

Abstract
Cervical cancer is the most common gynecological malignancy. In recent years, the incidence of cervical cancer has had a younger trend. Cervical cancer morbidity and mortality rates have been significantly reduced due to recent decades of cervical cytology screening leading to the early detection and treatment of cervical cancer and precancerous lesions. There are a number of methods used to treat cervical cancer and improve the survival rate. However, the prevalence and recurrence rates of cervical cancer are increasing every year. There is an urgent requirement for a better understanding of the molecular mechanism cervical cancer development. The present study used scientific information retrieval from the Gene Expression Omnibus database to download the GSE26511 dataset, which contained 39 samples, including 19 cervical cancer lymph node-positive samples and 20 cervical cancer lymph node-negative samples. Using Gene Ontology analysis, Kyoto Encyclopedia of Genes and Genomes analysis, and weighted gene co-expression network analysis, 1,263 differentially expressed genes were found that affected the biological processes, including ‘cell cycle process’, ‘signaling pathways’, ‘immune response’, ‘cell activation’, ‘regulation of immune system process’ and ‘inflammatory response’. These areas should be the focus of study for cervical cancer in the future.
Keywords: bioinformatics analysis, differentially expressed gene, cervical cancer
Introduction
Cervical cancer is the most common gynecological malignancy, which causes harm to the health of women globally (1). The mortality rate of cervical cancer is the fourth highest among all cancer types (2). In China, there is a significant regional difference in the incidence of cervical cancer. Cervical cancer is prominent in poorer provinces, such as Hubei and Shanxi, due to unsatisfactory sanitary conditions. High-risk human papillomavirus infection is a major risk factor for cervical cancer, with multiple sexual partners being a secondary risk factor for the disease. Other risk factors for cervical cancer also include smoking, malnutrition and poor health conditions (3). An appropriate individual treatment program is developed according to the clinical stage, the patient's age, fertility requirements, general condition and access to medical technology, and other comprehensive considerations. The current treatment mainly includes surgery, radiotherapy and chemotherapy (4,5). Hysterectomy and bilateral lymphadenectomy are standard treatments for cervical stage I and stage IIA cancer patients (6). Radical trachelectomy has been demonstrated to be an effective treatment for early cervical cancer and is associated with an acceptable live birth rate (7–10). The majority of cervical cancer cases can be successfully treated if diagnosed at an early stage (11). However, the 5-year survival rate of patients with cervical cancer is remains low, particularly in advanced cervical cancer. There are a number of clear clinical signs in the early stage of some patients with cervical cancer, but they can be easily ignored by patients. So some patients lose better treatment opportunities. Therefore, there is a requirement to elucidate the molecular mechanisms underlying cervical cancer development and progression, providing a basis for finding potential drug targets and diagnosing biomarkers of cervical cancer (12,13).
Gene expression microarrays are a frontier biotechnology. In the current data age, they combine a high throughput with simultaneous detection of thousands of genes. The features of gene expression microarrays are automated, integrated and miniaturized. In the present study, the original GSE26511 dataset was downloaded from the Gene Expression Omnibus (GEO) database to identify downregulated or upregulated genes in cervical cancer compared with non-malignant controls. Using gene chip technology, the analysis found a number of important key roles of differentially expressed genes (DEGs), which are important in the initiation and development of cervical cancer. DEGs are the first choice for the study of molecular targets and diagnostic markers. In the present study, key DEGs associated with cervical cancer in tumors and normal samples were identified, and then these genes were screened according to statistical methods. The study benefited from current powerful analysis software and statistical methods, including Gene Ontology (GO) terminology, enrichment analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, protein-protein interaction (PPI) network analysis and weighted gene co-expression network analysis (WGCNA).
Materials and methods
Microarray data
The gene expression profiles of the GSE26511 dataset (14) were downloaded from the GEO database. The GPL570 [HG-U133 Plus 2] Affymetrix Human Genome U133 Plus 2.0 Array platform was used. The GSE26511 dataset contained 39 samples, including 19 cervical cancer samples and 20 normal samples.
Screening for DEGs
The statistical software R and packages from Bioconductor (http://www.bioconductor.org/biocLite.R) were used to analyze the DEGs between the cervical cancer and normal samples. The AffyPLM (http://www.bioconductor.org/packages/release/bioc/html/affyPLM.html) package was used to fit the original data of the chip, obtaining the weights and residuals diagram, the relative log expression and the relative standard deviation (normalized unscaled standard errors) box diagram. Prior to analysis of the data, the microarray data was quality tested. In this process, R packets, including packages of affyPLM, packages of Affy and packages of RColorBrewer (http://www.bioconductor.org/packages/release/bioc/html/affy.html), were used. Subsequent to removing unqualified samples, a reasonable and useful sample was obtained. According to the Limma (http://www.bioconductor.org/packages/release/bioc/html/limma.html) package of Bioconductor, key DEGs were determined. P<0.05 was considered to indicate a statistically significant difference.
Functional and pathway enrichment analysis
GO (http://www.geneontology.org/) analysis was extensively used for a short list of genes with statistically significant differences in expression. GO terms are divided into three categories: Biological process, cellular component and molecular function. The KEGG (http://www.kegg.jp/) website is an online database of genomes, enzymatic pathways and biochemical that acts as a freely accessible gene database. Different input data methods are based on different analyses. Compared with other databases, KEGG has a powerful graphics function, using graphics rather than harassment of the text to introduce a large number of metabolic pathways and the association between the various pathways. P<0.05 was considered to indicate a statistically significant difference.
PPI network analysis
PPI information of DEGs was acquired from Search Tool for the Retrieval of Interacting Genes (database (http://www.stringdb.org/). The PPI network assisted with identifying the key genes and important gene modules, and then used the network visualization tools, such as Cytoscape (http://www.cytoscape.org/), drawing network diagram. The PPI network is an experimental network for biological network visualization. The gene co-expression network attempted to reconstruct the biological network through the expression of genes. In the PPI network, nodes and edges (lines) represent proteins and their interactions (P<0.05).
WGCNA
As a system biology method, gene co-expression network analysis was performed by the WGCNA package (http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/Rpackages/WGCNA) to investigate the association between gene expression patterns (15). The clustering criteria of WGCNA were biologically significant, and the consequence of this method is a higher degree of credibility. These results were obtained in order to complete further investigations, such as those into association traits, metabolic pathway modeling and the establishment of gene interaction networks. In a co-expression network, the expression of each gene at a particular time or space was treated as a node. WGCNA uses a soft threshold based on the determination method. Commonly used weighting functions included sigmoid functions and power functions. The similarity and proximity of gene co-expression were calculated using soft threshold power. Analysis of network topology confirmed the final structure of soft threshold power.
Patient samples
A total of 5 cervical cancer specimens and 5 non-tumor cervical cancer epithelial tissues were collected from the Cancer Hospital of Hunan Province, Central South University (Changsha, China). The average age of the patients was 45±5.5 years. Biopsy sample were collected from March 17, 2017 to May 17, 2017. Written informed consent was obtained from each patient, and the experimental protocols were approved by the Institutional Review Board of the Cancer Hospital of Hunan Province. Each biopsy sample was divided into two sections: One section was submitted for routine histological diagnosis.
The biopsy sample was fixed with 4% paraformaldehyde for 24 h at 4°C and then was placed in processing cassettes. The samples were dehydrated through a serial alcohol gradient at room temperature (50% alcohol for 1 h, 70% alcohol overnight and then 80% alcohol for 1 h, then 95% alcohol for 1 h, and then dehydrated twice in 100% alcohol for 30 min), and embedded in paraffin wax blocks. Prior to H&E staining, the 4-µm thick tissue sections were heated at 60°C for 1 h, dewaxed twice in 100% xylene for 10 min, and then rehydrated through decreasing concentrations of ethanol (twice in 100% alcohol for 5 min, followed by 95% alcohol for 5 min and then 80% alcohol for 5 min, then 70% alcohol for 5 min, and then in distilled water for 5 min), and washed in 1X PBS for 3 min twice at room temperature. The sections were then stained with hematoxylin for 20 min, washed in distilled water for 5 min and then stained with eosin for 3 min. The sections were dried in a fume cupboard at room temperature. The thickness of the sections was 4 µm. A light microscope was used to observe the sections at ×200 magnification while the other was freshly stored in RNALater reagent (Qiagen GmbH, Hilden, Germany) at −80°C for the following experiments.
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the biopsy samples with the RNeasy® kit (Qiagen GmbH) according to the manufacturer's protocols. A total of 1 µg of RNA samples were reverse transcribed into cDNA using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (Vazyme Biotech Co., Ltd., Nanjing, China) with oligo-dT primer (R223-01, Vazyme Biotech Co., Ltd.; primer sequences unavailable), according to the manufacturer's protocols. RT-qPCR was performed using TransStart Top Green qPCRSuperMix (Vazyme Biotech Co., Ltd.) on a Mastercyclerep realplex4 (Eppendorf, Hamburg, Germany). The PCR conditions included an initial step at 95°C for 30 sec, followed by 40 cycles of amplification and quantification (95°C for 15 sec, 60°C for 15 sec, and 68°C for 20 sec). GAPDH was used as an endogenous control for normalization. The sequences of the primers used for RT-qPCR were as follows: Mucin 1 cell surface associated (MUC1) forward, 5′-agacgtcagcgtgagtgatg-3′ and reverse, 5′-cagctgcccgtagttctttc-3′; fucosyltransferase 3 (FUT3; Lewis blood group) forward, 5′-gcaaggcttagaccagttcg-3′ and reverse, 5′-caccagcagctgaaatagca-3′; notch 3 (NOTCH3) forward, 5′-gtcgtggctacactggacct-3′ and reverse, 5′-aatgtccacctcgcaatagg-3′; fibroblast growth factor 2 (FGF2) forward, 5′-ggtgaaaccccgtctctaca-3′ and reverse, 5′-tctgttgcctaggctggact-3′; insulin-like growth factor 1 (IGF1) forward, 5′-ggctgaccaagctgaaactc-3′ and reverse, 5′-atcgcttaaacccaggaggt-3′; hepatocyte growth factor (HGF) forward, 5′-ctggttccccttcaatagca-3′ and reverse, 5′-ctccagggctgacatttgat-3′; and GAPDH forward, 5′-accacagtccatgccatcac-3′ and reverse, 5′-tccaccaccctgttgctgta-3′. The expression of mRNA was assessed by evaluated quantitation cycle (Cq) values. The Cq values were normalized with the expression levels of GAPDH and the relative amount of mRNA specific to each of the target genes was calculated using the 2−ΔΔCq method (16).
Statistical analysis
All results were presented as the mean ± standard error of three independent experiments. Unpaired t-test was used for statistical analysis. All statistical tests were performed with GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA). All statistical tests were two tailed, and P<0.05 was considered to indicate a statistically significant difference.
Results
Identification of DEGs
The GSE26511 expression profile shows clear distinction between the tumor and normal healthy epithelium. Isolation and identification of DEGs was not only used to assess the function of the genes, but also assisted in revealing the pathogenesis of the disease. Gene chip raw data quality detection lead to 8 low-quality samples being removed from the 20 normal samples and 6 low-quality samples being removed from the 19 cervical cancer samples. The remaining samples required integration and processing. Preprocessing with statistical analysis software R was necessary and important prior to analyzing the microarray data of gene differential expression. Altogether there were 1,263 DEGs, of which 568 DEGs were selected (fold change >2) (Fig. 1), consisting of 402 significantly downregulated and 166 significantly upregulated DEGs, for the subsequent bioinformatics analysis (fold change >2) (Fig. 2).
Figure 1.
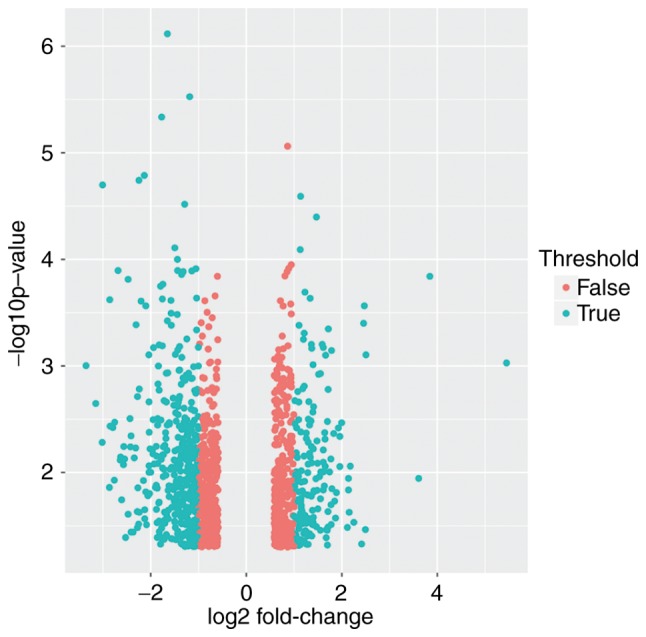
Volcano plot of 1,263 DEGs. Turquoise dots represent DEGs with a fold-change value of >2 and red dots represent DEGs with a fold-change value of <2. P<0.05. DEGs, differentially expressed genes.
Figure 2.

Heat map for cervical cancer DEGs. Red represents upregulated DEGs and green represents downregulated DEGs. DEGs, differentially expressed genes.
GO term enrichment analysis of DEGs
Noordhuis et al (14) performed research mainly concerned with the molecular mechanism of lymph node metastasis in cervical cancer. Pathway analysis of microarray expression profiles suggests that transforming growth factor-β (TGF-β) and p120-associated non-canonical β-catenin pathways are important in pelvic lymph node metastasis of early cervical cancer (14). However, the present results showed that the acquired clean reads were of a sufficiently high quality for subsequent analysis. The upregulated DEGs were primarily enriched in ‘cell adhesion molecule binding’, ‘plasma membrane’, ‘tissue development’, ‘DNA repairing’ and ‘epithelial cell differentiation’. Downregulation of the DEGs was mainly enriched by ‘signal transduction’, ‘cell surface receptor signaling pathway’, ‘positive regulation of biological process’, ‘receptor binding’ and ‘signal transducer activity’. For cellular component, to analyze the biological functions, biological processes and subcellular localization of these DEGs, the upregulated DEGs were enriched in ‘cell periphery’, ‘plasma membrane’ and ‘extracellular exosome’. Subsequent to the scientific analysis, it was found that downregulated genes were mainly found in ‘cell periphery’, ‘extracellular region’ and ‘cell junction’. For biological cell molecular function, the DEGs that were upregulated were significantly enriched in ‘cell adhesion molecule binding’ and ‘cadherin binding’. The DEGs of cervical cancer that were downregulated were significantly enriched in ‘receptor binding’, ‘signal transducer activity’ and ‘molecular transducer activity’. With regard to biological process, it was found that downregulated genes were mainly found in ‘signal transduction’, ‘positive regulation of cellular process’, ‘cell surface receptor signaling pathway’, ‘regulation of multicellular organismal process’ and ‘cell migration’. The upregulated DEGs were primarily enriched in ‘epidermal cell differentiation’, ‘tissue development’ and ‘O-glycan processing’. The analysis clearly and intuitively observed the changes in the enrichment of the DEGs (Figs. 3 and 4).
Figure 3.
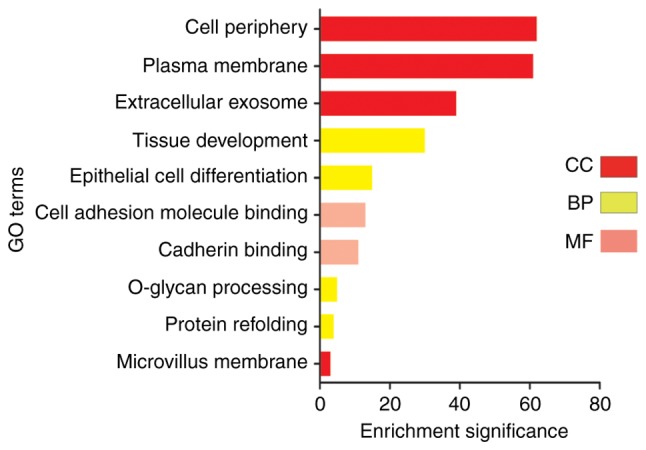
GO terms: Upregulation. Red coloration represents the BP of DEGs, coral coloration represents the CC of DEGs and aqua coloration represents the MF of DEGs (fold-change >2). GO, Gene Ontology; DEGs, differentially expressed genes; BP, biological process; CC, cellular component; MF, molecular function.
Figure 4.
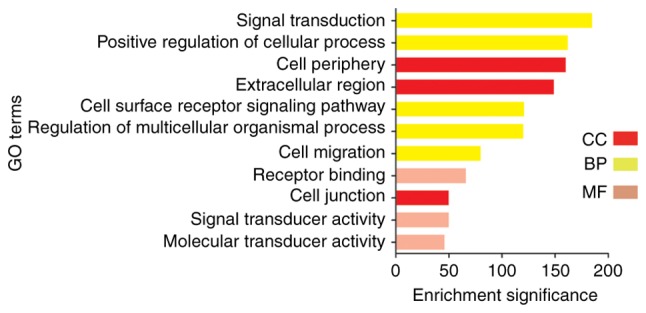
GO terms: Downregulation. Red coloration represents the BP of DEGs, coral coloration represents the CC of DEGs and aqua coloration represents the MF of DEGs (fold-change >2). GO, Gene Ontology; DEGs, differentially expressed genes; BP, biological process; CC, cellular component; MF, molecular function.
KEGG pathway analysis of cervical DEGs
By analyzing the KEGG and Biocarta databases (https://cgap.nci.nih.gov/Pathways/BioCarta_Pathways), Noordhuis et al (14) proved that the five pathways are the TGF-β, nuclear factor of activated T cells (NFAT), anaplastic lymphoma kinase (ALK), Bcl-2-associated death promoter (BAD) and protease-activated receptor-1 (PAR1) pathways. The present study found five significantly enriched pathways by KEGG pathway analysis (Fig. 5). First, the enrichment aspect of ‘aldosterone-regulated sodium reabsorption’ included 4 DEGs that were downregulated and 2 that were upregulated. Second, the enrichment aspect of ‘melanoma’ included 1 upregulated DEG and 8 that were downregulated. Third, the enrichment aspect of ‘gap junction’ included 9 downregulated DEGs. Fourth, the enrichment aspect of ‘cytokine-cytokine receptor interaction’ included 21 downregulated DEGs. Fifth, the enrichment aspect of ‘focal adhesion’ included 1 upregulated DEG and 13 downregulated DEGs. Through the analysis, the molecular mechanism of DEGs was observed in more detail (Fig. 6).
Figure 5.

Heat map for cervical cancer differentially expressed genes in Kyoto Encyclopedia of Genes and Genomes pathways. Red represents upregulation and blue represents downregulation.
Figure 6.

KEGG pathway analysis determined five significantly enriched pathways. Yellow represents the pathways. Red circles represent upregulated DEGs and blue circles represent downregulated DEGs (fold-change >2). DEGs, differentially expressed genes.
Analysis of DEGs by PPI network
Gene co-expression and the PPI network have a guiding role in the study of cervical cancer target genes and proteins, and have a prominent function in the future study of cervical cancer. Based on the Search Tool for the Retrieval of Interacting Genes/Proteins database, upregulated and downregulated genes were selected to observe the association between protein molecules. The associations between various protein molecules were highlighted, aiding in the observation and understanding of the molecular mechanism of cervical cancer. The upregulation of genes such as FGF2 is associated with tumor formation, and IL6 is associated with immunity (Fig. 7). Downregulated genes such as MUC1 are associated with tumor migration, and FUT3 is associated with the degree of tissue differentiation (Fig. 8).
Figure 7.

Upregulated differentially expressed genes with the highest connectivity degree in the protein-protein interaction network. Red represents upregulation, the difference in the red shade is to indicate level of connectivity.
Figure 8.

Downregulated differentially expressed genes with the highest connectivity degree in the protein-protein interaction network. Blue represents downregulation, the difference in the blue shade is to indicate level of connectivity.
WGCNA of DEGs
WGCNA, as an efficient and complex biology method, is widely used in biological information analysis. Through correlation analysis, the gene modules associated with specific sample traits were quickly screened from the data in the present study. More results are shown in Fig. 9. Interaction-associated genes frequently exhibit similar expression patterns. Consequently, the DEGs co-expression network was constructed to screen gene modules with similar expression profile. The DEGs that were in the blue module were significantly enriched in ‘enzyme-linked receptor protein signaling pathway’, ‘cell motility’, ‘embryonic skeletal system development’ and ‘embryo development’; the DEGs that were in the brown module were significantly enriched in ‘B cell activation’, ‘immune response’, ‘leukocyte activation’, ‘humoral immune response’ and ‘G-protein coupled receptor signaling pathway’; the DEGs that were in the grey module were significantly enriched in ‘spermatid development’, ‘spermatid differentiation’, ‘inorganic cation transmembrane transporter activity’, ‘metal ion transmembrane transporter activity’ and ‘regulation of phosphatase activity’; and the DEGs that were in the turquoise module were significantly enriched in ‘mammary gland epithelium development’, ‘mitotic cell cycle process’, ‘development of primary sexual characteristics’, ‘mitotic nuclear division’ and ‘negative regulation of NF-κB transcription factor activity’.
Figure 9.
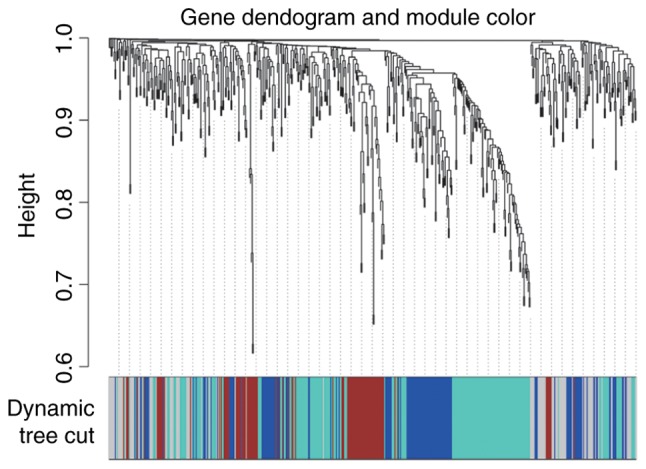
Differentially expressed genes in cervical cancer clustering and module screening based on gene expression pattern. Different modules have different colors.
Validation of differential genes by RT-qPCR
The present study aimed to determine whether the DEGs identified in the microarray analysis were upregulated or downregulated in a sample of clinical cervical cancer patients. Cervical cancer specimens and non-tumor cervical cancer epithelial tissues were obtained, and the differential genes validated with RT-qPCR (Fig. 10). The experimental results showed that MUC1, FUT3 and NOTCH3 gene expression was significantly downregulated, which was consistent with the result of the microarray analysis. However, among the genes found to be upregulated on microarray analysis, IGF1 and HGF were downregulated in the clinical samples. Additionally, FGF2 was upregulated, which confirmed the result of the microarray analysis. The results demonstrated that microarray analysis may provide reference for the identification of molecular markers and therapeutic targets for cervical cancer.
Figure 10.
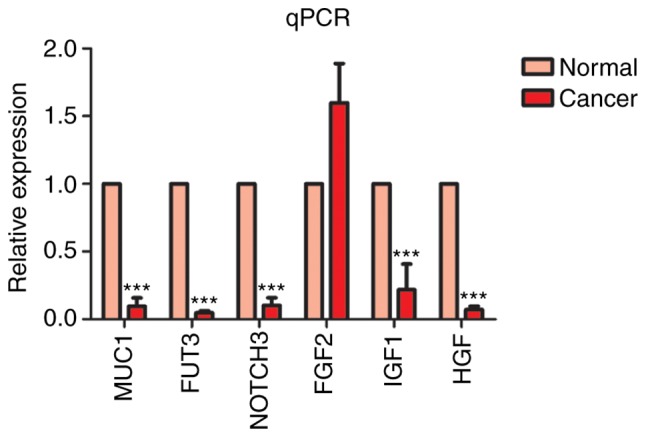
MUC1, FUT3, NOTCH3, FGF2, IGF1 and HGF gene validation with qPCR. ***P<0.001 vs. normal. qPCR, quantitative polymerase chain reaction. MUC1, mucin 1 cell surface-associated; FUT3, fucosyltransferase 3 (Lewis blood group); NOTCH3, notch 3; FGF2, fibroblast growth factor 2; IGF1, insulin-like growth factor 1; HGF, hepatocyte growth factor.
Discussion
Cervical cancer is one of the most common malignancies in women worldwide. There are ~490,000 new cases of cervical cancer in the world each year. In China, 150,000 novel cases of cervical cancer occur and ~20,000 cervical cancer-associated mortalities occur each year (17–19). Previous studies determined that cervical cancer cells were sensitive to radiotherapy and chemotherapy, and used the two treatment techniques to improve the survival rate of patients with cervical cancer (4,20–22). Simultaneously, radiotherapy was determined to have the better curative effect on cervical cancer (23–25). A comprehensive understanding of the molecular mechanism of cervical cancer is essential to the diagnosis and treatment of the disease. Since microarrays achieve rapid detection of gene information by detecting the corresponding position hybridization probes, in the present study, bioinformatics analysis was used to obtain DEGs from the GSE26511 dataset of cervical cancer and normal samples. The function of these differential genes was mainly concentrated in the ‘cell cycle’, ‘biosynthetic process’ and ‘immune response’. Hispolon induces apoptosis and antitumor effects in cervical cancer (26), indicating that drugs can affect the occurrence and development of cervical cancer. In order to identify the cell tumor pathways associated with early cervical cancer pelvic lymph node metastasis, GSE26511 was investigated and it was found that five of the 285 pathways (TGF-β, NFAT, ALK, BAD and PAR1) were dysregulated in cervical cancer-lymph node-negative samples, and two pathways (β-catenin and glycosphingolipid biosynthesis neo lactoseries) were dysregulated in cervical cancer-lymph node-positive samples (14). A large number of DEGs are enriched on these pathways. Genes associated with β-catenin, TGF-β, NFAT, ALK and BAD were found amongst the DEGs.
By observing the results of the bioinformatics analysis in the present study, a number of genes were found in cervical cancer that regulated signaling pathways. In the analysis of the results of the DEGs, transcription factor 4 (TCF4) expression was observed to confer a significant effect on the proliferation, apoptosis and radiosensitivity of cervical cancer cells. It is known that the β-catenin/TCF-4 pathway serves an important role in promoting the metastasis of gastric cancer (27). In early-stage cervical cancer, β-catenin pathways are important in pelvic lymph node metastasis. RASSF1 is also a member of the DEGs.RASSF1, which gene encodes a protein similar to the RAS effector proteins. Hypermethylation of RASSF1A gene is associated with colon cancer (28).
FGF2 and IGF1 are also members of the upregulated DEGs. High FGF2 expression is associated with colon cancer metastasis (29). FGF2 protects the tumor cells from the antiproliferative effect of gefitinib and largely prevents reprogramming of the proteome and phosphoproteom. The process of promoting angiogenesis between IGF1 and cocultured cells is through the phosphoinositide3 kinase/protein kinase B (Akt) signaling pathway (30). MUC1 and FUT3 are also members of the downregulated DEGs. MUC1 induces tamoxifen resistance in estrogen receptor-positive breast cancer (31). MUC1 expression is associated with metastatic recurrence in postoperative patients with esophageal squamous cell cancer (32). Experimental results show that microRNA expression vector targeting of the FUT3 gene can effectively inhibit cell proliferation, migration and invasion ability differences.
By observing the results of the present bioinformatics analysis, it was found that there were numerous genes in cervical cancer regulating the cell cycle. In the analysis of the KEGG results, PAK6 and serine/threonine-protein kinase PAK6 expression clearly demonstrated a significant effect on the proliferation, apoptosis and radiosensitivity of cervical cancer cells. PAK6 is specifically required for carcinoma cell-cell dissociation downstream of HGF for DU145 prostate cancer and HT29 colon cancer cells. In previous studies, PAK6 inhibition resulted in a significant reduction in the survival of prostate cancer cells (33,34). However, the high expression of PAK6 has tumor suppressor abilities and is a potential mechanism in the pathogenesis of hepatocellular carcinoma (35,36). By contrast, the DEGs CLCX12 and BCL2 were downregulated in Fig. 6 in the present study. The BCL2 marker is overexpressed in uterine smooth muscle tumors, and CXCL12 (37–39) overexpression improves neurobehavioral recovery following ischemic stroke through a variety of mechanisms, including the promotion of endothelial progenitor cell function in animal models (40).
By observing the results of the present bioinformatics analysis, immune response-related genes and pathways from WGCNA were found. The DEGs that were in the brown module, including AKT, cluster of differentiation (CD)46, MYC proto-oncogene bHLH transcription factor (MYC) and LIF receptor α (LIFR), were significantly enriched in ‘immune response’. These genes are necessary for the immune response to maintain cell stability. The high expression of LIFR (41) is an independent prognostic marker for primary melanoma (41,42). MYC regulation in the cervical cancer immune response has an irreplaceable role (43–47). The protein encoded by the CD46 gene is a type I membrane protein that is part of the complement system. The encoded protein has cofactor activity to protect the host cell from complement damage. CD46 is a membrane cofactor expressed on all nuclear human cells and serves an important role in inhibiting autoimmune responses and protecting host cells from complement-mediated attacks (48).
In conclusion, in the present study, DEGs of the GSE26511 dataset were assessed by systematic bioinformatics analysis followed by RT-qPCR validation of a number of the key genes in clinical samples. The study was successfully able to reveal biomarkers, indicating that DEGs have important effects for the development and progress of cervical cancer. However, further experiments are required to confirm the function of the identified genes in cervical cancer.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- DEGs
differentially expressed genes
- GEO
gene expression omnibus
- GO
gene ontology
- KEGG
Kyoto encyclopedia of genes and genomes
- PPI
protein-protein interaction
- WGCNA
weighted gene co-expression network analysis
Funding
The present study was supported by the National Natural Sciences Foundation of China (grant nos. 81672685, 81402270, 81272975 and 81672993) and the Beijing Xisike Clinical Oncology Research Foundation (grant no. Y-HR2016096).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
YG and YZ designed the study. CZ, SX, LL, SL, YX, KC and HC conducted the experiments. YX, KC and HC analyzed the data. The manuscript was drafted by YG and YZ. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Written informed consent was obtained from each patient, and experimental protocols were approved by the Institutional Review Board of the Cancer Hospital of Hunan Province.
Consent for publication
All participants provided written informed consent for publication.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Yang M, Wang M, Li X, Xie Y, Xia X, Tian J, Zhang K, Tang A. Wnt signaling in cervical cancer? J Cancer. 2018;9:1277–1286. doi: 10.7150/jca.22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. Lancet. 2013;382:889–899. doi: 10.1016/S0140-6736(13)60022-7. [DOI] [PubMed] [Google Scholar]
- 4.Shelley C, Gore M, Tan S, Thomas K, Eeles R. Cervical spine fractures in patients undergoing palliative radiotherapy to the cervical spine: Iimplications for Practice. Clin Oncol (R Coll Radiol) 2018 Apr 10; doi: 10.1016/j.clon.2018.02.068. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 5.Isono-Nakata R, Tsubamoto H, Ueda T, Inoue K, Shibahara H. Bevacizumab with metronomic chemotherapy of low-dose oral cyclophosphamide in recurrent cervical cancer: Four cases. Gynecol Oncol Rep. 2018;24:57–60. doi: 10.1016/j.gore.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sevin BU, Nadji M, Lampe B, Lu Y, Hilsenbeck S, Koechli OR, Averette HE. Prognostic factors of early stage cervical cancer treated by radical hysterectomy. Cancer. 1995;76(10 Suppl):S1978–S1986. doi: 10.1002/1097-0142(19951115)76:10+<1978::AID-CNCR2820761313>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 7.Einstein MH, Park KJ, Sonoda Y, Carter J, Chi DS, Barakat RR, Abu-Rustum NR. Radical vaginal versus abdominal trachelectomy for stage IB1 cervical cancer: A comparison of surgical and pathologic outcomes. Gynecol Oncol. 2009;112:73–77. doi: 10.1016/j.ygyno.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plante M, Gregoire J, Renaud MC, Roy M. The vaginal radical trachelectomy: An update of a series of 125 cases and 106 pregnancies. Gynecol Oncol. 2011;121:290–297. doi: 10.1016/j.ygyno.2010.12.345. [DOI] [PubMed] [Google Scholar]
- 9.Kim CH, Abu-Rustum NR, Chi DS, Gardner GJ, Leitao MJ, Jr, Carter J, Barakat RR, Sonoda Y. Reproductive outcomes of patients undergoing radical trachelectomy for early-stage cervical cancer. Gynecol Oncol. 2012;125:585–588. doi: 10.1016/j.ygyno.2011.12.150. [DOI] [PubMed] [Google Scholar]
- 10.Bentivegna E, Gouy S, Maulard A, Chargari C, Leary A, Morice P. Oncological outcomes after fertility-sparing surgery for cervical cancer: A systematic review. Lancet Oncol. 2016;17:e240–e253. doi: 10.1016/S1470-2045(16)30032-8. [DOI] [PubMed] [Google Scholar]
- 11.Baiocchi G, de Brot L, Faloppa CC, Mantoan H, Duque MR, Badiglian-Filho L, Da CA, Kumagai LY. Is parametrectomy always necessary in early-stage cervical cancer? Gynecol Oncol. 2017;146:16–19. doi: 10.1016/j.ygyno.2017.03.514. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Yao T, Lin Z, Gao Y. Aberrant methylation of MEG3 functions as a potential Plasma-based biomarker for cervical cancer. Sci Rep. 2017;7:6271. doi: 10.1038/s41598-017-06502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen AH, Qin YE, Tang WF, Tao J, Song HM, Zuo M. MiR-34a and miR-206 act as novel prognostic and therapy biomarkers in cervical cancer. Cancer Cell Int. 2017;17:63. doi: 10.1186/s12935-017-0431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noordhuis MG, Fehrmann RS, Wisman GB, Nijhuis ER, van Zanden JJ, Moerland PD, Ver Loren van Themaat E, Volders HH, Kok M, Ten Hoor KA, et al. Involvement of the TGF-beta and beta-catenin pathways in pelvic lymph node metastasis in early-stage cervical cancer. Clin Cancer Res. 2011;17:1317–1330. doi: 10.1158/1078-0432.CCR-10-2320. [DOI] [PubMed] [Google Scholar]
- 15.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. Bmc Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Zeng H, Chen W, Zheng R, Zhang S, Ji JS, Zou X, Xia C, Sun K, Yang Z, Li H, et al. Changing cancer survival in China during 2003–15: A pooled analysis of 17 population-based cancer registries. Lancet Glob Health. 2018;6:e555–e567. doi: 10.1016/S2214-109X(18)30127-X. [DOI] [PubMed] [Google Scholar]
- 18.Chen JG, Chen HZ, Zhu J, Yang YL, Zhang YH, Huang PX, Chen YS, Zhu CY, Yang LP, Shen K, et al. Cancer survival in patients from a hospital-based cancer registry, China. J Cancer. 2018;9:851–860. doi: 10.7150/jca.23039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu T, Zhao D, Li B, Wang Y, Liu S, Li P, Zuo J, Bai P, Zhang R, Wu L. Prognostic evaluation of postoperative adjuvant therapy for operable cervical cancer: 10 years' experience of national cancer center in China. Chin J Cancer Res. 2017;29:510–520. doi: 10.21147/j.issn.1000-9604.2017.06.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardoso MFS, Castelletti CHM, Lima-Filho JL, Martins DBG, Teixeira JAC. Putative biomarkers for cervical cancer: SNVs, methylation and expression profiles. Mutat Res. 2017;773:161–173. doi: 10.1016/j.mrrev.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Wei LC, Li X, Zhang Y, Dang YZ, Li WW, Li JP, Zhao LN, Liu SJ, Li X, Shi M. Individualized pelvic lymphadenectomy should follow neoadjuvant concurrent chemoradiotherapy for locally advanced cervical cancer. Medicine (Baltimore) 2018;97:e0331. doi: 10.1097/MD.0000000000010331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li F, Guo H, Qiu H, Liu S, Wang K, Yang C, Tang C, Zheng Q, Hou Y. Urological complications after radical hysterectomy with postoperative radiotherapy and radiotherapy alone for cervical cancer. Medicine (Baltimore) 2018;97:e0173. doi: 10.1097/MD.0000000000010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Kong W, Lv N, Li F, Chen J, Jiao S, Ding D, Zhao H, Song D. Incidence of radiation enteritis in cervical cancer patients treated with definitive radiotherapy versus adjuvant radiotherapy. J Cancer Res Ther. 2018;14(Suppl):S120–S124. doi: 10.4103/0973-1482.163762. [DOI] [PubMed] [Google Scholar]
- 24.Yang S, Xing L, Gu L, Cheng H, Feng Y, Zhang Y. Combination of RIZ1 overexpression and radiotherapy contributes to apoptosis and DNA damage of HeLa and SiHa cervical cancer cells. Basic Clin Pharmacol Toxicol. 2018 Mar 25; doi: 10.1111/bcpt.13008. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 25.Moelle U, Mathewos A, Aynalem A, Wondemagegnehu T, Yonas B, Begoihn M, Addissie A, Unverzagt S, Jemal A, Thomssen C, et al. Cervical cancer in Ethiopia: The effect of adherence to radiotherapy on survival. Oncologist. 2018 Mar 22; doi: 10.1634/theoncologist.2017-0271. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsin MC, Hsieh YH, Wang PH, Ko JL, Hsin IL, Yang SF. Hispolon suppresses metastasis via autophagic degradation of cathepsin S in cervical cancer cells. Cell Death Dis. 2017;8:e3089. doi: 10.1038/cddis.2017.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhuang K, Yan Y, Zhang X, Zhang J, Zhang L, Han K. Gastrin promotes the metastasis of gastric carcinoma through the β-catenin/TCF-4 pathway. Oncol Rep. 2016;36:1369–1376. doi: 10.3892/or.2016.4943. [DOI] [PubMed] [Google Scholar]
- 28.Chen HC, Huang HY, Chen YL, Lee KD, Chu YR, Lin PY, Hsu CC, Chu PY, Huang TH, Hsiao SH, Leu YW. Methylation of the tumor suppressor genes HIC1 and RassF1A clusters independently from the methylation of polycomb target genes in colon cancer. Ann Surg Oncol. 2017;24:578–585. doi: 10.1245/s10434-015-5024-z. [DOI] [PubMed] [Google Scholar]
- 29.Sako A, Kitayama J, Yamaguchi H, Kaisaki S, Suzuki H, Fukatsu K, Fujii S, Nagawa H. Vascular endothelial growth factor synthesis by human omental mesothelial cells is augmented by fibroblast growth factor-2: Possible role of mesothelial cell on the development of peritoneal metastasis. J Surg Res. 2003;115:113–120. doi: 10.1016/S0022-4804(03)00307-X. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Zhang F, Hong Y. Blocking of autocrine IGF-1 reduces viability of human umbilical cord mesenchymal stem cells via inhibition of the Akt/Gsk-3β signaling pathway. Mol Med Rep. 2018;17:4681–4687. doi: 10.3892/mmr.2018.8445. [DOI] [PubMed] [Google Scholar]
- 31.Merikhian P, Ghadirian R, Farahmand L, Mansouri S, Majidzadeh AK. MUC1 induces tamoxifen resistance in estrogen receptor-positive breast cancer. Expert Rev Anticancer Ther. 2017;17:607–613. doi: 10.1080/14737140.2017.1340837. [DOI] [PubMed] [Google Scholar]
- 32.Ye Q, Yan Z, Liao X, Li Y, Yang J, Sun J, Kawano T, Wang X, Cao Z, Wang Z, Huang L. MUC1 induces metastasis in esophageal squamous cell carcinoma by upregulating matrix metalloproteinase 13. Lab Invest. 2011;91:778–787. doi: 10.1038/labinvest.2011.12. [DOI] [PubMed] [Google Scholar]
- 33.Morse EM, Sun X, Olberding JR, Ha BH, Boggon TJ, Calderwood DA. PAK6 targets to cell-cell adhesions through its N-terminus in a Cdc42-dependent manner to drive epithelial colony escape. J Cell Sci. 2016;129:380–393. doi: 10.1242/jcs.177493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang M, Siedow M, Saia G, Chakravarti A. Inhibition of p21-activated kinase 6 (PAK6) increases radiosensitivity of prostate cancer cells. Prostate. 2010;70:807–816. doi: 10.1002/pros.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang ZP, Jiang BG, Gu XF, Zhao B, Ge RL, Zhang FB. P21-activated kinase 5 plays essential roles in the proliferation and tumorigenicity of human hepatocellular carcinoma. Acta Pharmacol Sin. 2014;35:82–88. doi: 10.1038/aps.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu W, Liu Y, Liu H, Zhang W, Fu Q, Xu J, Gu J. Tumor suppressive function of p21-activated kinase 6 in hepatocellular carcinoma. J Biol Chem. 2015;290:28489–28501. doi: 10.1074/jbc.M115.658237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng W, Xue S, Chen Y. The role of CXCL12 in tumor microenvironment. Gene. 2018;641:105–110. doi: 10.1016/j.gene.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Goffart N, Lombard A, Lallemand F, Kroonen J, Nassen J, Di Valentin E, Berendsen S, Dedobbeleer M, Willems E, Robe P, et al. CXCL12 mediates glioblastoma resistance to radiotherapy in the subventricular zone. Neuro Oncol. 2017;19:66–77. doi: 10.1093/neuonc/now136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collins PJ, McCully ML, Martinez-Muñoz L, Santiago C, Wheeldon J, Caucheteux S, Thelen S, Cecchinato V, Laufer JM, Purvanov V, et al. Epithelial chemokine CXCL14 synergizes with CXCL12 via allosteric modulation of CXCR4. FASEB J. 2017;31:3084–3097. doi: 10.1096/fj.201700013R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang S, Li Y, Yuan F, Qu M, Song Y, Zhang Z, Yang GY, Wang Y. Monomeric CXCL12 outperforms its dimeric and wild type variants in the promotion of human endothelial progenitor cells' function. Biochem Biophys Res Commun. 2017;488:303–310. doi: 10.1016/j.bbrc.2017.03.172. [DOI] [PubMed] [Google Scholar]
- 41.LIFR functions as a metastasis suppressor in hepatocellular carcinoma by negatively regulating phosphoinositide 3-kinase/AKT pathway. Carcinogenesis. 2015;36:1201–1212. doi: 10.1093/carcin/bgv108. [DOI] [PubMed] [Google Scholar]
- 42.Ma D, Jing X, Shen B, Liu X, Cheng X, Wang B, Fu Z, Peng C, Qiu W. Leukemia inhibitory factor receptor negatively regulates the metastasis of pancreatic cancer cells in vitro and in vivo. Oncol Rep. 2016;36:827–836. doi: 10.3892/or.2016.4865. [DOI] [PubMed] [Google Scholar]
- 43.Xu Y, Jin Y, Liu L, Zhang X, Chen Y, Wei J. Study of circulating IgG antibodies to peptide antigens derived from BIRC5 and MYC in cervical cancer. FEBS Open Bio. 2015;5:198–201. doi: 10.1016/j.fob.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan X, Karner CM, Carroll TJ. Myc cooperates with beta-catenin to drive gene expression in nephron progenitor cells. Development. 2017;144:4173–4182. doi: 10.1242/dev.153700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiu S, Liu PY, Liu T. Up-regulation of LYAR blocks Myc-induced cell death. Cell Cycle. 2017;16:1857–1858. doi: 10.1080/15384101.2017.1374682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JE, Rayyan M, Liao A, Edery I, Pletcher SD. Acute dietary restriction acts via TOR, PP2A, and Myc signaling to boost innate immunity in drosophila. Cell Rep. 2017;20:479–490. doi: 10.1016/j.celrep.2017.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim EY, Kim A, Kim SK, Chang YS. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer. 2017;110:63–67. doi: 10.1016/j.lungcan.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Zhang D, Sjölinder M, Wan Y, Sjölinder H. CD46 accelerates macrophage-mediated host susceptibility to meningococcal sepsis in a murine model. Eur J Immunol. 2017;47:119–130. doi: 10.1002/eji.201646397. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.