K., and Hendrickson W. A. (1992) Structure of a C-type mannose-binding protein complexed with an oligosaccharide. Nature 360, 127–134 10.1038/360127a0 [DOI] [PubMed] [Google Scholar]
- 24. Clark H. W., Mackay R.-M., Deadman M. E., Hood D. W., Madsen J., Moxon E. R., Townsend J. P., Reid K. B. M., Ahmed A., Shaw A. J., Greenhough T. J., and Shrive A. K. (2016) Crystal structure of a complex of surfactant protein D (SP-D) and Haemophilus influenzae lipopolysaccharide reveals shielding of core structures in SP-D-resistant strains. Infect. Immun. 84, 1585–1592 10.1128/IAI.01239-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crouch E., Hartshorn K., Horlacher T., McDonald B., Smith K., Cafarella T., Seaton B., Seeberger P. H., and Head J. (2009) Recognition of mannosylated ligands and influenza A virus by human surfactant protein D: contributions of an extended site and residue 343. Biochemistry 48, 3335–3345 10.1021/bi8022703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goh B. C., Rynkiewicz M. J., Cafarella T. R., White M. R., Hartshorn K. L., Allen K., Crouch E. C., Calin O., Seeberger P. H., Schulten K., and Seaton B. A. (2013) Molecular mechanisms of inhibition of influenza by surfactant protein D revealed by large-scale molecular dynamics simulation. Biochemistry 52, 8527–8538 10.1021/bi4010683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seaton B. A., Crouch E. C., McCormack F. X., Head J. F., Hartshorn K. L., and Mendelsohn R. (2010) Review: structural determinants of pattern recognition by lung collectins. Innate Immun. 16, 143–150 10.1177/1753425910368716 [DOI] [PubMed] [Google Scholar]
- 28. Crouch E., Nikolaidis N., McCormack F. X., McDonald B., Allen K., Rynkiewicz M. J., Cafarella T. M., White M., Lewnard K., Leymarie N., Zaia J., Seaton B. A., and Hartshorn K. L. (2011) Mutagenesis of surfactant protein D informed by evolution and x-ray crystallography enhances defenses against influenza A virus in vivo. J. Biol. Chem. 286, 40681–40692 10.1074/jbc.M111.300673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Job E. R., Deng Y. M., Tate M. D., Bottazzi B., Crouch E. C., Dean M. M., Mantovani A., Brooks A. G., and Reading P. C. (2010) Pandemic H1N1 influenza A viruses are resistant to the antiviral activities of innate immune proteins of the collectin and pentraxin superfamilies. J. Immunol. 185, 4284–4291 10.4049/jimmunol.1001613 [DOI] [PubMed] [Google Scholar]
- 30. Qi L., Kash J. C., Dugan V. G., Jagger B. W., Lau Y. F., Sheng Z. M., Crouch E. C., Hartshorn K. L., and Taubenberger J. K. (2011) The ability of pandemic influenza virus hemagglutinins to induce lower respiratory pathology is associated with decreased surfactant protein D binding. Virology 412, 426–434 10.1016/j.virol.2011.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tate M. D., Job E. R., Deng Y.-M., Gunalan V., Maurer-Stroh S., and Reading P. C. (2014) Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses 6, 1294–1316 10.3390/v6031294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hillaire M. L., van Eijk M., van Trierum S. E., van Riel D., Saelens X., Romijn R. A., Hemrika W., Fouchier R. A., Kuiken T., Osterhaus A. D., Haagsman H. P., and Rimmelzwaan G. F. (2011) Assessment of the antiviral properties of recombinant porcine SP-D against various influenza A viruses in vitro. PLoS One 6, e25005 10.1371/journal.pone.0025005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haagsman H. P., Elfring R. H., van Buel B. L., and Voorhout W. F. (1991) The lung lectin surfactant protein A aggregates phospholipid vesicles via a novel mechanism. Biochem. J. 275, 273–276 10.1042/bj2750273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leth-Larsen R., Holmskov U., and Højrup P. (1999) Structural characterization of human and bovine lung surfactant protein D. Biochem. J. 343, 645–652 10.1042/bj3430645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Munakata H., Nimberg R. B., Snider G. L., Robins A. G., Van Halbeek H., Vliegenthart J. F., and Schmid K. (1982) The structure of the carbohydrate units of the 36K glycoprotein derived from the lung lavage of a patient with alveolar proteinosis by high resolution 1H-NMR spectroscopy. Biochem. Biophys. Res. Commun. 108, 1401–1405 10.1016/S0006-291X(82)80062-4 [DOI] [PubMed] [Google Scholar]
- 36. Berg T., Leth-Larsen R., Holmskov U., and Højrup P. (2000) Structural characterisation of human proteinosis surfactant protein A. Biochim. Biophys. Acta 1543, 159–173 10.1016/S0167-4838(00)00184-9 [DOI] [PubMed] [Google Scholar]
- 37. van Eijk M., Bruinsma L., Hartshorn K. L., White M. R., Rynkiewicz M. J., Seaton B. A., Hemrika W., Romijn R. A., van Balkom B. W., and Haagsman H. P. (2011) Introduction of N-linked glycans in the lectin domain of surfactant protein D: impact on interactions with influenza A viruses. J. Biol. Chem. 286, 20137–20151 10.1074/jbc.M111.224469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crouch E. C., Smith K., McDonald B., Briner D., Linders B., McDonald J., Holmskov U., Head J., and Hartshorn K. (2006) Species differences in the carbohydrate binding preferences of surfactant protein D. Am. J. Respir. Cell Mol. Biol. 35, 84–94 10.1165/rcmb.2005-0462OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Varki A., Cummings R. D., Aebi M., Packer N. H., Seeberger P. H., Esko J. D., Stanley P., Hart G., Darvill A., Kinoshita T., Prestegard J. J., Schnaar R. L., Freeze H. H., Marth J. D., Bertozzi C. R., et al. (2015) Symbol nomenclature for graphical representations of glycans. Glycobiology 25, 1323–1324 10.1093/glycob/cwv091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Appelmelk B. J., Negrini R., Moran A. P., and Kuipers E. J. (1997) Molecular mimicry between Helicobacter pylori and the host. Trends Microbiol. 5, 70–73 10.1016/S0966-842X(96)10084-6 [DOI] [PubMed] [Google Scholar]
- 41. Hirabayashi J., Hashidate T., Arata Y., Nishi N., Nakamura T., Hirashima M., Urashima T., Oka T., Futai M., Muller W. E., Yagi F., and Kasai K. (2002) Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim. Biophys. Acta 1572, 232–254 10.1016/S0304-4165(02)00311-2 [DOI] [PubMed] [Google Scholar]
- 42. Morlot C., Hemrika W., Romijn R. A., Gros P., Cusack S., and McCarthy A. A. (2007) Production of Slit2 LRR domains in mammalian cells for structural studies and the structure of human Slit2 domain 3. Acta Crystallogr. D Biol. Crystallogr. 63, 961–968 10.1107/S0907444907035470 [DOI] [PubMed] [Google Scholar]
- 43. Wang H., Head J., Kosma P., Brade H., Müller-Loennies S., Sheikh S., McDonald B., Smith K., Cafarella T., Seaton B., and Crouch E. (2008) Recognition of heptoses and the inner core of bacterial lipopolysaccharides by surfactant protein D. Biochemistry 47, 710–720 10.1021/bi7020553 [DOI] [PubMed] [Google Scholar]
- 44. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 45. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L.-W., Jain S., Kapral G. J., Grosse Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., et al. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 10.1016/j.ymeth.2011.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 47. Hartshorn K. L., Sastry K., Brown D., White M. R., Okarma T. B., Lee Y. M., and Tauber A. I. (1993) Conglutinin acts as an opsonin for influenza A viruses. J. Immunol. 151, 6265–6273 [PubMed] [Google Scholar]
- 48. Hartshorn K. L., Sastry K., White M. R., Anders E. M., Super M., Ezekowitz R. A., and Tauber A. I. (1993) Human mannose-binding protein functions as an opsonin for influenza A viruses. J. Clin. Invest. 91, 1414–1420 10.1172/JCI116345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hartshorn K. L., Holmskov U., Hansen S., Zhang P., Meschi J., Mogues T., White M. R., and Crouch E. C. (2002) Distinctive anti-influenza properties of recombinant collectin-43 (CL-43). Biochem. J. 366, 87–96 10.1042/bj20011868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gonzalez J., Takao T., Hori H., Besada V., Rodriguez R., Padron G., and Shimonishi Y. (1992) A method for determination of N-glycosylation sites in glycoproteins by collision-induced dissociation analysis in fast atom bombardment mass spectrometry: identification of the positions of carbohydrate-linked asparagine in recombinant α-amylase by treatment with peptide-N-glycosidase F in 18O-labeled water. Anal. Biochem. 205, 151–158 10.1016/0003-2697(92)90592-U [DOI] [PubMed] [Google Scholar]
- 51. Reiding K. R., Blank D., Kuijper D. M., Deelder A. M., and Wuhrer M. (2014) High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification. Anal. Chem. 86, 5784–5793 10.1021/ac500335t [DOI] [PubMed] [Google Scholar]
- 52. Head J. F., Mealy T. R., McCormack F. X., and Seaton B. A. (2003) Crystal structure of trimeric carbohydrate recognition and neck domains of surfactant protein A. J. Biol. Chem. 278, 43254–43260 10.1074/jbc.M305628200 [DOI] [PubMed] [Google Scholar]
- 53. Roepstorff P., and Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601 10.1002/bms.1200111109 [DOI] [PubMed] [Google Scholar]
- 54. Johnson R. S., Martin S. A., Biemann K., Stults J. T., and Watson J. T. (1987) Novel fragmentation process of peptides by collision-induced decomposition in a tandem mass spectrometer: differentiation of leucine and isoleucine. Anal. Chem. 59, 2621–2625 10.1021/ac00148a019 [DOI] [PubMed] [Google Scholar]
- 55. Conboy J. J., and Henion J. D. (1992) The determination of glycopeptides by liquid chromatography/mass spectrometry with collision-induced dissociation. J. Am. Soc. Mass Spectrom. 3, 804–814 10.1016/1044-0305(92)80003-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.