

Abstract
Gastric cancer is a prevalent, malignant tumor that frequently escapes treatment. Histidine triad nucleotide-binding protein 1 (Hint1) is a haploinsufficient tumor suppressor gene which contributes to intercellular communication, helps to regulate cell proliferation and survival, and is frequently underexpressed in gastric cancer. To examine the involvement of Hint1 in gastric cancer, small interfering RNA was used to knock down Hint1 expression in the human gastric cancer cell line SGC-7901. The data revealed that Hint1 inhibited cell proliferation, reduced radiation-induced DNA damage repair and caused G1 phase arrest, which increased the radiosensitivity of gastric cancer cells. Further mechanistic studies revealed a novel function of Hint1, whereby it acted as a negative regulator of extracellular signal-regulated kinase. These results demonstrated the critical function of Hint1 in the biology of human gastric cancer. Acting as a tumor growth suppressor and a radiosensitive agent, this protein is a potential biomarker and may be an attractive target for specific therapeutic interventions against gastric cancer.
Keywords: histidine triad nucleotide binding protein 1, gastric cancer, proliferation, radiosensitivity, extracellular signal-regulated kinases
Introduction
Gastric cancer (GC) is the most prevalent digestive system carcinoma (1). In 2012, it was diagnosed in 950,000 patients and caused 723,000 mortalities (1). Its malignance is due to its capacity for rapid proliferation and resistance to chemoradiotherapy. While the biological events and key signaling pathway disruptions that drive its growth are being studied at present, the underlying mechanisms of resistance to conventional DNA-damaging agents and ionizing radiation (IR) remain largely unknown.
Histidine triad nucleotide binding protein 1 (Hint1) is a haploinsufficient tumor suppressor gene which contributes to intercellular communication, helps to regulate cell proliferation and survival, and is frequently underexpressed in the early stages of oncogenesis (2–5). Hint1 knockout mice demonstrated a marked increase in susceptibility to colorectal, mammary and ovarian tumors (2,3). In 2011, our team first reported that Hint1 was underexpressed in human GC tissues at the protein and gene level. Its downregulation was associated with poor tumor cell differentiation and bacterial or viral infection, including infection by Helicobacter pylori or the Epstein-Barr virus, suggesting patients with Hint1 underexpression may present with biologically aggressive tumors and poor prognosis (6).
The tumor suppressive effects of Hint1 protein primarily serve an inhibitory function in a number of gene transcription control pathways. For instance, Hint1 promotes apoptosis via upregulation of p53 and downregulation of B cell lymphoma-2 in the SW480 human colon cancer cell line and the MCF7 breast cancer cell line (7). Upon association with the plenty of SH3 domains protein and mitogen activated protein kinase 9 complex, Hint1 inhibits activity of the activator protein-1 transcription factor responsible for the proliferation and angiogenesis of colon cancer cells (8). In addition, Hint1 enhances cellular responses to DNA damage by regulating the functions of γ-H2A histone family member X and ATM serine/threonine kinase (ATM) in normal cells (9). However, little is known regarding the effect of Hint1 on radiotherapy in cancer, even though a relationship between Hint1 and DNA damage repair was reported previously (9).
The present study analyzed the tumor suppressive effects of Hint1 in the SGC7901 gastric cancer cell line. Its inhibition of cell viability in this cell line was demonstrated, and the involved signaling cascades were investigated. Hint1 may negatively regulate extracellular signal-regulated kinase (ERK), which is involved in gastric carcinogenesis. In addition, Hint1 prevented IR-induced DNA damage repair in SGC7901 cells via the repression of Cyclin D1-dependent retinoblastoma protein (Rb protein) phosphorylation, which induced G1 arrest and cell death.
Materials and methods
Cell culture and treatment
The SGC7901 and AGS human gastric cancer cell lines were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were maintained in RPMI-1640 medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), and incubated in a 100% moist incubator with 5% CO2 at 37°C. A Varian medical linear accelerator (Varian Medical Systems, Palo Alto, CA, USA), offered by the Department of Oncology, Nanjing First Hospital (Nanjing, China) was used to treat the cells. Gastric cancer cells were plated at a density of 1×104 cells/well into 96-well plates, and were incubated overnight. Then, cells were treated with 0, 1, 2, 4 or 6 Gy X-ray or 50 µM PD98059 for 24 h at 37°C (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), a specific ERK inhibitor (ERKi), for further experiments.
RNA interference
RNA interference was used to selectively knock down Hint1 in SGC7901 cells. The sequence of pGPU6/green fluorescent protein/Neo-short hairpin RNA (shRNA)-Hint1 (Sangon Biotech Co., Ltd., Shanghai, China) was 5′-CCGGCGACACGATCTTTGGGAAGATCTCGAGATCTTCCCAAAGATCGTGTCGTTTTTG-3′. Cells were transfected with the aforementioned shRNA using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) for 24–72 h. Then cells were re-seeded in RPMI-1640 medium containing G418 (400 µg/ml; Gibco; Thermo Fisher Scientific, Inc.) to enrich the culture for cells that were successfully transfected. Following 120 h transfection, the cells were harvested to determine knockdown efficiency by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analysis. A non-targeting shRNA vector (cat. no. E-07/F-07; Shanghai GenePharma Co., Ltd., Shanghai, China.) was used as a negative control for all experiments.
Colony survival assay
A colony survival assay was performed to determine the influence of Hint1 on SGC7901 cell proliferation. Exponentially growing cells were seeded at a low density in a 6-well plate (80 cells/well plate; Corning Incorporated, Corning, NY, USA) and allowed to grow for 7–10 days in RPMI-1640 medium. The media was then removed and replaced with 0.1% crystal violet dye. The size of live colonies which contained >50 cells was evaluated using a fluorescence microscope (Olympus IX71; Olympus Corporation, Tokyo, Japan) at magnification, −200. The number of colonies were then counted and the proliferation ratio was calculated as the ratio of the number of colonies in Hint1 silenced cells formed to those formed by the vector control group.
MTT assay
SGC7901 and AGS cells (1×104) were seeded in 96-well plates and then treated with 0, 1, 2, 4 or 6 Gy X-ray or PD98059 (50 µm). MTT reagent (10 µl/well; Trivegen, Gaithersburg, MD, USA) was added and cells were left inside the incubator for a further 4 h at 37°C, followed by the addition of 100 µl detergent reagent (Trivegen) according to the manufacturer's protocol. Absorbance of the colored solution was measured by a fully automated multi-detection microplate reader (POLARstar Optima; BMG Labtech GmbH, Ortenberg, Germany) at 570 nm.
Comet assay
The alkaline comet assay was performed in SGC7901 cells either directly following irradiation or following a recovery of 24 h in RPMI-1640 medium supplemented with 10% fetal bovine serum using the OxiSelect Comet Assay kit (Cell Biolabs, Inc., San Diego, CA, USA). All steps were performed on ice or in the cold to minimize repair processes. Images were recorded using a fluorescence microscope (Olympus IX71; Olympus Corporation, Tokyo, Japan) at magnification, −200. For each data point, 2–3 areas on parallel slides were scored with 51–60 cells each, and DNA in tail (%) was calculated for each cell using the image-analysis software CaspLab (version 1.2.3 beta1; http://casplab.com/download). The median of DNA in tail (%) was calculated for each area, and the presented values are the means of the medians of each data point.
Protein extraction and western blot
A total of 1×107 SGC7901 or AGS cells were collected and lysed using ice-cold lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% SDS, 1 mM EDTA, 1% NP-40) containing 1 mM protein inhibitor and 1 mM phenylmethylsulfonyl fluoride for 30 min on ice. The lysates were centrifuged at 10,000 × g at 4°C for 10 min and the supernatants were collected. Protein concentration was measured using a bicinchoninic acid protein assay (Pierce; Thermo Fisher Scientific, Inc.). Equal amounts of 30 µg total protein for each sample was loaded and separated by 7.5–12.5% SDS-PAGE and then transferred onto polyvinylidene difluoride membranes (Amersham Biosciences). Membranes were blocked with 5% skim milk in TBST (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20) for 1 h and incubated with specific antibodies diluted as needed at 4°C overnight. β-actin was used as the loading control. Then, membranes were incubated with goat anti-rabbit IgG (cat. no. sc-2040; 1:400) or goat anti-mouse IgG (cat no. sc-2039; 1:400; both Santa Cruz Biotechnology, Inc., Dallas, TX, USA) secondary antibodies for 1 h at room temperature. Target proteins were visualized with an Amersham Enhanced Chemiluminesence Western Blotting Detection kit following the manufacturer's protocol. (cat. no. RPN2108, GE Healthcare Life Sciences, Chalfont, UK). The primary antibodies included anti-Hint1 (1:500; ab64071; Abcam, Cambridge, UK), anti-phosphorylated (p-)ERK1/2 (1:250; cat. no. 4370), anti-ERK1/2 (1:1,000; cat. no. 9107; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-p-IκB kinase (1:500; IκB; Ser32; ab92700), anti-nuclear factor-κB (NF-κB) p65 (1:1,000; Ser536; cat no. ab86299), anti-ATM (1:300; cat no. ab17995; all Abcam, Cambridge, UK), anti-p-ATM (1:1,000; Ser1981; cat. no. 13050), anti-p-ATR serine/threonine protein kinase (1:500; ATR; Ser428; cat. no. 2853), anti-ATR (1:500; cat. no. 2790; all Cell Signaling Technology, Inc.), anti-Cyclin D1 (1:500; ab137875; Abcam), anti-p-Rb (1:1,000; Ser780; cat. no. 5225), anti-BCL2 associated X, apoptosis regulator (1:1,000; Bax; cat. no. 2772; both Cell Signaling Technology, Inc.) and anti-β-actin antibody (1:1,000; A1978, Sigma-Aldrich; Merck KGaA).
RT-qPCR
Hint1 mRNA levels were determined in SGC7901 cells using RT-qPCR. Total RNA was extracted from cells using an RNeasy kit (Qiagen, Inc., Valencia, CA, USA) and RT-qPCR was performed using a TaqMan reverse transcription kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. The PCR primers were as follows: Human Hint1 forward, 5′-ATTTCCCCTCAAGCACCAACA-3′ and reverse, 5′-ATCAGCAGCACATTTCTTGCC-3′; β-actin forward, 5′-CCCATCTATGAGGGTTACGC-3′ and reverse, 5′-TTTAATGTCACGCACGATTTC-3′ (Invitrogen; Thermo Fisher Scientific, Inc.). The thermocycler conditions were as follows: Initial denaturation at 95°C for 15 sec, then 40 cycles at 95°C for 5 sec and 60°C for 30 sec. Relative expression to the control gene was determined using the ΔΔCq method (10). The experiments were repeated three times.
NF-κB transcription factor DNA binding activity
SGC7901 cells were treated as described previously. Nuclear extracts were prepared by a nuclear extract kit (Active Motif, Carlsbad, CA, USA) and subsequently analyzed for NF-κB activity using the NF-κB p65 Transcription Factor Assay kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer's protocol. Briefly, cellular nuclear extracts were purified and plated in a 96-well plate. Then, samples were incubated with NF-κB (p65) primary antibody and secondary antibody successively. Absorbance of each well measured by a fully automated multi-detection microplate reader (POLARstar OPTIMA) at 450 nm.
Flow cytometric analysis of the cell cycle
SGC7901 cells (2×105) were seeded in 6-well plates with RPMI-1640 medium supplemented with 10% fetal bovine serum and then irradiated with X-rays (6 Gy). The cells were harvested either prior to irradiation or following a recovery period of 24 h. Then, cells were washed three times with cold PBS and resuspended in 1 ml staining solution (50 µg/ml propidium iodide, 20 µg/ml RNase A). Following incubation for 30 min at room temperature, samples were analyzed using a fluorescence-activated cell sorting flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). The percentage of cells in G0/G1, S, and G2-M phases was calculated and compared with ModFit LT software (version 3.2; Verity Software House, Topsham, ME, USA).
Statistical analysis
All experiments were performed at least in triplicate on separate experimental days. Statistical differences between the values obtained in different experimental settings were evaluated by the means of analysis of variance (two-way or one-way, as appropriate) with post hoc Duncan's range tests, or unpaired Student's t-tests, using IBM SPSS 21.0 software (IBM SPSS, Armonk, NY, USA) for Windows. P<0.05 was considered to indicate a statistically significant difference.
Results
Growth inhibitory effects of Hint1 in SGC7901 cells
To analyze the involvement of Hint1 in gastric cancer, a shRNA-Hint1 vector was transfected into SGC7901 gastric cancer cells. Transfection with the shHint1 vector resulted in significantly reduced Hint1 mRNA expression and visibly reduced Hint1 protein expression (Fig. 1A and B, respectively). In the subsequent MTT assay, cells transfected with shHint1 demonstrated significantly increased viability compared with the vector control group (Fig. 1C). Meanwhile, a colony survival assay was performed to verify the results of this test. There was a ~1.5 fold increase in the number of colonies observed in the shHint1 group compared with the vector control under the same culture conditions and time (Fig. 1D). These data demonstrated an inhibitory function of Hint1 on gastric cancer cell viability.
Figure 1.

Effect of transfection with shHint1 on Hint1 mRNA and protein expression levels, and SGC7901 cell viability and proliferation. (A) Reverse transcription-quantitative polymerase chain reaction was performed to measure Hint1 mRNA expression in control vector cells and shHint1 cells. (B) Western blot analysis was performed to measure Hint1 protein expression in control vector cells and shHint1 cells. (C) Cell viability was assessed by MTT assay. (D) Cell proliferation ratio was determined by colony survival assay. Data are presented as the mean ± standard error of the mean. *P<0.05 vs. vector control. si, small interfering RNA; Hint1, histidine triad nucleotide-binding protein 1.
Suppression of ERK-dependent NF-κB activation is involved in Hint1-mediated retardation of cell viability
The ERK signaling cascade is a central mitogen-activated protein kinase (MAPK) pathway that regulates various oncogenic responses, including proliferation and survival (11,12). To further investigate the mechanisms by which Hint1 inhibited the viability of gastric cancer cells, the interactions between Hint1 and ERK were determined. The results revealed that ERK phosphorylation activity was significantly increased following knockdown of Hint1 in SCG7901 cells (Fig. 2A). Activated ERK subsequently induced IκB phosphorylation and activated the NF-κB p65 signal pathway when lacking regulation from Hint1 (Fig. 2A and B). These cascades were repressed by ERKi, demonstrating that the activation of NF-κB p65 following Hint1 knockdown was ERK dependent. To verify this result, the effect on cell viability in the shHint1 group was analyzed following treatment with ERKi. ERKi efficiently reversed the increased viability induced by Hint1 deficiency (Fig. 2C). These data indicated that Hint1 may be a negative regulator of ERK, and further prevent activation of its downstream signaling pathways, including NF-κB, thus inhibiting cell viability.
Figure 2.

Effect of Hint1 interference on the ERK signaling pathway. (A) Western blot analysis was performed to measure ERK, IκB and NF-κB protein expression. (B) An NF-κB transcription assay was performed to measure NF-κB p65 DNA binding activity. (C) The effect of Hint1 on cell viability was tested by MTT assay. *P<0.05 vs. vector control and shHint1+ERKi groups. Hint1, histidine triad nucleotide-binding protein 1; ERK, extracellular signal-regulated kinase; IκB, IκB kinase; NF-κB, nuclear factor-κB; si, small interfering RNA; ERKi, ERK inhibitor; p-, phosphorylated.
Hint1 increased radiosensitivity in gastric cancer cells
Due to the deleterious influence of Hint1 on cancer cell viability, the present study investigated whether Hint1 modulated the radiation resistance of gastric cancer. Two human gastric cancer cell lines with 0, 1, 2, 4 or 6 Gy X-ray. Prior to the treatment, the protein expression of Hint1 was examined in these cell lines (Fig. 3A). Notably, differences in cell survival were observed between the gastric cancer cells lines following radiation. SGC7901 cells, which demonstrated higher Hint1 protein expression, exhibited higher sensitivity to radiation, whereas AGS cells, with lower Hint1 protein expression, were relatively resistant to treatment (Fig. 3A). shHint1-transfected SGC7901 cells were then treated with radiation, with ERKi or without. Radiation resistance was induced in the shHint1 SGC7901 cells, while ERKi overcame this (Fig. 3B).
Figure 3.
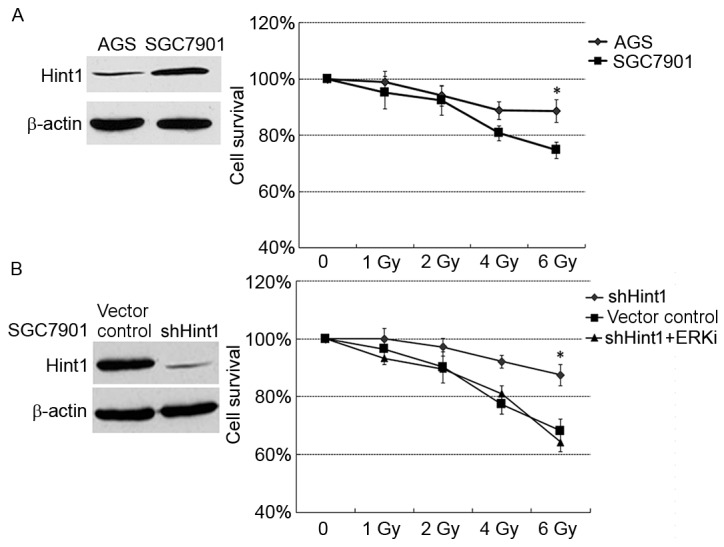
Effect of Hint1 on radiosensitivity. (A) Radiosensitivity was tested by MTT in two gastric cancer cell lines with different Hint1 expression levels. (B) Changes in radiosensitivity induced by Hint1 silencing and ERKi treatment were assessed in SGC7901 cells by MTT assay. Data are presented as the mean ± standard error of the mean. *P<0.05 vs. vector control and shHint1+ERKi groups. Hint1, histidine triad nucleotide-binding protein 1; ERKi, ERK inhibitor; si, small interfering RNA.
Hint1 delayed radiation-induced DNA damage repair in gastric cancer cells
A previous study demonstrated that Hint1 is involved in DNA damage repair in normal cells (9). The influence of Hint1 expression on DNA damage, a hallmark of radiation-induced cytotoxicity, was explored on SGC7901 cells using single cell gel electrophoresis, also known as a comet assay. shHint1 cells demonstrated visibly smaller DNA tails than the vector control 24 h following radiation treatment of 6 Gy, suggesting enhanced DNA repair in Hint1-deficient cells (Fig. 4A). There was no significant difference in the tail DNA fraction between 0 and 24 h in the vector control and ERKi treatment group (Fig. 4B). In this experiment, ERKi treatment did not affect the formation of DNA damage, instead retarding DNA damage repair.
Figure 4.
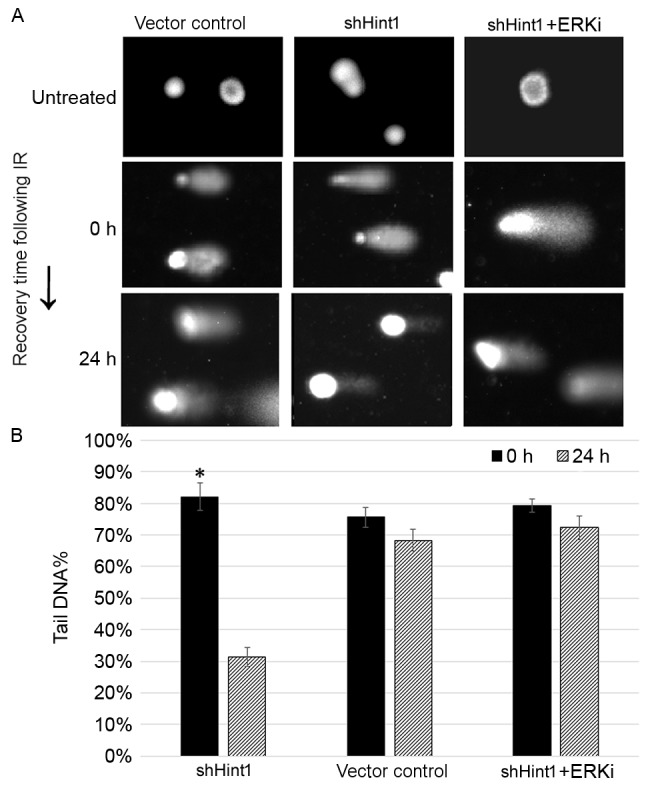
Results of the comet assay. (A) Representative images of comet assay induction prior to and following 6 Gy radiation treatment (magnification, −200). (B) The median of DNA in tail (%) was calculated using Casplab software. Data are presented as the mean ± standard error of the mean. *P<0.05 vs. 0 h. si, small interfering RNA; Hint1, histidine triad nucleotide-binding protein 1; ERKi, ERK inhibitor.
Hint1-mediated Cyclin D1 suppression results in persistent G1 arrest in gastric cancer cells following IR
The mechanisms underlying the involvement of Hint1 in the radiation response remain unclear. First, the effect of Hint1 expression on the cellular DNA damage response signal pathway was examined. As reported by a previous study, p-ATM levels increase in SGC7901 cells following radiation treatment, but this increase is partially suppressed in shHint1 cells (13). Notably, the activation of ATR, the other important DNA damage sensor, did not seem to be altered by Hint1 expression, but it was not possible to explain why Hint1 increased radiosensitivity with enhanced ATM activation (13). Similar results were observed in the present study (Fig. 5A). Therefore, the present study focused on the downstream effectors of the DNA damage response. Cyclin D1, a cell-cycle regulator essential for the G1 phase, was downregulated by Hint1 (Fig. 5B). During the G1 phase, Cyclin D1 increases DNA synthesis via Rb phosphorylation and promotes S phase entry (14). Decreased Cyclin D1 levels in SGC7901 cells significantly inhibited p-Rb activation, resulting in a persistent G1 arrest following IR treatment (Fig. 5B and C). Accordingly, the pro-apoptotic protein Bax was upregulated in vector control cells following IR compared with shHint1 cells following IR (Fig. 5B).
Figure 5.

(A) Expression of the DNA damage response sensors, ATM and ATR, was assessed by Western blot analysis. (B) Cyclin D1, p-Rb and Bax protein expression was assessed by Western blot. (C) G1 arrest was observed via flow cytometry in gastric cancer cells following IR. Data are presented as the mean ± standard error of the mean. *P<0.05 vs. other groups. ATM, ATM serine/threonine kinase; ATR, ATR serine/threonine protein kinase; p-, phosphorylated; Rb, retinoblastoma protein; Bax, BCL2 associated X, apoptosis regulator; IR, ionizing radiation; si, small interfering RNA.
Discussion
Previous investigations using genetically engineered mice have demonstrated that Hint1 is a novel haploinsufficient tumor suppressor gene (2,3). It demonstrates a strong analogy with fragile histidine triad, a known tumor suppressor gene on common fragile site fragile site, aphidicolin type, common, fra(3)(p14.2) (2), but its functions are yet to be completely understood. A previous study analyzed clinical samples and reported a potential tumor suppressor role for Hint1 in human gastric cancer (6). In the present study, the function of Hint1 was analyzed in human gastric cancer cells. The results obtained demonstrated, for the first time, that Hint1 decreased gastric cancer cell proliferation in vitro by decreasing cell viability. These observations were consistent with the proposed tumor suppressor function of Hint1 and with previous findings demonstrating that Hint1 inhibited human colon and breast cancer cell growth (2,3,8).
Mechanistic studies subsequently demonstrated the negative regulation of Hint1 on ERK signal pathway activation. ERK1/2 are associated protein-serine/threonine kinases that participate in the Ras-Raf-mitogen-activated protein kinase kinase-ERK signal transduction cascade. This cascade participates in the regulation of a number of processes, including cell adhesion, cell cycle progression, cell migration, cell survival, differentiation, metabolism, proliferation and transcription (11,12). ERKs directly phosphorylate multiple transcription factors, including the ETS transcription factor family, c-Jun and c-Myc (15–17). ERK also phosphorylates and activates the 90 kDa ribosomal S6 kinase (p90Rsk), which, in turn, leads to the activation of the transcription factor cAMP response element binding protein (18). Furthermore, ERK results in activation of the NF-κB transcription factor by phosphorylating and activating IκB kinase (19). Deregulated activation of NF-κB, which induces uncontrolled cell growth, is a central signature of multiple types of epithelial cancer. For instance, over-activation of NF-κB, regulated by ERK, promoted malignant transformation in endometrial epithelia cells (20). In the present study, silencing Hint1 resulted in the activation of the ERK-NF-κB signal pathway in SGC7901 cells. This demonstrated that Hint1 acted as an ERK-control factor in stomach carcinogenesis, resulting in tumor suppression.
As radiation therapy has become a pillar of the therapeutic approach to gastric cancer, the study of the molecular mechanisms underlying the radiation resistance of gastric cancer cells has become important. A previous study indicated that Hint1−/− mouse embryonic fibroblasts cells had defects in the double-stranded break (DSB) repair pathway, including loss of ATM activation and the presence of unrepaired DSB, while another study demonstrated that Hint1-deficient cells exhibited resistance to IR-induced apoptosis (2,9). Therefore, the exact function of Hint1 in response to radiation in gastric cancer cells was worthy of investigation. In the present study, Hint1-deficient SGC7901 cells also exhibited resistance to IR. Although Hint1 deficiency affected ATM activation, DNA damage repair was not retarded in Hint1-deficient SGC7901 cells in the present study. On the contrary, vector control cells demonstrated repressed DNA damage repair functions. One reason for this may be that cancer cells have more inordinate or defective DNA damage repair functions than normal cells. The loss of critical tumor suppressor genes, including Hint1, may activate other pro-survival signal pathways which enhance DNA repair in cancer cells. In addition, ATM activation is not completely inhibited following Hint1 silencing or in Hint1−/- cells, as previously described (9). In addition, Hint1 deficiency did not affect the activation of ATR, another important DNA damage sensor, in the present study.
As Hint1 was demonstrated to inhibit ERK activation, its downstream effectors, which may be involved in DNA repair, were investigated. Cyclin D1, a component of the core cell cycle machinery, was revealed to be significantly upregulated following silencing of Hint1. Cyclin D1 phosphorylates and inactivates Rb protein and promotes progression through the G1-S phase of the cell cycle (21). A number of studies have demonstrated that endogenous high levels of cyclin D1 promote carcinogenesis and radioresistance in the majority of malignant tumors (22–24). Downregulation of Cyclin D1 induces G1 arrest, followed by apoptosis, in human skin cancer A431 cells following UV exposure (25). Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA DSBs, which may gradually induce formation of acquired radioresistance during long term fractionated radiotherapy (26,27). In the present study, the negative regulation of ERK mediated by Hint1 may suppress Cyclin D1 expression, which leads to persistent G1 arrest followed by apoptosis in SGC7901 cells following IR exposure.
In summary, the results of the present study first indicated that Hint1 was involved in the regulation of cell proliferation and radioresponse via negative control of the ERK signal pathway. These results demonstrated the critical function of Hint1 in the biology of human gastric cancers. Acting as a tumor growth suppressor and a radiosensitive agent, this protein is a potential biomarker and an attractive target for specific therapeutic interventions against gastric cancer.
Acknowledgements
The authors would like to thank Dr Hong Fan (Key Laboratory of Developmental Genes and Human Diseases, Ministry of Education, Southeast University, Nanjing, China) for her help and advice.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 81201882), the Natural Science Foundation of Jiangsu Province, China (grant no. BK2012076) and the Key Project supported by the Medical Science and Technology Development Foundation, Nanjing Department of Health, China (grant no. ZKX14039).
Availability of data and materials
All data generated or analyzed during the present study are included in this published article.
Authors' contributions
The present study was concieved and designed by XWW and JFC. The methodology was developed by XMW, LZH and ZX. The data was aquired by XWW, JZ, HYZ and XWW. Analysis and interpretation was performed by XMW and XWW. The manuscript was written and revised by XMW and JFC. Administrative and technical support was provided by XWW and ZX. The study was overseen by JFC.
Ethics approval and consent to participate
The present study was approved by Nanjing Medical University (Nanjing, China) Ethics Committee in 2012.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Stewart BW, Wild CP. World cancer report 2014. World Health Organization. 2014. Chapter 1.1 ISBN 9283204298.
- 2.Su T, Suzui M, Wang L, Lin CS, Xing WQ, Weinstein IB. Deletion of histidine triad nucleotide-binding protein 1/PKC-interacting protein in mice enhances cell growth and carcinogenesis. Proc Natl Acad Sci USA. 2003;100:7824–7829. doi: 10.1073/pnas.1332160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H, Zhang Y, Su T, Santella RM, Weinstein IB. Hint1 is a haplo-insufficient tumor suppressor in mice. Oncogene. 2006;25:713–721. doi: 10.1038/sj.onc.1209111. [DOI] [PubMed] [Google Scholar]
- 4.Cen B, Li H, Weinstein IB. Histidine triad nucleotide-binding protein 1 up-regulates cellular levels of p27KIP1 by targeting ScfSKP2 ubiquitin ligase and Src. J Biol Chem. 2009;284:5265–5276. doi: 10.1074/jbc.M804531200. [DOI] [PubMed] [Google Scholar]
- 5.Weiske J, Huber O. The histidine triad protein Hint1 interacts with pontin and reptin and inhibits TCF-beta-catenin-mediated transcription. J Cell Sci. 2005;118:3117–3129. doi: 10.1242/jcs.02437. [DOI] [PubMed] [Google Scholar]
- 6.Huang H, Wei X, Su X, Qiao F, Xu Z, Gu D, Fan H, Chen J. Clinical significance of expression of Hint1 and potential epigenetic mechanism in gastric cancer. Int J Oncol. 2011;38:1557–1564. doi: 10.3892/ijo.2011.994. [DOI] [PubMed] [Google Scholar]
- 7.Weiske J, Huber O. The histidine triad protein Hint1 triggers apoptosis independent of its enzymatic activity. J Biol Chem. 2006;281:27356–27366. doi: 10.1074/jbc.M513452200. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Zhang Y, Li H, Xu Z, Santella RM, Weinstein IB. Hint1 inhibits growth and activator protein-1 activity in human colon cancer cells. Cancer Res. 2007;67:4700–4708. doi: 10.1158/0008-5472.CAN-06-4645. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Balajee AS, Su T, Cen B, Hei TK, Weinstein IB. The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage. J Cell Biol. 2008;183:253–265. doi: 10.1083/jcb.200711150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 11.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 12.Huang G, Tang B, Tang K, Dong X, Deng J, Liao L, Liao Z, Yang H, He S. Isoquercitrin inhibits the progression of liver cancer in vivo and in vitro via the MAPK signalling pathway. Oncol Rep. 2014;31:2377–2384. doi: 10.3892/or.2014.3099. [DOI] [PubMed] [Google Scholar]
- 13.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 14.Cheng YH, Li LA, Lin P, Cheng LC, Hung CH, Chang NW, Lin C. Baicalein induces G1 arrest in oral cancer cells by enhancing the degradation of cyclin D1 and activating AhR to decrease Rb phosphorylation. Toxicol Appl Pharmacol. 2012;263:360–367. doi: 10.1016/j.taap.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Duan Z, Zheng H, Hu D, Li M, Tao Y, Bode AM, Dong Z, Cao Y. EBV-encoded LMP1 upregulates Igκ 3′enhancer activity and Igκ expression in nasopharyngeal cancer cells by activating the Ets-1 through ERKs signaling. PLoS One. 2012;7:e32624. doi: 10.1371/journal.pone.0032624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park KH, Shin KS, Zhao TT, Park HJ, Lee KE, Lee MK. L-DOPA modulates cell viability through the ERK-c-Jun system in PC12 and dopaminergic neuronal cells. Neuropharmacology. 2016;101:87–97. doi: 10.1016/j.neuropharm.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Kwon YW, Jang S, Paek JS, Lee JW, Cho HJ, Yang HM, Kim HS. E-Ras improves the efficiency of reprogramming by facilitating cell cycle progression through JNK-Sp1 pathway. Stem Cell Res. 2015;15:481–494. doi: 10.1016/j.scr.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Chuang JI, Huang JY, Tsai SJ, Sun HS, Yang SH, Chuang PC, Huang BM, Ching CH. FGF9-induced changes in cellular redox status and HO-1 upregulation are FGFR-dependent and proceed through both ERK and AKT to induce CREB and Nrf2 activation. Free Radic Biol Med. 2015;89:274–286. doi: 10.1016/j.freeradbiomed.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 19.Tsao GSW, Zhu DD, Zhang J, Deng W. Abstract 1046: The role of NF-kB activation in the immortalization of nasopharyngeal epithelial cells. Cancer Res. 2015;75 doi: 10.1158/1538-7445.AM2015-1046. [DOI] [Google Scholar]
- 20.Mizumoto Y, Kyo S, Kiyono T, Takakura M, Nakamura M, Maida Y, Mori N, Bono Y, Sakurai H, Inoue M. Activation of NF-kappaB is a novel target of KRAS-induced endometrial carcinogenesis. Clin Cancer Res. 2011;17:1341–1350. doi: 10.1158/1078-0432.CCR-10-2291. [DOI] [PubMed] [Google Scholar]
- 21.Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510:547–551. doi: 10.1038/nature13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 23.Peurala E, Koivunen P, Haapasaari KM, Bloigu R, Jukkola-Vuorinen A. The prognostic significance and value of cyclin D1, CDK4 and p16 in human breast cancer. Breast Cancer Res. 2013;15:R5. doi: 10.1186/bcr3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, Sicinski P. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22:438–451. doi: 10.1016/j.ccr.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim AL, Athar M, Bickers DR, Gautier J. Ultraviolet-B-induced G1 arrest is mediated by downregulation of cyclin-dependent kinase 4 in transformed keratinocytes lacking functional p53. J Invest Dermatol. 2002;118:818–824. doi: 10.1046/j.1523-1747.2002.01734.x. [DOI] [PubMed] [Google Scholar]
- 26.Shimura T, Ochiai Y, Noma N, Oikawa T, Sano Y, Fukumoto M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle. 2013;12:773–782. doi: 10.4161/cc.23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimura T, Fukumoto M, Kunugita N. The role of cyclin D1 in response to long-term exposure to ionizing radiation. Cell Cycle. 2013;12:2738–2743. doi: 10.4161/cc.25746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during the present study are included in this published article.