

Abstract
A compounding feature of greater than 50% of all cancers is the high incidence of the cachexia syndrome, a complex metabolic disorder characterized by extreme weight loss due mainly to the gross depletion of skeletal muscle tissue. Although studies into the cause of cancer cachexia has spanned over multiple decades, little is known about the effects of various cancer treatments themselves on cachexia. For example, chemotherapy agents induce side effects such as nausea and anorexia, but these symptoms do not fully account for the changes seen with cancer cachexia. In this study we examine the effects of chemotherapeutic compounds, specifically, cisplatin in the colon-26 adenocarcinoma model of cancer cachexia. We find that although cisplatin is able to reduce tumor burden as expected, muscle wasting in mice nevertheless persists. Strikingly, cisplatin alone was seen to regulate muscle atrophy, which was independent of the commonly implicated ubiquitin proteasome system. Finally, we show that cisplatin is able to induce NF-κB activity in both mouse muscles and myotube cultures, suggesting that an additional side effect of cancer treatment is the regulation of muscle wasting that may be mediated through activation of the NF-κB signaling pathway.
Key Words: Cancer, cachexia, NF-κB, chemotherapy, skeletal muscle, wasting
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Patients with cancer often suffer from extensive and progressive muscle wasting, weight loss, and associated weakness, a constellation of symptoms referred to collectively as cancer cachexia. Progressive wasting of a patient’s lean body mass is one of the most prominent aspects of this syndrome, with muscle loss having a much greater impact than loss of adipose tissue in the body.1 The vast majority of patients with advanced cancer suffer from cancer cachexia, with a large number of these patients’ deaths being directly attributable to their cachexia.2,3 Cancer cachexia invariably implies a poor prognosis, contributes to reduced response and tolerance to therapy, confers a decline in quality of life, and is inversely correlated with patient survival.1
While the cause of this well-established syndrome has been studied for some time, little is known about the effects of various cancer treatments on cancer cachexia itself. It has been suggested in past studies that chemotherapy itself may be a major contributor to the development and sustainment of cancer cachexia.29=4 While many chemotherapy agents have known side effects of nausea, diarrhea, and anorexia, these symptoms do not fully account for the changes seen with cachexia. It has been postulated that many of these chemotherapeutic agents cause direct host cell modification to the processes of protein metabolism.4 Chemotherapy does cause negative nitrogen balance,5 yet the mechanism(s) for this catabolic effect has yet to be fully elucidated. The aim in this study was to further delineate the effects of chemotherapy on the process of cancer cachexia, with specific focus on skeletal muscle wasting. Since chemotherapy is known to induce NF-κB activity,6-8 and NF-κB has been implicated in regulating skeletal muscle wasting,9-11we reasoned that one of the effects of chemotherapy on cancer patients is the promotion of muscle wasting through the activation of NF-κB. To test this we examined the effects of a standard chemotherapeutic agent, cisplatin, on the established colon-26 (C-26) murine cancer cachexia model.12 Our results demonstrate that cisplatin, as predicted, is able to strongly reduce tumor burden, but strikingly this agent can also promote severe muscle wasting that is associated with activation of NF-κB. These findings have important clinical implications, for if chemotherapeutic agents can be shown to contribute to not only anorexia, but progressive muscle wasting and associated metabolic changes, these agents can be avoided when patients are at high risk or when clinical situations appropriately apply.
Materials and Methods
Animal Model
Male CD2F1 mice, at 7-8 weeks of age were housed in accordance with the regulations of the Institutional Animal Care and Use Committee (IACUC) of The Ohio State University. All mice were fed and watered ad libitum. Mice were weighed on day 8 and then every 3 days thereafter. Mice were also monitored for tumor occurrence, growth, and progression. Tumor weight and muscle weights were recorded at the time of sacrifice and surgical excision.
Tumor cell line and inoculation
The transplantable C-26 adenocarcinoma cell line was utilized. The cells were maintained as monolayers in culture flasks at 37º C with 5% CO2 in culture medium consisting of Roswell Park Memorial Institute (RPMI) 1640 (Gibco, Rockville, MD) supplemented with 5 % fetal bovine (Hyclone, Logan, UT) and 1% penicillin/streptomycin (Gibco, Rockville, MD). Immediately before injection, cells were harvested at approximately 80% confluence, washed, and resuspended in phosphate-buffered saline (PBS) at a concentration of 1 x 107 cells/ml. A volume of 100 l of the cell solution was then injected subcutaneously into the right flank of the mice. Mice not challenged with tumor inoculation were given a vehicle control injection of 100 µl PBS alone.
Chemotherapy inoculation
Cisplatin (5 mg/Kg) was administered intravenously starting at day 7 post tumor cell or saline administration, and injections were repeated at days 10 and 14, and at day 17 skeletal muscles were harvested for analysis.
Muscle preparation / immunohistochemistry / fiber diameter measurements
Muscles were sectioned at 10μM on a cryostat (Leica) and stained with H&E. Fiber measurements were obtained from a minimum of 1500 fibers per 5-6 randomly chosen fields and recorded from the shortest diameter using Axiovision 3.1 software (Zeiss). Immunofluorescence was performed as previously described, respectively.13
Northern Blots and Electrophoretic mobility shift analysis (EMSA)
Northern blots were performed as in.14 EMSA analysis was performed as previously described.15
Results
To initiate this study we sought to determine the effects of treating mice bearing C-26 tumors with cisplatin (3 doses at 5 mg/Kg). Consistent with previous reports,14 mice bearing C-26 tumors alone underwent substantial weight loss that was associated with pronounced reduction in skeletal muscle mass (Figures 1A and 1B). Interestingly, although cisplatin treatment decreased tumor load nearly 4-fold compared to untreated C-26 bearing mice (Figures. 1A-C), it did little to restore body weight and muscle mass. In fact, compared to healthy controls, mice treated with cisplatin alone exhibited similar reduction in body weight and muscle loss as tumor bearing mice. Hematoxylin and eosin (H&E) staining of cross- sections of the resected gastrocnemius muscles revealed visibly smaller caliber muscle fibers in all treatment groups compared to control (Figure 1D). No obvious differences were appreciated when comparing the three treatment groups to each other. Figure 1E represents these findings graphically, showing a left shift toward smaller diameter muscle fibers in the treatment groups compared to the control group centered around a median diameter range of 40-49 μm. In contrast, tumor-bearing mice, cisplatin-treated tumor-bearing mice, and healthy cisplatin-treated mice all showed median muscle fiber diameter ranges of 20-29 μm. Taken together, these findings suggest that cisplatin treatment alone is capable of inducing certain features of cachexia. To delineate the mechanism of cisplatin action on muscle wasting, Northern blot assays were carried out to address the potential regulation on the ubuiquitin proteasome system. Expression of muscle RING finger protein 1 (MuRF1), a muscle-specific E3 ligase, is a known marker of proteolysis.16 Consistent with previous findings,13,14 MuRF1 expression was highly elevated in untreated C-26 tumor-bearing mice (Figure 2A). In striking contrast, tumor-bearing mice treated with cisplatin reduced MuRF1 expression (Figure 2A) as well as the other classic muscle specific E3 ubiquitin ligase, MAFBx/Atrogin1 (data not shown).17,18 In addition, cisplatin treatment of healthy mice, which was observed to cause equivalent reduction of muscle mass as tumor-bearing mice (Figure 1), showed no significant increase in E3 ligase expression. Thus, these data indicates that cisplatin-induced muscle wasting functions independently of the ubiquitin-proteasome system. Findings in Figure 1 showed that cisplatin treated C-26 bearing mice significantly reduced tumor burden that corresponded to a decrease in E3 ubiquitin ligase expression. Based on these findings, we hypothesized that muscle E3 ubiquitin ligase activity was dependent on the C-26 tumor burden. To test this hypothesis, we induced mice with C-26 tumors for 15 days, and subsequently surgically removed the tumor and re-analyzed MuRF1 expression at various days post-surgery. As seen in Figure 2B, MuRF1 levels were elevated, as expected, in mice bearing C-26 tumors for 15 days. However, when the tumor was removed, even as early as 1 day post-resection, MuRF1 expression was reduced to basal levels (Figure 2B). This reduction in MuRF1 was also maitained after 7 days post operation, which coincided with the continued absence of tumor burden. When examining the fiber diameter of gastrocnemius muscles resected from tumor-bearing mice that underwent surgical resection of their tumor, we observed a shift back towards larger diameter muscle fibers, suggesting that when the E3 ligases are no longer actively promoting proteolysis, muscle fiberscan be restored to their nearly normal size (Figure 2C). Together, these results demonstrate that tumor factors are directly responsible for regulating E3 ligase expression in skeletal muscle. These findings also reaffirm the notion that cisplatin-induced muscle wasting is not ubiquitin proteasome dependent. Since it is thought that cisplatin, along with many other chemotherapeutic agents, functions by inducing cytotoxicity or apoptosis of the cancer cells,19,20 and given that apoptosis has been proposed as a mechanism of skeletal muscle atrophy,21,22 we addressed whether cisplatin induced effects on muscle mass and abnormalities in fiber diameter occurred by direct cytotoxicity to the myocytes. However, when comparing gastrocnemius muscle sections of healthly control mice, untreated tumor-bearing mice, and healthy cisplatin-treated mice, we were unable to detect any differences in TUNEL staining (data not shown), suggesting that myocytes from cisplatin treated mice do not undergo atrophy due to chemotherapy-induced toxicity. In addition, previous results have shown that NF-κB activity is upregulated in cancer cachexia as well as other models of atrophy,23,24 and this activity is required to mediate muscle loss.9,10 Since NF-κB is activated by chemotherapeutic compounds, we asked whether cisplatin-induced muscle wasting might be associated with NF-κB. By immunohistochemical staining, activation of NF-κB, via phosphorylation of its classic p65 subunit, was clearly observed in healthy cisplatin-treated mice (Figure 3A). To further explore the regulation of NFκB by cisplatin in muscle cells, we treated cultured C2C12 myotubes with cisplatin for various times. By electrophoretic mobility shift analysis (EMSA), results showed that cisplatin maximally increased NF-κB DNA binding activity in myotubes 4-fold. Supershift EMSAs further confirmed that the p65 subunit was contained in this binding complex. Since p65 has previously been demonstrated to be a major regulator of muscle wasting in cachexia and well as in muscular dystrophy,9,25 the data imply that cisplatin-induced regulation of muscle wasting may occur through an NF-κB signaling pathway. In the last part of this study, we sought to investigate whether the effects that we showed on muscle atrophy with chemotherapy was specific to cisplatin. Healthy mice were therefore treated with common chemotherapeutic agents, CPT-11 (Irinotecan), Adriamycin, or Etoposide. Results showed that each of these compounds promoted muscle loss when compared to control mice (Figure 4A). Shown graphically, a similar left shift toward smaller diameter muscle fibers was seen when any of the three chemotherapeutic agents were administered. Similar to cisplatin, the median fiber diameter range was centered approximately 20-29μm. This suggests that chemo-therapeutic agents possess the capacity to promote cachexia via their regulation of muscle atrophy.
Fig 1.
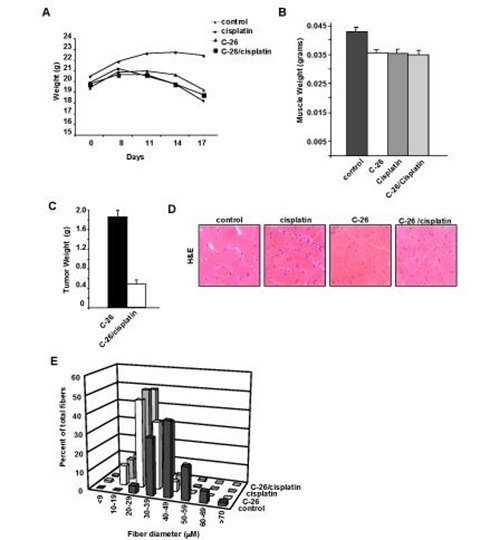
Cisplatin induces weight loss and muscle atrophy in mice. Four sets of treatment group were used with CD2F1 mice. In group 1, mice were injected with saline (control); in group 2, mice were injected with saline and then treated with cisplatin at days 7, 10, and 14 post-injection (cisplatin); in group 3, mice were injected with C-26 tumor cells (C-26); in group 4, mice were injected with C-26 cells then treated with cisplatin at days 7, 10, and 14 post injection (C-26/cisplatin). (A) Following treatments, body weights (n=4, each group) were recorded at indicated days. (B) Gastrocnemius muscle weight (n=6 each group) were recorded in grams on day 17 post injection. (C) C-26 tumors were removed from untreated and cisplatin treated mice and weighed. (D) H&E stained cryosections of gastrocnemius muscles were analyzed (n=4 per group). (E) Fiber diameter was measured from H&E muscle sections from(D).
Fig 2.
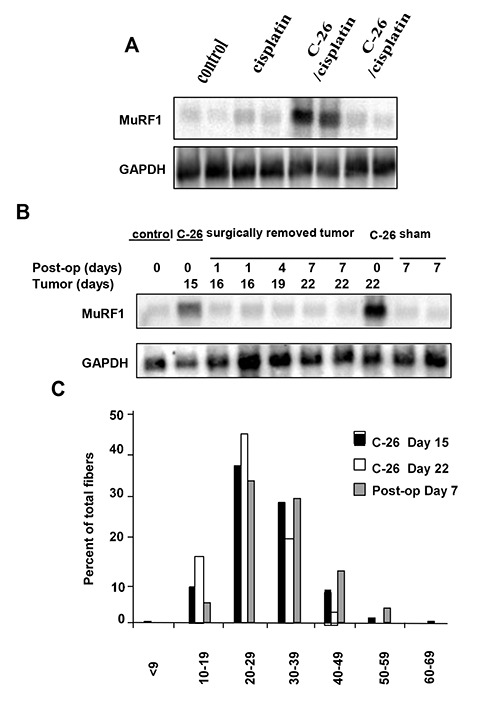
Expression of muscle E3 ubiquitin ligase is dependent on C-26 tumors. (A) Muscles were harvested 17 days post-injection in each of the 4 treatment groups (n=2). Total RNA was prepared and Northerns were performed probing for the muscle ubiqutin ligase gene, MuRF1. To confirm equal loading, blots were stripped and reprobed with GAPDH. (B) MuRF1 Northern was performed with RNA prepared from muscles harvested at various times either from C-26 tumor bearing mice alone (days 15 and 22), or from mice where C-26 tumors were surgically removed (post-op, days 1, 4, or 7). Control designates basal level of MuRF1, while Sham is level of MuRF1 following surgery in mice without tumors. GAPDH was also determined to control for loading control. (C) At indicated days and conditions, gastrocnemius muscles were harvested and fiber diameters were calculated from H&E sections.
Fig 3.
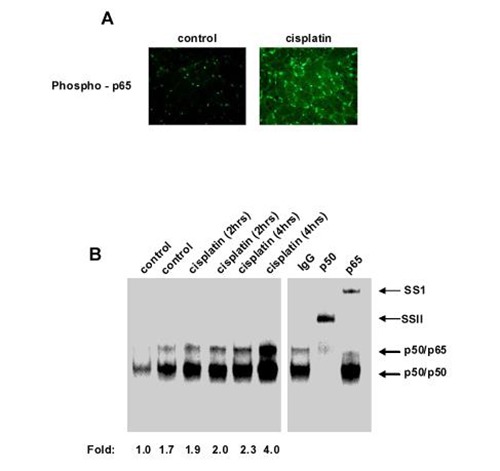
Cisplatin is an inducer of NF-κB in muscle cells. (A) CD2F1 mice were treated with cisplatin alone as described above in legend for Figure 1. At day 17 post-injection, muscle sections were prepared and immunostained for activated p65 (phospho-p65). (B) C2C12 myotube cultures were either left alone (control) or treated with cisplatin for indicated times. Nuclear extracts were prepared and EMSA was performed. Intensity of binding complexes were quantitated by phosphoimaging and recorded underneath each lane. Fold designates the amount of binding complex over that of untreated myotubes, which was set to a value of 1.0. Separate Supershift EMSA were preformed with p50 and p65 antibodies. SS refers to supershift complexes.
Fig 4.
Chemotherapy-induced muscle wasting is not specific to cisplatin. CD2F1 mice were treated with either saline (PBS) or different chemotherapeutic compounds, similar to the regimen described in legends for Figure 1. Following treatment, gastrocnemius muscle sections were prepared and stained for H&E (A) and calculated for fiber diameters (B).
Discussion
Muscle mass is determined by the relative balance between its synthesis and degradation. In healthy individuals, these two processes are in equilibrium, contributing to stable muscle protein turnover and renewal, with little change in overall muscle mass.2 It is imperative to discern the processes that are occurring in the muscle wasting observed in cancer cachexia. The therapeutic interventions for decreased protein synthesis would be very different from the therapy for increased protein breakdown.2 According to Baracos, the pathophysiology of cancer cachexia has two main components: a failure of food intake and a systemic hypermetabolism state.26 Decreased protein synthesis has a direct relationship to this failure of food intake, as sufficient nitrogen and calories are necessary in order for the body to promote anabolism. Yet, in the cachectic cancer patient, even with adequate nutrient and calorie loads, muscle protein synthesis does not occur with the same efficiency as seen in equally malnourished patients without cancer.27 In regards to catabolism, there is a prevailing consensus that it has a central role in many of the effects seen in cancer cachexia. This accelerated muscle proteolysis occurs mostly via activation of ATP- and ubiquitin-dependent pathways.28-30 Specifically, E3 ligases in these pathways control polyubiquitination, a rate-limiting step in the proteolysis that occurs with the ubiquitin-proteasome system. These E3 ligases have direct involvement in the skeletal muscle protein breakdown that is seen in cancer cachexia.31 MuRF1 is a muscle-specific E3 ligase that was utilized in this study as an indirect marker for muscle proteolysis. The relative contribution that increased protein catabolism and decreased anabolism play in cancer cachexia is complex, with temporal relationships of the two playing roles in the initiation and progression of muscle wasting.22 It has clearly been shown that pro-inflammatory cytokines are produced at accelerated rates in patients and animals that have actively growing cancers.27 Cytokines have a key role as the main humoral factors involved in cancer Muscle mass is determined by the relative balance between its synthesis and degradation. In healthy individuals, these two processes are in equilibrium, contributing to stable muscle protein turnover and renewal, with little change in overall muscle mass.2 It is imperative to discern the processes that are occurring in the muscle wasting observed in cancer cachexia. The therapeutic interventions for decreased protein synthesis would be very different from the therapy for increased protein breakdown.2 According to Baracos, the pathophysiology of cancer cachexia has two main components: a failure of food intake and a systemic hypermetabolism state.26 Decreased protein synthesis has a direct relationship to this failure of food intake, as sufficient nitrogen and calories are necessary in order for the body to promote anabolism. Yet, in the cachectic cancer patient, even with adequate nutrient and calorie loads, muscle protein synthesis does not occur with the same efficiency as seen in equally malnourished patients without cancer.27 In regards to catabolism, there is a prevailing consensus that it has a central role in many of the effects seen in cancer cachexia. This accelerated muscle proteolysis occurs mostly via activation of ATP- and ubiquitin-dependent pathways.28-30 Specifically, E3 ligases in these pathways control polyubiquitination, a rate-limiting step in the proteolysis that occurs with the ubiquitin-proteasome system. These E3 ligases have direct involvement in the skeletal muscle protein breakdown that is seen in cancer cachexia.31 MuRF1 is a muscle-specific E3 ligase that was utilized in this study as an indirect marker for muscle proteolysis. The relative contribution that increased protein catabolism and decreased anabolism play in cancer cachexia is complex, with temporal relationships of the two playing roles in the initiation and progression of muscle wasting.22 It has clearly been shown that pro-inflammatory cytokines are produced at accelerated rates in patients and animals that have actively growing cancers.27 Cytokines have a key role as the main humoral factors involved in cancer 37 it would appear that a similar regulation does not occur in intact skeletal muscles. Le Bricon and colleagues showed in a Morris hepatoma murine model that chemotherapy treatment with either methotrexate or cisplatin caused significant anorexia and poor nitrogen balance, which likely played a key role in the development of cancer cachexia.4 Samuels and colleagues also found that the administration of cystemustine, another chemo-therapeutic agent, transiently and acutely reduced the levels of protein synthesis in both healthy and tumor-bearing mice and did not significantly affect protein degradation.22 In a similar study examining the effects of cystemustine on small intestine mass, it was also shown that with chemotherapy treatment and decrease in tumor burden, small intestinal mass recovered to near-baseline levels and this process was due to an increase in protein synthesis. Conversely, it was shown that a single intraperitoneal injection of the chemotherapeutic cystemustine to C-26 tumor-bearing mice caused a down-regulation of proteolysis and subsequent reversal of the cachexia process once tumor burden was significantly decreased.30 It has also been shown that both healthy and tumor-bearing mice treated with cystemustine chemotherapy eventually undergo significant weight gain after initial decreases. However, the mice never achieve weights that were comparable to their baseline, pre-treatment weights, indicating there may be a long-term change in body metabolism that is caused by cystemustine.22,38 Cisplatin has been shown in this model to regulate muscle atrophy independent of the ubiquitin proteasome system through activation of the NF-κB signaling pathway.39,40 Along with this information, further studies need to be carried out to further delineate this complex process. In terms of current therapeutic agents being utilized to treat cancer cachexia today, a wide range of agents exist, including nutritional supplements, appetite stimulants, anti- inflammatory mediators, anabolic agents, and inhibitors of proteolytic pathways.26With the information gained in this study, novel therapies in the future may be able to provide more specific and targeted interactions that can help treat cancer cachexia.
Acknowledgments
We thank J. Wang and M. Carathers for their technical expertise as well as other members of the Guttridge laboratory for helpful discussions throughout the course of this study. Funding: This work was supported by an OSU Undergraduate Fellowship to J.S.D and by an NIH grant, RO1 CA098466 to D.C.G.
Reprinted with permission in the Eur J Transl Myol 28(2): 158-166, 2018.
References
- 1.Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer 2002;2:862-71. [DOI] [PubMed] [Google Scholar]
- 2.Baracos VE. Management of muscle wasting in cancer-associated cachexia: understanding gained from experimental studies. Cancer 2001;92(6 Suppl):1669-77. [DOI] [PubMed] [Google Scholar]
- 3.Moore-Carrasco R, Busquets S, Almendro V, et al. The AP-1/NF-kappaB double inhibitor SP100030 can revert muscle wasting during experimental cancer cachexia. Int J Oncol 2007;30:1239-45. [PubMed] [Google Scholar]
- 4.Le Bricon T, Gugins S, Cynober L, Baracos VE. Negative impact of cancer chemotherapy on protein metabolism in healthy and tumor-bearing rats. Metabolism 1995;44:1340-8. [DOI] [PubMed] [Google Scholar]
- 5.Samuels SE, Knowles AL, Tilignac T, et al. Protein metabolism in the small intestine during cancer cachexia and chemotherapy in mice. Cancer Res 2000;60:4968-74. [PubMed] [Google Scholar]
- 6.Camp ER, Li J, Minnich DJ, et al. Inducible nuclear factor- kappaB activation contributes to chemotherapy resistance in gastric cancer. J Am Coll Surg 2004;199:249-58. [DOI] [PubMed] [Google Scholar]
- 7.Cusack JC Jr, Liu R, Houston M, et al. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Cancer Res 2001;61:3535-40. [PubMed] [Google Scholar]
- 8.Hellin AC, Calmant P, Gielen J, et al. Nuclear factor - kappaB- dependent regulation of p53 gene expression induced by daunomycin genotoxic drug. Oncogene 1998;16:1187-95. [DOI] [PubMed] [Google Scholar]
- 9.Cai D, Frantz JD, Tawa NE Jr, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004;119:285-8. [DOI] [PubMed] [Google Scholar]
- 10.Guttridge DC, Mayo MW, Madrid LV, et al. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 2000;289(5488):2363-6. [DOI] [PubMed] [Google Scholar]
- 11.Hunter RB, Kandarian SC. Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J Clin Invest 2004;114:1504-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka Y, Eda H, Tanaka T, et al. Experimental cancer cachexia induced by transplantable colon 26 adenocarcinoma in mice. Cancer Res 1990;50:2290-5. [PubMed] [Google Scholar]
- 13.Acharyya S, Butchbach ME, Sahenk Z, et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005;8:421-32. [DOI] [PubMed] [Google Scholar]
- 14.Acharyya S, Ladner KJ, Nelsen LL, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 2004;114:370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hertlein E, Wang J, Ladner KJ, et al. RelA/p65 regulation of IkappaBbeta. Mol Cell Biol 2005;25:4956-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 2005;37:1974-84. [DOI] [PubMed] [Google Scholar]
- 17.Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704-8. [DOI] [PubMed] [Google Scholar]
- 18.Gomes MD, Lecker SH, Jagoe RT, et al. Atrogin-1, a muscle specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A 2001;98:14440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol 2001;59:657-63. [DOI] [PubMed] [Google Scholar]
- 20.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 2005;4:307-20. [DOI] [PubMed] [Google Scholar]
- 21.Dupont-Versteegden EE. Apoptosis in skeletal muscle and its relevance to atrophy. World J Gastroenterol 2006;12:7463-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samuels SE, Knowles AL, Tilignac T, et al. Higher skeletal muscle protein synthesis and lower breakdown after chemotherapy in cachectic mice. Am J Physiol Regul Integr Comp Physiol 2001;281:R133-9. [DOI] [PubMed] [Google Scholar]
- 23.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 2006;33:155-65. [DOI] [PubMed] [Google Scholar]
- 24.Wyke SM, Tisdale MJ. NF-kappaB mediates proteolysis-inducing factor induced protein degradation and expression of the ubiquitin-proteasome system in skeletal muscle. Br J Cancer 2005;92:711-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Acharyya S, Villalta SA, Bakkar N, et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest 2007;117:889-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baracos VE. Cancer-associated cachexia and underlying biological mechanisms. Annu Rev Nutr 2006;26:435-61. [DOI] [PubMed] [Google Scholar]
- 27.Espat NJ, Moldawer LL, Copeland EM. 3rd, Cytokine-mediated alterations in host metabolism prevent nutritional repletion in cachectic cancer patients. J Surg Oncol 1995;58:77-82. [DOI] [PubMed] [Google Scholar]
- 28.Breen HB, Espat NJ. The ubiquitin-proteasome proteolysis pathway: potential target for disease intervention. J Parenter Enteral Nutr 2004;28:272-7. [DOI] [PubMed] [Google Scholar]
- 29.Camps C, Iranzo V, Bremnes RM, Sirera R. Anorexia-Cachexia syndrome in cancer: implications of the ubiquitin-proteasome pathway. Support Care Cancer 2006;14:1173-83. [DOI] [PubMed] [Google Scholar]
- 30.Tilignac T, Temparis S, Combaret L, et al. Chemotherapy inhibits skeletal muscle ubiquitin-proteasome-dependent proteo-lysis. Cancer Res 2002;62:2771-7. [PubMed] [Google Scholar]
- 31.Kwak KS, Zhou X, Solomon V, et al. Regulation of protein catabolism by muscle-specific and cytokine-inducible ubiquitin ligase E3alpha-II during cancer cachexia. Cancer Res 2004;64:8193-8. [DOI] [PubMed] [Google Scholar]
- 32.Argiles JM, Busquets S, Lopez-Soriano FJ. Cytokines in the pathogenesis of cancer cachexia. Curr Opin Clin Nutr Metab Care 2003;6:401-6. [DOI] [PubMed] [Google Scholar]
- 33.Koyama S, Hata S, Witt CC, et al. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol 2008;376:1224-36. [DOI] [PubMed] [Google Scholar]
- 34.Emery PW. Cachexia in experimental models. Nutrition 1999;15:600-3. [DOI] [PubMed] [Google Scholar]
- 35.Donaldson SS, Lenon RA. Alterations of nutritional status: impact of chemotherapy and radiation therapy. Cancer 1979;43(5 Suppl):2036-52. [DOI] [PubMed] [Google Scholar]
- 36.McAnena OJ, Daly JM. Impact of antitumor therapy on nutrition. Surg Clin North Am 1986;66:1213-28. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto Y, Hoshino Y, Ito T, et al. Atrogin-1 ubiquitin ligase is upregulated by doxorubicin via p38-MAP kinase in cardiac myocytes. Cardiovasc Res 2008;79:89-96. [DOI] [PubMed] [Google Scholar]
- 38.Guttridge DC, Albanese C, Reuther JY, et al. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 1999;19: 5785-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuroda K, Horiguchi Y, Nakashima J, et al. , Prevention of cancer cachexia by a novel nuclear factor {kappa}B inhibitor in prostate cancer. Clin Cancer Res 2005;11:5590-4. [DOI] [PubMed] [Google Scholar]
- 40.Sandri M. Apoptotic signaling in skeletal muscle fibers during atrophy. Curr Opin Clin Nutr Metab Care 2002;5:249-53. [DOI] [PubMed] [Google Scholar]