

Abstract
Protein post-translational modifications (PTMs) are chemical modifications of a protein after its translation. Owing to its play an important role in deep understanding of various biological processes and the development of effective drugs, PTM site prediction have become a hot topic in bioinformatics. Recently, many online tools are developed to prediction various types of PTM sites, most of which are based on local sequence and some biological information. However, few of existing tools consider the relations between different PTMs for their prediction task. Here, we develop a web server called PTM-ssMP to predict PTM site, which adopts site-specific modification profile (ssMP) to efficiently extract and encode the information of both proximal PTMs and local sequence simultaneously. In PTM-ssMP we provide efficient prediction of multiple types of PTM site including phosphorylation, lysine acetylation, ubiquitination, sumoylation, methylation, O-GalNAc, O-GlcNAc, sulfation and proteolytic cleavage. To assess the performance of PTM-ssMP, a large number of experimentally verified PTM sites are collected from several sources and used to train and test the prediction models. Our results suggest that ssMP consistently contributes to remarkable improvement of prediction performance. In addition, results of independent tests demonstrate that PTM-ssMP compares favorably with other existing tools for different PTM types. PTM-ssMP is implemented as an online web server with user-friendly interface, which is freely available at http://bioinformatics.ustc.edu.cn/PTM-ssMP/index/.
Keywords: Post-translational modifications, web server, site-specific modification profile, prediction
Introduction
Protein post-translational modifications (PTMs) are chemical modifications of a protein after its translation, which regulate variety of critical cellular processes such as cell cycle control, DNA repair, signal transduction and protein-protein interactions. 1-3. Currently, many types of PTMs have been experimentally found and they play a critical role in various biological processes. For example, as a reversible PTM, phosphorylation not only functions significantly in cell signaling but also related to many diseases 4-6. Moreover, lysine acetylation plays an important role in regulating gene expression and cellular signaling 7. Ubiquitination is also implicated in the regulation of a variety of cellular processes, such as budding of retroviral virions, regulation of transcription factor activity and receptor endocytosis 8. Besides these PTMs, there are many studies describing experimental validated PTMs, such as sumoylation, methylation, sulfation, O-linked glycosylation and proteolytic cleavage 9-14, which also affect many aspects of cellular functionalities and relate to various diseases. Therefore, PTM site identification in proteomes is important to study and analyze the underlying molecular mechanisms, which also provide useful information for drug discovery 15-17.
Due to the biological importance of protein PTM, many conventional experimental methods have been used to identify potential PTM sites, such as mass spectrometry (MS) and Chip-Chip. Since experimental methods are high-cost and time-consuming, many studies are dedicated to developing efficient and reliable theoretical computation method for the PTM site prediction. Most of computation methods use local sequence information in PTM site prediction because the local sequence of the PTM site is generally conserved 18-20. For example, a number of methods are proposed to predict phosphorylation sites, such as PhosphoSVM 21, Musite 22, GPS 3.0 23, KinasePhos 2.0 24, PPSP 25 and NetPhos 9. Besides phosphorylation, many bioinformatics tools have been developed to identify other PTM sites. For example, Ubipred 26 and Ubisite 27 are ubiquitination site prediction server which build prediction model using feature extraction method based on the local sequences. In addition, Shao et al. provide a online service called BRABSB 28 for identification of lysine acetylation site. Despite the success achieved by above sequence based methods, using sequence alone may not provide sufficient information for good prediction performance as PTM is a complex process that involves various biological mechanisms 8, 29-31. Therefore, additional biological information, such as protein-protein interaction (PPI) 29, 30, 32 and protein structure information 8, 19, 31, has been introduced by different approaches in the task of PTM site prediction. An interesting phenomenon about PTM is that functional associations may exist among proximal PTMs in the protein sequence, i.e., PTM crosstalk 33-37. It has been shown that phosphorylation can influence and regulate other PTMs such as sumoylation 38, 39 and O-linked glycosylation 40, 41. Meanwhile, various crosstalks between lysine acetylation and other PTMs including phosphorylation 42-44 and methylation 45-47 have also been validated. For example, previous studies 44, 48 report that lysine acetylation inhibits phosphorylation of its upstream + 3 position when peptides containing the KXXS motifs. On the other side, some studies 37, 49 show that there are differences in proximal PTM information for different type of PTM sites due to the existence of intrinsic functional PTM crosstalks. Accordingly, we speculate that nearby PTM sites may be helpful to determine a candidate PTM site. More generally, it may be worthy of bringing the information of proximal PTMs into the task of PTM site prediction, and this idea has been supported by our previous work 50 by using information of in situ PTMs that occurs on the same site.
Inspired by aforementioned research, in this study, we develop a web server called PTM-ssMP for PTM site prediction, which adopts site-specific modification profile (ssMP) to efficiently extract and encode the information of both proximal PTMs and local sequence simultaneously. To assess the performance of PTM-ssMP, a large amount of experimentally verified PTM data are collected from several sources and used to train and test the prediction models. Our results suggest that ssMP consistently contributes to remarkable improvement of prediction performance. In addition, independent tests demonstrate that PTM-ssMP compares favorably with other existing tools for different PTM types. For example, the performance of PTM-ssMP obtain more than 10% improvement compares with other exist tools for lysine acetylation and ubiquitination site prediction. Another advantage of ssMP is that it can be easily applied to various kinds of PTM types, therefore in PTM-ssMP we provide efficient predictions of multiple types of PTM sites including phosphorylation, lysine acetylation, ubiquitination, sumoylation, methylation, sulfation, proteolytic cleavage, O-GalNAc and O-GlcNAc. Therefore, PTM-ssMP is a comprehensive web server and it would provide effective assistance to researchers who are interested in proteome data.
Methods and materials
Overall framework
PTM-ssMP is an online web server that can predict nine types of PTM site by using ssMP. There are four main procedures in our work (Figure 1): (i) constructing a valid dataset to train and test the SVM predictive models for multiple PTM types separately, (ii) selecting local sequence containing 10 residues up- and downstream of candidate sites, (iii) extracting ssMP for each candidate site (iv) and developing a user-friendly web server called PTM-ssMP that is accessible to the public.
Figure 1.

The overall framework of PTM-ssMP
Data collection and pre-processing
The experimentally determined phosphorylation, lysine acetylation, ubiquitination, sumoylation, methylation, sulfation, proteolytic cleavage, O-GalNAc and O-GlcNAc sites are extracted from several public databases including dbPTM version 3.0 51, SysPTM 52, Phospho.ELM 53, HPRD 54, PhosphoSitePlus 55 and dbOGAP 56. We then extract all of the experimentally verified PTM sites in human protein from these databases for further analysis. To eliminate sequence redundancy and avoid overestimation of the prediction performance, we use CD-HIT 57 to ensure that none of the protein sequences show a sequence similarity more than 40% for dataset of each PTM type 49, 58. Subsequently, we totally obtain 7866 protein sequences with at least one of the nine PTM types investigated in this study, and experimentally determined PTM sites with proximal PTM information are extracted as positive samples for each PTM type, respectively. We use a local sliding window that comprised 21 residues 27, 59 to extract the local sequence and proximal PTM information around candidate sites, where the candidate sites are located at the center with ten neighboring residues upstream and downstream. We find that more than 70% of the PTM sites have at least one proximal PTM site. For negative samples, since it is difficult to verify that a particular residue is a not PTM site under any conditions 60, we extract negative sites for each PTM type with a common requirement 60, 61 that for a specific PTM type, the residue is not verified as a modification site of such PTM type in any database and that protein contains at least one residue known to be modified by such PTM type. To precisely assess the prediction performance, only negative sites with proximal PTMs are used for further analysis. To address a common issue in PTM site prediction that the dataset is unbalanced with much more negative samples than positive ones 21, 62, 63, we follow a widely used procedure by randomly selecting negative samples to match the number of positive ones 21, 62, 63 and the final datasets are outlined in Table S1. Furthermore, in consistent with previous studies 61, 64, we randomly select ∼20% sample as the independent test datasets and the remaining part as the benchmark datasets. The numbers of samples in the benchmark datasets and independent test datasets are outlined in Table S2 and Table S3 respectively, and their detailed sequences and positions information in the proteins are given in download page of web server (http://bioinformatics.ustc.edu.cn/PTM-ssMP/download/). Moreover, to reduce the bias of randomness, we follow the previous study 63 to repeat selection procedure of negative sample10 times for benchmark dataset.
Construction of site-specific modification profile
We denote a specific amino acid R0 as a valuable of attribute data with Na types of values which represent the Na types of amino acids (here Na = 20), i.e. R0 ∈ Sa = {Aj | j = 1,…, Na} where Aj is the j-th type of amino acid. To predict the PTM types on residue R0, we define Ri as the i-th downstream amino acid of the R0. The set {Ri | 1 ≤ i ≤ L, i ∈N} is the 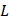 -th order downstream residue set, which contains the downstream amino acid residue neighbors of R0. Similarly, R-i denotes the i-th upstream amino acid of the R0, and the set {R-i | 1 ≤ i ≤ L, i ∈N} is the L-th order upstream residues set of R0. We then define the local sequences of R0 as a set SeqL, i.e.
-th order downstream residue set, which contains the downstream amino acid residue neighbors of R0. Similarly, R-i denotes the i-th upstream amino acid of the R0, and the set {R-i | 1 ≤ i ≤ L, i ∈N} is the L-th order upstream residues set of R0. We then define the local sequences of R0 as a set SeqL, i.e.
| SeqL = {R-i | -L ≤ i ≤ L, i ∈N} | (1) |
where the cardinality of PL is 2L+1 and L is set to 10 in this study. Specifically, when the length of the upstream sequence Lu or the length of the downstream sequence Ld is less than L, we use '*' to represent the not-exist amino acid, and the values of these not-exist amino acid is the dummy attribute. We further define another attribute variable Qi to denote known PTM types on Ri that have been previously validated, of which the value represents the NP types of PTMs. The variable space of Qi is a set whose cardinality is NP (here NP = 14), which is defined as SP = {Pj | j = 1, …, NP} and Pj is the j-th type of PTM.
To encode the information of proximal PTMs, we introduce a NP × (2L+1) dimension matrix Yij to represent the profile of proximal PTMs for the sites where -L ≤ i ≤ L. Here NP represents the totally number of PTM types for attribute variable Qi. The (i,j) -th element of the matrix Yij is:
| Yij = Ind (Qi = Pj), for -L ≤ i ≤ L and 1 ≤ j ≤ NPTM, i, j ∈N | (2) |
where Pj is j-th type of PTM that is represented by the j-th feature of the vector Yi. The function Ind (·) is the indicator function, which gives an output of 1 when the input equation is true and 0 otherwise. Since there may be more than one type of known PTM occurring on a same site, the matrix Yij for representing known PTM types is not orthogonal. For example, there is a local sequence in Figure 1B, which consist of 21 amino acids (including a candidate site at the center and its local sequence at both upstream and downstream). In this local sequence, acetylation and methylation occurred on upstream 6 position, correspondingly, these PTM information are represented as (0,1,0,1,0,0,0,0,0,0,0,0,0,0). Likewise, we encode the local sequence information as a NR × (2L+1) dimension matrix (Xij), which is also the numeric representation of Ri for -L ≤ i ≤ L. The entry xij of the matrix is calculated as
| Xij = Ind (Ri = Aj), for -L ≤ i ≤ L and 1 ≤ j ≤ NR, i, j ∈N | (3) |
where Aj is the j-th type of amino acid in set SR that is represented by the j-th feature of the vector. Note that Xij is an orthogonal binary matrix with only one entry of each row assigned with 1 and others assigned with 0. For example, a local sequence like 'LSTEKVMKLHKSYRSMTPAQY', which will be encoded into a 21 by 21 matrix. Specifically, the first site of this local sequence is 'L', and it will be encoded as (0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,0,0), correspondingly, the first row of the matrix is (0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,0,0).
Finally, we define the ssMP matrix Zij as Zij = Xij ⊕ Yij, where the symbol ⊕ is the concatenation operation of matrices Xij and Yij. The variable space is the Cartesian product of the spaces for the two variables SZ = SR ⨂ SP (the symbol ⨂ denotes to Cartesian product). For all the entries Zij in the ssMP matrix, we can extract the features contained in SZ of the sample for each site in the local sequence and the related PTMs. To develop the classification models for PTM site prediction, a ssMP is generated for each sample in the training data and the entries of the ssMP are then adopted as the input features to train a SVM classifier with LibSVM 65 for each PTM type. These classifiers are then used to make perditions with the ssMP features extracted from candidate PTM sites.
Implementation and web interface
PTM-ssMP is implemented by the python on a Linux machine with Apache 2.2.3 HTTP Server. Web server consists of six sections, including Home, Server, Tutorial, Result, Download and Contact. Home page include a brief introduction about PTM-ssMP. In Server page (Figure 2), users can submit the data of proteins with known PTM information and select the task-related prediction models and thresholds. For the convenience of most users, we present a user guide in Tutorial page and our contact information in contact page. Furthermore, PTM-ssMP allows users to download datasets of all PTM types, including corresponding Protein ID, Position and protein local sequences from Download page. After the prediction task, PTM-ssMP generates a Result page that contains prediction results, where each row of the result represents a candidate site. The running time required for a prediction task depends on the selected models and the length of the query sequence. In PTM-ssMP we provide a test example, which have two query sequences with the total length more than 500 amino acid. The prediction task requires 10 s to generate and return the results if select all predictive models.
Figure 2.

A semi-screenshot to show the server-page of the PTM-ssMP web-server at http://bioinformatics.ustc.edu.cn/PTM-ssMP/server/
Performance evaluation
In this study, by following previous studies we perform a 10-fold cross-validation 2, 25 (based on the benchmark datasets) to evaluate the importance of ssMP and independent tests 27, 61(based on the independent test datasets) to compare with other existing PTM prediction tools. To evaluate the performance of each prediction model, six standard measurements are adopted, including accuracy (Acc), specificity (Sp), sensitivity (Sn), Matthew's correlation coefficient (MCC), precision (Pre) and F1 score (F1). They are defined as follows:
| Acc = (TN+TP)/(TN+TP+FN+FP) | (4) |
| Sn = TP/(TP+FN) | (5) |
| Sp = TN/(TN+FP) | (6) |
| Pre = TP/(TP+FP) | (7) |
| F1 = 2×Pre×Sn/(Pre_Sn) | (8) |
| MCC = (TP×TN-FP×FN)/[(TP+FN) ×(TP+FP) ×(TN+FN) ×(TN+FP)]1/2 | (9) |
TP and TN are abbreviations of true positives and true negatives, which indicate the number of positive and negative sites that are correct predicted. FP and FN are abbreviations false positives and false negatives, which indicate the number of positive and negative sites that are predicted falsely. Sn and Sp are abbreviations of sensitivity and specificity, and they measure the proportion of positives and negatives that are correctly identified. MCC is used to reflect the balance quality when the numbers of negative and positive data are significant imbalance.
Results
Statistical difference of proximal PTMs in positive and negative samples
We first investigate the statistical difference of proximal PTMs in positive and negative samples. Use lysine acetylation as example, previous studies 44, 48 report that lysine acetylation inhibits phosphorylation in +3 positions. Consequently, we calculate the number of phosphorylation of the +3 positions in ssMP of positive and negative samples respectively, and the statistical significance is examined by using Fisher exact test. The results (Figure 3A) show that phosphorylation in +3 positions of acetylation-modified lysine is lower (p = 7.73 × 10-7), which indicate that ssMP can reflect the proximal PTMs difference between positive and negative samples and it could be a helpful feature for PTM site prediction. Furthermore, many studies report that there is mutual exclusion between lysine acetylation and its upstream phosphorylation. 66-68. Correspondingly, we perform the same analysis on all upstream phosphorylation between lysine acetylation positive and negative samples, and we find a significant difference between the numbers of upstream phosphorylation in positive and negative samples (p = 4.05 × 10-44) (Figure 3B). The above analysis shows that there are clear differences between the proximal PTMs of positive and negative samples, which might contribute to the PTM site prediction.
Figure 3.

Comparison of proximal PTMs between positive and negative data. The gray bar represents the proportion of lysine acetylation proximal phosphorylation of positive samples and the black bar represents the negative ones.
Evaluation of the prediction model
In this section, we adopt ten-fold cross-validation to evaluate the performance of ssMP in PTM site prediction based on benchmark dataset, and we assess the prediction models trained by both local sequences and ssMP are assessed. In Figure S1 we show the ROC curves of ubiquitination, acetylation, O-GlcNAc and phosphorylation in AGC, CK1, STE kinase groups. Furthermore, in Table 1 we list the corresponding AUC values in terms of mean and variance. As can be seen from Figure S1, our proposed method that are trained with ssMP has better prediction performance in predicting multiple types PTM sites. For phosphorylation kinase group AGC, STE and CK1 the AUC values of method trained with only local sequences are 0.822, 0.728 and 0.852, respectively. In our proposed method, the AUC values of AGC, STE and CK1 are increased to 0.873, 0.806 and 0.901. In addition, for ubiquitination, the AUC value of method trained with local sequences only is 0.668. With ssMP, the corresponding AUC value is 0.752, which is 8.4% higher than the method trained with local sequences. Similarly, for lysine acetylation, and O-GlcNAc, the AUC value of our proposed method with ssMP also obtain 9.5%, 5.8% improvement than those methods that only use local sequences. The AUC value of the other PTM types are listed in Table S4, which further suggests that our proposed method is superior to the method that only consider local sequences. Therefore, we believe that ssMP are very useful features which can significant improve the performance of PTM site prediction for multiple types.
Table 1.
AUC values comparison of ssMP and local sequences of ubiquitination, lysine acetylation, O-GlcNAc and phosphorylation kinase groups AGC, CK1 and STE.
| PTM | method | AUC | PTM | method | AUC |
|---|---|---|---|---|---|
| Phosphorylation (AGC) |
SEQ | 0.822±1.96e-5 | Ubiquitination | SEQ | 0.668±3.00e-6 |
| ssMP | 0.873±2.76e-5 | ssMP | 0.752±1.90e-6 | ||
| Phosphorylation (CK1) |
SEQ | 0.852±7.49e-5 | Lysine acetylation | SEQ | 0.649±1.32e-5 |
| ssMP | 0.901±4.70e-5 | ssMP | 0.744±7.14e-6 | ||
| Phosphorylation (STE) |
SEQ | 0.728±3.19e-4 | O-GlcNAc | SEQ | 0.772±1.98e-4 |
| ssMP | 0.806±3.33e-4 | ssMP | 0.830±6.12e-5 |
In addition to the AUC value, we also adopt other six measurements including Sn, Sp, Acc, F1, Pre and MCC to verify the reliability of ssMP. By following the study of Wang et al. 69, we set the threshold of specificity equal to 95.0% (high stringency levels) or 90.0% (medium stringency levels). Take two phosphorylation kinase groups AGC, STE and another two PTM ubiquitination, lysine acetylation as examples, the corresponding measurements in high stringency levels are computed and reported in Table S3. By incorporating ssMP, the values of Acc, Sn, F1, Pre and MCC for AGC are 0.743, 0.536, 0.675, 0.915 and 0.534, respectively, whereas the Acc, Sn, F1, Pre and MCC values with only local sequences are 0.706, 0.461, 0.610, 0.903 and 0.472, respectively. And for STE, the corresponding measurements are also improved by 7.0%, 14.0%, 15.9%, 6.0% and 13.3%, respectively. In addition to phosphorylation, we can find that our proposed method also have better performance for ubiquitination and acetylation. Taken ubiquitination as instance, the values of Acc, Sn, F1, Pre and MCC values are 0.588, 0.227, 0.355, 0.819 and 0.256, which are increased by 6.2%, 12.4%, 17.7%, 14.7% and 15.7% compared with the method using only local sequences. Similarly, as shown in Table S5, for other PTM types and phosphorylation groups our proposed method is consistently better than the method that using only local sequences. We also compute such measurements at medium stringency level, the detailed results of all PTM and phosphorylation groups are listed in Table S6. These results suggest that ssMP are efficient features which can significant improve the prediction of multiple PTMs.
Comparison with existing tools
In this section, base on independent dataset we compare the prediction performance of PTM-ssMP and several other existing tools. Firstly, two common and efficient phosphorylation prediction methods, PPSP and GPS 3.0 are used to make comparison. To illustrate the prediction performance, we take four kinase groups Atypical, CK1, AGC and TK as examples and plot the ROC curves for three methods (Figure 4). As shown in Figure 4, for four kinase groups PTM-ssMP achieves better performance than other prediction methods. Moreover, for each method we also calculate the corresponding AUC value for each phosphorylation kinase group and displayed in Figure 4. For Atypical, CK1, AGC and TK, the AUC values of PTM-ssMP are 0.924, 0.880, 0.894 and 0.821 , which is 10.8%, 25.5%, 23.1% and 8.4% higher than GPS, and the AUC value of PPSP only have 0.813, 0.808, 0.833 and 0.765, respectively. Therefore, PTM-ssMP outperformed other prediction methods in predicting multiple kinase groups of phosphorylation. Meanwhile, the ROC curves for other kinase groups are also plotted (Figure S3), and we find that PTM-ssMP consistently better than other methods.
Figure 4.

Performance of phosphorylation ROC curves in kinase group Atypical CK1, AGC and TK with different methods. The red lines represent the performance of PTM-ssMP, the blue and purple lines represent the GPS, PPSP respectively.
Additionally, we also plot the Acc-Sn-MCC-Pre-F1 bar graph of three methods to assess the detailed performance for phosphorylation kinase group AGC, CK1, STE and Atypical according to the high and medium stringency levels, as shown in Figure 5. It is obvious that PTM-ssMP achieves the best prediction performance in most of kinase groups. For instance, at high stringency level (Sp =95.0%), the Acc, Sn, MCC, Pre, F1 values of AGC are increased by 8.5%, 17.0%, 14.2%, 2.5%, 14.1% compared with PPSP. For STE, the Acc, Sn, MCC, Pre, F1 values are improved by 15.0%, 30.0%, 27.6%, 11.1%, 30.8% compared with PPSP. Similarly, PTM-ssMP has better or comparable performance than GPS in most of kinase groups. In addition, PTM-ssMP also obtain better performance at medium stringency (Sp=90%). Taking CK1 as an example, PTM-ssMP outperforms PPSP with 5.5%, 11.0%, 9.8%, 1.8%, 7.6% higher Acc, Sn, MCC, Pre, F1 values. The detailed results other groups at high stringency level and medium stringency level are listed in Table S7 and Table S8 respectively. As can be seen from the results, PTM-ssMP has better performance than other prediction method in almost all kinase groups.
Figure 5.

The Acc, Sn, MCC, Pre, F-score value comparison with different methods for kinase group AGC, CK1, STE and Atypical at two stringency levels. The left part is at specificity of 95.0%, the measurements in the right one are at specificity of 90.0%. The horizontal axis represents accuracy, sensitivity, Matthew correlation coefficient, precision and F-score respectively.
In order to assess the prediction performance of PTM-ssMP more comprehensively, besides phosphorylation, we compare PTM-ssMP with other PTM type prediction tools. For ubiquitination, we compare PTM-ssMP with two common ubiquitination prediction methods Ubipred 26 and Ubisite 27, and the ROC curves of this methods are plotted and shown in Figure S4A. PTM-ssMP achieves AUC value of 75.0%, and the corresponding AUC values of Ubisite and Ubipred are 58.4% and 54.2% (Figure S4A). For lysine acetylation, PAIL 70 and BRABSB-PHKA 28 are applied to compare the prediction performance. As shown in Figure S4B, the AUC values are increased by 15.4% compared with BRABSB-PHKA and 21.8% with PAIL.
As we all known, the prediction top-ranked results are very important in practice, which is used for proteomic-wide screening and systematic examination 71. This requires computational method with both low false positive rate and ability to predict potential PTM site 71, 72. Hence, we follow previous studies 71, 73 to compare the number of correctly retrieved PTM sites in top-ranked results. For each percentile k%, we count the number of true PTM site in the top ranked k%*total samples predictions and their proportion. Here, we take lysine acetylation and ubiquitination as example, results of six percentiles 1%, 2%, 5%, 10%, 15% and 20% of the total corresponding PTM sites number are compared, as shown in Figure 6. It is observed that, for ubiquitination, almost at all percentiles PTM-ssMP have more true positive prediction than Ubisite and Ubipred, and for lysine acetylation, our method also consistently better than BRABSB-PHKA and PAIL. In conclusion, aforementioned analyses suggest that PTM-ssMP obtain better performance than the other prediction tools in predicting multiple types of PTM.
Figure 6.

The fraction of retrieved sites for lysine acetylation and ubiquitination (A) The left part represents the performance of Lysine Acetylation, and (B) the right part represents the performance of Ubiquitination. The horizontal axis represents five top 1, 2, 5, 10, 15 and 20 percent of the total samples.
Usage
For the convenience of most users, a detailed user guide is provided below.
(1) Opening the web-server of PTM-ssMP at http://bioinformatics.ustc.edu.cn/PTM-ssMP/server/, users can see the server page of PTM-ssMP on their computer screen. (2) On the server page, users can input the query protein sequences into the input box at the center of Figure 2 or choose the batch prediction by upload they desired batch input file via the 'Browse' button. PTM-ssMP requires FASTA format inputs, users can click the 'Example' button left below the input box to obtain the examples of sequences with FASTA format. (3) Select PTM type and specificity threshold, click on the 'Submit' button to submit the prediction task. (4) After the prediction task, PTM-ssMP will generate a result page that contains prediction results, each row of the result represents a candidate site (Figure 7). Users can export prediction result in JSON, TXT, XML, CSV, SQL and MS-EXCEL format by click 'export data' button. (5) Users can click the Download button at the top of the page to download all types of PTM datasets that is used to train and test the current predictors.
Figure 7.

A semi-screenshot to show an example of PTM-ssMP prediction result
Conclusions
PTMs play a critical role in various cellular processes, including maintain protein structure and integrity, regulate metabolism and defense processes. However, experimental methods are high-cost and time-consuming, it is urgent to develop effective tools to predict PTM site. Many PTM prediction tools only adopt the local sequence information or functional information for candidate site without considering the relations between different PTMs, which may limit the prediction performance. In this work, we develop a novel web server called PTM-ssMP to predict multiple PTM sites, which adopts the ssMP that can efficiently incorporate the relationships between substrate sites and PTMs. PTM-ssMP allows users submit multiple query protein sequences simultaneously and export prediction result in a variety of format. In addition, as can be seen from the results, the performance of PTM-ssMP is better than other existing tools. Overall, we believe PTM-ssMP will be very helpful for the identification of multiple types of PTM site.
Supplementary Material
Supplementary figures and tables.
Acknowledgments
This work is supported by National Natural Science Foundation of China (No. 61471331, No. 61571414 and No. 61101061), University of Science and Technology of China, USTC. We appreciate the valuable suggestions from any reviewers.
Author contributions
All of the authors listed made substantial contributions to the manuscript and qualify for authorship, and no authors have been omitted. Conception and design: Yu Liu; development of methodology and acquisition of data: Yu Liu, Minghui Wang, Fenglin Luo, Ao Li; analysis and interpretation of data: Yu Liu, Jianing Xi, Fenglin Luo, Ao Li; writing and revision of the manuscript: Yu Liu, Jianing Xi, Minghui Wang, Fenglin Luo Ao Li.
References
- 1.Song C, Ye M, Liu Z, Cheng H, Jiang X, Han G. et al. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Molecular & Cellular Proteomics. 2012;11:1070–83. doi: 10.1074/mcp.M111.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu X, Li A, Zou L, Shen Y, Fan W, Wang M. Improving the performance of protein kinase identification via high dimensional protein-protein interactions and substrate structure data. Molecular bioSystems. 2014;10:694–702. doi: 10.1039/c3mb70462a. [DOI] [PubMed] [Google Scholar]
- 3.Zhu L, Li N. Quantitation, networking, and function of protein phosphorylation in plant cell. Frontiers in plant science. 2012;3:302. doi: 10.3389/fpls.2012.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ubersax JA, Ferrell Jr JE. Mechanisms of specificity in protein phosphorylation. Nature reviews Molecular cell biology. 2007;8:530–41. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 5.Matthews HR. Protein kinases and phosphatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: a possible regulator of the mitogen-activated protein kinase cascade. Pharmacology & therapeutics. 1995;67:323–50. doi: 10.1016/0163-7258(95)00020-8. [DOI] [PubMed] [Google Scholar]
- 6.Li L, Shakhnovich EI, Mirny LA. Amino acids determining enzyme-substrate specificity in prokaryotic and eukaryotic protein kinases. Proceedings of the National Academy of Sciences. 2003;100:4463–8. doi: 10.1073/pnas.0737647100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T. et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–4. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW. et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins: Structure, Function, and Bioinformatics. 2010;78:365–80. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–49. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 10.Hortin G, Folz R, Gordon JI, Strauss AW. Characterization of sites of tyrosine sulfation in proteins and criteria for predicting their occurrence. Biochemical and biophysical research communications. 1986;141:326–33. doi: 10.1016/s0006-291x(86)80372-2. [DOI] [PubMed] [Google Scholar]
- 11.Shen Z, Pardington-Purtymun PE, Comeaux JC, Moyzis RK, Chen DJ. UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics. 1996;36:271–9. doi: 10.1006/geno.1996.0462. [DOI] [PubMed] [Google Scholar]
- 12.Clarke S. Protein methylation. Current opinion in cell biology. 1993;5:977–83. doi: 10.1016/0955-0674(93)90080-a. [DOI] [PubMed] [Google Scholar]
- 13.Xue Y, Zhou F, Fu C, Xu Y, Yao X. SUMOsp: a web server for sumoylation site prediction. Nucleic acids research. 2006;34:W254–W7. doi: 10.1093/nar/gkl207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou F, Olman V, Xu Y. Large-scale analyses of glycosylation in cellulases. Genomics, proteomics & bioinformatics. 2009;7:194–9. doi: 10.1016/S1672-0229(08)60049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y, Hao W, Gu J, Liu D, Fan C, Chen Z. et al. PredPhos: an ensemble framework for structure-based prediction of phosphorylation sites. Journal of Biological Research-Thessaloniki. 2016;23:12. doi: 10.1186/s40709-016-0042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krueger KE, Srivastava S. Posttranslational protein modifications current implications for cancer detection, prevention, and therapeutics. Molecular & Cellular Proteomics. 2006;5:1799–810. doi: 10.1074/mcp.R600009-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Audagnotto M, Dal Peraro M. Protein post-translational modifications: In silico prediction tools and molecular modeling. Computational and structural biotechnology journal. 2017;15:307–19. doi: 10.1016/j.csbj.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenhaber B, Eisenhaber F. Prediction of posttranslational modification of proteins from their amino acid sequence. Methods in molecular biology. 2010;609:365–84. doi: 10.1007/978-1-60327-241-4_21. [DOI] [PubMed] [Google Scholar]
- 19.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. Journal of molecular biology. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 20.Miller ML, Blom N. Kinase-specific prediction of protein phosphorylation sites. Phospho-Proteomics: Methods and Protocols; 2009. pp. 299–310. [DOI] [PubMed] [Google Scholar]
- 21.Dou Y, Yao B, Zhang C. PhosphoSVM: prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino acids. 2014;46:1459–69. doi: 10.1007/s00726-014-1711-5. [DOI] [PubMed] [Google Scholar]
- 22.Gao J, Thelen JJ, Dunker AK, Xu D. Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Molecular & Cellular Proteomics. 2010;9:2586–600. doi: 10.1074/mcp.M110.001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & cellular proteomics. 2008;7:1598–608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong Y-H, Lee T-Y, Liang H-K, Huang C-M, Wang T-Y, Yang Y-H. et al. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic acids research. 2007;35:W588–W94. doi: 10.1093/nar/gkm322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue Y, Li A, Wang L, Feng H, Yao X. PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC bioinformatics. 2006;7:163. doi: 10.1186/1471-2105-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tung C-W, Ho S-Y. Computational identification of ubiquitylation sites from protein sequences. BMC bioinformatics. 2008;9:310. doi: 10.1186/1471-2105-9-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang C-H, Su M-G, Kao H-J, Jhong J-H, Weng S-L, Lee T-Y. UbiSite: incorporating two-layered machine learning method with substrate motifs to predict ubiquitin-conjugation site on lysines. BMC systems biology. 2016;10(suppl 1):S6. doi: 10.1186/s12918-015-0246-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao J, Xu D, Hu L, Kwan Y-W, Wang Y, Kong X. et al. Systematic analysis of human lysine acetylation proteins and accurate prediction of human lysine acetylation through bi-relative adapted binomial score Bayes feature representation. Molecular BioSystems. 2012;8:2964–73. doi: 10.1039/c2mb25251a. [DOI] [PubMed] [Google Scholar]
- 29.Fan W, Xu X, Shen Y, Feng H, Li A, Wang M. Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino acids. 2014;46:1069–78. doi: 10.1007/s00726-014-1669-3. [DOI] [PubMed] [Google Scholar]
- 30.Du Y, Zhai Z, Li Y, Lu M, Cai T, Zhou B. et al. Prediction of Protein Lysine Acylation by Integrating Primary Sequence Information with Multiple Functional Features. Journal of proteome research. 2016;15:4234–44. doi: 10.1021/acs.jproteome.6b00240. [DOI] [PubMed] [Google Scholar]
- 31.Li T, Du P, Xu N. Identifying human kinase-specific protein phosphorylation sites by integrating heterogeneous information from various sources. PloS one. 2010;5:e15411. doi: 10.1371/journal.pone.0015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang B, Wang M, Li A. Prediction of post-translational modification sites using multiple kernel support vector machine. PeerJ. 2017;5:e3261. doi: 10.7717/peerj.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minguez P, Parca L, Diella F, Mende DR, Kumar R, Helmer-Citterich M. et al. Deciphering a global network of functionally associated post-translational modifications. Molecular systems biology. 2012;8:599. doi: 10.1038/msb.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J. et al. Systematic functional prioritization of protein posttranslational modifications. Cell. 2012;150:413–25. doi: 10.1016/j.cell.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schweiger R, Linial M. Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biology direct. 2010;5:6. doi: 10.1186/1745-6150-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minguez P, Letunic I, Parca L, Bork P. PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic acids research. 2013;41:D306–D11. doi: 10.1093/nar/gks1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Z, Cheng Z, Zhao Y, Volchenboum SL. Bioinformatic analysis and post-translational modification crosstalk prediction of lysine acetylation. PloS one. 2011;6:e28228. doi: 10.1371/journal.pone.0028228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuo C-Y, Shieh C, Cai F, Ann DK. Coordinate to guard: crosstalk of phosphorylation, sumoylation, and ubiquitylation in DNA damage response. Frontiers in oncology. 2012;1:61. doi: 10.3389/fonc.2011.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wimmer P, Blanchette P, Schreiner S, Ching W, Groitl P, Berscheminski J. et al. Cross-talk between phosphorylation and SUMOylation regulates transforming activities of an adenoviral oncoprotein. Oncogene. 2013;32:1626–37. doi: 10.1038/onc.2012.187. [DOI] [PubMed] [Google Scholar]
- 40.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annual review of biochemistry. 2011;80:825–58. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao H, Li A, Wang M. Systematic analysis and prediction of in situ cross talk of O-GlcNAcylation and phosphorylation. BioMed research international. 2015; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Molecular cell. 2000;5:905–15. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 43.Yang X-J, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Molecular cell. 2008;31:449–61. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C. et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Human molecular genetics. 2013;23:104–16. doi: 10.1093/hmg/ddt402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daujat S, Bauer U-M, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Current Biology. 2002;12:2090–7. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 46.Xu L, Chen J, Gao J, Yu H, Yang P. Crosstalk of homocysteinylation, methylation and acetylation on histone H3. Analyst. 2015;140:3057–63. doi: 10.1039/c4an02355b. [DOI] [PubMed] [Google Scholar]
- 47.Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H. et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Molecular cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 48.Parker BL, Shepherd NE, Trefely S, Hoffman NJ, White MY, Engholm-Keller K. et al. Structural basis for phosphorylation and lysine acetylation cross-talk in a kinase motif associated with myocardial ischemia and cardioprotection. Journal of Biological Chemistry. 2014;289:25890–906. doi: 10.1074/jbc.M114.556035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan Z, Liu Z, Cheng H, Wang Y, Gao T, Ullah S. et al. Systematic analysis of the in situ crosstalk of tyrosine modifications reveals no additional natural selection on multiply modified residues. Scientific reports. 2014;4:7331. doi: 10.1038/srep07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang M, Jiang Y, Xu X. A novel method for predicting post-translational modifications on serine and threonine sites by using site-modification network profiles. Molecular BioSystems. 2015;11:3092–100. doi: 10.1039/c5mb00384a. [DOI] [PubMed] [Google Scholar]
- 51.Lu CT, Huang KY, Su MG, Lee TY, Bretana NA, Chang WC. et al. DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic acids research. 2013;41:D295–305. doi: 10.1093/nar/gks1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H, Xing X, Ding G, Li Q, Wang C, Xie L. et al. SysPTM: a systematic resource for proteomic research on post-translational modifications. Molecular & Cellular Proteomics. 2009;8:1839–49. doi: 10.1074/mcp.M900030-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diella F, Cameron S, Gemünd C, Linding R, Via A, Kuster B. et al. Phospho. ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC bioinformatics. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peri S, Navarro JD, Kristiansen TZ, Amanchy R, Surendranath V, Muthusamy B. et al. Human protein reference database as a discovery resource for proteomics. Nucleic acids research. 2004;32:D497–D501. doi: 10.1093/nar/gkh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B. et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic acids research. 2012;40:D261–70. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Torii M, Liu H, Hart GW, Hu Z-Z. dbOGAP-an integrated bioinformatics resource for protein O-GlcNAcylation. BMC bioinformatics. 2011;12:91. doi: 10.1186/1471-2105-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–2. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao X, Zhang W, Xu X, Ma Z, Yin M. Prediction of protein phosphorylation sites by using the composition of k-spaced amino acid pairs. PloS one. 2012;7:e46302. doi: 10.1371/journal.pone.0046302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Z, Chen Y-Z, Wang X-F, Wang C, Yan R-X, Zhang Z. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PloS one. 2011;6:e22930. doi: 10.1371/journal.pone.0022930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neuberger G, Schneider G, Eisenhaber F. pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biology direct. 2007;2:1. doi: 10.1186/1745-6150-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li F, Li C, Wang M, Webb GI, Zhang Y, Whisstock JC. et al. GlycoMine: a machine learning-based approach for predicting N-, C-and O-linked glycosylation in the human proteome. Bioinformatics. 2015;31:1411–9. doi: 10.1093/bioinformatics/btu852. [DOI] [PubMed] [Google Scholar]
- 62.Gnad F, Ren S, Choudhary C, Cox J, Mann M. Predicting post-translational lysine acetylation using support vector machines. Bioinformatics. 2010;26:1666–8. doi: 10.1093/bioinformatics/btq260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hou T, Zheng G, Zhang P, Jia J, Li J, Xie L. et al. LAceP: lysine acetylation site prediction using logistic regression classifiers. PloS one. 2014;9:e89575. doi: 10.1371/journal.pone.0089575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang J-R, Huang W-L, Tsai M-J, Hsu K-T, Huang H-L, Ho S-Y. ESA-UbiSite: accurate prediction of human ubiquitination sites by identifying a set of effective negatives. Bioinformatics. 2016;33:661–8. doi: 10.1093/bioinformatics/btw701. [DOI] [PubMed] [Google Scholar]
- 65.Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. ACM transactions on intelligent systems and technology (TIST) 2011;2:27. [Google Scholar]
- 66.Cui Y, Zhang M, Pestell R, Curran EM, Welshons WV, Fuqua SA. Phosphorylation of estrogen receptor α blocks its acetylation and regulates estrogen sensitivity. Cancer Research. 2004;64:9199–208. doi: 10.1158/0008-5472.CAN-04-2126. [DOI] [PubMed] [Google Scholar]
- 67.Kassardjian A, Rizkallah R, Riman S, Renfro SH, Alexander KE, Hurt MM. The transcription factor YY1 is a novel substrate for Aurora B kinase at G2/M transition of the cell cycle. PloS one. 2012;7:e50645. doi: 10.1371/journal.pone.0050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gu B, Zhu W-G. Surf the post-translational modification network of p53 regulation. International journal of biological sciences. 2012;8:672. doi: 10.7150/ijbs.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang M, Wang T, Wang B, Liu Y, Li A. A Novel Phosphorylation Site-Kinase Network-Based Method for the Accurate Prediction of Kinase-Substrate Relationships. BioMed Research International. 2017;2017:1826496.. doi: 10.1155/2017/1826496. doi: 10.1155/2017/1826496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li A, Xue Y, Jin C, Wang M, Yao X. Prediction of N ε-acetylation on internal lysines implemented in Bayesian Discriminant Method. Biochemical and biophysical research communications. 2006;350:818–24. doi: 10.1016/j.bbrc.2006.08.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu X, Wang M. Inferring Disease Associated Phosphorylation Sites via Random Walk on Multi-Layer Heterogeneous Network. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2016;13:836–44. doi: 10.1109/TCBB.2015.2498548. [DOI] [PubMed] [Google Scholar]
- 72.Li H, Wang M, Xu X. Prediction of kinase-substrate relations based on heterogeneous networks. Journal of bioinformatics and computational biology. 2015;13:1542003. doi: 10.1142/S0219720015420032. [DOI] [PubMed] [Google Scholar]
- 73.Peng C, Li A. A heterogeneous network based method for identifying GBM-related genes by integrating multi-dimensional data. IEEE/ACM transactions on computational biology and bioinformatics. 2017;14:713–20. doi: 10.1109/TCBB.2016.2555314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures and tables.