

Abstract
Endothelium-derived epoxyeicosatrienoic acids (EETs) have numerous vascular activities mediated by G protein–coupled receptors. Long-chain free fatty acids and EETs activate GPR40, prompting us to investigate the role of GPR40 in some vascular EET activities. 14,15-EET, 11,12-EET, arachidonic acid, and the GPR40 agonist GW9508 increase intracellular calcium concentrations in human GPR40–overexpressing HEK293 cells (EC50 = 0.58 ± 0.08 μm, 0.91 ± 0.08 μm, 3.9 ± 0.06 μm, and 19 ± 0.37 nm, respectively). EETs with cis- and trans-epoxides had similar activities, whereas substitution of a thiirane sulfur for the epoxide oxygen decreased the activities. 8,9-EET, 5,6-EET, and the epoxide hydrolysis products 11,12- and 14,15-dihydroxyeicosatrienoic acids were less active than 11,12-EET. The GPR40 antagonist GW1100 and siRNA-mediated GPR40 silencing blocked the EET- and GW9508-induced calcium increases. EETs are weak GPR120 agonists. GPR40 expression was detected in human and bovine endothelial cells (ECs), smooth muscle cells, and arteries. 11,12-EET concentration-dependently relaxed preconstricted coronary arteries; however, these relaxations were not altered by GW1100. In human ECs, 11,12-EET increased MAP kinase (MAPK)-mediated ERK phosphorylation, phosphorylation and levels of connexin-43 (Cx43), and expression of cyclooxygenase-2 (COX-2), all of which were inhibited by GW1100 and the MAPK inhibitor U0126. Moreover, siRNA-mediated GPR40 silencing decreased 11,12-EET–induced ERK phosphorylation. These results indicated that GPR40 is a low-affinity EET receptor in vascular cells and arteries. We conclude that epoxidation of arachidonic acid to EETs enhances GPR40 agonist activity and that 11,12-EET stimulation of GPR40 increases Cx43 and COX-2 expression in ECs via ERK phosphorylation.
Keywords: endothelial cell, cyclooxygenase (COX), connexin, smooth muscle, fatty acid
Introduction
Epoxyeicosatrienoic acids (EETs)2 are cytochrome P450 metabolites of arachidonic acid (1, 2). Four regioisomeric EETs (14,15-, 11,12-, 8,9-, and 5,6-EETs) are synthesized by cytochrome P450. In the vasculature, EETs are synthesized by the endothelium with 14,15- and 11,12-EETs representing the predominant isomers (3, 4). The EETs have actions on both endothelial cells (ECs) and smooth muscle cells (SMCs) (5). Specifically, the four regioisomeric EETs function as endothelium-derived hyperpolarizing factors (3, 6, 7). In response to acetylcholine, bradykinin, or shear stress, endothelium-derived EETs act on smooth muscle cells to activate large conductance, calcium-activated potassium (BKCa) channels causing membrane hyperpolarization and relaxation. Additionally, 11,12- and 14,15-EETs inhibit smooth muscle cell migration and aromatase expression (8, 9). On endothelial cells, 11,12-EET promotes cell growth, decreases apoptosis, migration, tube formation, and angiogenesis, increases tissue plasminogen activator (tPA) release, increases cyclooxygenase-2 (COX-2) expression, and alters connexin-43 (Cx43) expression and gap junctions (10–17). EET isomers also inhibit platelet adhesion to the endothelium, and 11,12- and 8,9-EETs inhibit inflammation by decreasing leukocyte adhesion to the endothelium (18, 19). Interestingly, some of these actions occur with nanomolar concentrations of the EETs (BKCa channel activation, relaxation, tPA release, and decreasing leukocyte adhesion), whereas other activities require micromolar concentrations (inhibition of smooth muscle cell growth and aromatase expression, COX-2 expression, gap junction communication, stimulation of endothelial growth, and inhibition of platelet adhesion) (4). These differences in EET concentrations for particular activities and differences in activities of EET regioisomers suggest that multiple mechanisms and/or receptors are involved.
Several lines of evidence indicate that some of the actions of the EETs require a guanine nucleotide (G)–binding protein–coupled receptor (GPCR) (3, 4). Using radioligand binding, a high-affinity, specific, and saturable binding site was characterized for 14,15-EET in U937 cells and membranes (20–22). This binding was reversible and G protein–dependent. Biologically active 14,15-EET and EET analogs inhibited the specific binding, whereas inactive 14,15-EET analogs and other eicosanoids did not. The G protein Gαs is required for EETs to activate SMC BKCa channels and cause relaxation (23). Similarly, Gαs and increases in cellular cAMP are involved in release of tPA from ECs (13). In other cells, EETs do not increase cAMP (24, 25). These studies indicate that there is a high-affinity GPCR for the EETs in some cells. The identity of this GPCR is not known. However, experiments using a 14,15-EET photoaffinity probe indicate that the EET receptor is a 47-kDa protein (26).
EETs may interact with several known receptors and possibly ion channels. EETs bind to PPARγ in μm concentrations and activate PPARγ-mediated transcription (27). In rodent arteries, EETs inhibit binding of a thromboxane (TP) receptor agonist in micromolar concentrations and block the relaxations to TP agonists (28). EET also activates prostaglandin E type-2 (EP2) receptors in micromolar concentrations (28, 29). The transient receptor potential vanilloid-1 (TRPV1) channel has been proposed to represent the vascular EET receptor; however, there is no evidence for an EET-binding site on this channel (30).
GPR40 or free fatty acid receptor-1 (FFAR-1) is a member of a family of receptors that have short- and long-chain fatty acids as ligands (31–33). Other members of this family include GPR41 (FFAR3), GPR43 (FFAR2), and GPR120 (FFAR4) (34–37). Specifically, GPR40 is activated by micromolar concentrations of fatty acids of 12–22 carbons in chain length and is coupled to the G proteins Gs, Gq/G11, and Gi (31–33). In cells overexpressing GPR40, 11,12- and 8,9-EET also stimulate an increase in intracellular calcium in micromolar concentrations as do long chain free fatty acids (32, 38). GPR40 is highly expressed in the pancreas and central nervous system. On pancreatic β cells, activation of GPR40 with free fatty acids increases intracellular calcium and enhances glucose-stimulated insulin release (32, 33, 39–41). Specific agonists and antagonists of GPR40 have been synthesized. GW9508 activates GPR40 (42). GW1100 and DC260126 are GPR40 antagonists that block the actions of free fatty acids in cells overexpressing GPR40 (42, 43). GW1100 attenuated linoleic acid-induced increase in insulin release.
In this study, we investigated the activation of GPR40 by EET regioisomers and analogs and the role of GPR40 in some vascular effect of EETs. The vascular expression of GPR40 and the effects of EETs on endothelial potassium currents, mitogen-activated protein kinase (MAPK) activity, and Cx43 and COX-2 expression were determined. These studies suggest that GPR40 represents a low-affinity EET receptor in the vasculature and mediates some of the vascular actions of the EETs.
Results
Expression of GPR40 and GPR120 in vascular cells and/or arteries
A stable HEK293 cell line overexpressing human GPR40 was produced and termed HEK293 + GPR40. Using PCR with human GPR40 primers, a 317-bp product was detected in HEK293 + GPR40 cells but not nontransfected HEK293 cells (Fig. 1A). Identical results were observed with a second set of GPR40 primers producing a 170-bp product (data not shown). Sequencing the band from HEK293 + GPR40 cells confirmed it as GPR40. Also, a 31-kDa band corresponding to GPR40 was detected by immunoblotting in HEK293 + GPR40 cell lines. A weak 31-kDa band was detected in nontransfected HEK293 cells (Fig. 1C). Because EETs affect ECs and SMCs, we determined the expression of GPR40 in human and bovine vascular cells. Using PCR primers for human GPR40, the predicted 317-bp product was observed in HUVECs, HCaECs, and HAoSMC (Fig. 1A). Similarly, a 315-bp product was observed in bovine coronary arteries, ECs, and SMCs with bovine GPR40 primers (Fig. 1B). The expression of GPR40 was confirmed by Western immunoblotting (Fig. 1C). A 31-kDa immunoreactive band was detected in lysates of HUVECs, HAoSMCs, HCaECs, and BCAECs. Faint bands were in BCASMCs and BCA. The 31-kDa protein was same size as the band in HEK293 + GPR40 cells. Thus, vascular cells express GPR40. A 251-bp product corresponding to hGPR120 was also detected by PCR in HUVECs, HAoSMCs, and HCaECs (Fig. 1D). The same product was obtained in HEK293 cells overexpressing GPR120 (HEK293 + GPR120(s)) and PanC-1 cells, a cell known to express GPR120 and GPR40 (44).
Figure 1.

Expression of GPR40 and GPR120 in human (h) and bovine (b) endothelial cells and smooth muscle cells and bovine coronary arteries. A, expression of human GPR40 measured by RT-PCR in human vascular cells. B, expression of bovine GPR40 measured by RT-PCR in bovine vascular cells and arteries. C, expression of GPR40 measured by immunoblotting in human and bovine vascular cells and bovine arteries. D, expression of human GPR120 measured by RT-PCR in human vascular cells. Nontransfected HEK293 cells (HEK), HEK293 cells stably expressing human GPR40 (HEK + GPR40), pancreatic cancer cells-1 (PanC-1), and HEK293 cells transiently expressing human GPR120 (S) (HEK + GPR120) were used as controls.
Effect of EETs, EET analogs, and GW9508 on [Ca2+]i in HEK293 and HEK293 + GPR40 cells
A stable HEK293 cell line overexpressing human GPR40 and untransfected HEK293 cells was used to evaluate the effect of GPR40 agonists and antagonists by measuring [Ca2+]i as an indication of GPR40 activity. 14,15-, 11,12-, 8,9-, and 5,6-EET (10−7–10−5 m) stimulated an increase in [Ca2+]i in HEK293 + GPR40 cells (Fig. 2B). Although nontransfected HEK293 may express low levels of GPR40 protein but not mRNA (Fig. 1, C and A), this is apparently not of functional consequence. EETs did not alter [Ca2+]i in nontransfected HEK293 cells (Fig. 2A). Thus, the overexpression of GPR40 was required for EET activity. The increases in [Ca2+]i by the EETs were concentration-related. 11,12- and 14,15-EET were similar in activity and potency (EC50 = 0.91 ± 0.08 and 0.58 ± 0.08 μm, respectively) and more potent than 8,9- and 5,6-EET. 17,18-Epoxyeicosatetraenoic acid (17,18-EEQ), the epoxide of the ω-3 fatty acid eicosapentaenoic acid, also increased [Ca2+]i in GPR40-expressing cells; however, it was less active than the EETs (data not shown). 11,12-EET (Fig. 2D) and 14,15-EET (Fig. 2F) caused transient increases in [Ca2+]i in HEK293 + GPR40 cells. The rapid increases in [Ca2+]i were followed by a decline to baseline over the following 150 s. The heights of the transients increased with concentration of the EETs, but the patterns of the transients were the same. Vehicle was without effect. The EETs did not produce transient changes in [Ca2+]i in nontransfected HEK293 cells over the same concentration range (Fig. 2, C and E).
Figure 2.

Effect of EETs on [Ca2+]i in HEK293 cells and HEK293 cells stably expressing human GPR40 (HEK293 + GPR40). A and B, effect of various concentrations of the EETs on [Ca2+]i in HEK293 cells (A) and HEK293 + GPR40 cells (B) (data expressed in Δ relative fluorescence units (ΔRFU) per μg of protein). C and D, effect of 11,12-EET on [Ca2+]i fluorescence over time in HEK293 cells (C) and HEK293 + GPR40 cells (D) (data shown in RFU × 10−3). E and F, effect of 14,15-EET on [Ca2+]i fluorescence over time in HEK293 cells (E) and HEK293 + GPR40 cells (F) (data shown in RFU × 10−3). Each value represents the mean ± S.E. for n = 4. veh, vehicle.
In the presence of 1.26 mm calcium in the incubation buffer, 14,15-, 11,12-, and 8,9-EET increased [Ca2+]i in a concentration-related manner in HEK293 + GPR40 cells (Fig. 3A). Similar increases were observed with GW9508, a GPR40 agonist. GW9508 was more potent than the EETs, but the maximal effects were similar. When the buffer was free of calcium (0 mm calcium), the [Ca2+]i responses to the EETs and GW9508 were reduced when compared with cells in the 1.26 mm calcium buffer (Fig. 3B). The concentration responses were shifted to the right, and the maximal increases were reduced with 0 mm calcium by ∼20%. The increases in [Ca2+]i with the EETs and GW9508 were inhibited further with 0 mm calcium containing 50 μm EGTA (Fig. 3C). 11,12-EET (1 μm) produced transient increases in [Ca2+]i with time (Fig. 3D). The baseline fluorescence and maximal height of the [Ca2+]i increase with 11,12-EET were reduced in 0 mm calcium and further reduced with 0 mm calcium plus EGTA compared with 1.26 mm calcium. The [Ca2+]i responses to EETs and GW9508 are influenced by extracellular [Ca2+].
Figure 3.

Effect of extracellular [Ca2+]i on EET activity in HEK293 cells expressing human GPR40. A–C, comparison of the activity of various concentrations of EET regioisomers and GPR40 agonist GW9508 in the presence of 1.26 mm extracellular calcium (A), in the absence of extracellular calcium (B), and in the absence of extracellular calcium plus 50 μm EGTA (C). D, effect of 1 μm 11,12-EET on [Ca2+]i with time in the presence of 1.26 mm extracellular Ca, in the absence of extracellular Ca, and in the absence of extracellular calcium plus 50 μm EGTA. Each value represents the mean ± S.E. for n = 4. veh, vehicle.
The EETs undergo hydrolysis to their respective vic-dihydroxyeicosatrienoic acids (DHETs) by soluble epoxide hydrolase (sEH) (45). However, unlike the EETs, 11,12- and 14,15-DHET were without GPR40 activity at 10−7–5 × 10−5 m (Fig. 4A). There was a small increase with 10−5 m DHETs. Pretreatment of the cells with the sEH inhibitor EH1555 (1 μm) did not alter the increase in [Ca2+]i to 11,12- or 14,15-EET (data not shown). Thus, HEK293 cells did not metabolize EETs to DHETs within the time frame of these experiments. Substitution of sulfur for the epoxide oxygen giving a thiirane results in a reduction in activity. Unlike the EETs, 14,15-thiirane did not increase [Ca2+]i in HEK293 + GPR40 cells, and the activity of 11,12-thiirane was greatly reduced (Fig. 4B). GPR40 is stimulated by fatty acids with cis- and trans-double bonds (46). We compared the activity of EETs with cis- and trans-epoxides (Fig. 4C). The trans-11,12- and -14,15-EETs and the cis-11,12- and -14,15-EETs increased [Ca2+]i to a similar extent. Thus, the configuration of the epoxide is not critical for GPR40 activation. Arachidonic acid also stimulated GPR40 to increase [Ca2+]i (EC50 = 3.9 ± 0.06 μm), but it was less potent than the EETs (Fig. 4D). Additionally, a low concentration of 11,12-EET (0.1 μm) did not enhance the increase in [Ca2+]i by arachidonic acid, and a low concentration of arachidonic acid (1 μm) did not enhance the effect of 11,12-EET. Thus, 11,12-EET and arachidonic acid are not synergistic in their action on GPR40. In contrast to EETs and arachidonic acid, 20-HETE was without GPR40 activity in HEK293 + GPR40 cells (data not shown). Thus, the epoxy group is important for EET stimulation of GPR40 activity; however, the cis or trans configuration of the epoxide is not critical. Locating the epoxide in the middle of the molecule results in greater GPR40 activity. Substitution of a diol, hydroxyl, or thiirane for the epoxide reduces or eliminates activity. Importantly, conversion of arachidonic acid to 11,12- or 14,15-EET increases the potency on GPR40.
Figure 4.

Effect of EETs, DHETs, EET isomers, and EET analogs on [Ca2+]i in HEK293 cells expressing human GPR40. A, comparison of the activity of 11,12-, 14,15-EETs, and -DHETs. B, comparison of 11,12- and 14,15-EET and their thiirane analogs. C, comparison of the cis- and trans-isomers of 11,12- and 14,15-EETs. D, effect of 11,12-EET in the absence and presence of 1 μm arachidonic acid and effect of arachidonic acid in the absence and presence of 0.1 μm 11,12-EET. Each value represents the mean ± S.E. for n = 4. veh, vehicle.
GW9508, a GPR40 agonist, stimulated an increase in [Ca2+]i in HEK293 + GPR40 cells (10−9–10−5 m) (EC50 = 19 ± 0.37 nm) (Figs. 3A and 5A), but not in nontransfected HEK293 cells (data not shown). The GPR40 antagonists GW1100 (10 μm) (Fig. 5A) and DC260126 (30 μm) (data not shown) inhibited the stimulation by GW9508. Because of the chemical instability of DC260126, subsequent studies utilized GW1100 only. GW1100 (10 μm) also inhibited the stimulation of GPR40 by 11,12- and 14,15-EET (Fig. 5C). GW1100 at 1 μm failed to inhibit the EETs (data not shown). In nontargeting siRNA-treated HEK293–GPR40 cells, GW9508, 11,12-EET, and 14,15-EET increased [Ca2+]i as in vehicle-treated cells (Fig. 5, B and D). As with GW1100, the concentration-response curves for the three agonists were shifted to the right in GPR40 targeted siRNA-treated cells. Fig. 5, E and F, shows GPR40 immunoblots of HEK293–GPR40 cells treated with nontargeted and GPR40-targeted siRNAs. GPR40 targeted siRNA reduced expression of GPR40. These data further indicate the role of GPR40 in EET-induced [Ca2+]i increases.
Figure 5.

Effect of the GPR40 antagonist GW1100 (10 μm) and GPR40 siRNA on the increase in [Ca2+]i by 11,12-EET, 14,15-EET, and the GPR40 agonist GW9508 in human GPR40-expressing HEK293 cells. A, effect of GW1100 on the activity of GW9508. B, effect of GPR40 siRNA on the activity of GW9508. C, effect of GW1100 on the activity of EETs. D, effect of GPR40 siRNA on the activity of EETs. E and F, Western blotting (E) and densitometry (F) of GPR40 protein levels and knockdown in human GPR40-expressing HEK293 cells treated with control and GPR40 siRNA for 48 h. A and C, each value represents the mean ± S.E. for n = 4. B and D, each value represents the mean ± S.E. for n = 8. F, each value represents the mean ± S.E. for n = 4. * indicates p < 0.05, compared with control siRNA. veh, vehicle.
Effect of EETs on [3H]TAK-875 binding to HEK293–GPR40 cell membranes
TAK-875, like GW9508, is a GPR40 agonist (47). It stimulated a concentration-related increase in [Ca2+]i in HEK293 + GPR40 cells (10−9–10−5 m) (EC50 = 30 ± 0.11 nm) (Fig. 6A) and had no effect on [Ca2+]i in HEK293 cells (data not shown). The increase in [Ca2+]i with TAK-875 was inhibited by GW1100. [3H]TAK-875 has been used as a high-affinity GPR40 radioligand (Kd = 4.8 nm) to evaluate binding of agonists to the receptor (47–49). Specific binding of [3H]TAK-875 occurred in membranes of HEK293 + GPR40 cells but not in membranes of nontransfected HEK293. 11,12-EET and 14,15-EET inhibited the specific binding to HEK293 + GPR40 membranes with Ki values of 2.7 and 6.4 μm, respectively (Fig. 6B).
Figure 6.
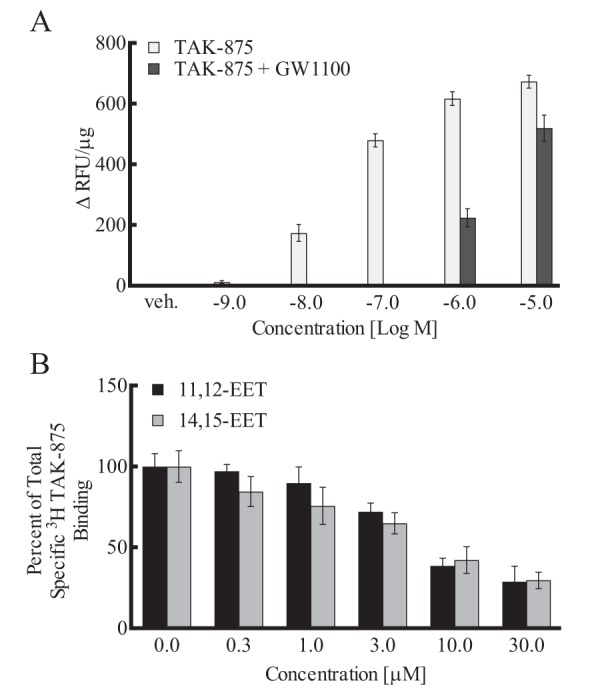
Effects of the GPR40 agonist TAK-875 on [Ca2+]i and EETs on [3H]TAK-875–specific binding in HEK293 + GPR40 cells. A, effect of the GPR40 agonist TAK-875 on [Ca2+]i in the presence and absence of the GPR40 antagonist GW1100 (10 μm). B, effect of 11,12- and 14,15-EET on the specific binding of [3H]TAK-875 to HEK293 + GPR40 membranes. Each value represents the mean ± S.E. for n = 6–9. veh, vehicle.
Effect of EETs and GW9508 on [Ca2+]i in HEK293–GPR120 cells
GPR120 is another long chain free fatty acid receptor (FFAR4) that occurs in two forms, a long and short (50). These forms were transiently expressed in HEK293 cells and tested as described for GPR40. GW9508 stimulated an increase in [Ca2+]i in cells overexpressing the short form of GPR120 (Fig. 7A) but not the long form (Fig. 7B). GW9508 was without effect in nontransfected HEK293 cells. GW9508 was ∼100-fold less potent in stimulating the GPR120 short form than GPR40. 14,15-, 11,12-, and 8,9-EETs only stimulated the short form of GPR120 at 10−5 m (Fig. 7). 17,18-EEQ was without effect (data not shown). Thus, GW9508 and EETs are weaker agonists for GPR120 than GPR40. GW1100 is not an antagonist for GPR120 (42).
Figure 7.
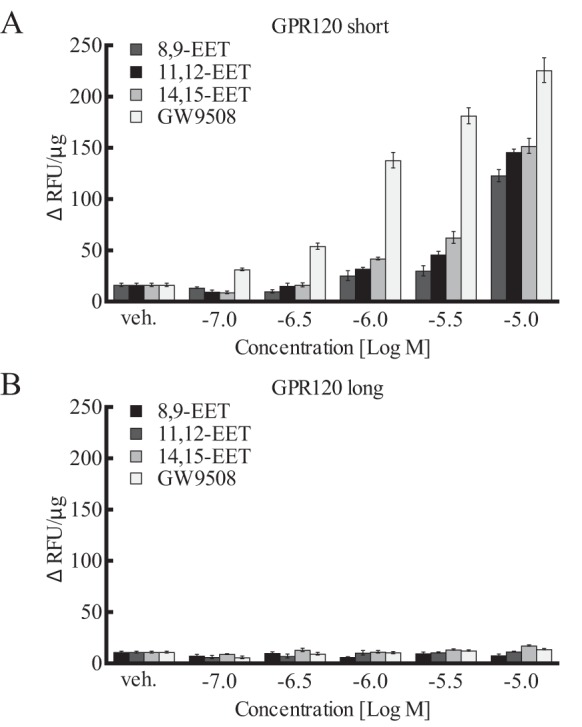
Effect of EETs and GW9508 on [Ca2+]i in HEK293 cells expressing GPR120. Two splice variants for GPR120 were tested: short form (A) and long form (B). Each value represent the mean ± S.E. for n = 4. veh, vehicle.
Effect of EETs on GPR40-mediated [Ca2+]i increase in INS-1 832/13 insulinoma cells
In the remaining studies, we examined the role of GPR40 in some of the actions of the EETs. GPR40 is expressed in pancreatic β cells and the rat insulinoma INS-1E cell line (39, 41). In this cell line, palmitic acid increased [Ca2+]i and insulin release via GPR40. Using a related insulinoma cell line, INS-1 832/13 cells, a 31-kDa protein corresponding to GPR40 was detected by immunoblotting (Fig. 8A, inset). In addition, 11,12- and 14,15-EET increased [Ca2+]i in these cells, and the increase in [Ca2+]i was inhibited by GW1100 (Fig. 8A).
Figure 8.

GPR40-mediated actions of the EETs. A, role of GPR40 in the increase in [Ca2+]i in INS-1 832/13 insulinoma cells by EETs. Inset represents a GPR40 immunoblot comparing HEK293 + GPR40 and INS-1 832/13 cells. B, role of GPR40 on the relaxation of bovine coronary arteries by 11,12-EET. Arteries were precontracted with U46619. 11,12-EET was tested in the presence and absence of the GPR40 antagonist GW1100 (10 μm), the BKCa channel inhibitor iberiotoxin (100 nm), or the combination of both inhibitors. C and D, effect of 11,12-EET on whole-cell K+ currents in HUVECs: role of GPR40 and calcium. 11,12-EET (1 μm) increased whole-cell K+ currents that were inhibited by GW1100 (10 μm). Studies were repeated in the absence (C) and presence (D) of EDTA in the patch pipette. Each value represents the mean ± S.E. for the stated N values. veh, vehicle.
Role of GPR40 in 11,12-EET relaxation of BCAs
In arteries preconstricted with U46619, 11,12-EET causes concentration-related relaxations with an EC50 = 0.46 ± 0.14 μm (Fig. 8B). Pretreatment with the GPR40 antagonist GW1100 did not affect vascular tone and did not significantly alter the relaxations to 11,12-EET (EC50 = 0.63 ± 0.37 μm). Relaxations were repeated in arterial rings pretreated with 100 nm iberiotoxin to block the BKCa channel component of 11,12-EET relaxations (4). The potency and maximal relaxation to 11,12-EET were reduced by iberiotoxin indicating BKCa-dependent and -independent components. The iberiotoxin-resistant component of relaxation was not significantly altered by GW1100. Thus, the relaxations to 11,12-EET are not mediated through GPR40.
Effect of EETs on K+ channel activity in HUVECs
Stepwise increases in the holding potential in vehicle-treated HUVECs increased the whole-cell K+ currents (Fig. 8C). 11,12-EET (1 μm) increased the K+ currents when compared with vehicle. The increase by 11,12-EET was blocked by GW1100. GW1100 had no effect on K+ currents when tested alone. The stimulation of K+ currents by 11,12-EET was attenuated by the presence of EDTA in the patch pipette solution (Fig. 8D). The attenuated effect of 11,12-EET was also blocked by GW1100. GPR40 mediates the ability of 11,12-EET to increase K+ currents in HUVECs.
Effect of 11,12-EET on ERK phosphorylation in HUVECs
Because GPR40 agonists activate MAPKs, such as ERK and p38 MAPK (38, 51, 52), we investigated whether EETs increase MAPK signaling via GPR40 activation. The ERK and phospho-ERK antibodies detected 42- and 44-kDa proteins in untreated and treated HUVECs. 11,12-EET (1 μm) increased phospho-ERK without changing the amount of total ERK1/2 (Fig. 9). The increase in ERK phosphorylation by 11,12-EET was inhibited by GW1100 pretreatment (Fig. 9B), whereas the MEK inhibitor U0126 (1 μm) inhibited control and EET-induced ERK phosphorylation (Fig. 9A). HUVECs were transfected with control, nontargeting siRNA or GPR40-targeted siRNA. By immunoblot, the GPR40 siRNA treatment reduced the expression of the 31-kDa GPR40 protein in HUVECs compared with control siRNA treatment (Fig. 9C). As with GW1100 treatment, knockdown of GPR40 with siRNA reduced 11,12-EET-stimulated ERK phosphorylation (Fig. 9D). Control ERK phosphorylation was not changed. These data indicate that 11,12-EET–induced ERK phosphorylation requires GPR40. 11,12-EET did not alter phosphorylation of p38 MAPK (data not shown).
Figure 9.

Effect of 11,12-EET on ERK phosphorylation in HUVECs: role of GPR40. Cells were pretreated with vehicle, MEK inhibitor U0126 (1 μm) (A), or GPR40 antagonist GW1100 (10 μm) (B). 11,12-EET (1 μm) was then added and incubation continued for 10 min. C, GPR40 protein level and knockdown in HUVEC treated with control and GPR40 siRNA for 48 h. D, effect of GPR40 knockdown on 11,12-EET-stimulated phospho-p42/44 (P-ERK). Both phospho-p42/44 (P-ERK) and total p42/44 (ERK) were measured by immunoblotting. Each value represent mean ± S.D. of three independent experiments. ** and * indicate p < 0.001 and p < 0.05, respectively, compared with the untreated control. ## indicates p < 0.005, compared with 11,12-EET alone. § indicates p < 0.05, compared with control siRNA with 11,12-EET.
Effect of 11,12-EET on Cx43 expression in HUVECs
11,12-EET alters intercellular gap junction communication by activating Cx43 in ECs (12). Gap junctions are in a constant state of assembly and disassembly (53). Phosphorylation of Cx43 by MAPK leads to disassembly and internalization of Cx43 from membrane gap junctions (Triton-insoluble fraction) to the cytosolic compartment (Triton-soluble fraction). We also found that 11,12-EET (1 μm) increased Cx43 in the Triton-soluble fraction, and this increase was inhibited by pretreatment with U0126 (Fig. 10A) and GW1100 (Fig. 10B). There were several Cx43 bands detected indicating various levels of phosphorylation (12, 53). These data indicate that 11,12-EET–induced phosphorylation and internalization of Cx43 involves GPR40-activated ERK phosphorylation.
Figure 10.

Effect of 11,12-EET on Cx43 expression and phosphorylation in HUVECs: role of GPR40. Cells were pretreated with vehicle, MEK inhibitor U0126 (1 μm) (A), or GPR40 antagonist GW1100 (10 μm) (B) for 20 min. 11,12-EET (1 μm) was then added and incubation continued for 10 min. Cx43 was measured by immunoblotting, and β-actin was a loading control. Each value represent mean ± S.D. of three independent experiments. ** and * indicate p < 0.001 and p < 0.05, respectively, compared with the untreated control. ## and # indicate p < 0.005 and p < 0.05, respectively, compared with 11,12-EET alone.
Effect of 11,12-EET on COX-2 expression in HUVECs
Previous studies have demonstrated that 11,12-EET increases the expression of COX-2 in ECs (11). A 72-kDa protein corresponding to COX-2 was detected in HUVECs. Treatment of HUVECs with 11,12-EET for 24 h increased the expression of COX-2 (Fig. 11). This COX-2 increase by 11,12-EET was inhibited by U0126 (Fig. 11A), GW1100 (Fig. 11B), and the NF-κB inhibitor andrographolide (Fig. 11C). Thus, 11,12-EET stimulation of COX-2 expression involves GPR40 signaling through the MAPK and NF-κB pathways. Pretreatment of the cells with the sEH inhibitor EH1555 increased COX-2 expression by 11,12-EET to a greater extent than 11,12-EET alone (Fig. 11D). Thus, with prolonged 24-h incubation, EH-1555 enhances the COX-2 expression by 11,12-EET, whereas it was without effect in shorter incubations such as increases in [Ca2+]i.
Figure 11.

Effect of 11,12-EET on COX-2 expression in HUVECs: role of GPR40, MAPK, NF-κB, and soluble epoxide hydrolase. Cells were pretreated with vehicle, MEK inhibitor U0126 (1 μm) (A), or GPR40 antagonist GW1100 (10 μm) (B), NF-κB inhibitor andrographolide (Andro)(40 μm) (C), or sEH inhibitor EH1555 (1 μm) (D) for 20 min. 11,12-EET (1 μm) was then added and incubation continued for 24 h. COX-2 was measured by immunoblotting with β-actin as a loading control. Each value represent mean ± S.D. of three independent experiments. ** and * indicate p < 0.001 and p < 0.05, respectively, compared with the untreated control. # indicates p < 0.05, respectively, compared with 11,12-EET alone.
Discussion
EETs have diverse biological actions, including vasodilation, inhibition of inflammation, promoting angiogenesis, and organ protection (5, 6, 10, 18). These actions and the cellular components to these actions occur over a wide concentration range from 10−9–10−5 m (4). Compelling evidence indicates that 14,15-EET acts via a GPCR (4, 21, 23). In cells and cell membranes, 14,15-EET displays high affinity, specific, saturable binding that is reversible and G protein-dependent. The Kd for binding is 11 nm. Receptors for the other EET regioisomers have not been characterized. This high-affinity GPCR explains 14,15-EET actions occurring in nanomolar concentrations, but not those requiring micromolar concentrations. Therefore, we hypothesized that low-affinity receptor(s) for the EETs mediate these actions. Previous studies indicate that 11,12- and 8,9-EET stimulate GPR40 with EC50 values of 1.4 and 6.1 μm, respectively (32). We screened long-chain free fatty acid receptors, GPR40 and GPR120, as possible low-affinity EET receptors. 14,15-, 11,12-, and 8,9-EETs and GW9508 stimulated increases in [Ca2+]i in GPR40- and GPR120-expressing HEK293 cells. GPR40 stimulation by EETs required high nanomolar concentrations, but micromolar concentrations were required for GPR120. The EC50 for 14,15-EET was 6.7-fold lower than the EC50 for arachidonic acid on GPR40 indicating epoxygenation confers increased potency. In a broader context, EETs have the lowest EC50 of any of the naturally occurring GPR40 ligands, including free fatty acids. Thus, GPR40 is a low-affinity EET receptor. This may represent a more appropriate name for GPR40 than FFAR1. In support of this suggestion, 14,15- and 11,12-EET inhibited [3H]TAK-875 binding to GPR40 indicating a direct interaction of the receptor with EETs. The existence of low- and high-affinity receptors for endogenous agonists is not unique. Adenosine, for example, acts on high-affinity A1, A2A, and A3 receptors as well low-affinity A2B receptors (54, 55).
In rodent arteries, EP receptors are low-affinity receptors for 14,15-EET; however, the Ki values for 14,15-EET on the four EP receptors range from 11 to 40 μm (28, 29, 56). Thus, 20–70-fold more 14,15-EET is required to activate EP receptors than GPR40. Thus, GPR40 is the more relevant low-affinity EET receptor.
The EET regioisomers and analogs differed in their ability to stimulate GPR40. 14,15- and 11,12-EET have similar activities; however, 5,6- and 8,9-EET were less potent. When binding of 14,15-EET to the high-affinity GPCR was determined by radioligand binding or photoaffinity labeling, the EET regioisomers displaced the radioligand and reduced the labeling with the same order of potency, 11,12- > 14,15- ≫ 8,9-EET (21, 26). Also, like 5,6- and 8,9-EET, 17,18-EEQ, the epoxide of eicosapentaenoic acid, was a weak agonist. GPR40 is activated equally by long chain fatty acids with cis- and trans-double bonds (46). As a result, we tested the activity of 11,12- and 14,15-EETs with cis- and trans-epoxides. The cis- and trans-11,12- and -14,15-EETs were equally potent in stimulating GPR40. The cis- and trans-EET have been measured in the plasma and red blood cells, and they relax rat renal arcuate arteries equally (57, 58). In contrast, the cis-14,15-EET was more potent than the trans-isomer in relaxing bovine coronary arteries (59). GPR40 may be the receptor for trans-EETs as it is for trans-fatty acids. Whereas the configuration of the epoxy group may not be critical for GPR40 activity, modification of the epoxide structure is critical. Substitution of a thiirane sulfur for the epoxy oxygen of 11,12- or 14,15-EET results in a decrease in GPR40 activity. Similarly, replacement of the epoxide with a vicinal diol, e.g. DHETs, or single hydroxyl, e.g. 20-HETE, results in loss of GPR40 activity. Similarly, EET analogs with these modifications of the epoxide did not displace 14,15-EET from its high-affinity receptor (21). Thus, the epoxy group and the placement of the epoxide in the arachidonic acid backbone are critical for GPR40 activity.
Studies of the GPR40 crystal structure indicate the presence of three ligand-binding sites (60). Agonists may stimulate GPR40 by binding to different sites on GPR40 and coupling to different signaling mechanisms, Gq, Gi, or Gs (32, 60). Long-chain fatty acids, TAK-875, and GW9508 increase [Ca2+]i but do not increase intracellular cAMP (60). Slight decreases in forskolin-stimulated cAMP have been reported with fatty acids (32). Thus, fatty acid activation of GPR40 may be Gq- and Gi-coupled. In contrast, the synthetic GPR40 agonists, AM1638 and AM5262, increase both [Ca2+]i and cAMP (60). EETs, TAK-875, and GW9508 act similarly to stimulate a Gq-mediated signaling pathways to increase [Ca2+]i in GPR40-expressing HEK293 cells, and both agonists are inhibited by the GPR40 antagonist GW1100 and GPR40 siRNA. Because GW1100 inhibits GPR40, but not GPR120, it provides an important pharmacological tool to assess the role of GPR40 in EET actions (42).
GPR120 occurs as two splice variants, a long and short form (50). Activation of the short form with fatty acids is coupled to Gq, whereas the long form acts via β-arrestin. GW9508 and the EETs increased [Ca2+]i in HEK293 cells expressing the GPR120 short form but not GPR120 long form. Higher concentrations, 100-fold, of both GW9508 and EETs were required to activate GPR120 short form than for GPR40. Thus, GPR40 is selectively stimulated by EET concentrations less than 5 × 10−6 m. As a result, studies of the endothelial effects of GPR40 used 10−6 m 11,12-EET. The EETs and GW9508 were not tested on β-arrestin translocation in cells expressing GPR120 splice variants. 14,15-, 11,12-, and 8,9-EETs were equally active in stimulating GPR120, whereas 14,15- and 11,12-EET were more active than 8,9-EET on GPR40. GPR120 is a receptor for long chain ω-3 fatty acids (36); however, the ω-3 epoxide, 17,18-EEQ, was without effect on this receptor. In contrast, 17,18-EEQ was a weak GPR40 agonist. Thus, the concentration and order of potency of EETs and activity of 17,18-EEQ can be used to distinguish activities mediated by GPR40 and GPR120.
14,15- and 11,12-EET are metabolized by sEH to 14,15- and 11,12-DHET, respectively, and this metabolism results in a loss of biological activity (45). 14,15- and 11,12-DHET were less active than the EETs in stimulating GPR40. Similarly, 14,15-EET displaced the 14,15-EET radioligands in U937 cell membranes with an EC50 of 40 nm compared with 8.8 μm for 14,15-DHET (21). Thus, the DHETs are weak agonists for the high- and low-affinity EET receptors. Both cis- and trans-EETs are substrates for sEH (45, 57). Thus, inhibition of sEH will increase both cis- and trans-EETs and decrease the DHETs. Because both cis- and trans-EETs are equally active as GPR40 agonists, whereas cis-EETs are better agonists than trans-EET for the high-affinity receptor, it is possible that GPR40 activation may contribute to some of the biological effects of the sEH inhibitors. For example, GPR40 activation by fatty acids enhances glucose-dependent insulin release from β-cells and β-cell lines (32, 33, 39–41). GPR40 activation may contribute to the blood glucose lowering in sEH inhibitor-treated rats (61, 62). Along these lines, 11,12- and 14,15-EET also increase [Ca2+]i in INS-1 832/13 insulinoma cells, and the effect is blocked by the GPR40 antagonist. Whereas insulin release was not measured, these data suggest that EET may regulate insulin release.
GPR40 is predominantly expressed in the pancreas and brain with lower expression in heart, lung, and kidney (31, 32). Similarly, GPR40 expression was detected in rat insulinoma INS-1 832/13 cells and pancreatic carcinoma PanC-1 cells. Monocytes express GPR40; however, GPR40 was not detected in lymphocytes or neutrophils. Because EETs activate GPR40 and have multiple actions in the vasculature, we examined the expression of GPR40 in vascular cells and arteries. Using PCR and immunoblotting, the expression of GPR40 was identified in human and bovine SMCs and ECs. GPR40 has also been detected in microvessels of brain sections (46). The presence of GPR40 in vascular cells and arteries led us to examine the role of the receptor in some previously documented vascular actions of the EETs.
EETs are endothelium-derived hyperpolarizing factors that mediate a portion of the vasodilation to shear stress, bradykinin, and acetylcholine (3, 6, 7). They relax coronary arteries by opening BKCa channels in vascular SMCs causing membrane hyperpolarization. 11,12-EET activation of the BKCa channel is Gs-mediated and has a nanomolar EC50 similar to the Kd value for 14,15-EET binding to the high-affinity EET receptor (21, 23). In bovine coronary arteries, 11,12-EET caused concentration-related relaxations that were inhibited by pretreatment with the BKCa channel blocker iberiotoxin (63). The GPR40 antagonist, GW1100, did not alter EET relaxations or the component of EET relaxation that was resistant to iberiotoxin inhibition. Thus, the high-affinity EET receptor, and not GPR40, mediates 11,12-EET–induced relaxation. With EET and fatty acid activation, GPR40 is Gq-coupled, which promotes increases in [Ca2+]i (32). Thus, in vascular SMCs, EET activation of GPR40 would be expected to cause contractions, and inhibition of GPR40 with GW1100 would block the contractions and enhance the relaxations to EETs (64). However, this was not the case. In ECs, activation of KCa channels by EETs would result in endothelial hyperpolarization, and myoendothelial gap junctional transfer of the hyperpolarization to smooth muscle would cause relaxation (4). This pathway has been described in porcine coronary arteries (65), but not bovine coronary arteries (66). This species difference is likely due to the presence of myoendothelial gap junction in porcine, but not bovine, coronary arteries (67). If myoendothelial gap junctions were present, simultaneous activation of endothelial and smooth muscle GPR40 would be antagonistic, and the effect on vascular tone would depend on whether one or the other predominates. These studies indicate that vascular GPR40 does not contribute to or antagonize the relaxations to 11,12-EET in bovine coronary arteries.
In HUVECs, 11,12-EET increased whole-cell outward K+ currents. This increase was inhibited by the presence of EDTA in the patch pipette solution indicating a calcium-dependent effect. The K+ channel activation by 11,12-EET was also GPR40-dependent and inhibited by GW1100. The loss of K+ ions would result in endothelial hyperpolarization and enhance the electrochemical gradient for the influx of cations such as calcium (68). Besides regulating vascular tone, EET activation of endothelial KCa channels also increases permeability of the endothelial monolayer (69).
The MAPK pathways mediate many of the actions of the EETs. In endothelial and epithelial cells, EETs stimulate ERK phosphorylation and cell proliferation (14, 38, 70). MEK inhibitors block EET-induced cell proliferation. Additionally, EET regulation of Cx43 and COX-2 requires ERK phosphorylation (11, 12). Activation of GPR40 also stimulates ERK phosphorylation in several cell types (38, 51, 52). We observed that the GPR40 antagonist GW1100 and GPR40 targeted siRNA, and the MEK inhibitor U0126 blocked 11,12-EET–induced ERK phosphorylation in HUVECs. Thus, it is likely that some or all of the ERK-mediated activities of the EETs such as COX-2 and Cx43 are GPR40-coupled. These possibilities were tested experimentally.
Gap junction channels are composed of connexin subunits and are in a constant state of assembly and disassembly (53, 71). This process is regulated by phosphorylation of Cx43. PKA phosphorylation promotes assembly of gap junctions, and PKC and MAPK phosphorylation promotes disassembly. As a result, the half-life of Cx43 is 1–3 h. In ECs, Cx43 contributes to gap junctions (12, 72). 11,12-EET has a biphasic effect on gap junctions and Cx43 localization and phosphorylation over time (12). Within the 1st min, 11,12-EET increased dye transfer between ECs through gap junctions. This effect is mediated by increases in cAMP and translocation of Cx43 to the membrane (Triton-insoluble fraction). However, after 5–10 min of exposure to 11,12-EET, Cx43 and phospho-Cx43 increased in the Triton-soluble fraction indicating gap junction disassembly. This resulted in a decrease in dye transfer. These effects were associated with ERK phosphorylation and blocked by MEK inhibitors. Thus, phosphorylation of Cx43 by ERK was associated with disassembly and uncoupling of gap junctions (12, 53, 71). 11,12-EET increased Cx43 and the presence of high-mobility bands attributed to phosphorylated Cx43 in the Triton-soluble fraction. In addition, GW1100 inhibited the 11,12-EET–induced increase in Cx43 and Cx43 phosphorylation as did MEK inhibition with U0126. As a result, these Cx43 events are dependent on ERK phosphorylation. Because GPR40 siRNA silencing also inhibits ERK phosphorylation by 11,12-EET, it would be expected to inhibit EET-dependent Cx43 changes as well. Thus, 11,12-EET activates GPR40 to promote ERK phosphorylation of Cx43 increasing cytosolic Cx43 and gap junction disassembly. These changes in Cx43 will have physiological consequences. In HUVECs and other ECs, TNFα, IL-1β, or oxidized low-density lipoprotein promote apoptosis, inhibit cell growth, and inhibit barrier function; however, they also promote Cx43 expression and formation of gap junctions (73–75). Inhibition of Cx43 expression and gap junctions inhibit apoptosis, promote proliferation, and maintain barrier function. Thus, GPR40-mediated gap junction disassembly by 11,12-EET would be expected to reduce apoptosis, promote cell growth, and maintain barrier function. Consistent with this conclusion, EETs inhibit apoptosis and promote proliferation in ECs (15–17).
COX-2 is expressed in ECs in response to inflammatory cytokines and other stimuli (76, 77). 11,12-EET and overexpression of cytochrome P450 2C9 epoxygenase increase COX-2 expression and tube formation in ECs (11). Inhibition of COX-2 with celecoxib prevents EET-induced tube formation. This work and other studies indicate that EETs promote angiogenesis, and a component of EET-induced angiogenesis requires COX-2 expression (11, 70, 78). In neutrophils, activators of GPR40 increase COX-2 expression by decreasing the endogenous NF-κB inhibitor IκB (51). The increase in COX-2 and decrease in IκB were blocked by GW1100 and inhibitors of MEK. Additionally, an NF-κB inhibitor blocked the increase in COX-2. We therefore wondered whether GPR40 mediated the EET-induced COX-2 expression in ECs. 11,12-EET increased endothelial COX-2 expression after 24 h, and this increase was enhanced with sEH inhibition. GW1100 blocked the EET-induced increase in COX-2 expression. The MEK inhibitor U0126 and the NF-κB inhibitor andrographolide also inhibited COX-2. Thus, 11,12-EET activation of GPR40 promotes ERK phosphorylation and NF-κB activation to increase COX-2 expression. GPR40 activation by EETs would be expected to promote endothelial tube formation and angiogenesis, and inhibition of GPR40 may represent a therapeutic approach to reducing angiogenesis (11, 70).
In summary, GPR40 is a low-affinity receptor for 14,15-, 11,12-, 8,9-, and 5,6-EET and mediates some of the vascular actions of the EETs that occur at micromolar concentrations. Whether these concentrations are achieved locally under physiological or pathological conditions or whether these concentrations evoke pharmacological effects warrants further study. Importantly, EETs are more potent GPR40 agonists than free fatty acids and thus represent the best endogenous GPR40 agonists reported to date. A high-affinity receptor for 14,15-EET has been described (20, 21); however, its identity is currently unknown. Thus, like adenosine, 14,15-EET has both high- and low-affinity receptors that may mediate its actions. High-affinity receptors for 11,12-, 8,9-, or 5,6-EET have not been characterized, but these EETs activate GPR40. GPR40 is expressed in human and bovine vascular cells and mediates the increase in Cx43 phosphorylation and COX-2 expression in human ECs. Because Cx43 phosphorylation promotes gap junction disassembly and COX-2 mediates endothelial tube formation (11, 12), GPR40 has a pivotal role in endothelial proliferation, inhibition of apoptosis, and tube formation that contribute to angiogenesis and barrier function.
Materials and methods
Cell culture
HUVECs, HCaECs, and HAoSMCs were purchased from Lonza, Ltd. (Walkersville, MD) or ThermoFisher Scientific and cultured as described previously (26, 79). BCAECs and BCASMCs were prepared and cultured as described previously (26, 79–81). HEK293 cells were grown in phenol red-free DMEM (Invitrogen) containing 25 mm d-glucose, 10% fetal bovine serum (Hyclone), 100 units/ml penicillin, 100 μg/ml streptomycin, 4 mm l-glutamine, and 1 mm sodium pyruvate (Invitrogen) (82). INS-1 832/13 cells were grown in phenol red-free RPMI 1640 medium (Invitrogen) containing 11.1 mm d-glucose, 10% fetal bovine serum (Hyclone), 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm l-glutamine, 10 mm HEPES, 1 mm sodium pyruvate (Invitrogen) and 50 μm β-mercaptoethanol.
DNA constructs and transfection of GPCRs
The plasmid pcDNA3.1(+), including the coding region of human GPR40 (hGPR40), cDNA was purchased from cDNA Resource Center (Rolla, MO). Expression plasmids for GPR120 short chain (S) and long chain (L) inserted into pCMV6 were purchased from OriGene (Rockville, MD) (50). The hGPR40-coding region was digested from the plasmid described above at the BamHI and XhoI sites and then religated into a pcDNA3.1/Hygro(+) vector. This generated pcDNA3.1/Hygro(+)-hGPR40 that codes for the hGPR40 protein expression system with the hygromycin resistance gene. The plasmid sequences were confirmed and used for stable transfection experiments.
HEK293 cells were stably transfected with pcDNA3.1(+)/Hygro-hGPR40 (5 × 105/35-mm dish) using 3 μg of the plasmid DNA and Lipofectamine 2000 (Invitrogen). After 48 h, the cells were then trypsinized and plated into 100-mm dishes and selected with 100 μg/ml hygromycin for 10 days. Single colonies were isolated, and the expression of hGPR40 was identified by RT-PCR and immunoblot analysis (Fig. 1). For the transient transfection of GPR120 (S) and GPR120 (L), HEK293 cells (3 × 106/100-mm dish) were transfected with one of the plasmids using Lipofectamine 2000. The transfected cells were cultured for 24 h, and expression was confirmed by RT-PCR or re-plated into 96-well plates.
[Ca2+]i assay in HEK293 cells using a fluorescence imaging plate reader (FLIPR)
The assay was performed in a black-walled clear-bottomed poly-d-lysine coated 96-well plate (Corning). Briefly, HEK293 cells and GPR40-transfected HEK293 cells were seeded (50,000 cells/well) in phenol-red free DMEM growth media and allowed to adhere (82). INS-1 832/13 cells were seeded (80,000 cells/well) in phenol-red free RPMI 1640 growth media. After 24 h, growth medium was removed, and 0.1 ml of 1× Fluo-4 NW dye loading solution (proprietary concentration of Fluo-4 NW dye and 2.5 mm probenecid in 1× Hanks' balanced salt solution (HBSS) with Ca2+, Mg2+, and 20 mm HEPES) was added to each well. Cells were incubated with dye for 30 min at 37 °C in 5% CO2 in air followed by an additional 30-min incubation at room temperature. The dye loading of 96-well plates was staggered so calcium assays could be performed within 5 min or less of completing the dye loading. EETs and EET analogs were synthesized in the laboratory of J. R. Falck (59, 83). Dilutions of these test compounds were prepared in assay buffer, without probenecid and dye, in V-shape 96-well plates (Greiner Bio-one, Germany). The calcium assay was performed at room temperature using a FLIPR-3 (Molecular Devices) as described (82). Basal fluorescence was monitored for 20 s, then 25 μl of each compound dilution was added, and the signal was monitored over 200 s. Relative changes in [Ca2+]i were calculated by subtracting the average basal fluorescence (average fluorescence from 0 to 20 s) from the maximum fluorescence (peak fluorescence value) and normalizing to total protein in each well. Results were expressed as change in relative fluorescence units (ΔRFU)/μg protein. Protein concentrations were measured using the Pierce BCA protein assay kit. Cells were examined at the end of [Ca2+]i measurements for morphological changes. No changes were observed following treatment with EETs, EET analogs, GPR40 agonists or antagonists, or vehicle. ATP stimulation of [Ca2+]i was included as a positive control in each plate, and each inhibitor was tested for nonspecific inhibition of ATP.
[3H]TAK-875 radioligand binding in HEK293 and HEK293 + GPR40 cells
[3H]TAK-875 is a high-affinity GPR40 radioligand used to assess binding of agonist (48, 49). HEK293 and HEK293 + GPR40 cells were rinsed twice and scraped into phosphate-buffered saline (PBS). Cells were collected by centrifugation at 735 × g for 5 min, and the supernatant was discarded. The cell pellets were resuspended in HBSS (without Ca2+ and Mg2+) containing protease inhibitors (EDTA-free, Roche Diagnostics) and sonicated for five 20-s bursts on ice with an ultrasonic homogenizer. The homogenate was centrifuged at 1310 × g for 10 min at 4 °C to remove intact cells. The supernatant was then centrifuged at 110,000 × g for 45 min at 4 °C. The pellet representing the membrane fraction was resuspended in binding buffer (10 mm HEPES, 5 mm CaCl2, 5 mm MgCl2, and 5 mm EDTA, pH 7.4). HEK293 + GPR40 or HEK293 membranes (50 μg) were incubated in a total volume of 0.25 ml with 2.5 nm [3H]TAK-875 (80 Ci/mmol, American Radiolabeled Chemicals) in the presence of 10 μm TAK-875 (nonspecific binding), vehicle (total binding), or various concentrations of 11,12-EET or 14,15-EET (0.3–30 μm) on ice for 30 min with shaking. Incubations were terminated by filtration through a GF/B glass filter on a Brandel harvester. Filters were washed three times with 4 ml of ice-cold 50 mm Tris-HCl buffer, pH 7.5. Filter disks were transferred to scintillation vials, and radioactivity was determined by liquid scintillation counting.
Total RNA isolation and RT-PCR
Total RNA was isolated from human and bovine vascular cells using TRIzol reagent (Invitrogen). The RNA was then treated with DNase I (Invitrogen) at 2 units for 30 min at 37 °C and further purified using RNeasy mini kit (Qiagen, Valencia, CA). The total RNA was used for cDNA synthesis using the SuperScript first-strand synthesis system (Invitrogen). A standard 10-μl reaction contained 1 μg of RNA, 50 ng of random hexamers, and 0.5 mm dNTPs. The mixture was incubated at 65 °C for 5 min, on ice for 1 min, and then supplemented with 10× RT buffer (1× final concentration), 5 mm MgCl2, 10 mm dithiothreitol (DTT), RNase inhibitor, and 50 units of SuperScript III RT. The reaction was incubated first at 25 °C for 10 min, then at 50 °C for 50 min, and finally at 85 °C for 10 min. For RT-PCR analysis, 1 μl of the synthesized cDNA was used as the template in 25-μl reactions each containing 0.2 mm dNTPS, 0.4 μm primers, 1.2 m betaine, and 1 unit of Phusion DNA polymerase (Invitrogen) in 1× reaction buffer, including 1.5 mm MgCl2. The primer sequences are indicated in Table 1. The reactions were incubated at 98 °C for 1 min, followed by 35 amplification cycles each including incubations at 98 °C for 10 s, 58 °C for 30 s, 72 °C for 30 s, and then additional extension time at 72 °C for 5 min. The RT-PCR products were then resolved in 2% agarose gels.
Table 1.
Primers used for PCR
| Gene name | Accession no. | Forward primer | Reverse primer | Amplification size |
|---|---|---|---|---|
| bp | ||||
| Human GPR40 | AF024687 | 5′-TGGCCCACTTCTTCCCACTC-3′ | 5′-CAGGAGAGAGAGGCTGAAGC-3′ | 317 |
| Bovine GPR40 | NM001309646 | 5′-TCCACTTCGCTCCACTCTAT-3′ | 5′-GCAGAAGCGAGAGGCTAAA-3′ | 315 |
| Human GPR120 | NM181745 | 5′-AGATCTCGTGGGATGTCTCT-3′ | 5′-GATGAGGAGGATGGTGATGATG-3′ | 251 |
Cell and tissue lysates preparation
Cell and tissue lysates for immunoblotting were prepared with 1× cell lysis buffer (Cell Signaling Technology, Beverly, MA). In brief, the cells were washed twice with ice-cold PBS, harvested into 1 ml of PBS, and pelleted at 1000 × g for 5 min. The cell pellet was then lysed by resuspension in 30–50 μl of the lysis buffer containing complete protease inhibitor mixture (Roche Applied Science) and incubated on ice for 5 min with brief sonication. The sample was then centrifuged at 1000 × g for 5 min, and the supernatant used for immunoblotting. Tissues were frozen in liquid nitrogen, ground using a mortar and pestle, sonicated, and centrifuged at 1000 × g for 10 min. The supernatants were removed and analyzed.
Western blot analysis
The lysates were boiled for 5 min in SDS sample buffer, and equal amounts of protein (20–30 μg) were loaded in each lane of a 10% Tris-glycine gel (Bio-Rad) as described previously (82, 84). After gel electrophoresis, proteins were transferred to a nitrocellulose membrane. Membranes were incubated for 1 h in 0.1% Tween/TBS (TBST) containing 5% nonfat milk, then incubated with primary rabbit polyclonal anti-GPR40 antibody (ThermoFisher Scientific) at 1:1000 dilution in fresh blocking solution overnight at 4 °C, and finally incubated with horseradish peroxidase-conjugated secondary antibody (Invitrogen) for 1 h at room temperature. Blots were developed using the enhanced chemiluminescence Western blotting detection system (ThermoFisher Scientific) and quantified with an ImageQuant LAS4000 system. Blots were then stripped using 1 m glycine, washed with TBST, and immunoblotted and visualized as described above using an anti-β-actin antibody (1:2000).
Isometric tension measurements in BCAs
Measurement of isometric tension was determined as described previously (6). The BCAs were obtained from a local slaughterhouse, and rings of the left anterior descending coronary arteries were used for tension measurements. The thromboxane mimetic U46619 (20 nm) was applied to preconstrict the rings. Relaxations to increasing concentrations of 11,12-EET were recorded in the presence and absence of GW1100 (10 μm). These experiments were repeated in arteries pretreated with iberiotoxin (100 nm) to eliminate the contribution of smooth muscle cell BKCa channels to 11,12-EET relaxations. Results were expressed as percent of relaxation with the basal tension represented as 100%.
Whole-cell patch-clamp measurements of potassium currents in HUVECs
K currents were obtained in HUVECs using standard voltage-clamp pulse protocols as described previously (85, 86). Patched cells were dialyzed with a pipette solution with and without 1 mm EDTA. Vehicle, 11,12-EET (1 μm) with vehicle, GW1100 (10 μm), or 11,12-EET plus GW1100 were added to the bath solution. Macroscopic K+ currents were generated by progressive, stepwise 10-mV depolarizing pulses from a constant holding potential of 60 mV. Currents were sampled at 3 kHz and filtered at 1 kHz. Membrane capacitance was estimated, and currents were expressed in picoamperes/picofarads.
11,12-EET stimulation of ERK phosphorylation and Cx43 and COX-2 expression in HUVECs
HUVECs were grown to 90% confluency in 20% FBS media and serum-starved overnight in 0.5% FBS media (79). The cells were treated with vehicle or 11,12-EET (1 μm) in serum-free media. This concentration of 11,12-EET stimulates GPR40 but not GPR120. Vehicle and U0126 (1 μm), GW1100 (10 μm), andrographolide (40 μm), or EH1555 (1 μm) (provided by Dr. Bruce D. Hammock, University of California, Davis) were added 20 min before 11,12-EET, and the incubation was continued for an additional 30 min for phospho-p42/44 (pp42/44) and Cx43 and 24 h for COX-2 studies. The cells were washed twice in cold PBS and lysed in cell lysis buffer (Cell Signaling Technologies) containing 1× protease inhibitor mixture (Roche Applied Science), 1× PhosStop mixture (Roche Applied Science), and 1 mm PMSF. Cx43 samples were separated by centrifugation into Triton-soluble and Triton-insoluble fractions. The Triton-soluble fraction was analyzed for Cx43. Western blotting was performed using anti-pp42/44 and (Cell Signaling Technologies) in 5% BSA as blocking reagent. When using anti-p44/42 (Cell Signaling Technologies), anti-Cx43 (BD Biosciences), or anti-COX-2 (Cayman Chemical, Ann Arbor, MI) monoclonal antibodies, 5% nonfat milk was the blocking reagent (77). Detection was by chemiluminescence as described above.
Transfection of HEK293 + GPR40 cells and HUVECs with GPR40-targeted siRNA
HEK293–GPR40 cells and HUVECs were grown on 60-mm dishes to 70–90% confluence in 20% FBS media and serum-starved overnight in 0.1% FBS media. The cells were then transfected with GPR40 targeted or control nontargeting siRNAs (Dharmacon, Denver, CO) at a final concentration of 30 nm for HEK293 + GPR40 cells or 10 nm for HUVECs using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. After 24 h, the transfection solution was replaced with fresh 0.5% FBS media and incubated for an additional 24 h. HEK293 + GPR40 cells were replated in 96-well plates and analyzed for [Ca2+]i by FLIPR as described above. HUVECs were treated with vehicle or 11,12-EET (1 μm) in serum-free media for 30 min and then lysed in Cell Lysis buffer (Cell Signaling Technologies) containing 1× proteinase inhibitor mixture (Roche Applied Science), 1× PhosStop (Roche Applied Science), and 1 mm PMSF for Western blot analysis as described above.
Statistical analysis
The data were expressed as means ± S.E. and evaluated statistically by the Student's t test and Student-Newman-Keuls multiple comparison test and followed by one-way analysis of variance. p < 0.05 was considered significant.
Author contributions
S. K. P., A. H., S. L. P., and K. M. G. data curation; S. K. P., A. H., S. L. P., K. M. G., B. A. F., J. R. F., and W. B. C. formal analysis; S. K. P., A. H., S. L. P., J. R. F., and W. B. C. methodology; S. K. P., A. H., S. L. P., J. R. F., and W. B. C. writing-review and editing; B. A. F. and J. R. F. resources; W. B. C. conceptualization; W. B. C. supervision; W. B. C. writing-original draft.
Acknowledgments
We thank Gretchen Barg for secretarial assistance, Dr. John Corbett (Medical College of Wisconsin) for the INS-1 832/13 cells, and Dr. Bruce Hammock (University of California, Davis) for the sEH inhibitor EH1555.
This work was supported by National Institutes of Health Grants HL-83297 and HL-111392 from NHLBI, the Ralph and Marian Falk Medical Research Trust Bank of America, N.A., Trustee Grant, and Robert A. Welch Foundation Award I-0011. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EET
- epoxyeicosatrienoic acid
- EC
- endothelial cell
- GPCR
- G protein–coupled receptor
- SMC
- smooth muscle cell
- HUVEC
- human umbilical vein endothelial cell
- HAoSMC
- human aortic smooth muscle cell
- HCaEC
- human coronary artery endothelial cell
- BCAEC
- bovine coronary artery endothelial cell
- BCASMC
- bovine coronary artery smooth muscle cell
- DMEM
- Dulbecco's modified Eagle's medium
- PMSF
- phenylmethylsulfonyl fluoride
- DHET
- vic-dihydroxyeicosatrienoic acid
- sEH
- soluble epoxide hydrolase
- RFU
- relative fluorescence unit
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- tPA
- tissue plasminogen activator
- TP
- thromboxane
- EEQ
- epoxyeicosatetraenoic acid
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- PPAR
- peroxisome proliferator-activated receptor
- FLIPR
- fluorescence imaging plate reader
- HBSS
- Hanks' balanced salt solution
- BCA
- bovine coronary artery.
References
- 1. Capdevila J., Chacos N., Werringloer J., Prough R. A., and Estabrook R. W. (1981) Liver microsomal cytochrome P450 and the oxidative metabolism of arachidonic acid. Proc. Natl. Acad. Sci. U.S.A. 78, 5362–5366 10.1073/pnas.78.9.5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Capdevila J., Marnett L. J., Chacos N., Prough R. A., and Estabrook R. W. (1982) Cytochrome P450-dependent oxygenation of arachidonic acid to hydroxyieosatetraenoic acids. Proc. Natl. Acad. Sci. U.S.A. 79, 767–770 10.1073/pnas.79.3.767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campbell W. B., and Falck J. R. (2007) Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension 49, 590–596 10.1161/01.HYP.0000255173.50317.fc [DOI] [PubMed] [Google Scholar]
- 4. Campbell W. B., and Fleming I. (2010) Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 459, 881–895 10.1007/s00424-010-0804-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larsen B. T., Campbell W. B., and Gutterman D. D. (2007) Beyond vasodilation: non-vasomotor roles of epoxyeicosatrienoic acids in the cardiovascular system. Trends Pharmacol. Sci. 28, 32–38 10.1016/j.tips.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 6. Campbell W. B., Gebremedhin D., Pratt P. F., and Harder D. R. (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 78, 415–423 10.1161/01.RES.78.3.415 [DOI] [PubMed] [Google Scholar]
- 7. Fisslthaler B., Popp R., Kiss L., Potente M., Harder D. R., Fleming I., and Busse R. (1999) Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 401, 493–497 10.1038/46816 [DOI] [PubMed] [Google Scholar]
- 8. Sun J., Sui X., Bradbury J. A., Zeldin D. C., Conte M. S., and Liao J. K. (2002) Inhibition of vascular smooth muscle cell migration by cytochrome P450 epoxygenase-derived eicosanoids. Circ. Res. 90, 1020–1027 10.1161/01.RES.0000017727.35930.33 [DOI] [PubMed] [Google Scholar]
- 9. Snyder G. D., Krishna U. M., Falck J. R., and Spector A. A. (2002) Evidence for a membrane site of action for 14,15-EET on expression of aromatase in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 283, H1936–H1942 10.1152/ajpheart.00321.2002 [DOI] [PubMed] [Google Scholar]
- 10. Ding Y., Fromel T., Popp R., Falck J. R., Schunck W.-H., and Fleming I. (2014) The biological actions of 11,12-epoxyeicosatrienoic acid in endothelial cells are specific to the R/S-enantiomer and require the Gs protein. J. Pharmacol. Exp. Ther. 350, 14–21 10.1124/jpet.114.214254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michaelis U. R., Falck J. R., Schmidt R., Busse R., and Fleming I. (2005) Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce the expression of cyclooxygenase-2 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 25, 321–336 [DOI] [PubMed] [Google Scholar]
- 12. Popp R., Brandes R. P., Ott G., Busse R., and Fleming I. (2002) Dynamic modulation of interendothelial gap junctional communication by 11,12-epoxyeicosatrienoic acid. Circ. Res. 90, 800–806 10.1161/01.RES.0000015328.20581.D6 [DOI] [PubMed] [Google Scholar]
- 13. Node K., Ruan X.-L., Dai J., Yang S.-X., Graham L., Zeldin D. C., and Liao J. K. (2001) Activation of Gαs mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem. 276, 15983–15989 10.1074/jbc.M100439200 [DOI] [PubMed] [Google Scholar]
- 14. Wang Y., Wei X., Xiao X., Hui R., Card J. W., Carey M. A., Wang D.-W., and Zeldin D. C. (2005) Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J. Pharmacol. Exp. Ther. 314, 522–532 10.1124/jpet.105.083477 [DOI] [PubMed] [Google Scholar]
- 15. Yang S., Lin L., Chen J.-X., Lee C. R., Seubert J. M., Wang Y., Wang H., Chao Z.-R., Tao D.-D., Gong J.-P., Lu Z.-Y., Wang D.-W., and Zeldin D. C. (2007) Cytochrome P450 epoxygenase protects endothelial cells from apoptosis induced by tumor necrosis factor-α via MAPK and PI3K/Akt signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 293, H142–H151 10.1152/ajpheart.00783.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dhanasekaran A., Al-Saghir R., Lopez B., Zhu D., Gutterman D. D., Jacobs E. R., and Medhora M. (2006) Protective effects of epoxyeicosatrienoic acids on human endothelial cells from the pulmonary and coronary vasculature. Am. J. Physiol. Heart Circ. Physiol. 291, H517–H531 10.1152/ajpheart.00953.2005 [DOI] [PubMed] [Google Scholar]
- 17. Chen J. K., Capdevila J., and Harris R. C. (2001) Cytochrome P450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Mol. Cell. Biol. 21, 6322–6331 10.1128/MCB.21.18.6322-6331.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Node K., Huo Y., Ruan X., Yang B., Spiecker M., Ley K., Zeldin D. C., and Liao J. K. (1999) Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285, 1276–1279 10.1126/science.285.5431.1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krötz F., Riexinger T., Buerkle M. A., Nithipatikom K., Gloe T., Sohn H. Y., Campbell W. B., and Pohl U. (2004) Membrane potential-dependent inhibition of platelet adhesion to endothelial cells by epoxyeicosatrienoic acids. Arterioscler. Thromb. Vasc. Biol. 24, 595–600 [DOI] [PubMed] [Google Scholar]
- 20. Wong P. Y., Lin K. T., Yan Y. T., Ahern D., Iles J., Shen S. Y., Bhatt R. K., Falck J. R. (1993) 14(R),15(S)-Epoxyeicosatrienoic acid receptor in guinea pig mononuclear cell membranes. J. Lipid Mediat. 6, 199–208 [PubMed] [Google Scholar]
- 21. Yang W., Tuniki V. R., Anjaiah S., Falck J. R., Hillard C. J., and Campbell W. B. (2008) Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20–125I-14,15-epoxyeicosa-8(Z)-enoic acid. J. Pharmacol. Exp. Ther. 324, 1019–1027 [DOI] [PubMed] [Google Scholar]
- 22. Yang W., Holmes B. B., Gopal V. R., Kishore R. V., Sangras B., Yi X.-Y., Falck J. R., and Campbell W. B. (2007) Characterization of 14,15-epoxyeicosatrienoyl-sulfonamides as 14,15-epoxyeicosatrienoic acid agonists: use for studies of metabolism and ligand binding. J. Pharmacol. Exp. Ther. 321, 1023–1031 10.1124/jpet.107.119651 [DOI] [PubMed] [Google Scholar]
- 23. Li P.-L., and Campbell W. B. (1997) Epoxyeicosatrienoic acids activate potassium channels in coronary smooth muscle through guanine nucleotide binding protein. Circ. Res. 80, 877–884 10.1161/01.RES.80.6.877 [DOI] [PubMed] [Google Scholar]
- 24. Van Voorhis B. J., Dunn M. S., Falck J. R., Bhatt R. K., VanRollins M., and Snyder G. D. (1993) Metabolism of arachidonic acid to epoxyeicosatrienoic acids by human granulosa cells may mediate steroidogenesis. J. Clin. Endocrinol. Metab. 76, 1555–1559 10.1210/jcem.76.6.8388882 [DOI] [PubMed] [Google Scholar]
- 25. Henrich W. L., Falck J. R., and Campbell W. B. (1990) Inhibition of renin release by 14,15-epoxyeicosatrienoic acid in renal cortical slices. Am. J. Physiol. 258, E269–E274 [DOI] [PubMed] [Google Scholar]
- 26. Chen Y., Falck J. R., Manthati V. L., Jat J. L., and Campbell W. B. (2011) 20-Iodo-14,15-epoxyeicosa-8(Z)-enoyl-3-azidophenylsulfonamide: photo affinity labeling of a 14,15-epoxyeicosatrienoic acid receptor. Biochemistry 50, 3840–3848 10.1021/bi102070w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y., Zhang Y., Schmelzer K., Lee T.-S., Fang X., Zhu Y., Spector A. A., Gill S., Morisseau C., Hammock B. D., and Shyy J. Y. (2005) The antiinflammatory effect of laminar flow: the role of PPARγ, epoxyeicosatrienoic acids and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 102, 16747–16752 10.1073/pnas.0508081102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Behm D. J., Ogbonna A., Wu C., Burns-Kurtis C. L., and Douglas S. A. (2009) Epoxyeicosatrienoic acids function as selective, endogenous antagonists of native thromboxane receptors: identification of a novel mechanism of vasodilation. J. Pharmacol. Exp. Ther. 328, 231–239 10.1124/jpet.108.145102 [DOI] [PubMed] [Google Scholar]
- 29. Yang C., Kwan Y. W., Au A. L., Poon C. C., Zhang Q., Chan S. W., Lee S. M., and Leung G. P. (2010) 14,15-Epoxyeicosatrienoic acid induces vasorelaxation through prostaglandin EP(2) receptors in rat mesenteric artery. Prostaglandins Other Lipid Mediat. 93, 44–51 10.1016/j.prostaglandins.2010.06.004 [DOI] [PubMed] [Google Scholar]
- 30. Earley S., Heppner T. J., Nelson M. T., and Brayden J. E. (2005) TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BK Ca channels. Circ. Res. 97, 1270–1279 10.1161/01.RES.0000194321.60300.d6 [DOI] [PubMed] [Google Scholar]
- 31. Briscoe C. P., Tadayyon M., Andrews J. L., Benson W. G., Chambers J. K., Eilert M. M., Ellis C., Elshourbagy N. A., Goetz A. S., Minnick D. T., Murdock P. R., Sauls H. R. Jr., Shabon U., Spinage L. D., Strum J. C., et al. (2003) The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 278, 11303–11311 10.1074/jbc.M211495200 [DOI] [PubMed] [Google Scholar]
- 32. Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S., Ogi K., Hosoya M., Tanaka Y., Uejima H., Tanaka H., Maruyama M., Satoh R., Okubo S., Kizawa H., Komatsu H., et al. (2003) Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422, 173–176 10.1038/nature01478 [DOI] [PubMed] [Google Scholar]
- 33. Mancini A. D., and Poitout V. (2013) The fatty acid receptor FFA1/GPR40 a decade later: how much do we know? Trends Endocrinol. Metab. 24, 398–407 10.1016/j.tem.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 34. Stoddart L. A., Smith N. J., and Milligan G. (2008) International Union of Pharmacology. LXXI. Free fatty acid receptors FFA-1.-2 and -3: Pharmacology and pathophysiological functions. Pharmacol. Rev. 60, 405–417 10.1124/pr.108.00802 [DOI] [PubMed] [Google Scholar]
- 35. Vangaveti V., Shashidhar V., Jarrod G., Baune B. T., and Kennedy R. L. (2010) Free fatty acid receptors: emerging targets for treatment of diabetes and its complications. Ther. Adv. Endocrinol. Metab. 1, 165–175 10.1177/2042018810381066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oh D. Y., Talukdar S., Bae E. J., Imamura T., Morinaga H., Fan W., Li P., Lu W. J., Watkins S. M., and Olefsky J. M. (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698 10.1016/j.cell.2010.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Talukdar S., Olefsky J. M., and Osborn O. (2011) Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol. Sci. 32, 543–550 10.1016/j.tips.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma S. K., Wang Y., Chen J., Zhang M.-Z., Harris R. C., and Chen J.-K. (2015) Overexpression of G-protein-coupled receptor 40 enhances the mitogenic response to epoxyeicosatrienoic acids. PLoS ONE 10, e0113130 10.1371/journal.pone.0113130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shapiro H., Shachar S., Skekler I., Hershfinkel M., and Walker M. D. (2005) Role of GPR40 in fatty acid action in the beta cell ine INS-1E. Biochem. Biophys. Res. Commun. 335, 97–104 10.1016/j.bbrc.2005.07.042 [DOI] [PubMed] [Google Scholar]
- 40. Yamada H., Yoshida M., Ito K., Dezaki K., Yada T., Ishikawa S. E., and Kakei M. (2016) Potentiation of glucose-stimulated insulin secretion by the GPR40-PLC-TRPC pathway in pancreatic beta-cells. Sci. Rep. 6, 25912 10.1038/srep25912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schnell S., Schaefer M., and Schöfl C. (2007) Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from beta-cells through activation of GPR40. Mol. Cell. Endocrinol. 263, 173–180 10.1016/j.mce.2006.09.013 [DOI] [PubMed] [Google Scholar]
- 42. Briscoe C. P., Peat A. J., McKeown S. C., Corbett D. F., Goetz A. S., Littleton T. R., McCoy D. C., Kenakin T. P., Andrews J. L., Ammala C., Fornwald J. A., Ignar D. M., and Jenkinson S. (2006) Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br. J. Pharmacol. 148, 619–628 10.1038/sj.bjp.0706770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu H., He L. Y., Gong Z., Li N., Lu Y. N., Zhai Q. W., Liu H., Jiang H. L., Zhu W. L., and Wang H. Y. (2009) A novel class of antagonists for the FFAs receptor GPR40. Biochem. Biophys. Res. Commun. 390, 557–563 10.1016/j.bbrc.2009.10.004 [DOI] [PubMed] [Google Scholar]
- 44. Fukushima K., Yamasaki E., Ishii S., Tomimatsu A., Takahashi K., Hirane M., Fukushima N., Honoki K., and Tsujiuchi T. (2015) Different roles of GPR120 and GPR40 in the acquisition of malignant properties in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 465, 512–515 10.1016/j.bbrc.2015.08.050 [DOI] [PubMed] [Google Scholar]
- 45. Imig J. D., and Hammock B. D. (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 8, 794–805 10.1038/nrd2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Honoré J.-C., Kooli A., Hamel D., Alquier T., Rivera J.-C., Quiniou C., Hou X., Kermorvant-Duchemin E., Hardy P., Poitout V., and Chemtob S. (2013) Fatty acid receptor GPR40 mediates neuromicrovascular degeneration induced by transarachidonic acid in rodents. Arterioscler. Thromb. Vasc. Biol. 33, 954–961 10.1161/ATVBAHA.112.300943 [DOI] [PubMed] [Google Scholar]
- 47. Negoro N., Sasaki S., Mikami S., Ito M., Suzuki M., Tsujihata Y., Ito R., Harada A., Takeuchi K., Suzuki N., Miyazaki J., Santou T., Odani T., Kanzaki N., Funami M., et al. (2010) Discovery of TAK-875: a potent, selective and orally bioavailable GPR40 agonist. ACS Med. Chem. Lett. 1, 290–294 10.1021/ml1000855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Y., Song M., Riley J. P., Hu C. C., Peng X., Scheuner D., Bokvist K., Maiti P., Kahl S. D., Montrose-Rafizadeh C., Hamdouchi C., and Miller A. R. (2016) A selective GPR40 (FFAR1) agonist LY2881835 provides immediate and durable glucose control in rodent models of type 2 diabetes. Pharmacol. Res. Perspect. 4, e00278 10.1002/prp2.278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bertrand R., Hamp I., Brönstrup M., Weck R., Lukacevic M., Polyak A., Ross T. L., Gotthardt M., Plettenburg O., and Derdau V. (2016) Synthesis of GPR40 targeting 3H- and 18F-probes towards selective beta cell imaging. J. Labelled Comp. Radiopharm. 59, 604–610 10.1002/jlcr.3412 [DOI] [PubMed] [Google Scholar]
- 50. Watson S. J., Brown A. J., and Holliday N. D. (2012) Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol. Pharmacol. 81, 631–642 10.1124/mol.111.077388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mena S. J., Manosalva C., Carretta M. D., Teuber S., Olmo I., Burgos R. A., and Hidalgo M. A. (2016) Differential free fatty acid receptor-1 (FFAR1/GPR40) signalling is associated with gene expression or gelatinase granule release in bovine neutrophils. Innate Immun. 22, 479–489 10.1177/1753425916656765 [DOI] [PubMed] [Google Scholar]
- 52. Zhang Y., Xu M., Zhang S., Yan L., Yang C., Lu W., Li Y., and Cheng H. (2007) The role of G protein-coupled receptor 40 in lipoapoptosis in mouse beta-cell line NIT-1. J. Mol. Endocrinol. 38, 651–661 10.1677/JME-06-0048 [DOI] [PubMed] [Google Scholar]
- 53. Solan J. L., and Lampe P. D. (2014) Specific Cx43 phosphorylation events regulate gap junction turnover. FEBS Lett. 588, 1423–1429 10.1016/j.febslet.2014.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fredholm B. B., IJzerman A. P., Jacobson K. A., Linden J., and Müller C. E. (2011) International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol. Rev. 63, 1–34 10.1124/pr.110.003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fredholm B. B. (2014) Adenosine–a physiological or pathophysiological agent? J. Mol. Med. 92, 201–206 10.1007/s00109-013-1101-6 [DOI] [PubMed] [Google Scholar]
- 56. Yang C., Yang J., Xu X., Yan S., Pan S., Pan X., Zhang C., and Leung G. P. (2014) Vasodilatory effect of 14,15-epoxyeicosatrienoic acid on mesenteric arteries in hypertensive and aged rats. Prostaglandins Other Lipid Mediat. 112, 1–8 10.1016/j.prostaglandins.2014.05.001 [DOI] [PubMed] [Google Scholar]
- 57. Jiang H., Zhu A. G., Mamczur M., Morisseau C., Hammock B. D., and McGiff J. C. (2008) Hydrolysis of cis- and trans-epoxyeicosatrienoic acids by rat red blood cells. J. Pharmacol. Exp. Ther. 326, 330–337 10.1124/jpet.107.134858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang H., Quilley J., Doumand A. B., Zhu A. G., Falck J. R., Hammock B. D., Stier C. T. Jr., and Carroll M. A. (2011) Increases in plasma trans-EETs and blood pressure reduction in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 300, H1990–H1996 10.1152/ajpheart.01267.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Falck J. R., Krishna U. M., Reddy Y. K., Kumar P. S., Reddy K. M., Hittner S. B., Deeter C., Sharma K. K., Gauthier K. M., and Campbell W. B. (2003) Comparison of the vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am. J. Physiol. Heart Circ. Physiol. 284, H337–H349 10.1152/ajpheart.00831.2001 [DOI] [PubMed] [Google Scholar]
- 60. Hauge M., Vestmar M. A., Husted A. S., Ekberg J. P., Wright M. J., Di Salvo J., Weinglass A. B., Engelstoft M. S., Madsen A. M., Lückmann M., Miller M. W., Trujillo M. E., Frimurer T. M., Holst B., Howard A. D., and Schwartz T. W. (2015) GPR40 (FFAR1)-Combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol. Metab. 4, 3–14 10.1016/j.molmet.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Luria A., Bettaieb A., Xi Y., Shieh G. J., Liu H. C., Inoue H., Tsai H. J., Imig J. D., Haj F. G., and Hammock B. D. (2011) Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in the model of insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 9038–9043 10.1073/pnas.1103482108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Luther J. M., and Brown N. J. (2016) Epoxyeicosatrienoic acids and glucose homeostasis in mice and men. Prostaglandins Other Lipid Mediat. 125, 2–7 10.1016/j.prostaglandins.2016.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Larsen B. T., Miura H., Hatoum O. A., Campbell W. B., Hammock B. D., Zeldin D. C., Falck J. R., and Gutterman D. D. (2006) Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BKca channels: implications for soluble epoxide hydrolase inhibition. Am. J. Physiol. Heart Circ. Physiol. 290, H491–H499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fang X., Weintraub N. L., Stoll L. L., and Spector A. A. (1999) Epoxyeicosatrienoic acid increases intracellular calcium concentration in vascular smooth muscle. Hypertension 34, 1242–1246 10.1161/01.HYP.34.6.1242 [DOI] [PubMed] [Google Scholar]
- 65. Weston A. H., Félétou M., Vanhoutte P. M., Falck J. R., Campbell W. B., and Edwards G. (2005) Bradykinin-induced, endothelium-dependent responses in porcine coronary arteries: involvement of potassium channel activation and epoxyeicosatrienoic acids. Br. J. Pharmacol. 145, 775–784 10.1038/sj.bjp.0706256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Campbell W. B., Falck J. R., and Gauthier K. (2001) Role of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factor in bovine coronary arteries. Med. Sci. Monit. 7, 578–584 [PubMed] [Google Scholar]
- 67. van Kempen M. J., and Jongsma H. J. (1999) Distribution of connexin 37, connexin 40 and connexin 43 in the aorta and coronary artery of several mammals. Histochem. Cell Biol. 112, 479–486 10.1007/s004180050432 [DOI] [PubMed] [Google Scholar]
- 68. Fleming I., Rueben A., Popp R., Fisslthaler B., Schrodt S., Sander A., Haendeler J., Falck J. R., Morisseau C., Hammock B. D., and Busse R. (2007) Epoxyeicosatrienoic acids regulate Trp-channel-dependent Ca signaling and hyperpolarization in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 27, 2612–2618 10.1161/ATVBAHA.107.152074 [DOI] [PubMed] [Google Scholar]
- 69. Lin M. T., Jian M. Y., Taylor M. S., Cioffi D. L., Yap F. C., Liedtke W., and Townsley M. I. (2015) Functional coupling of TRPV4, IK, and SK channels contributes to Ca2+-dependent endothelial injury in rodent lung. Pulm. Circ. 5, 279–290 10.1086/680166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pozzi A., Macias-Perez I., Abair T., Wei S., Su Y., Zent R., Falck J. R., and Capdevila J. (2005) Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids as potent in vivo angiogenic lipids. J. Biol. Chem. 280, 27138–27146 10.1074/jbc.M501730200 [DOI] [PubMed] [Google Scholar]
- 71. Saez J. C., Berthoud V. M., Branes M. C., Martinez A. D., and Beyer E. C. (2003) Plasma membrane channels formed by connexins: their regulation and function. Physiol. Rev. 83, 1359–1400 10.1152/physrev.00007.2003 [DOI] [PubMed] [Google Scholar]
- 72. de Wit C., Roos F., Boltz S.-S., Kirchhoff S., Krüger O., Willecke K., and Pohl U. (2000) Impaired conduction of vasodilation along arterioles in connexin 40-deficient mice. Circ. Res. 86, 649–655 10.1161/01.RES.86.6.649 [DOI] [PubMed] [Google Scholar]
- 73. Yin G., Yang X., Li B., Yang M., and Ren M. (2014) Connexin43 siRNA promotes HUVEC proliferation and inhibits apoptosis induced by ox-LDL: an involvement of ERK signaling pathway. Mol. Cell. Biochem. 394, 101–107 10.1007/s11010-014-2085-4 [DOI] [PubMed] [Google Scholar]
- 74. Kameritsch P., Khandoga N., Pohl U., and Pogoda K. (2013) Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis. 4, e584 10.1038/cddis.2013.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang J., O'Carroll S. M., Henare K., Ching L. M., Ormonde S., Nicholson L. F., Danesh-Meyer H. V., and Green C. R. (2014) Connexin hemichannel induced vascular leak suggests a new paradigm for cancer therapy. FEBS Lett. 588, 1365–1371 10.1016/j.febslet.2014.02.003 [DOI] [PubMed] [Google Scholar]
- 76. Davidge S. T. (2001) Prostaglandin H synthase and vascular function. Circ. Res. 89, 650–660 10.1161/hh2001.098351 [DOI] [PubMed] [Google Scholar]
- 77. Wohlfeil E. R., and Campbell W. B. (1997) 25-Hydroxycholesterol enhances eicosanoid production in cultured bovine coronary artery endothelial cells by increasing prostaglandin G/H synthase-2. Biochim. Biophys. Acta 1345, 109–120 10.1016/S0005-2760(96)00170-1 [DOI] [PubMed] [Google Scholar]
- 78. Fleming I. (2007) Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediat. 82, 60–67 10.1016/j.prostaglandins.2006.05.003 [DOI] [PubMed] [Google Scholar]
- 79. Revtyak G. E., Hughes M. J., Johnson A. R., and Campbell W. B. (1988) Histamine stimulation of prostaglandin and HETE synthesis in human endothelial cells. Am. J. Physiol. 255, C214–C225 10.1152/ajpcell.1988.255.2.C214 [DOI] [PubMed] [Google Scholar]
- 80. Revtyak G. E., Johnson A. R., and Campbell W. B. (1988) Cultured bovine coronary arterial endothelial cells synthesize HETEs and prostacyclin. Am. J. Physiol. 254, C8–C19 10.1152/ajpcell.1988.254.1.C8 [DOI] [PubMed] [Google Scholar]
- 81. Campbell W. B., Holmes B. B., Falck J. R., Capdevila J. H., and Gauthier K. M. (2006) Adenoviral expression of cytochrome P450 epoxygenase in coronary smooth muscle cells: regulation of potassium channels by endogenous 14(S),15(R)-EET. Am. J. Physiol. Heart Circ. Physiol. 290, H64–H71 10.1152/ajpheart.00516.2005 [DOI] [PubMed] [Google Scholar]
- 82. Siangjong L., Gauthier K. M., Pfister S. L., Smyth E. M., and Campbell W. B. (2013) Endothelial 12(S)-HETE vasodilation is mediated by thromboxane receptor inhibition in mouse mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 304, H382–H392 10.1152/ajpheart.00690.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Falck J. R., Yadagiri P., and Capdevila J. (1990) Synthesis of epoxyeicosatrienoic acids and heteroatom analogs. Methods Enzymol. 187, 357–364 10.1016/0076-6879(90)87042-2 [DOI] [PubMed] [Google Scholar]
- 84. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 85. Gauthier K. M., Liu C., Popovic A., Albarwani S., and Rusch N. J. (2002) Freshly isolated bovine coronary endothelial cells do not express the BKCa channel gene. J. Physiol. 545, 829–836 10.1113/jphysiol.2002.029843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Marchenko S. M., and Sage S. O. (1993) Electrical properties of resting and acetylcholine-stimulated endothelium in intact rat aorta. J. Physiol. 462, 735–751 10.1113/jphysiol.1993.sp019579 [DOI] [PMC free article] [PubMed] [Google Scholar]