

Abstract
The proto-oncogene Akt/protein kinase B (PKB) is a pivotal signal transducer for growth and survival. Growth factor stimulation leads to Akt phosphorylation at two regulatory sites (Thr-308 and Ser-473), acutely activating Akt signaling. Delineating the exact role of each regulatory site is, however, technically challenging and has remained elusive. Here, we used genetic code expansion to produce site-specifically phosphorylated Akt1 to dissect the contribution of each regulatory site to Akt1 activity. We achieved recombinant production of full-length Akt1 containing site-specific pThr and pSer residues for the first time. Our analysis of Akt1 site-specifically phosphorylated at either or both sites revealed that phosphorylation at both sites increases the apparent catalytic rate 1500-fold relative to unphosphorylated Akt1, an increase attributable primarily to phosphorylation at Thr-308. Live imaging of COS-7 cells confirmed that phosphorylation of Thr-308, but not Ser-473, is required for cellular activation of Akt. We found in vitro and in the cell that pThr-308 function cannot be mimicked with acidic residues, nor could unphosphorylated Thr-308 be mimicked by an Ala mutation. An Akt1 variant with pSer-308 achieved only partial enzymatic and cellular signaling activity, revealing a critical interaction between the γ-methyl group of pThr-308 and Cys-310 in the Akt1 active site. Thus, pThr-308 is necessary and sufficient to stimulate Akt signaling in cells, and the common use of phosphomimetics is not appropriate for studying the biology of Akt signaling. Our data also indicate that pThr-308 should be regarded as the primary diagnostic marker of Akt activity.
Keywords: Akt/protein kinase B (PKB), protein phosphorylation, cell biology, cell signaling, enzyme, aminoacyl tRNA synthetase, transfer RNA (tRNA), genetic code expansion, phosphomimetics, phosphoseryl-tRNA synthetase (SepRS), tRNASep, activation loop, hydrophobic motif
Introduction
The proto-oncogene Akt/protein kinase B (PKB)6 is a central transducer of growth and survival signaling (1). There are three isozymes of Akt in mammals. Akt1, Akt2, and Akt3 include kinase domains with extensive homology to those of protein kinases A, G, and C, defining them as members of the AGC family of Ser/Thr protein kinases (2). Akt transduces signals in the phosphoinositide 3-kinase (PI3K) signaling cascade, which is one of the most commonly deregulated pathways in human cancer (3, 4). Thus, enormous efforts are directed at understanding the mechanisms of activation of Akt and how to target these enzymes therapeutically (5).
Akt activity in the cell is acutely controlled by growth factor-dependent phosphorylation mechanisms (6). Following activation by agonist-bound receptor tyrosine kinases at the plasma membrane, PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the lipid second messenger phosphatidylinositol 3,4,5-triphosphate (PIP3) (7). This second messenger engages Akt at the plasma membrane by binding its autoinhibitory pleckstrin homology (PH) domain, resulting in a conformational change that exposes the kinase domain for phosphorylation (8). The phosphoinositide-dependent kinase 1 (PDK1) phosphorylates a conserved Thr on the activation loop (Thr-308 in Akt1), leading to partial activation of Akt (9). Full activation of Akt1 results from a second phosphorylation event in the C-terminal tail, a regulatory region referred to as the “hydrophobic motif” (Ser-473 in Akt1), that was originally identified in protein kinase C and S6 kinase (10–12). Phosphorylation at Ser-473 in Akt1 depends on the mTOR complex 2 (mTORC2) (13). In addition, Akt is co-translationally and constitutively phosphorylated at another C-terminal site termed the “turn motif” (Thr-450 in Akt1) by mTORC2, a modification that regulates the stability of the enzyme (14). Given that Akt is activated by phosphorylation of Thr-308 and Ser-473, phospho-specific antibodies to these sites are widely used as diagnostic markers (15–17). To a striking degree, many clinical (18–21) and biochemical (22) studies rely solely on Ser-473 phosphorylation as a proxy for Akt activity.
The activation of Akt by phosphorylation is well-established (8), yet the contribution of each specific phosphorylation site toward the maximal Akt activity and pathogenesis is less well-defined. This knowledge gap resulted from the inability previously to prepare Akt variants in site-specifically phosphorylated forms. Earlier work established production of partially active and truncated Akt1 in Escherichia coli. The attempt was unsuccessful in producing a sufficient amount of full-length Akt1 to determine activity (23). Instead, this study relied on a construct lacking the PH domain with Ser-473 substituted by glutamate, and the authors were unable to show how the phosphomimetic compared with pSer at position 473 (23). A protocol for active Akt1 production in insect (Sf9) cells has been established (24). The ability to generate ppAkt1 required a complex and low-yield in vitro procedure to phosphorylate Akt with two additional purified upstream kinases in the presence of lipid vesicles (24). Protein production in Sf9 cells fails to produce Akt1 with site-specific or programmed phosphorylation. The resulting protein is a mixture of singly and doubly phosphorylated species (25) and includes phosphorylation at Thr-450 in addition to potentially other modifications, so the ability to isolate the activity of each regulatory site individually and in precise combinations has remained elusive. As demonstrated below, we have developed a facile and efficient approach that is a novel combination of in vivo enzymatic phosphorylation with genetic code expansion to produce pAkt1 and ppAkt1 variants with specifically programmed phosphorylation (Fig. 1A).
Figure 1.

A novel route to doubly phosphorylated and active Akt1. A, schematic representation of recombinant Akt1 biosynthesis with pSer-473 genetically encoded in response to the UAG codon and pThr-308 enzymatically phosphorylated in vivo in E. coli. Genetically encoded pSer incorporation requires phosphoseryl-tRNA synthetase (SepRS), a UAG-decoding tRNASep, and the elongation factor mutant (EFSep). B, enzyme activity of differentially phosphorylated Akt1 variants with a GSK-3β substrate peptide. Akt1 quantitatively phosphorylated at both 308 and 473 (ppAktS473,T308, blue diamonds) showed maximal activity compared with the unphosphorylated Akt1 (gray circles) and singly phosphorylated Akt1 variants: pAktT308 (black cross) and pAktS473 (brown diamonds). The reported values represent the mean of triplicate experiments with error bars indicating S.D. Lower-activity variants show above-background kinase activity (inset).
Because of the previous technological barriers to producing specifically phosphorylated kinase variants, a significant literature continues to accumulate for Akt (23, 26–32) and other kinases (33), in which “phosphomimetic” substitutions are used as a genetic tool to interrogate the biological consequences of phosphate on a site of interest (33). The rationale behind these experiments is that the acidic residues, Asp and Glu, are negatively charged like phosphate. Yet the carboxylate of an acidic amino acid has considerably less electronegativity than a phosphate and a significantly smaller hydration sphere and volume (34, 35). A complementary approach involves nullifying the effect of a particular phosphorylation site by introducing a nonphosphorylatable Ala mutation (26); however, an Ala is considerably smaller and less polar than a Ser or Thr, and Ala lacks the capacity to form hydrogen bonds. In light of these observations, the widespread use of Ala and phosphomimetic mutants in signaling studies begs the question of how appropriate these mutations are to interrogate the cellular function of Akt or other phosphorylated proteins.
Here we provide, for the first time, a quantitative analysis of the contribution of phosphate at positions 308 and 473 to the catalytic activity of Akt1 and, additionally, examine the effects of substitutions of acidic residues or Ala at these positions. To do this, we developed an optimal strategy to produce recombinant full-length human Akt1 in E. coli with genetically encoded pSer introduced at specific phosphorylation sites. We measured the activity of singly and doubly phosphorylated Akt1 variants with phosphate at positions 308 (pAkt1S308, pAkt1T308) and/or 473 (pAkt1S473) compared with inactive and unphosphorylated Akt1 as well as Akt1 variants with phosphomimetic Glu or Asp mutations. With a FRET-based Akt activity sensor (BKAR) (36), we conducted complementary experiments of specific Akt1 variants in live cells. Our data reveal that, compared with unphosphorylated enzyme, phosphorylation of Thr-308 alone increases the apparent catalytic rate by nearly 400-fold, which is sufficient to observe maximal signaling in cells. Phosphorylation of only Ser-473 boosts Akt1 activity by ∼80-fold over unphosphorylated enzyme; however, our data suggest that phosphorylation at Ser-473 alone may not be sufficient to elicit Akt1 signaling in cells. Thus, phosphorylation of Thr-308 is necessary and sufficient for the activity of Akt. Importantly, our cell-based observations and biochemical data confirm that Ala does not mimic a nonphosphorylated Thr-308 and further that acidic residues fail to activate Akt1. Phosphomimetic substitutions, therefore, do not mimic phosphorylation in Akt1 at either of its two key regulatory phosphorylation sites.
Results
Activation of full-length pAkt1 variants by genetic code expansion
We used in vitro radioactive kinase assays with [γ-32P]ATP to test the activity of full-length Akt1 variants that were made with site-specifically incorporated pSer residues (Figs. S1–S3). The assays quantify the ability of each Akt1 variant to phosphorylate an Akt1 substrate peptide (CKRPRAASFAE) that is derived from the natural Akt1 target glycogen synthase kinase 3β (GSK-3β). Using genetic code expansion (37, 38), we reassigned the UAG stop codon to genetically encode pSer in recombinant Akt1 proteins produced in E. coli. For example, we expressed pAkt1S473 with a genetically encoded pSer residue inserted in response to a UAG codon at position 473 in the Akt1 construct. We used multiple-reaction monitoring MS/MS to unambiguously identify pSer at position 473 (Fig. S3). Genetically encoded pAkt1S473 showed a clear signal for phosphorylation at position 473 with no evidence of dephosphorylation (Fig. S3, A and B). In WT or unphosphorylated Akt1, Ser-473 is readily detected with no evidence of pSer-473 (Fig. S3, C and D).
For pThr incorporation at position 308, we relied on enzymatic phosphorylation by the upstream kinase PDK1 (23). We produced phosphorylated pAkt1T308 as well as the doubly phosphorylated ppAkt1T308,S473 by co-expressing PDK1 in E. coli with either the WT Akt1 or pAkt1S473 constructs, respectively (Fig. 1A). We confirmed phosphorylation at Thr-308 (Fig. S4) by parallel reaction–monitoring MS/MS (PRM-MS/MS). The MS/MS data identified an insignificant level of Thr-308 in the pAkt1T308 sample, indicating essentially quantitative phosphorylation of Thr-308 by PDK1 in E. coli. Finally, we confirmed site-specific phosphorylation of the ppAkt1T308,S473 variant using PRM-MS/MS (Fig. S2). In the MS/MS analysis, we achieved up to 90% coverage of the full-length Akt1 (Fig. S2D), and we were unable to identify significant peptides with modifications other than the anticipated and programmed phosphorylations. The data indicate that both genetic code expansion and PDK1 phosphorylation are highly site-specific and that endogenous modification or dephosphorylation by E. coli enzymes is absent or minimal.
We first measured the activity of Akt phosphorylated at both positions (ppAkt1T308/S473). Doubly phosphorylated Akt1 (Fig. S4) had an apparent reaction rate of 44 ± 6 fmol/min/pmol of enzyme (Fig. 1, Table 1, and Figs. S5 and S6). We performed these kinase assays in conditions of subsaturating ATP. We chose these conditions so that Akt1 protein variants with a wide range of activities could be assayed using the same experimental conditions. To compare the relative activities of Akt1 and pAkt1 variants, we determined an apparent catalytic rate (kapp = vo/[Akt1]) based on the initial velocity (vo) observed in the kinase assays. Under subsaturating ATP, our reaction velocities are far below Vmax for Akt and related kinases (24, 39); kapp is not intended to estimate kcat. Interestingly, the bacterially expressed ppAkt1 construct consistently migrated slightly faster than the other pAkt1 variants and Akt1 mutants on SDS-PAGE (Figs. S1 and S2A). We determined that this was not a result of premature termination of the translation product because PRM-MS/MS unambiguously identified intact full-length and doubly phosphorylated ppAktT308,S473 (Fig. S2, B–D). We found no evidence of truncation or stopping at the UAG473 codon or dephosphorylation according to MS. Phosphorylation at Thr-450 is known to specifically decrease the electrophoretic mobility of Akt1 expressed in mammalian cells (40). Our bacterially expressed pAkt1 variants show no evidence of phosphorylation at Thr-450. These observations together suggest that phosphorylation status impacts the mobility of Akt1 on SDS-PAGE.
Table 1.
Specific activity of Akt1 variants
Initial velocities (v0) and apparent catalytic rates (kapp; v0/[Akt1]) of each enzyme variant were calculated using linear regression analysis of the activity plots.
| Akt1 variant | Akt1 amount | vo | kapp | Activation |
|---|---|---|---|---|
| pmol | fmol/min | fmol/min/pmol Akt1 | fold increase | |
| Akt1 (unphosphorylated) | 18 | 0.6 ± 0.2 | 0.03 ± 0.01 | 1 |
| pAkt1 | ||||
| pSer-473 | 18 | 46 ± 5 | 2.5 ± 0.3 | 83 |
| pSer-308 | 6 | 2.9 ± 0.2 | 0.48 ± 0.03 | 16 |
| pThr-308a | 1.8 | 22 ± 4 | 12 ± 2 | 400 |
| ppAkt1 (pThr-308, pSer-473)a | 1.8 | 79 ± 11 | 44 ± 6 | 1467 |
| Phosphomimetics | ||||
| Glu-473 | 18 | 1.5 ± 0.1 | 0.083 ± 0.006 | 2.7 |
| Glu-308 | 18 | 1.0 ± 0.1 | 0.056 ± 0.006 | 1.9 |
| Asp-473 | 18 | 0.40 ± 0.03 | 0.022 ± 0.002 | 0.7 |
| Asp-308 | 18 | 2.6 ± 0.1 | 0.14 ± 0.01 | 5 |
| pAkt1S473 mutants | ||||
| Asp-308 | 18 | 83 ± 1 | 4.6 ± 0.1 | 153 |
| Ala-308 | 18 | 5.4 ± 0.3 | 0.30 ± 0.02 | 10 |
| pAkt1T308 mutants | ||||
| Asp-473 | 18 | 188 ± 11 | 10.4 ± 0.6 | 348 |
| Glu-473 | 18 | 120 ± 16 | 6.6 ± 0.8 | 222 |
| Ala-473 | 18 | 74 ± 3 | 4.1 ± 0.2 | 137 |
a To measure the initial velocity accurately, the most active variants were assayed at a 10-fold reduced enzyme concentration (Figure S5).
Akt1 variants that contain pThr-308 were so highly active under the conditions of our assays that we reduced the Akt1 concentration 10-fold to establish a linear range of activity to determine vo (Fig. S5). Unphosphorylated Akt1 was essentially inactive, showing a low basal activity of 0.03 ± 0.01 fmol/min/pmol of enzyme (Table 1). Thus, compared with unphosphorylated enzyme, phosphorylation at both activating sites increased the catalytic rate of Akt1 by 1500-fold. We note this is the first time that the activity of Akt1 has been definitively measured and reported for homogeneously pure variants with site-specifically programmed phosphorylation at both key regulatory sites and without phosphate occupancy at other sites (e.g. pThr-450).
Phosphorylation of Thr-308 is necessary and sufficient for Akt1 activation
We next examined the contribution of phosphate at each site individually to the maximal activity of Akt1. Based on measurements of the initial velocity, the activity of monophosphorylated pAkt1T308 corresponded to ∼27% of that of the doubly phosphorylated species (Fig. 1 and Table 1). Thus, phosphorylation at the PDK1 site alone results in an over 400-fold increase in activity compared with the unphosphorylated enzyme. These data establish that phosphorylation at Thr-308 alone is sufficient for robust activation of Akt. Purified, monophosphorylated pAkt1S473 was considerably less active than protein with phosphate at Thr-308 (Fig. 1). pAkt1S473 had activity that corresponded to about 5% of the activity of the doubly phosphorylated species (Table 1). Although significantly reduced relative to that of the doubly phosphorylated species, activity from pAkt1S473 still represents an 80-fold increase over the unphosphorylated Akt1 enzyme. Thus, genetic code expansion with pSer enables production of active Akt1 without the need to purify and activate upstream Akt1 kinases (24). In the absence of substrate peptide, we were able to detect low, but above-background, phosphorylation that may be attributed to autophosphorylation by pAkt1S473 (Fig. 2).
Figure 2.
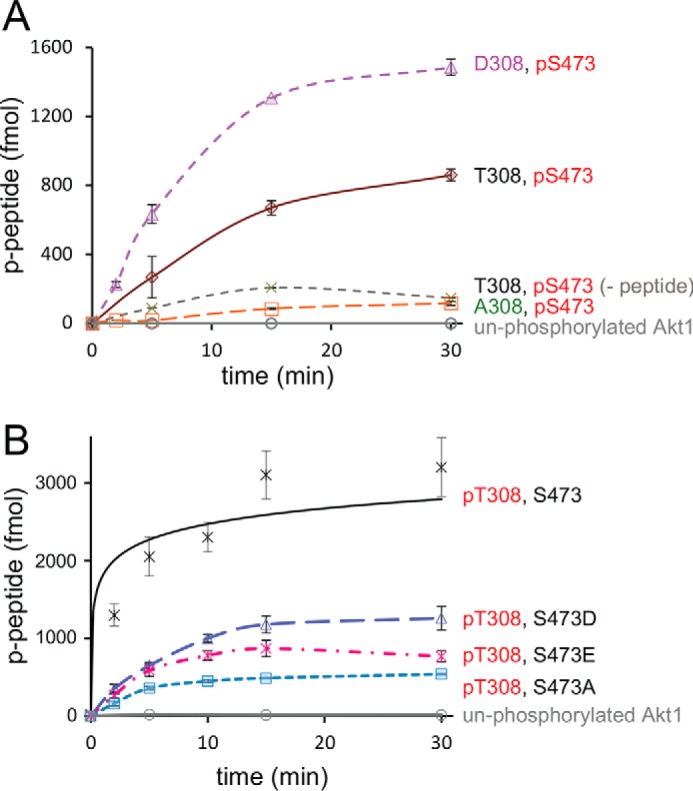
Activity of singly phosphorylated Akt1 variants with regulatory site mutations. A, Akt1 enzyme activity is shown for variants with Ser-473 phosphorylated and Thr-308 unphosphorylated (brown diamond) or mutated: T308D (purple triangles) and T308A (orange squares). Controls include unphosphorylated Akt1 (gray circles), and pAkt1S473 in the absence of substrate peptide (−peptide, green crosses). B, compared with pAkt1T308 activity (black crosses) and in the context of Akt1 phosphorylated at Thr-308, mutations S473D (blue triangles), S473E (magenta crosses), or S473A (cyan squares) resulted in marginally reduced activity. All reported values represent the mean of triplicate experiments with error bars indicating S.D.
Genetic code expansion was also used to produce pAkt1S308. The Akt enzyme containing pSer-308 had above-background but low activity (Fig. 3A). Western blot analysis revealed a band co-migrating with full-length Akt but also a species with an apparent molecular mass of ∼35 kDa (Fig. S7). This could result either from degradation or premature truncation from stopping at UAG308, which would produce a protein of 37.6 kDa. We adjusted our estimated rate for this enzyme by accounting for the fraction of full-length pAkt1S308 in our preparation. Although it is possible that we underestimated the activity Akt1 with pSer-308 (Fig. 3A), the data show that pSer cannot substitute for pThr-308 to produce optimally active Akt1.
Figure 3.
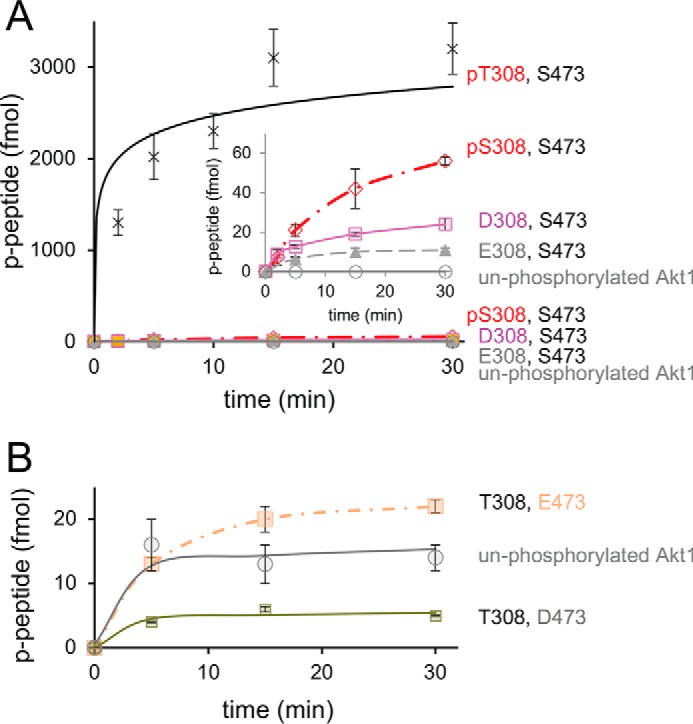
Activity of T308S and phosphomimetic Akt1 variants. A, compared with pAkt1T308 activity (black crosses), mutations of Thr-308 to either Asp (pink squares), Glu (green triangles), or pSer (red diamonds) resulted in low but above-background kinase activity (inset). B, kinase activity was also measured in the context of an unphosphorylated Akt1 (gray circles) with phosphomimetic mutations S473D (green squares) and S473E (peach squares). All reported values represent the mean of triplicate experiments with error bars indicating S.D.
Enzymatic activity of phosphomimetic Akt1 variants
We next tested the activity of purified phosphomimetic Akt1 mutants (Fig. S6). In these experiments, we programmed either Ser-473 phosphorylation (Fig. 2A) or unphosphorylated Ser-473 (Fig. 3A) and examined the effect of residue substitutions at position 308. Replacement of Thr-308 with Asp increased the activity of pAkt1S473 by a modest 2-fold (from 2.5 to 4.6 fmol/min/pmol Akt), resulting in enzyme that displayed only 10% of the activity of doubly phosphorylated enzyme (Fig. 2A, Fig. S6, and Table 1). Surprisingly, replacing the Thr-308 with an Ala eliminated the activity of the monophosphorylated pAkt1S473 (Fig. 2A), demonstrating that Ala-308 is not an appropriate substitute for unphosphorylated Thr.
In the context of Akt1 that was not phosphorylated at Ser-473 (Fig. 3B), Akt1D308 demonstrated 5-fold higher enzyme activity compared with unphosphorylated Akt1, but this was still ∼100 times lower than that of pAkt1T308 (Fig. 3A, Fig. S6, and Table 1). Akt1E308 was >200-fold less active than pAkt1T308 (Fig. 3A and Fig. S6). Interestingly, production of pAkt1S308 by our genetic code expansion approach resulted in an enzyme with only 3-fold more activity than Akt1D308 (Fig. 3A), suggesting that Asp-308 may, to some extent, mimic pSer-308, but not the natural pThr-308 residue in Akt1.
Phosphomimetic substitution at Ser-473 also failed to activate Akt1. We tested Ser-473 mutations in the context of unphosphorylated Akt1 (Fig. 3B and Fig. S6) and in Akt1 variants phosphorylated at Thr-308 (Fig. 2B). For unphosphorylated Akt1, we examined the effect of Asp and Glu substitutions at position 473 (Fig. S6). Substitution with Asp at position 473 led to essentially inactive enzyme (Fig. 3B, Fig. S6, and Table 1). Replacement of Ser-473 with Glu resulted in an insignificant increase above the activity of unphosphorylated Akt1 enzyme (Fig. 2C, Fig. S6, and Table 1). In the context of pAkt1T308 (Fig. 2B), phosphomimetic mutations (S473D and S473E) failed to stimulate activity above pAkt1T308 with Ser-473. Indeed, all Ser-473 variants reduced pAkt1T308 activity with S473A showing the most significant reduction (Tables 1 and 2).
Table 2.
Akt enzyme activity comparison in vitro and in COS-7 cells
Enzyme activity determined biochemically is given as a percentage of the maximally active ppAkt1. +, agonist-evoked enzyme activity in cells; 0, no detectable activity; ND, not determined.
| Akt1 sites |
Akt1 activity |
||
|---|---|---|---|
| 308 | 473 | In vitro | In cell |
| % | % | ||
| pThr | pSer | 100 | + |
| Thr | Ser | <1 | ND |
| pThr | Ser | 27 | ND |
| pThr | Asp | 24 | + |
| pThr | Glu | 15 | + |
| pThr | Ala | 9 | + |
| Thr | pSer | 5 | ND |
| Asp | pSer | 10 | 0 |
| Glu | pSer | ND | 0 |
| Ala | pSer | <0.001 | 0 |
| pSer | pSer | ND | +a |
| pSer | Ser | 1 | ND |
| pSer | Ala | ND | +a |
| Thr | Asp | <1 | ND |
| Thr | Glu | <1 | ND |
| Asp | Ser | <1 | ND |
| Glu | Ser | <1 | ND |
a A reduced response was observed compared with WT when the Akt1 mutant indicated was expressed at a reduced level, similar to the level of endogenous Akt (Fig. 3B).
Akt1 mutant activity in live cells
Having assessed the activity of Akt mutants in vitro, we next examined the activity of Akt1 phosphorylation site mutants in live cells. To this end, we utilized our quantitative FRET-based kinase activity reporter, BKAR, and examined Akt signaling (36). Co-expression of kinase constructs with FRET-based activity sensors affords a sensitive assay for examining agonist-evoked signaling in a cellular context (41). As the response to endogenous Akt activity is so low, it provides an ideal system to evaluate signaling by introduced Akt variants, as a significant response can be observed from the overexpressed kinase.
Serum-starved COS-7 cells expressing BKAR and mCherry-tagged Akt variants were stimulated with epidermal growth factor (EGF) followed by Akt inhibition with GDC 0068. In real time, we monitored resulting changes in the BKAR FRET ratio (cyan fluorescent protein (CFP)/FRET) that are reflective of cellular Akt activity (Fig. 4A). For these experiments, cells selected for analysis had comparable levels of Akt1 expression, as assessed by quantifying mCherry levels; thus, in this system, one can clearly compare the relative signaling competence between WT and mutant Akt. We have documented previously that the mCherry tag does not significantly impact Akt1 activity (41).
Figure 4.
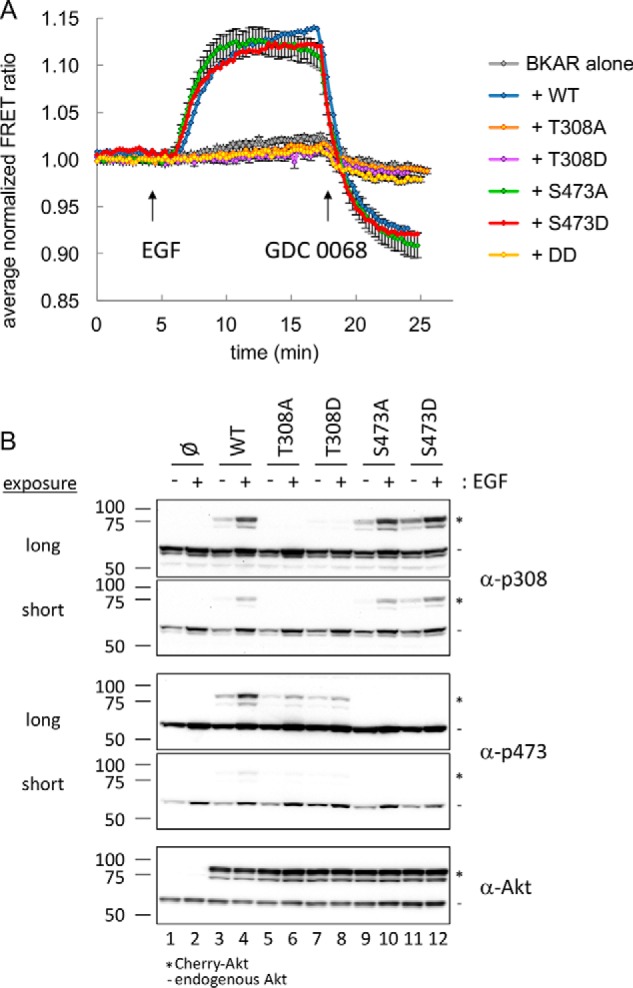
Cellular activity of Akt1 variants. A, serum-starved COS-7 cells expressing BKAR alone (gray) or BKAR with Cherry-tagged Akt WT (blue), T308A (orange), T308D (purple), S473A (green), S473D (red), or T308D/S473D (DD, yellow) were imaged, stimulated with EGF, and then treated with the Akt inhibitor GDC 0068. Multiple cells were included from at least two independent experiments for analysis. FRET ratios from each cell were normalized, and their average was plotted over time. Error bars, S.E. B, following a 10-min treatment with EGF, lysates from serum-starved COS-7 cells expressing the indicated Cherry-tagged Akt were analyzed by Western blotting for Akt activation using phospho-specific antibodies toward the activation loop (α-p308) and hydrophobic motif (α-p473). *, migration of Cherry-Akt; −, endogenous Akt.
Overexpression of WT Akt1 resulted in a significant BKAR response to EGF treatment compared with the response observed from endogenous Akt (Fig. 4A, blue versus gray trace). Interestingly, despite the level of overexpressed Akt in these experiments being in vast excess compared with endogenous Akt, substitution of either the nonphosphorylatable Ala or the “phosphomimetic” Asp or Glu at the Thr-308 site yielded an Akt1 enzyme that displayed no signaling in the cell (Fig. 4A (orange and purple traces) and Fig. S8). These data establish the requirement of pThr-308 for detectable Akt1 activation in cells and show that T308D is not a constitutively active Akt1 variant.
In contrast to the strict requirement for phosphorylation at Thr-308, either Ala, Asp, or Glu substitution at the Ser-473 site were tolerated without detrimental effects on cellular Akt activity, as assessed using BKAR (Fig. 4A (green and red trace) and Fig. S8). Importantly, a double mutation of both 308 and 473 to Asp resulted in an inactive enzyme. Thus, the activity of Akt monophosphorylated at Thr-308 is necessary and sufficient for maximal signaling in the context of cells.
Mutant Akt1 kinases are phosphorylated following EGF stimulation
We next examined the phosphorylation state of Thr-308 and Ser-473 of the mutant Akt kinases in our cell-based assay (Fig. 4B). In experiments in Fig. 5, we reduced the expression of the tagged Akt variants to be equivalent to the endogenous Akt expression level as determined by Western blotting. EGF stimulation (all even-numbered lanes) induced phosphorylation of both endogenous and WT Akt1 (lane 4) as assessed by Western blotting (Fig. 5). Importantly, phosphorylation of Thr-308 in the Ser-473 mutants (lanes 10 and 12) was at the same level as the WT Akt1 (lane 4), demonstrating that PDK1 phosphorylation downstream of PI3K signaling was intact. Analysis of Ser-473 phosphorylation revealed that, whereas this site was phosphorylated downstream of PI3K signaling, the level of phosphorylation at Ser-473 was reduced in the T308A and T308D mutants (lanes 6 and 8) compared with WT Akt1 (lane 4). This suggests that lack of phosphate on Thr-308 impairs phosphorylation of Ser-473. Although the level of Ser-473 phosphorylation was reduced in these mutants, the level of total overexpressed Akt protein was saturating in Fig. 4A. Given the excessive amount of Akt overexpressed in the experiments in Fig. 4A, this lack of activity observed using BKAR indicates that T308A, T308D, and T308E (Fig. S8) are clearly not able to signal, and any catalytic activity observed in these variants is below the threshold to result in detectable cellular activity.
Figure 5.
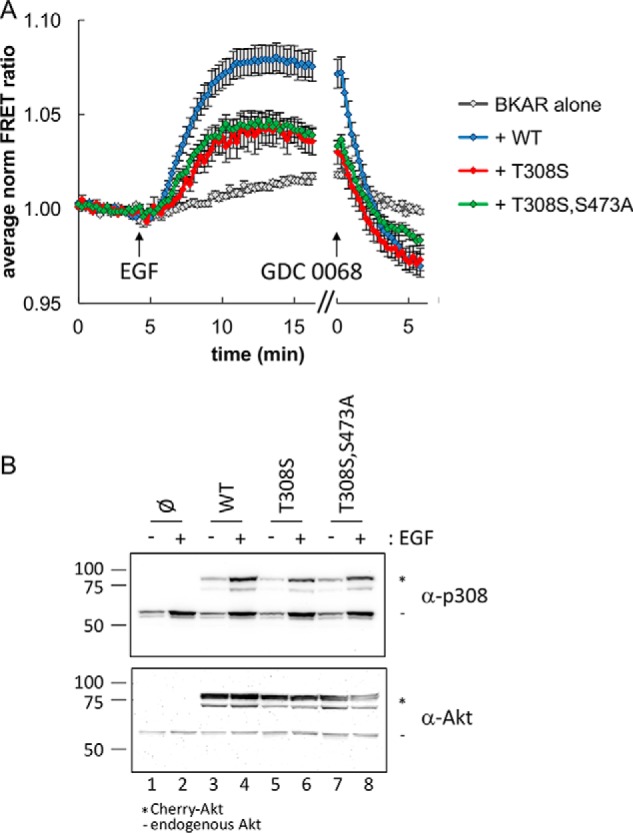
Reduced, but active, signaling from Akt T308S. A, serum-starved COS-7 cells expressing BKAR with or without minimally detectable levels of Cherry-tagged Akt were imaged during stimulation with EGF followed by treatment with the Akt inhibitor GDC 0068. Normalized average FRET ratios for WT (blue), T308S (red), and T308S/S473A (green) compared with BKAR alone (gray) are shown. Data were analyzed from cells expressing equal low levels of Cherry-Akt. Multiple cells were included from at least two independent experiments. FRET ratios from each cell were normalized, and their average was plotted over time. Error bars, S.E. B, following a 10-min treatment with EGF, lysates from serum-starved COS-7 cells expressing the indicated Cherry-tagged Akt were analyzed by Western blotting for Akt activation using phospho-specific antibodies toward the activation loop (α-p308) and hydrophobic motif (α-p473). *, migration of Cherry-Akt; −, endogenous Akt.
Impact of Ser substitution at Thr-308 in cells
As the recombinant Akt1D308 had similar activity as the pAkt1S308 protein in vitro, we were interested to examine the impact of Ser at 308 in the overexpressed mCherry-Akt system using BKAR. Overexpression of mCherry-Akt-T308S resulted in a BKAR response similar to that of WT Akt1 (data not shown). As this imaging system is monitoring an excess of the overexpressed mutant Akt, we attempted to express reduced levels of the kinase to discern whether there was a difference in cellular activity between WT Akt1 and Akt1 with Ser at position 308. Indeed, titrating down the expression levels and selecting for the lowest-expressing cells based on mCherry intensity, we observed a reduction in Akt1 signaling output from the Ser-308 enzyme compared with the WT enzyme with Thr-308 (Fig. 5A, blue and red traces). In the impaired kinase (Akt1S308), we then substituted the 473 site with Ala (Ser-308 and Ala-473); however, even in the context of an impaired Akt kinase, mutation of the 473 site did not influence Akt signaling (Fig. 5A, green trace). Under the same conditions noted here to analyze minimal levels of overexpressed kinase, we were unable to detect a reduced level in signaling from the S473A mutant compared with WT Akt1 (Fig. S9). This result is in agreement with our observations that mutation of Ser-473 to Ala in the context of Akt1 phosphorylated at residue 308 either on Thr (Fig. 4, green trace) or Ser (Fig. 5, green trace) does not impact signaling in cells. Furthermore, the data indicate that the ability of pAkt1T308 S473A to signal is greater than that of Akt1 T308S.
Western blot analysis confirmed that phosphorylation of the 308 site (even with a Ser substitution) was intact following EGF stimulation (Fig. 5B). Our data in live cells indicate that phosphate, specifically, not an acidic residue, at position 308 is the most critical component to induce Akt activity. In the cell, as we found in vitro, phosphomimetic substitutions of Asp or Glu at the 308 site are unable to propagate Akt signaling, and, in agreement with our enzymatic data, substitution of Thr-308 to Ser leads to reduced Akt activity and signaling.
Discussion
Using genetic code expansion and in vivo enzymatic phosphorylation, we produced fully active Akt1 directly from E. coli for the first time. We validated our biochemical findings in COS-7 cells using a genetically encoded reporter for Akt signaling. This allowed us to 1) measure the specific activity of Akt1 site-specifically and exclusively phosphorylated at either or both regulatory sites, Thr-308 and Ser-473, 2) systematically examine the role of phosphate at each site in modulating the intrinsic catalytic activity of the pure enzyme, and 3) examine the effectiveness of amino acid substitutions to mimic phosphorylated or unphosphorylated residues in cells.
Generation of active Akt1
We demonstrated the compatibility of enzymatic phosphorylation with genetically encoded phosphoserine incorporation by producing site-specific and doubly phosphorylated Akt1 (Fig. 1A). This was achieved by co-expressing the upstream kinase PDK1, to induce Thr-308 phosphorylation, and simultaneously genetically encoded pSer-473 in Akt1. To our knowledge, this is the first demonstration of protein production in E. coli with programmed pSer and pThr residues in the same protein. Consistent with numerous studies (24), our in vitro kinase assays revealed robust activity of ppAkt1 activity in the absence of PIP3, suggesting that PIP3 is not required for Akt1 activity per se. This contradicts a recent study proposing that PIP3 binding may be essential for both Akt1 activation and activity in cells (42). As noted above, producing site-specifically and doubly phosphorylated, and thus fully activated, Akt1 has previously not been achieved via recombinant expression in vivo in E. coli, given the complicated nature of Akt activation by multiple kinases, chaperones, and lipid second messengers (24). Our experiments demonstrate a novel approach, taking advantage of genetic code expansion and enzymatic phosphorylation, that overcomes the existing limitations in both methods to produce fully active human kinases.
Role of phosphorylation in activating Akt1
The kinetic mechanisms of Akt activation by phosphorylation are less well-characterized relative to other kinases, such as protein kinase A (PKA) (43) and PKC (44), stemming in part from the difficulty in obtaining Akt in specifically phosphorylated forms. Most studies relied on immunoprecipitating Akt from unstimulated and growth factor–stimulated cells. Akt1 activity is then determined using a kinase activity assay in the immunoprecipitates. Such studies typically report a 10–50-fold stimulation of the activity of Akt immunoprecipitated from growth factor–treated compared with unstimulated cells (e.g. see Refs. 45–49). Alessi et al. (6) showed that Akt1 had 45-fold higher activity when immunoprecipitated from cells treated with IGF-1 compared with untreated cells. In the context of an Akt1 mutant with Ala present at position 473, the increase was only 5-fold, and if Ala was present at position 308, there was essentially no stimulated activity. Similar results were reported by Hemmings and co-workers for Akt3 (50). These data are qualitatively similar to the results from our study, yet our quantitative analysis defines Thr-308 as the critical regulator of Akt activity, and its phosphorylation alone increases activity to approximately one-third that of the doubly phosphorylated enzyme. Our unique ability to produce recombinant fully phosphorylated Akt1 from bacteria has revealed that specific phosphorylation at both regulatory sites increases the intrinsic catalytic activity by over 3 orders of magnitude, defining the stringency with which Akt signaling is “silent” in the absence of agonist stimulation and the resulting phosphorylation of Akt.
Several lines of evidence support our finding that phosphorylation of Thr-308, but not Ser-473, is necessary and sufficient for the activation of the intrinsic catalytic activity of Akt. First, Hart and Vogt (26) concluded that phosphorylation of this site, but not Ser-473, was sufficient for the oncogenic potential of Akt. Specifically, they showed that mutating Thr-308 prevented the phosphorylation of Akt substrates and transformation of chicken embryonic fibroblasts. Second, Jacinto et al. (14) showed that in cells lacking mTORC2, Akt was not phosphorylated on Ser-473, yet retained the ability to phosphorylate a subset of Akt substrates. Based on this and other reports from the literature, phosphorylation of Ser-473 was suggested to determine substrate specificity of Akt. Supporting a role of Ser-473 in the cellular function of Akt, loss of phosphorylation of this site by overexpressing the PH domain leucine-rich repeat protein phosphatase (PHLPP) (which dephosphorylates Ser-473) results in increased apoptosis (51), and conversely, enhancing phosphorylation at Ser-473 by depletion of PHLPP results in suppression of tumors in a xenograft model. Although these effects on apoptosis may arise from non-Akt functions of PHLPP (52), they support a role of Ser-473 phosphorylation in Akt biology. Phosphorylation at Ser-473 may function to tune rather than activate Akt signaling. However, our results establish that the phosphorylation state of Thr-308 is a superior readout for the activation state of Akt. Whereas phosphorylation at Thr-308 alone is able to activate Akt1, phosphorylation of Ser-473 alone leads to relatively weak activity in vitro and no detectable activity in the cell. Neither the Asp-308 nor Glu-308 Akt1 mutants had EGF-stimulated activity in cells. Although the pAkt1S473 T308D mutant had 10% of the activity of the doubly phosphorylated Akt1 in vitro (Table 2), we observed no cellular Akt1 activity and impaired Ser-473 phosphorylation in Akt1 variants with Asp, Glu, or Ala mutations at position 308. Even if these variants are active in the cell, the low intrinsic activity of the Asp-308 mutant, for example, may be too low to overcome the opposing dephosphorylation reaction catalyzed by cellular phosphatases acting on Akt substrates, including the BKAR reporter. Akt1 T308D mutants can no longer be regarded as constitutively active Akt1 variants (30–32).
The phosphorylation status of Akt1 is indeed clinically relevant. Whereas pSer-473 is the most commonly used biomarker (53, 54), pThr-308 was identified as a better prognostic biomarker in human non-small-cell lung cancer (55) and acute myeloid leukemia (56). Our study underscores the relevance of examining the phosphorylation state of Thr-308 as a general marker for the activation state of Akt in basic research and clinical settings.
Acidic residues do not function as phosphomimetics in Akt1
Thorsness and Koshland (57) first introduced the use of acidic amino acids to mimic phosphate, showing that an Asp mimicked the functional effect of phosphate to inactivate isocitrate dehydrogenase. There are other examples where the negative charge of the amino acid effectively mimics that of phosphate, and indeed, nature has used the trick in reverse; it is estimated that 5% of pSer sites may have evolved from an ancestor with Glu or Asp at the homologous position (34). However, acidic residues are not necessarily “phosphomimetics”; their negative charge, hydration sphere, and size are considerably different from those of a phosphorylated Ser or Thr residue. Acidic amino acids have a net negative charge of 1 compared with 2 for phosphate at neutral pH, the hydration sphere is 4 waters compared with 14 for phosphate, and their volume is considerably smaller than the phospho-amino acids they replace.
Because kinases are often activated by phosphorylation, Glu and Asp mutations are routinely used to mimic the function of phosphorylated serine or threonine residues in many kinases (33), including Akt1 (26, 29). Our current study reveals that acidic residues do not mimic phosphorylation in Akt1. This is an important finding, given the widespread and continued use of phosphomimetics in interrogating Akt signaling, and kinase signaling generally, in cells. The phosphomimetic variants that were active in our assays are 30–210-fold less active than their monophosphorylated counterparts. Based on our data in live cells, Thr-308 phosphomimetic variants fail to activate Akt signaling (Fig. 4A). Although Akt1 T308D was the most active of the phosphomimetic mutants in vitro, this level of activity is insufficient to produce a cellular response. This result is consistent with a study from Hart and Vogt (26), who reported that T308D was an “unsuitable” substitution for pThr-308. Their data showed that Akt T308D did not cause the phosphorylation of cellular Akt substrates such as GSK-3β, nor did it induce oncogenic transformation, functions that were robustly mediated by WT Akt. Nonetheless, phosphomimetic constructs of Akt pervade the literature on Akt-signaling mechanisms. Because phosphomimetics do not result in physiologically relevant activation of the kinase activity of Akt, it is possible that any biological effects of such mutations may arise from scaffolding functions or noncatalytic mechanisms, as increasingly described for protein kinase family members (58).
An interesting finding from our study is that pSer is not well-tolerated at position 308. According to the in vitro kinase assays, pAkt1S308 is more active than phosphomimetic 308 mutants, yet pAkt1T308 is ∼25-fold more active than pAkt1S308. Furthermore, when phosphorylated post-translationally in the cell, pAkt1S308 also showed reduced activity compared with that of pAkt1T308. To the best of our understanding, there is no literature-based evidence for the effect of pSer-308 on the activation of Akt1 in vivo or in vitro. Our results indicate that the γ-methyl group of Thr-308 optimally positions the phosphate to create the ordered hydrogen-bonding network between the phosphate of pThr-308, His-194, and Arg-273, which facilitates the binding to ATP and substrate at the Akt1 active site (26). Indeed, this observation is supported by crystallographic structures of Akt in partially active form and in complex with substrate peptide and an ATP analog (59, 60). The structure shows that pThr-308 is involved in salt bridge interactions with Arg-273 and Lys-297, and a water-mediated hydrogen bond with His-194 is also evident (Fig. 6). In the structure, steric complementarity can be seen between the γ-methyl of pThr-308 and Cys-310 (Fig. 6B). In agreement with our cell-based and biochemical data, the structure suggests that the interaction between the γ-methyl of pThr-308 and Cys-310 optimally orients the phosphate for interaction with its positively charged counterparts in the salt bridges. We hypothesize that pSer-308, which lacks this methyl group, will rotate more freely in the active site, perhaps shifting the enzyme in and out of its active confirmation.
Figure 6.
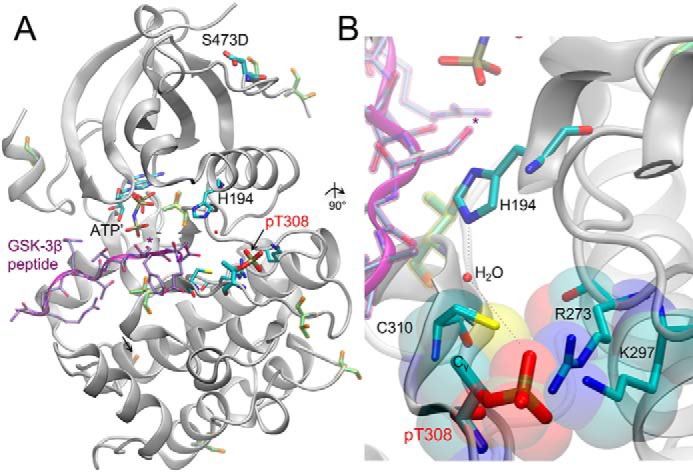
Structure of human Akt in complex with GSK-3β substrate peptide. A, structure of the active human pAktT308 (S473D) kinase domain (gray, cartoon) shown in complex with GSK-3β peptide (purple) and a nonhydrolyzable ATP analog (AMP-PNP (ATP′)). *, target of Akt1 phosphorylation on the GSK-3β substrate peptide. Key residues are labeled, and in addition to the regulatory phosphorylation sites (pThr-308 and phosphomimetic mutation S473D), other Ser/Thr phosphorylation sites are highlighted (yellow). B, a 90° rotated and close-up view of the Akt1 active site, focused on the position of pThr-308. The phosphate at position 308 makes extensive salt-bridge interactions with Arg-273 and Lys-297; pThr-308 also participates in a water-mediated hydrogen bond network with His-194. The Cγ methyl group on pThr-308 forms a hydrophobic interaction with Cys-310, as indicated in the van der Waals surface representation (transparent). The figure includes structural data from Protein Data Bank entries 3CQU (59) and 1O6K (60).
Our study also revealed that Ala is not a mimic of unphosphorylated Thr-308. Ala at position 308 resulted in a 10-fold reduction in the basal activity of Akt1 monophosphorylated at Ser-473. Taylor and colleagues (61, 62) also found that Ala at the activation loop of PKA results in a more severe kinetic defect compared with having an unphosphorylated Thr at the activation loop. The detrimental effect of Ala may arise from a loss of hydrogen-bonding ability. The Ala mutation favors a helical conformation of the activation loop that may also drive structural ensembles away from the active conformation, or it may favor an inhibited conformation due to interactions with other domains or proteins.7
Akt1 activation threshold in cells
Live cell imaging experiments revealed that all forms of Akt1 that had phosphorylation of Thr at position 308 had maximal signaling output following EGF stimulation of cells, regardless of the amino acid at position 473 (Table 2). In contrast, no activity was observed if the residue at position 308 was not phosphorylated, regardless of the amino acid at position 473 (Table 2). Based on in vitro data, we can estimate a threshold to observe Akt activity in cells; our in vitro studies reveal that phosphorylation of Ser-473 alone stimulated Akt1 activity 80-fold over the unphosphorylated enzyme, yet this level of Akt1 activity is apparently below the threshold required to elicit Akt1 signaling in the cell. In vitro, the activity of monophosphorylated Akt1 at Thr-308 was reduced by <3-fold compared with the maximally active and doubly phosphorylated enzyme, and this level of activity is sufficient for maximal signaling output in cells. In addition, pAkt1T308 S473A was 10-fold less active than the doubly phosphorylated enzyme, yet this enzyme was sufficient for robust Akt1 signaling in the cell. Taken together, our data suggest that Akt1 variants whose intrinsic catalytic activity is greater than ∼10% of that of doubly phosphorylated enzyme are robustly active in cells. This novel finding further suggests that in cells, only a fraction of Akt needs to be activated by phosphorylation for full Akt-dependent signaling.
Although previous reports have identified a 10-fold activity increase associated with Ser-473 phosphorylation in immunoprecipitates, the stoichiometry of phosphorylation was unknown (6). Furthermore, the phosphorylation state of Akt1 may differentially impact substrates based on the local concentration or abundance of the substrate. For example, if Akt1 is poised on a scaffold and co-localized with substrate, a lack of Ser-473 phosphorylation may be irrelevant, as the local concentration of substrate is high. For phosphorylation of an untethered or low abundant substrate, that 3-fold difference in activity may reduce or eliminate Akt1 signaling in a substrate-specific manner. Indeed, previous work suggested that phosphorylation at Ser-473 may increase the activity of Akt1 for particular substrates (e.g. FoxO1/3a) (14), yet significant future efforts are required to define the impact of Akt1 phosphorylation status on substrate selectivity.
Mutation and phosphorylation state can impact Akt1 activity directly, but also indirectly by altering the accessibility of phosphatases. For example, previous studies showed that Ala substitution in PKC at the hydrophobic motif increased phosphatase sensitivity of PKC at additional sites (63). Our finding that Akt1 mutants with very low activity did not signal in cells suggests that localized activation needs to exceed a threshold to outcompete phosphatases in the cellular environment. Thus, the dynamic range of Akt signaling in a cell is such that sufficient Akt needs to be activated to overcome phosphatase suppression, but additional changes in phosphorylation site occupancy may or may not be needed for maximal signaling, particularly if localized to protein scaffolds and microdomains (64).
Conclusion
We established a robust protocol to generate and characterize fully active and differentially active Akt1 variants. These Akt1 variants will next be employed as a unique set of tools to investigate Akt1 substrate specificity and to screen for potential drug candidates against the most potent and active forms of the oncogenic kinase. The ability to produce optimally active Akt1 will have broad implications for drug discovery efforts, as we hypothesize that drug screening against a fully active Akt1 enzyme will more likely produce a potent and selective inhibitor.
We presented a systematic study of the impact of phosphorylation and phosphomimetic substitutions on the activity of Akt1 in the test tube and in living cells. Our data suggest that phosphomimetic substitutions should be tested for their ability to mimic phosphorylation, and Ala substitutions should also be assessed for their ability to act as nonphosphorylatable counterparts. We found that these common assumptions simply do not hold for Akt1, and complementary experiments in vitro and in the cell were invaluable in reaching this conclusion. Viewing the cell-based assays in isolation from the kinase activity data may have led to the erroneous conclusion that phosphomimetic substitution at 473 supports full Akt1 activity. Rather, the situation in the cell is more complex. Phosphorylation at 308 alone is sufficient to observe maximal Akt1 signaling, and mutation of 473 to Ala, Asp, or Glu had no apparent impact on Akt1 activity in the cell. In the test tube, we determined that phosphomimetics fall far short of activating Akt1 to the same extent as true phosphorylation at both regulatory sites. Indeed and counterintuitively, we found that T308D is a good mimic of a lack of phosphorylation at 308, whereas T308A is a kinase-dead enzyme that is not active in vitro despite phosphorylation at 473. In the cell, only true phosphorylation at 308 led to observable activity, suggesting that mimics of pThr are particularly inappropriate at this site. Interestingly, pSer-308 shows reduced activity, which was still sufficient to observe Akt1 signaling in the cell. The data demonstrate conclusively that pSer is a far better mimic of pThr than Asp or Glu phosphomimetic substitutions.
The foregoing experiments reveal that phosphorylated Thr-308 is necessary, sufficient, and irreplaceable for activation of Akt in vitro and in cells. We showed that no other residue is tolerated at that site, including acidic amino acids, which are frequently used as “phosphomimetics.” Our findings also question the generally assumed validity of using Ala mutation to mimic a nonphosphorylated form of residues like Ser and Thr. In total, our results beg caution in using amino acid substitutions to examine the role of phosphorylation in protein function and in the biology of cell signaling.
Experimental procedures
Bacterial strains and plasmids
The full-length human AKT1 gene was cloned into a pUC18-derived vector (pDS1 (38)) and pCDFDuet1 vector (see supporting Experimental procedures). Ser-473 and Thr-308 sites were mutated by site-directed mutagenesis in E. coli DH5α to amber (TAG), Asp (GAC), or Glu (GAG) codons according to methods described previously (65). All clones were verified by DNA sequencing (London Genomics Research Center, Robarts Research Institute, London, Canada; Genewiz Inc., South Plainfield, NJ). Phosphoproteins were produced by genetically encoding pSer at UAG codons with the second-generation pSer incorporation system (pDS-pSer2, kanamycin-resistant) (38, 66). Recombinant Akt1 and pAkt1 variants were over expressed in E. coli BL21(DE3) (Invitrogen); exogenous pSer (5 mm) was added to the expression media for production of pAkt1 variants (see supporting Experimental procedures for a detailed protocol). For the imaging studies, the mouse Akt1 gene (a gift from A. Toker; 98.3% amino acid identity to human Akt1) was subcloned into pcDNA3 (Invitrogen) containing mCherry at its N terminus. Mutations were generated by QuikChange mutagenesis (Agilent Genomics) and confirmed by Sanger sequencing (Eton Bioscience). Generation of BKAR was described previously (36).
Protein production and purification
For phosphoprotein production, pDS1-AKT1 expression plasmid variants with TAG codons at the indicated phosphosite were co-transformed with pDS-pSer2 into E. coli BL21(DE3). For unphosphorylated and phosphomimetic Akt1 variants, the appropriate pDS1-AKT1 plasmid was transformed alone into BL21(DE3). Cells were grown and pelleted at 5000 × g as described in the supporting Experimental procedures. The cell pellets were resuspended in lysis buffer (20 mm Hepes, 150 mm NaCl, 3 mm β-mercaptoethanol, 3 mm DTT, 10 mm imidazole) at 10 ml/g of cells. Lysis buffer for phosphoproteins contained phosphatase inhibitors (1 mm Na3VO4 and 5 mm NaF). One tablet of EDTA-free mini protease inhibitor mixture (Roche Applied Science) and 1 mm phenylmethylsulfonyl fluoride were added to the cell suspension (typically 200 ml). Cells were treated with lysozyme (1 mg/ml) for 20 min, shaking at 4 °C, and lysed using a French pressure cell press (American Instrument Co. Inc.) at 1000 p.s.i. Cell lysates were centrifuged at 38,000 × g for 1 h at 4°C. The supernatant was filtered through a 1.2-μm filter, mixed with nickel-nitrilotriacetic acid affinity beads (Thermo Scientific), and pre-equilibrated with lysis buffer for 1 h. Finally, proteins bound to beads were purified under gravity flow. Elution fractions were further purified using a Superdex200 gel filtration column attached to an AKTA Pure L1 fast protein LC (FPLC) system (GE Healthcare, Little Chalfont, UK) (see supporting Experimental procedures). Protein yields ranged from 20 to 100 μg/liter of E. coli culture.
In vitro Akt1 kinase assay
For the kinase activity assay, we established a set of common conditions to assay a series of Akt1 variants of widely varying activity. To achieve this, we worked in a regime of subsaturating ATP; thus, our apparent reaction constants (kapp) serve well to compare the Akt1 variants with one another, but these rates are significantly lower than kcat. Our data represent single-turnover kinetics. Akt1 activity was determined using 200 μm substrate peptide CKRPRAASFAE (SignalChem, Vancouver, Canada) derived from the natural Akt1 substrate, GSK-3β. The reported Km of Akt1 for a similar substrate peptide is 18 μm (67), so the peptide concentration is in excess. Assays were performed in MOPS (25 mm, pH 7.0), β-glycerolphosphate (12.5 mm), MgCl2 (25 mm), EGTA (5 mm, pH 8.0), EDTA (2 mm), ATP (20 μm), and 0.4 μCi (33 nm) of [γ-32P]ATP in a 30-μl reaction volume, at 37 °C in 30-min time courses. Unless otherwise indicated, reactions were initiated by the addition of 18 pmol of the indicated Akt1 variant (to yield a concentration of 600 nm) and quenched by spotting 5 μl from each reaction on P81 paper at specified time points (68). Following washes with 1% phosphoric acid (3 × 10 min) and 95% ethanol (1 × 5 min), P81 paper was air-dried and exposed to a phosphor screen. The 32P-peptide products were imaged and quantitated using a Storm 860 molecular imager and ImageQuant TL software (Molecular Dynamics, Caesarea, Israel).
Cell culture media and conditions
COS-7 cells were maintained in Dulbecco's modified Eagle's medium (Cellgro) containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in 5% CO2. For cell imaging experiments, cells were plated onto sterilized glass coverslips in 35-mm dishes before transfection with 1 μg of pcDNA3-BKAR with or without 1 μg of pcDNA3-mCherry-Akt using Lipofectamine 3000 (Invitrogen). For experiments in which low expression levels of Cherry-Akt were desired, cells were transfected with 1 μg of pcDNA3-BKAR and 0.05 μg of pcDNA3-mCherry-Akt. Cells were imaged within 24 h following transfection. For Western blotting experiments, in which average overexpressed Akt levels were desired to be at a similar level to endogenous Akt, COS-7 cells were transfected with 0.05 μg of pcDNA3-mCherry-Akt using Lipofectamine 3000 (Invitrogen) in 6-well dishes.
Cell imaging
Transfected COS-7 cells were serum-starved at least 4 h (as long as overnight) before imaging. Cells were washed one time in Hanks' balanced salt solution (HBSS; Cellgro) supplemented with 1 mm CaCl2 and imaged in this HBSS in the dark at room temperature. Data were collected on a Zeiss Axiovert microscope (Carl Zeiss Microimaging, Inc.) using a MicroMax digital camera (Roper-Princeton Instruments) controlled by MetaFluor software (Universal Imaging Corp.). Optical filters were obtained from Chroma Technologies and Semrock. Data were collected through a 10% neutral density filter. CFP and FRET images were obtained every 15 s through a 420/20-nm excitation filter, a 450-nm dichroic mirror, and a 475/40- or 535/25-nm emission filter for CFP and FRET, respectively. Cherry images were acquired through a 560/25-nm excitation filter, a 593-nm dichroic mirror, and a 629/53-nm emission filter. Excitation and emission filters were switched in filter wheels (Lambda 10-2, Sutter). Integration times were 200 ms for CFP and FRET and 150 ms for mCherry. Cells were stimulated with 50 ng/ml EGF (PeproTech, Inc.) and then treated with 20 μm GDC 0068 (Selleckchem.com) at the indicated times. Data were normalized to the first 3 min of the experiment for each cell. Error bars represent S.E.
Western blotting
Within 24 h after COS-7 cell transfection, cells were serum-starved for at least 4 h. Cells were rinsed once in HBSS/1 mm Ca2+ and then left untreated or stimulated with 50 ng/ml EGF for 10 min at room temperature. Cells were lysed in 50 mm Na2HPO4, 1 mm Na4P2O7, 20 mm NaF, 2 mm EDTA, 2 mm EGTA, 1% Triton X-100 (supplemented with 1 mm DTT, 200 μm benzamidine, 40 μg/ml leupeptin, 300 μm phenylmethylsulfonyl fluoride, and 1 μm microcystin) and cleared by a high-speed centrifugation for 2.5 min. The cleared lysates were analyzed by Western blotting to determine the relative amounts of Akt-Thr-308 (phospho-Akt (Thr-308), Cell Signaling catalog no. 9275) or Akt-Ser-473 (phospho-Akt (Ser-473) (D9E) XP, Cell Signaling catalog no. 4060) phosphorylation. Total Akt levels were assessed with an antibody against all Akt isozymes (anti-AKT1/2/3, Abcam catalog no. 126811). Western blots were developed using chemiluminescence.
Author contributions
N. B., M. T. K., A. C. N., and P. O. conceptualization; N. B., M. T. K., K. K. B., A. C. N., and P. O. data curation; N. B., M. T. K., X. L., and K. K. B. formal analysis; N. B. and M. T. K. validation; N. B., M. T. K., X. L., S. S.-C. L., A. C. N., and P. O. investigation; N. B., M. T. K., X. L., K. K. B., and P. O. visualization; N. B., M. T. K., X. L., K. K. B., S. S.-C. L., A. C. N., and P. O. methodology; N. B., M. T. K., A. C. N., and P. O. writing-original draft; N. B., M. T. K., A. C. N., and P. O. writing-review and editing; S. S.-C. L., A. C. N., and P. O. supervision; M. T. K., S. S.-C. L., A. C. N., and P. O. funding acquisition; A. C. N. and P. O. resources; A. C. N. and P. O. project administration.
Supplementary Material
Acknowledgments
We are grateful to Ilka Heinemann and David Litchfield for critical discussions and suggestions on the manuscript and to Alexandr Kornev and Susan Taylor for helpful discussions.
This work was supported by Natural Sciences and Engineering Research Council of Canada Grant RGPIN 04282-2014 (to P. O.), Canada Foundation for Innovation Grant 229917 (to P. O.), Ontario Research Fund Grant 229917 (to P. O.), Canada Research Chairs 950-229917 (to P. O.), the Canadian Cancer Society Research Institute Innovation Grant 704324 (to P. O. and S. L.), the Canadian Breast Cancer Foundation (to S. L.), and National Institutes of Health Grants R03 CA178524 (to M. T. K.) and R35 GM122523 and R01 GM43154 (to A. C. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting Experimental procedures and Figs. S1–S9.
A. Kornev and S. Taylor, personal communication.
- PKB
- protein kinase B
- PI3K
- phosphoinositide 3-kinase
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PIP3
- phosphatidylinositol 3,4,5-triphosphate
- PH
- pleckstrin homology
- PDK1
- phosphoinositide-dependent kinase 1
- mTOR
- mechanistic target of rapamycin
- mTORC2
- mTOR complex 2
- BKAR
- B kinase activity reporter
- GSK-3β
- glycogen synthase kinase 3β
- PRM-MS/MS
- parallel reaction–monitoring MS/MS
- EGF
- epidermal growth factor
- CFP
- cyan fluorescent protein
- PHLPP
- PH domain leucine-rich repeat protein phosphatase
- PKA
- protein kinase A
- HBSS
- Hanks' balanced salt solution
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate.
References
- 1. Manning B. D., and Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanada M., Feng J., and Hemmings B. A. (2004) Structure, regulation and function of PKB/AKT–a major therapeutic target. Biochim. Biophys. Acta 1697, 3–16 10.1016/j.bbapap.2003.11.009 [DOI] [PubMed] [Google Scholar]
- 3. Altomare D. A., and Testa J. R. (2005) Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 10.1038/sj.onc.1209085 [DOI] [PubMed] [Google Scholar]
- 4. Dai D. L., Martinka M., and Li G. (2005) Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J. Clin. Oncol. 23, 1473–1482 10.1200/JCO.2005.07.168 [DOI] [PubMed] [Google Scholar]
- 5. Wong K. K., Engelman J. A., and Cantley L. C. (2010) Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 20, 87–90 10.1016/j.gde.2009.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., and Hemmings B. A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 7. Hassan B., Akcakanat A., Holder A. M., and Meric-Bernstam F. (2013) Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg. Oncol. Clin. N. Am. 22, 641–664 10.1016/j.soc.2013.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martini M., De Santis M. C., Braccini L., Gulluni F., and Hirsch E. (2014) PI3K/AKT signaling pathway and cancer: an updated review. Ann. Med. 46, 372–383 10.3109/07853890.2014.912836 [DOI] [PubMed] [Google Scholar]
- 9. Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R., Reese C. B., and Cohen P. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269 10.1016/S0960-9822(06)00122-9 [DOI] [PubMed] [Google Scholar]
- 10. Keranen L. M., Dutil E. M., and Newton A. C. (1995) Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol. 5, 1394–1403 10.1016/S0960-9822(95)00277-6 [DOI] [PubMed] [Google Scholar]
- 11. Tsutakawa S. E., Medzihradszky K. F., Flint A. J., Burlingame A. L., and Koshland D. E. Jr. (1995) Determination of in vivo phosphorylation sites in protein kinase C. J. Biol. Chem. 270, 26807–26812 10.1074/jbc.270.45.26807 [DOI] [PubMed] [Google Scholar]
- 12. Pearson R. B., Dennis P. B., Han J.-W., Williamson N. A., Kozma S. C., Wettenhall R. E. H., and Thomas G. (1995) The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 14, 5279–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- 14. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., and Su B. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 10.1016/j.cell.2006.08.033 [DOI] [PubMed] [Google Scholar]
- 15. Iacovides D. C., Johnson A. B., Wang N., Boddapati S., Korkola J., and Gray J. W. (2013) Identification and quantification of AKT isoforms and phosphoforms in breast cancer using a novel nanofluidic immunoassay. Mol. Cell. Proteomics 12, 3210–3220 10.1074/mcp.M112.023119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. LoRusso P. M. (2016) Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J. Clin. Oncol. 34, 3803–3815 10.1200/JCO.2014.59.0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Massihnia D., Avan A., Funel N., Maftouh M., van Krieken A., Granchi C., Raktoe R., Boggi U., Aicher B., Minutolo F., Russo A., Leon L. G., Peters G. J., and Giovannetti E. (2017) Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer. J. Hematol. Oncol. 10, 9 10.1186/s13045-016-0371-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liao Y., Yuan S., Chen X., Zhu P., Li J., Qin L., and Liao W. (2017) Up-regulation of BRCA1-associated RING domain 1 promotes hepatocellular carcinoma progression by targeting Akt signaling. Sci. Rep. 7, 7649 10.1038/s41598-017-07962-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao J., Yu W., Hu K., Li M., Chen J., and Li Z. (2017) miR-92a promotes tumor growth of osteosarcoma by targeting PTEN/AKT signaling pathway. Oncol. Rep. 37, 2513–2521 10.3892/or.2017.5484 [DOI] [PubMed] [Google Scholar]
- 20. Parker L., Levinger I., Mousa A., Howlett K., and de Courten B. (2016) Plasma 25-hydroxyvitamin D is related to protein signaling involved in glucose homeostasis in a tissue-specific manner. Nutrients 8, E631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang H., Wu Y., Liu M., Qin Y., Wang H., Wang L., Li S., Zhu H., He Z., Luo J., Wang H., Wang Q., and Luo S. (2016) SEMA3B improves the survival of patients with esophageal squamous cell carcinoma by upregulating p53 and p21. Oncol. Rep. 36, 900–908 10.3892/or.2016.4901 [DOI] [PubMed] [Google Scholar]
- 22. Yang Y., Huang Y., Wang Z., Wang H. T., Duan B., Ye D., Wang C., Jing R., Leng Y., Xi J., Chen W., Wang G., Jia W., Zhu S., and Kang J. (2016) HDAC10 promotes lung cancer proliferation via AKT phosphorylation. Oncotarget 7, 59388–59401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klein S., Geiger T., Linchevski I., Lebendiker M., Itkin A., Assayag K., and Levitzki A. (2005) Expression and purification of active PKB kinase from Escherichia coli. Protein Expr. Purif. 41, 162–169 10.1016/j.pep.2005.01.003 [DOI] [PubMed] [Google Scholar]
- 24. Zhang X., Zhang S., Yamane H., Wahl R., Ali A., Lofgren J. A., and Kendall R. L. (2006) Kinetic mechanism of AKT/PKB enzyme family. J. Biol. Chem. 281, 13949–13956 10.1074/jbc.M601384200 [DOI] [PubMed] [Google Scholar]
- 25. Fabbro D., Batt D., Rose P., Schacher B., Roberts T. M., and Ferrari S. (1999) Homogeneous purification of human recombinant GST-Akt/PKB from Sf9 cells. Protein Expr. Purif. 17, 83–88 10.1006/prep.1999.1102 [DOI] [PubMed] [Google Scholar]
- 26. Hart J. R., and Vogt P. K. (2011) Phosphorylation of AKT: a mutational analysis. Oncotarget 2, 467–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tobisawa T., Yano T., Tanno M., Miki T., Kuno A., Kimura Y., Ishikawa S., Kouzu H., Nishizawa K., Yoshida H., and Miura T. (2017) Insufficient activation of Akt upon reperfusion because of its novel modification by reduced PP2A-B55α contributes to enlargement of infarct size by chronic kidney disease. Basic Res. Cardiol. 112, 31 10.1007/s00395-017-0621-6 [DOI] [PubMed] [Google Scholar]
- 28. Warsi J., Fezai M., Fores M., Elvira B., and Lang F. (2015) Up-regulation of voltage gated K+ channels Kv1.3 and Kv1.5 by protein kinase PKB/Akt. Cell Physiol. Biochem. 37, 2454–2463 10.1159/000438598 [DOI] [PubMed] [Google Scholar]
- 29. Liu P., Begley M., Michowski W., Inuzuka H., Ginzberg M., Gao D., Tsou P., Gan W., Papa A., Kim B. M., Wan L., Singh A., Zhai B., Yuan M., Wang Z., et al. (2014) Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545 10.1038/nature13079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhuo D. X., Zhang X. W., Jin B., Zhang Z., Xie B. S., Wu C. L., Gong K., and Mao Z. B. (2013) CSTP1, a novel protein phosphatase, blocks cell cycle, promotes cell apoptosis, and suppresses tumor growth of bladder cancer by directly dephosphorylating Akt at Ser473 site. PLoS One 8, e65679 10.1371/journal.pone.0065679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Munoz C., Almilaji A., Setiawan I., Föller M., and Lang F. (2013) Up-regulation of the inwardly rectifying K+ channel Kir2.1 (KCNJ2) by protein kinase B (PKB/Akt) and PIKfyve. J. Membr. Biol. 246, 189–197 10.1007/s00232-012-9520-9 [DOI] [PubMed] [Google Scholar]
- 32. Berndt N., Yang H., Trinczek B., Betzi S., Zhang Z., Wu B., Lawrence N. J., Pellecchia M., Schönbrunn E., Cheng J. Q., and Sebti S. M. (2010) The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ. 17, 1795–1804 10.1038/cdd.2010.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dissmeyer N., and Schnittger A. (2011) Use of phospho-site substitutions to analyze the biological relevance of phosphorylation events in regulatory networks. Methods Mol. Biol. 779, 93–138 10.1007/978-1-61779-264-9_6 [DOI] [PubMed] [Google Scholar]
- 34. Pearlman S. M., Serber Z., and Ferrell J. E. Jr. (2011) A mechanism for the evolution of phosphorylation sites. Cell 147, 934–946 10.1016/j.cell.2011.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hunter T. (2012) Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2513–2516 10.1098/rstb.2012.0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kunkel M. T., Ni Q., Tsien R. Y., Zhang J., and Newton A. C. (2005) Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J. Biol. Chem. 280, 5581–5587 10.1074/jbc.M411534200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park H. S., Hohn M. J., Umehara T., Guo L. T., Osborne E. M., Benner J., Noren C. J., Rinehart J., and Söll D. (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333, 1151–1154 10.1126/science.1207203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. George S., Aguirre J. D., Spratt D. E., Bi Y., Jeffery M., Shaw G. S., and O'Donoghue P. (2016) Generation of phospho-ubiquitin variants by orthogonal translation reveals codon skipping. FEBS Lett. 590, 1530–1542 10.1002/1873-3468.12182 [DOI] [PubMed] [Google Scholar]
- 39. Adams J. A., and Taylor S. S. (1992) Energetic limits of phosphotransfer in the catalytic subunit of cAMP-dependent protein kinase as measured by viscosity experiments. Biochemistry 31, 8516–8522 10.1021/bi00151a019 [DOI] [PubMed] [Google Scholar]
- 40. Toker A., and Newton A. C. (2000) Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J. Biol. Chem. 275, 8271–8274 10.1074/jbc.275.12.8271 [DOI] [PubMed] [Google Scholar]
- 41. Kunkel M. T., and Newton A. C. (2009) Spatiotemporal dynamics of kinase signaling visualized by targeted reporters. Curr. Protoc. Chem. Biol. 1, 17–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ebner M., Lučić I., Leonard T. A., and Yudushkin I. (2017) PI(3,4,5)P3 engagement restricts Akt activity to cellular membranes. Mol. Cell 65, 416–431.e6 10.1016/j.molcel.2016.12.028 [DOI] [PubMed] [Google Scholar]
- 43. Walsh D. A., Perkins J. P., and Krebs E. G. (1968) An adenosine 3′,5′-monophosphate-depandant protein kinase from rabbit skeletal muscle. J. Biol. Chem. 243, 3763–3765 [PubMed] [Google Scholar]
- 44. Antal C. E., and Newton A. C. (2014) Tuning the signalling output of protein kinase C. Biochem. Soc. Trans. 42, 1477–1483 10.1042/BST20140172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andjelković M., Alessi D. R., Meier R., Fernandez A., Lamb N. J., Frech M., Cron P., Cohen P., Lucocq J. M., and Hemmings B. A. (1997) Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272, 31515–31524 10.1074/jbc.272.50.31515 [DOI] [PubMed] [Google Scholar]
- 46. Kohn A. D., Takeuchi F., and Roth R. A. (1996) Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J. Biol. Chem. 271, 21920–21926 10.1074/jbc.271.36.21920 [DOI] [PubMed] [Google Scholar]
- 47. Andjelković M., Maira S.-M., Cron P., Parker P. J., and Hemmings B. A. (1999) Domain swapping used to investigate the mechanism of protein kinase B regulation by 3-phosphoinositide-dependent protein kinase 1 and Ser473 kinase. Mol. Cell. Biol. 19, 5061–5072 10.1128/MCB.19.7.5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scheid M. P., Marignani P. A., and Woodgett J. R. (2002) Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol. Cell. Biol. 22, 6247–6260 10.1128/MCB.22.17.6247-6260.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hauge C., Antal T. L., Hirschberg D., Doehn U., Thorup K., Idrissova L., Hansen K., Jensen O. N., Jørgensen T. J., Biondi R. M., and Frödin M. (2007) Mechanism for activation of the growth factor-activated AGC kinases by turn motif phosphorylation. EMBO J. 26, 2251–2261 10.1038/sj.emboj.7601682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brodbeck D., Cron P., and Hemmings B. A. (1999) A human protein kinase Bγ with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J. Biol. Chem. 274, 9133–9136 10.1074/jbc.274.14.9133 [DOI] [PubMed] [Google Scholar]
- 51. Gao T., Furnari F., and Newton A. C. (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 10.1016/j.molcel.2005.03.008 [DOI] [PubMed] [Google Scholar]
- 52. Newton A. C., and Trotman L. C. (2014) Turning off AKT: PHLPP as a drug target. Annu. Rev. Pharmacol. Toxicol. 54, 537–558 10.1146/annurev-pharmtox-011112-140338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Slipicevic A., Holm R., Nguyen M. T., Bøhler P. J., Davidson B., and Flørenes V. A. (2005) Expression of activated Akt and PTEN in malignant melanomas: relationship with clinical outcome. Am. J. Clin. Pathol. 124, 528–536 10.1309/YT58WWMTA6YR1PRV [DOI] [PubMed] [Google Scholar]
- 54. Tsao A. S., McDonnell T., Lam S., Putnam J. B., Bekele N., Hong W. K., and Kurie J. M. (2003) Increased phospho-AKT (Ser473) expression in bronchial dysplasia: implications for lung cancer prevention studies. Cancer Epidemiol. Biomarkers Prev. 12, 660–664 [PubMed] [Google Scholar]
- 55. Vincent E. E., Elder D. J., Thomas E. C., Phillips L., Morgan C., Pawade J., Sohail M., May M. T., Hetzel M. R., and Tavaré J. M. (2011) Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 104, 1755–1761 10.1038/bjc.2011.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gallay N., Dos Santos C., Cuzin L., Bousquet M., Simmonet Gouy V., Chaussade C., Attal M., Payrastre B., Demur C., and Récher C. (2009) The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 23, 1029–1038 10.1038/leu.2008.395 [DOI] [PubMed] [Google Scholar]
- 57. Thorsness P. E., and Koshland D. E. (1987) Inactivation of isocitrate dehydrogenase by phosphorylation is mediated by the negative charge of the phosphate. J. Biol. Chem. 262, 10422–10425 [PubMed] [Google Scholar]
- 58. Hu J., Ahuja L. G., Meharena H. S., Kannan N., Kornev A. P., Taylor S. S., and Shaw A. S. (2015) Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol. 35, 264–276 10.1128/MCB.00943-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lippa B., Pan G., Corbett M., Li C., Kauffman G. S., Pandit J., Robinson S., Wei L., Kozina E., Marr E. S., Borzillo G., Knauth E., Barbacci-Tobin E. G., Vincent P., Troutman M., Baker D., Rajamohan F., Kakar S., Clark T., and Morris J. (2008) Synthesis and structure based optimization of novel Akt inhibitors. Bioorg. Med. Chem. Lett. 18, 3359–3363 10.1016/j.bmcl.2008.04.034 [DOI] [PubMed] [Google Scholar]
- 60. Yang J., Cron P., Good V. M., Thompson V., Hemmings B. A., and Barford D. (2002) Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 9, 940–944 10.1038/nsb870 [DOI] [PubMed] [Google Scholar]
- 61. Steichen J. M., Kuchinskas M., Keshwani M. M., Yang J., Adams J. A., and Taylor S. S. (2012) Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J. Biol. Chem. 287, 14672–14680 10.1074/jbc.M111.335091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adams J. A., McGlone M. L., Gibson R., and Taylor S. S. (1995) Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry 34, 2447–2454 10.1021/bi00008a007 [DOI] [PubMed] [Google Scholar]
- 63. Bornancin F., and Parker P. J. (1997) Phosphorylation of protein kinase C-alpha on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J. Biol. Chem. 272, 3544–3549; Correction (1997) J. Biol. Chem. 272, 13458 10.1074/jbc.272.6.3544 [DOI] [PubMed] [Google Scholar]
- 64. Antal C. E., and Newton A. C. (2013) Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Mol. Cell. Proteomics 12, 3498–3508 10.1074/mcp.R113.029819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Edelheit O., Hanukoglu A., and Hanukoglu I. (2009) Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9, 61 10.1186/1472-6750-9-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee S., Oh S., Yang A., Kim J., Söll D., Lee D., and Park H. S. (2013) A facile strategy for selective incorporation of phosphoserine into histones. Angew. Chem. Int. Ed. Engl. 52, 5771–5775 10.1002/anie.201300531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reuveni H., Livnah N., Geiger T., Klein S., Ohne O., Cohen I., Benhar M., Gellerman G., and Levitzki A. (2002) Toward a PKB inhibitor: modification of a selective PKA inhibitor by rational design. Biochemistry 41, 10304–10314 10.1021/bi0202530 [DOI] [PubMed] [Google Scholar]
- 68. Turowec J. P., Duncan J. S., French A. C., Gyenis L., St Denis N. A., Vilk G., and Litchfield D. W. (2010) Protein kinase CK2 is a constitutively active enzyme that promotes cell survival: strategies to identify CK2 substrates and manipulate its activity in mammalian cells. Methods Enzymol. 484, 471–493 10.1016/B978-0-12-381298-8.00023-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.