

Abstract
Posttraumatic epilepsy (PTE) is one of the most common and devastating complications of traumatic brain injury (TBI). Currently, the etiopathology and mechanisms of PTE are poorly understood and as a result, there is no effective treatment or means to prevent it. Antiepileptic drugs remain common preventive strategies in the management of TBI to control acute posttraumatic seizures and to prevent the development of PTE, although their efficacy in the latter case is disputed. Different strategies of PTE prophylaxis have been showing promise in preclinical models, but their translation to the clinic still remains elusive due in part to the variability of these models and the fact they do not recapitulate all complex pathologies associated with human TBI. TBI is a multifaceted disorder reflected in several potentially epileptogenic alterations in the brain, including mechanical neuronal and vascular damage, parenchymal and subarachnoid hemorrhage, subsequent toxicity caused by iron-rich hemoglobin breakdown products, and energy disruption resulting in secondary injuries, including excitotoxicity, gliosis, and neuroinflammation, often coexisting to a different degree. Several in vivo models have been developed to reproduce the acute TBI cascade of events, to reflect its anatomical pathologies, and to replicate neurological deficits. Although acute and chronic recurrent posttraumatic seizures are well-recognized phenomena in these models, there is only a limited number of studies focused on PTE. The most used mechanical TBI models with documented electroencephalographic and behavioral seizures with remote epileptogenesis include fluid percussion, controlled cortical impact, and weight-drop. This chapter describes the most popular models of PTE-induced TBI models, focusing on the controlled cortical impact and the fluid percussion injury models, the methods of behavioral and electroencephalogram seizure assessments, and other approaches to detect epileptogenic properties, and discusses their potential application for translational research.
Keywords: Posttraumatic epilepsy, Posttraumatic seizures, Epileptogenesis, Animal model, Rodents, CCI, TBI, Fluid percussion injury, Behavioral seizures, Electroencephalography (EEG)
1 Introduction
1.1 Development of Posttraumatic Epilepsy (PTE) Following Brain Injuries
Traumatic brain injury (TBI) afflicts over ten million people worldwide and around 1.7 million in the United States alone annually, and is also a hallmark injury of the wars in Iraq and Afghanistan [1]. TBI often results in the development of devastating chronic consequences that significantly affect the quality of life of survivors. PTE is one such common consequence of brain injury [2–5] and TBI is among the major causes of acquired epilepsy [6], accounting for about 5 % of the over three million all of epilepsy cases in the US. One remarkable feature of PTE is the variable, often very prolonged, latency from injury to epilepsy, which can range from weeks to years [2, 3, 7]. In general, according to the International League Against Epilepsy (ILAE), epilepsy is defined as a brain disorder that is characterized by either of the following conditions: (a) Operational (practical) clinical definition of epilepsy at least two unprovoked (or reflex) seizures occurring >24 h apart; (b) One unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60 %) after two unprovoked seizures occurring within the next 10 years, or (c) Diagnosis of an epilepsy syndrome [8]. The development of acute posttraumatic seizure (PTS) occurring within weeks after TBI is a common complication of TBI and is often thought to be associated with the progression of PTE further along in life. The reoccurrence of epileptic seizures within the next 2 years in TBI patients with a single acute PTS might be as high as 86 % and the remission rates with longer terms are 25–40 % [9]. The PTS resulting from brain injury or surgery often are categorized as “early” or “late” PTS, occurring within a week or after a week following injury, respectively. “Late” PTS may even occur months or years after initial injury; PTE is a disorder that is characterized by reoccurring “late” seizures.
The etiology of PTE, as well as other types of epilepsy in general, is currently poorly understood and there is little known about the pathophysiological processes involved in the generation of individual seizures or the progression of PTE. The need for better understanding of the detailed molecular mechanisms underlying these processes is among the major translational challenges for those who are developing treatment and prophylactic strategies [1, 10]. Treatment with antiepileptic drugs (AEDs) aimed at controlling seizures remains the principal option for most epilepsy patients, although almost 30 % of those who suffer from epilepsy are resistant to such medications [11–13].
1.1.1 Risk Factors for the Devel-opment of PTE
Understanding of the risk factors for the development of PTE after TBI is important so that preclinical research can allow for reliable modeling of the clinical features as well as better control of the conditions to assure the reproducibility of the results and decrease the variability of the outcome measures in animal models. Intensive studies performed over the past century documented that penetrating TBI is a well-established risk factor for PTE with significantly higher incidence compared to closed head injuries, including contusions and concussions, although these studies were focused mostly on severe brain injuries from wounds garnered in war, with up to 53 % of patients having penetrating wounds [3, 7], reviewed in [9]. Historically, anatomical brain injuries from a blast have not been commonly detected. However, in recent conflicts, blast exposure has been the most common mechanism of injury, accounting for over 80 % of TBI cases in veterans of the Iraq and Afghanistan wars. The risk factors and prevalence of PTE in blast-induced TBI is currently poorly understood, in part because the occurrence of PTE is normally observed over several years after initial injury and the latency may be even more prolonged with mild injuries. Blast-induced TBI is often associated with persistent post-concussive syndrome, posttraumatic stress disorder (PTSD), and chronic pain, and may pose a significant risk of PTE [14]. Limited clinical data suggest that the prevalence of psychogenic nonepileptic seizures is more characteristic in blast-induced TBI [15]. A small study including veterans who were assigned an outpatient diagnosis of both epilepsy and TBI has shown that the diagnosis of PTE was clinically confirmed in 18 % of the sample, whereas a diagnosis of nonepileptic, or psychogenic, seizures was suspected in 44 % of the patients [15]. Similar tendencies are observed in TBI patients from the civilian population [16]. In children, PTS following TBI affects 12–35 % of children after TBI and the occurrence of PTS is associated with worse cognitive and functional outcome after adjustment for injury severity [17].
Significant risk factors for the development of both “early” (within 1 week following TBI) and “late” (after 1 week following TBI) PTS include acute intracerebral and subdural hematomas, surgery for an intracerebral hematoma, increased injury severity (Glasgow Coma Scale in the severe range of 3–8), depressed skull fractures, and dural penetration [9, 16]. Younger age is a risk factor for the development of “early” PTS, whereas older age at the time of injury (over 65 years) is a risk factor for “late” PTS [9]. In addition, the risk factors include preexisting conditions such as chronic alcoholism. The age-related occurrence of the seizures and acquired epilepsies are also observed in other brain injuries. For example, post-stroke seizures and post-stroke epilepsy are widely recognized neurological consequences of stroke, especially in the elderly population [18].
1.1.2 Clinical Management of TBI and PTE
Despite extensive research and limited success with preclinical trials with numerous neuroprotective strategies, the translational viability of such approaches is still elusive [19]. To date, the reduction in elevated intracranial pressure using decompressive craniectomy remains one of the common symptomatic treatments with proven efficacy for reducing mortality following TBI. However, such lifesaving treatment is associated with acute post-craniectomy seizures with a high risk (76.2 %) of development of epilepsy [20]. Due to the increased risk of seizures after TBI and the possible association of acute seizures with the development of PTE, several common antiepileptic drugs (AEDs) are routinely used in clinical practice, although their efficacy in the prophylaxis of PTE is disputed [21–28]. Phenytoin and, most recently, levetiracetam are commonly used in the clinic to control PTS and as prophylaxis for PTE [21–23]. These AEDs have different mechanisms of action and, accordingly, differentially affect seizure outcomes in preclinical models. For example, a recent study showed that none of the drugs studied (phenytoin, phenobarbital, carbamazepine, valproate, or some combination therein) gave reliable evidence that it prevented, or even suppressed, epileptic seizures after TBI [25].
Phenytoin is considered a classic anticonvulsant, exhibiting an immediate effect on electrically or chemically induced seizures, whereas levetiracetam is not effective in such acute models but demonstrates antiepileptic properties in chronic seizure models. In addition, limited preclinical data suggest that chronic treatment with levetiracetam may also have neuroprotective properties [29], whereas chronic treatment with phenytoin is deleterious [30], although these studies neither assessed seizures nor other PTE outcomes. Interestingly, certain AEDs are commonly used in the clinic for treatment and prophylaxis of chronic headaches and migraine, suggesting the presence of common mechanisms involved in the progression of these disorders. In general, a relationship between epilepsy and migraine has long been recognized, but the nature of this interaction is still debated [31]. Chronic posttraumatic headaches or migraine occurs in up to 90 % of TBI patients [32], whereas PTE occurs in approximately 10 % of TBI patients with closed head trauma and in over 50 % in patients with penetrating TBI [2–5].
In addition, a significant proportion of TBI patients suffer “silent” non-convulsive seizures, including many without evident behavioral manifestations that can only be conclusively diagnosed by electroencephalogram (EEG) [33, 34]. The incidence of non-convulsive seizures has been reported to be from 18 to 38 % in neurointensive care patients [35–40], with the incidence in patients with brain lesions even higher (up to 55 %) [41].
1.2 Putative Mechanisms Underlying the Development of PTE
TBI causes potentially epileptogenic brain damage through several coexisting mechanisms, including mechanical damage, and primary and secondary excitotoxicity associated with ischemia, intra-cerebral and subdural hemorrhage, and neuroinflammation [12]. In addition, it is widely recognized that different acute brain injuries involving many common injury mechanisms such as ischemic and hemorrhage strokes are associated with increased prevalence of acute seizures and acquired epilepsy [18]. The anatomical brain pathologies resulting from these brain injuries often resemble brain pathologies observed in epilepsy patients, suggesting the existence of common mechanisms in these neurological disorders.
The clinical and pathophysiological features of PTE closely resemble many features of temporal lobe epilepsy. Well-recognized hallmarks of PTE that could be reproduced in animal TBI/PTE models include: (a) chronic seizure susceptibility; (b) persistent hyperexcitability in the dentate gyrus; (c) hippocampal sclerosis selective cell loss in the hilus of the dentate gyrus and CA3 area of the hippocampus, and (d) synaptic reorganization of mossy fiber.
1.2.1 Cortical and Hippocampal Damage After TBI Associated with Epileptogenesis
It is believed that one of the most important factors associated with acute seizures and the development of PTE is the level of mechanical brain damage and brain lesions resulting from secondary neurodegenerative processes. It is well recognized that seizure susceptibility is associated with the severity of TBI and that total brain volume loss predicts seizure frequency [2]. In addition, experimental and clinical data suggest that hippocampal damage resulting from TBI is also associated with recurrent seizure activity and might be involved in the etiology of PTE [42, 43]. In human TBI patients, reduction of hippocampal volume and alterations in cortico-subcortical connectivity are well-recognized phenomena following TBI and the levels of anatomical pathology associated with cognitive and neurological outcomes [44–47]. Importantly, hippocampal atrophy after TBI reported in humans resembles the hippocampal sclerosis often associated with temporal lobe epilepsy [44], and the presence of such hippocampal sclerosis is associated with poor prognosis and a significantly higher rate of AED resistance [48]. Selective hippocampal cell death has been reported in several common preclinical TBI models, including fluid percussion injury (FPI) controlled cortical impact (CCI; mouse) [49], and weight-drop impact acceleration injury [43]. Further, the data suggest that an early and selective cell loss in the hilus of the dentate gyrus and CA3 areas of the hippocampus is associated with enhanced susceptibility to pentylenetetrazole (PTZ)-induced seizures up to 15 weeks following experimental TBI [43].
1.2.2 Ischemia and Hypoxia Following TBI
Brain ischemia and hypoxia associated with primary vascular damage and secondary injuries are common features of TBI, affecting overall neurological outcomes and mortality [50]. It is well recognized that brain ischemia is associated with glutamate-induced excitotoxicity, and thus, the lowering of the seizure threshold resulting from depolarization is believed to be one of the major contributory factors to seizure. Although ischemia following TBI is commonly associated with severe head injury, there is also evidence of its association with moderate or mild head injuries [51]. Clinical and preclinical studies suggest that the persistent micro-vascular damage that occurs at chronic stages following TBI resembles microvascular pathologies associated with Alzheimer disease and might be involved in chronic neurodegenerative processes associated with these disorders [52]. On the other hand, the evidence indicates that different ischemic brain injuries are associated with the increased risk of the development of acute seizures and epilepsies. Acute seizures and the increased prevalence of epilepsy are widely recognized neurological consequences of cerebral ischemia. The stroke is also the primary cause of symptomatic epilepsy in the elderly population [18]. In hospital settings, up to 40 % of stroke patients experience seizures manifested as clinical motor convulsions [53, 54]. However, similar to TBI patients, a significant proportion of stroke patients suffer seizures that may or may not have evident behavioral manifestations and that could be conclusively diagnosed only by EEG [33, 34, 53, 54]. Convulsive and non-convulsive seizures are associated with poor outcome and increased mortality [33, 55]. In addition, non-convulsive seizures are also connected with a decline in patient outcome after ischemic brain injury, and are significantly more refractory to AED therapy as compared to convulsive seizures [28, 37, 56].
1.2.3 Hemorrhage and Hemoglobin Breakdown Products
In the hospital setting, the proportion of stroke patients experiencing seizures varies from 1 to 40 %, which may be due to differences in study design and diagnostic criteria [53, 54]. ICH and sub-arachnoid hemorrhage (SAH) are associated with increased risk for early offset seizures, as defined as 1–15 days after the hemorrhagic stroke [53, 55, 57]. Most seizures associated with ICH occur at onset or within the first 24 h [57, 58]. In about 18 % of patients with SAH, seizures occur before admission and in 4–10 % of patients after admission to hospital; in aneurysmal SAH, the percentage is higher (10–26 %) [59, 60]. The hemorrhagic component associated with acute stroke is associated with increased risk of development of epilepsy and poor quality of life [53, 54, 61, 62].
Although the detailed mechanisms involving in seizure initiation and epileptogenesis associated with ICH are not currently established, the data obtained in animal models suggest that hemolysis, the formation of reactive hemoglobin breakdown products, and accumulation of free iron and iron-rich compounds such as hemosiderin may play significant role [63–65]. Interestingly, recent preclinical data demonstrate chronic progressive punctate deposition of hemosiderin in the traumatic brain [66]. The experimental data suggest that in an iron-induced epilepsy model, which is often referred to as a PTE model, the main mechanisms underlying seizure activity involve N-methyl-d-aspartate (NMDA)-receptor-mediated glutamate excitotoxicity, formation of reactive oxygen species, and subsequent membrane lipid peroxidation, and neuronal cell death [64, 65, 67, 68].
1.2.4 Blood–Brain Barrier (BBB) Disruption in PTS and PTE
The role of BBB dysfunction has long been suggested as a factor of epileptogenesis [69, 70]. A concern for the possible role of the BBB in the various epilepsies has been based on ultrastructural studies that demonstrated increased micropinocytosis in cerebral capillaries during seizures. Preclinical results indicate that evolving white matter degeneration following experimental TBI is associated with significantly delayed microvascular damage and focal microbleeds that are temporally and regionally associated with the development of punctate BBB breakdown and progressive inflammatory responses [66]. Experimental and clinical evidence suggest that BBB pathologies are associated with several potentially epileptogenic abnormalities connected to brain injuries such as infiltration of erythrocytes and other blood cells and blood components resulting in hemoglobin-related products which collectively are associated with inflammatory mechanisms with augmented neuronal activity [70, 71]. Interestingly, acute and chronic BBB pathologies and neurodegeneration in TBI models are associated with biomarker responses similar to those observed in one of a most common seizure model induced by kainic acid neurotoxicity [66, 72, 73].
1.2.5 Neuroinflammation
Neuroinflammation is characterized by the activation of several neuronal and glial pathways in response to insult or injury, primarily involving microglia that are considered to be the resident innate immune cells in the brain, resulting in the release of proinflammatory cytokines and other mediators. Infiltrating macrophages could also trigger a parallel inflammatory cascade. A growing body of experimental and clinical evidence suggests that neuroinflammation involving the increased production of various proinfammatory molecules. Among them, the so-called proinflammatory prostaglandins results from the upregulation of cyclooxygenase (COX) and prostaglandin E synthase (PGES) enzymes in the head and the spinal cord after trauma [30, 32, 74–76] and these prostaglandins consequently affect related neurological disorders such as stroke, seizure disorders, and epilepsy [77, 78]. Several preclinical studies using different animal models demonstrated COX-2 upregulation during the seizures, mainly in hippocampal neurons and glia [79, 80], and this upregulation was associated with increased neuronal activity. There are two major isotypes of COX enzymes expressed in the brain: COX-1 and COX-2, often called constitutive and inducible, respectively, that are encoded by different genes and differently respond to traumatic insults or toxic stimuli. Although both COX-1 and COX-2 are constitutively expressed in the brain, the levels of their expression may change during pathological conditions [81]. Data suggest that the COX-2 expression is primarily increased in the brain in response to a variety of brain injuries and diseases involving excitotoxic brain damage, ischemic and hypoxic insults, and ICH, and increased COX-2 immunoreactivity has been demonstrated in both microglia and neurons [82–84]. A number of studies in rats and mice subjected to TBI demonstrated that COX-2 expression is increased mainly in ipsilateral cortex and hippocampus [74–76, 85, 86]. However, the level of COX-2 expression is more profound in the hippocampus, with significant increases starting within hours after injury and persisting up to 2 weeks [30, 32, 87]. The increased COX-2 mRNA immunoreactivity was localized through neuronal and astrocytic markers: MAP2 and GFAP, respectively [87]. Additionally, some preclinical data suggest that both the selective and non-selective COX-2 inhibitors currently in use clinically might also be used for the treatment of neurological disorders, notably ischemic stroke [88–92] and TBI [30, 32, 74, 75], though studies have also shown that COX-2 induction following TBI may play a protective role [75, 76].
However, one has to be consistently careful of well-documented adverse side effects of COX-2 inhibitors [93]. Similarly, targeting downstream COX-2 pathways such as selective prostaglandin receptors PGE 2 EP1 and PGF 2α FP receptors have been suggested as an alternative to COX-2 inhibitors affecting total prostaglandin production. Pharmacological blockade or genetic deletion of FP receptor improves some outcomes in mouse TBI model [94]. However, the role of the EP1 receptor is complex and its blockade differentially affects outcomes in different neurological conditions such as ischemia [88–92], hemorrhagic stroke [85, 95], excitotoxicity [88, 89, 91], epilepsy [96, 97], surgical brain injury [98], and TBI [49, 99]. Interestingly, blockade of the EP1 receptor was tested as a strategy in the treatment of epilepsy [96, 97], but to reach a desired effect, it required up to a 1000-fold higher dose of one of the commonly used antagonists (i.e., SC-51089) [97] than that used in models of stroke and NMDA-induced excitotoxicity [100].
Increased prostaglandin levels has been described in a wide range of animal models of epilepsy, including with both chemically (e.g., kainic acid [80, 101–103], NMDA [104], PTZ [105–110], picrotoxin [106], and pilocarpine [111]) or electrically induced seizures, as well as during seizures in epileptic and convulsion-susceptible strains [112, 113]. One difference between these models is PGF 2α, one of the major prostanoids increased during seizures, and the data indicate that this increase is due to augmented neuronal activity [106, 108, 113]. On the other hand, there is experimental evidence that in rodent PTZ and kainic acid mouse models, the increased levels of PGF 2α preceded epileptic activity [114], which might be considered a factor modulating seizure susceptibility. Although PGF 2α is elevated during seizures, its role has long been disputed. Interestingly, recent data suggested the differential roles of the PGF 2α cognate FP receptor in TBI and seizure models. The pharmacological blockade of the FP receptor after TBI with a selective antagonist (i.e., AL-8010) improves neurological outcomes and hippocampal swelling [94]. In contrast, the data obtained in immature and adult models of kainic acid-induced seizures demonstrated that the decrease in PGF 2α levels or blockade of FP receptors worsened seizure outcomes [115, 116].
1.3 Animal Models of PTE
Several animal models have been developed to reproduce the mechanics of TBI, reflect its anatomical pathologies, and replicate its neurological deficits. Historically, the development of preclinical models was focused on using larger animals, which were further adapted to rodent species. Although larger animals with gyrencephalic brains are likely more closely reflect neurophysiological and biomechanical aspects of human TBI, recent rodent models have become standard in TBI and epilepsy research because of their modest cost, small size, and accelerated development compared to larger animals, in addition to some of their well-characterized outcome measures. Currently, the most widely used preclinical models in TBI research include FPI [117], CCI injury [118, 119], weight-drop impact acceleration injury [120], and blast injury [121].
Acute and chronic recurrent PTS is a well-recognized and commonly observed phenomena observed in most of these models using different species except with blast injuries. However, there is only a limited number of studies using these models that are specifically focused on PTE, probably because of its chronic nature and complicated outcome assessment. The most commonly used mechanical TBI models with documented electroencephalographic and behavioral seizures and remote epileptogenesis include FPI, CCI, and weight-drop.
Recent clinical data suggest the utility of modern neuroimaging techniques for the prognosis of long-term TBI outcomes; that would likely also be useful for revealing the mechanisms underlying PTE. Nonetheless, prospective clinical studies employing advanced brain imaging and EEG monitoring, as well as preclinical research and development of improved animal models closely reflecting all features and aspects of human disease, are still warranted to better understand the phenotypes and etiopathology of PTE [122].
Preclinical models of brain contusion and concussion are among the most widely used models of PTE, as they represent the biomechanics, brain pathology, and neurological deficits characteristic of human TBI. In contrast, even some clinical and experimental data suggest epileptogenic alterations following blast injury preclinical studies focusing on PTS or PTE are still warranted.
1.3.1 PTS in Animal Models of Blast Injury
In blast animal models, the occurrence of PTS or EEG epileptiform is not generally assessed for reported outcomes [123]. There are several models of blast exposure-induced TBI often referred to as overpressure injury (OBI) that produce different severities of TBI. A lack of seizure data in blast models might be due to fewer blast studies compared to other models; it is also possible that the impact magnitudes used in most of these studies was insufficient to produce injury with obvious seizure outcomes.
Only a few studies employed EEG in the blast models and the data suggest that blast affects electrophysiological properties associated with brain injury at acute time points similar to those observed in contusive or concussive models of TBI, but provided no evidence of epileptiform activity observed in other models of TBI and stroke. For example, the exposure of goats to a biphasic blast waveform comprising incident and reflection peaks caused alterations in EEG waveforms including an increase in amplitude and a decrease in frequency reflecting brain depression that was associated with substantial changes in gross brain pathology assessed microscopically, including SAH and parenchymal hemorrhage, enlarged perivascular space, vascular dilatation, and congestion accompanied by elevated serum concentrations of S-100β and specific enolase at acute time points [124]. In rabbits, blast exposure caused characteristic systemic responses (e.g., mean arterial pressure, bradycardia, fast and shallow respiration) and significant increases in both frequency and amplitude of EEG activity, but there were no anatomical changes such as hemorrhage or brain lesions [125]. Acute PTSs were reported following severe blast and, based on our best knowledge, there are no published studies to date reporting the occurrence of chronic seizures or epileptogenic changes.
1.3.2 PTS and PTE in FPI Models
The FPI model is one of the most commonly used models of TBI and is one of the most commonly used models of PTS and PTE. The main advantages of this model are a relatively simple setup and a possibility for inducing pathophysiological features of human brain trauma (with the intrinsic caveats of the given species brain anatomy), including intracranial hemorrhage, brain swelling (edema), changes in intracranial pressure, and progressive grey matter lesions [126, 127]. It is also often considered as model of diffuse TBI. However, one of the disadvantages of this model is a requirement for precise surgical procedures and control of the injury parameters in the commonly used devices where even minor deviations may result in significant variability of the outcomes and relatively high mortality compared to other models. However, recently a pneumatic instrument with microprocessor-controlled pressure and dwell time has been developed to address operational concerns and reproducibility issues presented in standard FPI devices [128]. Initially, the FPI model was developed for use in cats and rabbits [129–131] and was soon adapted for use in rats [132, 133] and mice [134]. The FPI model produces a rapid controlled fluid pressure pulse applied directly to the dura mater through a craniotomy window, causing a brief deformation of brain tissue. The change in pressure is produced by moving a pendulum striking the plunger of a cylinder filled with a fluid (e.g., physiological solution, PBS, ACSF).
There are three major variations of this model based on the position of craniotomy: midline, parasagittal, and lateral. Of these three variations, the lateral FPI in rodents is currently the most commonly used for small animals [133, 134]. FPI model has been used in many different species, including cats [135], rabbits [130], dogs and sheep [136], rats [137], mice [134, 138], and pigs [137, 139].
The pressure is a variable that allows for control of injury severity in the FPI model, affecting overall neurological and anatomical outcomes. In rats, lateral FPI produces a combination of focal cortical contusion and diffuse subcortical neuronal injury extended to the hippocampus and thalamus, but the progression of delayed neuronal cell death following FPI brain injury is slower than in the CCI model and the tissue loss and the glial scarring progress within a year after initial impact [140, 141]. Similarly, cognitive dysfunction and neurological impairment persist for more than a year following severe FPI [132].
In midline FPI models, at acute tine points, hemorrhage is present in the corpus callosum, fimbria hippocampi, and thalamus, and to a lesser extent, in the brain stem [117]. The mossy fiber sprouting in the dentate gyrus revealed by Timm’s sulfide silver staining can be observed at both acute and chronic time points [142]. The anatomical pathology in the FPI model is associated with neurological locomotor deficits persistent within the first week of injury, even at moderate magnitudes [117].
An investigation of acute EEG changes after FPI with different pressure magnitudes reflecting injury severity has shown significant decreases in EEG amplitude immediately after initial impact injury, and decreases in delta frequency band (1–4 Hz) activity with more profound changes at higher injury severity [137]. In addition, the changes in waveform dynamics are important for acute outcomes. A study of the effects of peak pressure on neurological outcomes performed in juvenile rats using a programmable FPI device has shown that FPI with the parameter of fast-rising pressure, which could be translated as a primary concussive injury, caused less profound acute neurological outcomes (including fewer incidences of seizure) and decreased acute neurodegeneration of hilar neurons within hours after injury compared to a standard rise in injury pressure parameters [143]. However, one week after injury, the differences in hilar cell loss and dentate hyperexcitability between fast and standard peak pressure rates were no longer significant.
Alteration of Gene Expression After FPI Associated with Epilepsy and Seizures
Excitotoxic neurodegeneration in the hippocampus is evident within the first hours following FPI neuronal hilar cell loss and neuronal cell death has progressed within the first week or weeks after FPI; the pattern of cell death resembles, to a certain extent, the pattern of neuronal death observed in some patients with temporal lobe epilepsy [143, 144]. Lateral FPI injury leads to enhanced expression of the selected immediate early gene markers associated with neuronal hyperactivation within 24 h after injury, including brain-derived neurotrophic factor (BDNF) and c-fos in the ipsilateral hippocampus and Bax in both ipsilateral and contralateral hippocampi, and these changes in the immediate early gene responses are associated with overactivation of NMDA receptors [145]. The overactivation of the glutamate receptors seems to play a major role in the hyperexcitability characteristic of FPI, resulting in excitotoxic cell death. One week after FPI injury, there is neuronal hilar cell loss assessed by silver staining and increased dentate field excitability in response to stimulation of the perforant path [142, 143]. The electrophysiological changes recorded in vitro demonstrate a long-lasting increase in the frequency of spontaneous inhibitory postsynaptic currents (IPSCs) that were associated with the augmented activity of inotropic glutamate receptors [142].
Electrophysiological recordings from slices obtained from FPI-injured rats has demonstrated that the injury caused a persistent decrease in the threshold to induction of seizure-like electrical activity in response to high-frequency tetanic stimulation in the ipsilateral hippocampus [142]. In vitro studies have shown electro-physiological changes resulting in neuronal hyperexcitability and associated with reactive gliosis such as abnormalities in potassium conductance and local field potential recordings, suppressed paired-pulse facilitation, enhanced evoked and miniature glutamatergic excitatory postsynaptic potential, and decreased in the frequency of miniature inhibitory postsynaptic currents (IPSCs) [146–149]. When in vitro electrophysiological recordings from rat slices that underwent FPI were compared to the data obtained from the animals with chronic pilocarpine-induced epilepsy, the results revealed that the changes within the limbic system resulting from TBI are qualitatively similar but quantitatively less severe than changes in rats with chronic temporal lobe epilepsy [150].
Acute PTS and the Development of Epilepsy After FPI
In a midline FPI model, tonic–clonic seizure-like convulsions were noted in early studies, although the convulsions were evident only in animals injured at higher levels of pressure, and the occurrence of PTS was associated with increased mortality [117]. The seizures were observed in only in 11–15 % of surviving rats who underwent FPI, whereas the incidence of seizures was much greater in nonsurviving rats (33–80 %); the occurrence of convulsions was not related to the magnitude of injury [117]. Another study has shown that PTSs were observed in about 50 % of rats within 15 min after FPI [151].
In rats, single severe lateral FPI in rats is sufficient to induce spontaneous chronic seizures evident on intracortical EEG recordings associated with behavioral manifestations [146, 152]. Interestingly, experimental PTE outcomes in lateral FPI strongly depend on the location of injury [153]. Epileptiform activity without behavioral seizure manifestation occurs 5 weeks after lateral FPI and this activity progresses with time; by 33 weeks after impact, nearly all rats who underwent FPI exhibited spontaneous convulsive and non-convulsive seizures. In addition, the latter study reported the occurrence of non-convulsive seizures of lesser severity and duration in some control animals and in over 50 % of rats who underwent sham procedures [154].
PTZ Challenge Test as a Measure of Epileptogenesis After FPI
The occurrence of acute and chronic spontaneous behavioral seizures is strongly associated with injury severity. The seizure could be only observed following FPI with the pressure magnitudes corresponding to severe TBI, whereas in the models with moderate injuries, behavioral seizures are not evident [43, 142, 155]. In addition, chronic spontaneous seizures generally develop within several weeks after induction of FPI.
Thus, to study epileptogenic changes in mild or moderate FPI models or at subacute time points, surrogate tests of epileptogenesis employing induction of seizures with ether bolus injection of infusion of PTZ to assess seizure severity using the Racine’s scale or measure seizure threshold. The data using a bolus injection of PTZ at a dose below the seizure threshold in non-injured animals have shown that FPI causes an increase in seizure susceptibility [156]. Persistently enhanced susceptibility to PTZ-induced seizures was observed for at least 6 months after FPI, even in animals that had no spontaneous seizures [43]. However, seizure susceptibility in injured rats was comparable to that in control animals with the lowest number of neurons immunoreactive for parvalbumin, a calcium-binding albumin protein, in the reticular nucleus of the thalamus [157].
The dependence of seizure susceptibility on the magnitude of injury suggests that epileptogenesis is associated with anatomical damage and neuroprotective strategy following TBI and will be also beneficial against the development of PTE. Neuroprotective treatment with hypothermia for 4 h immediately after FPI significantly decreased PTZ-induced seizure susceptibility and attenuated mossy fiber sprouting, a hallmark of the epileptic brain compared with normothermic controls 12 weeks after recovery following experimental TBI [158]. In addition, increased seizure susceptibility in FPI-injured rats determined using the PTZ challenge was associated with enhanced neuronal activity detected on BOLD-fMRI, but not EEG abnormalities assessed 1 week before the PTZ test [159]. On the other hand, induction of seizures with PTZ in rats who underwent FPI resulted in worsened anatomical outcomes such as lesion size and neuronal cell death in the CA3 region of the hippocampus and mossy fiber sprouting [156].
Pharmacological Interventions Affecting TBI and PTE Outcomes in FPI Model
A limited number of studies have shown that some pharmacological interventions may ameliorate PTS and attempts were made to test whether AEDs used clinically could protect from experimental TBI. Interestingly, rats administered tacrolimus, a calcineurin inhibitor, acutely after TBI showed significantly fewer non-convulsive seizures than untreated rats months after FPI, but a similar degree of cortical atrophy [154]. In addition, attempts were made to use common AEDs for neuroprotection following FPI. A study performed with the slices from rats receiving treatment with felbamate after FPI exhibited long-term potentiation in the CA1 region of the hippocampus, which is suppressed in untreated animals, suggesting that the neuroprotective property of felbamate is neuroprotective against traumatic neuronal injury [160]. Another study has shown no significant effects of lacosamide on anatomical damage or functional recovery [161].
1.3.3 PTS and PTE in CCI Injury Model
CCI is one of the most commonly used TBI models, primarily due to the tight control of injury parameters and the resemblance of several anatomical and neurological outcomes with those observed in man following head trauma [119, 162–165]. Applying different CCI parameters allows for the production of experimental TBI with different magnitudes affecting a pattern of excitotoxic, ischemic, and hemorrhagic injury components that can differently affect epileptogenic cascades in the brain following TBI.
Thus, the CCI model allows for the reproduction, in an experimental animal, of several of the characteristics of human TBI, and anatomical and neurological deficits, to a certain degree in a controlled fashion [119, 162–165]. The CCI model study was initially developed as a TBI model in ferrets to reflect features of closed head trauma commonly observed in human TBI with distinctive characteristics of axonal injury caused by deformation of brain tissue that results in delayed axonal degeneration due to secondary injury that spreads over time via the white matter tract [162, 166]. Further CCI models were extended to rodent species and currently there is one of the most characterized and commonly used preclinical models for studying various secondary consequences of TBI such as gliosis and activation of neuroinflammatory cascades [119, 163–165]. To date, the CCI model has been adopted for many different species, including ferrets [118], rats [119, 167], mice [164], pigs [168, 169], and monkeys [170].
In the CCI model, the injury to the brain is induced by compression of brain tissues with a rigid impactor directly positioned to the cortical surface with intact dura through the craniectomy window using a specific pneumatically or electromagnetically driven impact device, allowing for the tight control of the velocity of impact, the dwelling distance, and the time of compression [119, 171]. However, there are studies suggesting some advantages of the electromagnetic versus pneumatic devices [171].
Furthermore, using impactor tips with different diameters allows researchers to add another variable to change the injury severity as well as to scale the magnitudes to animal size. Both types of device are widely used in TBI research and are commercially available. The shape of the impactor (flat of rounded) may also affect biomechanical features as well as outcomes of experimental TBI. The craniotomy (or craniectomy) procedure is crucial for the reproducibility of the injury as even minor damage to the dura mater or bleeding during the surgery may dramatically affect anatomical behavioral outcomes and increase the variability of quantitative measures. Two types of craniotomy surgeries are commonly performed in the CCI model: without closing the craniotomy window or covering the opening with the bone flap secured with bone wax. However, replacing the bone flap after craniotomy surgery even without induction of CCI can produce considerable brain lesions [172]. In contrast, the craniotomy in sham animals without bone flap replacement causes no detectable brain lesions [69, 168]. Furthermore, based on our unpublished observations in both rats and mice, a craniotomy site without bone flap replacement in sham animals starts to heel within 48 h after surgery and is completely healed with longer time points; the bone looks normal with no macroscopic changes within 1–3 months, whereas in animals who underwent CCI, the craniotomy site could be easily detected by lesser bone thickness.
The levels of anatomical alterations in the brain after CCI depends on the injury magnitude from no obvious or significant macroscopic or microscopic changes at low levels to significant cortical contusion and subdural and intraparenchymal hemorrhages that progress with time and complete loss of brain tissue within the injury site, often referred as to cavitation, due to delayed necrosis at higher injury levels [119, 164].
Distinctive anatomical hallmarks that mimic human TBI observed in rodents include concussion [119], diffuse axonal injury, acute subdural and intraparenchymal hematomas [119], loss of cortical tissue [164], delayed hippocampal atrophy, neuronal degeneration in the cortex, hippocampus, and thalamus [164, 173], vascular damage [174], disruption of the BBB and extravasation of blood content [164], and gliosis [66, 164]. However, diffuse axonal injury in rodent CCI is less distinctive than that described in ferrets [162]. The severity of histopathological alterations show correlation with incising in velocity and depth of cortical deformation [171, 172, 175].
The functional changes observed in CCI injury in rodents include both cognitive and motor deficits [171, 176–179]. The motor deficits are transient and most resolve within months after CCI induction, whereas cognitive deficits are persistent and could be observed even at 1 year after injury [176–179]. Cognitive deficits are commonly observed at chronic to subchronic time points and are commonly associated with anatomical changes in the brain such as cortical and hippocampal atrophy, progressive decrease in cerebral circulation [180], Aβ and Tau pathologies, and the level of impairment strictly dependents on the magnitude of the adjustable CCI parameters. In addition, recent behavioral studies suggest changes associated with emotional deficits [178, 180], which are also characteristic for human TBI; although changes in emotional behavior are less dependent on the CCI magnitude compared with cognitive outcomes [178].
Strain- and Gender-Dependent Differences in Outcomes in CCI Models
Strain-dependent differences in the rodent species have been extensively reported in many animal models of brain injuries, including models of TBI and seizures/epilepsy [176]. The mouse strain should be considered especially carefully as the mice from different strains have different sensitivities to excitotoxic injuries, reflecting the differences in seizure susceptibility in both chemically induced and reflex seizures models. However, in a mouse CCI model, the animals from different strains subjected to either to sham or experimental injury show significant differences in functional rather than anatomical outcomes. The differences are most evident in behavioral responses for performing cognitive tasks (e.g., learning in the Morris water maze and Barnes circular maze) [175, 181], whereas the differences in the performance of motor tasks are generally not significant among strains [175].
A study performed in three mouse strains commonly used as background in genetically altered mice (i.e., C57BL/6, FVB/N, and 129/SvEMS) showed that there was no significant difference in body weight between mice subjected to sham surgery or CCI injury or lesion volume induced by CCI within any of the three different strains [175], and these data are consistent with the results of several other studies. Importantly, the posttraumatic mossy fiber sprouting, an outcome implicated in epileptogenic alterations in the brain, was qualitatively similar among CD-1, C57BL/6, and FVB/N mouse strains [181].
Several studies performed using the CCI model suggest that gender does not critically affect several important CCI outcomes such as lesion severity, which is quantified using cresyl violet staining [168] and silver staining volume [168, 171, 177]. However, some gender differences were observed in temporal changes with silver staining following CCI, while the rate of resolution of staining between 48 h and 4 weeks was similar [177]. Interestingly, some outcomes following CCI showing no gender difference are in contrast to that seen in the weight-drop injury paradigm [177]. In addition, some gender-specific differences were observed in PTS activity after CCI where 33 % of male and 25 % of female wild type mice exhibited seizure activity within 2 h after CCI; also, some difference in seizure incidence was observed in A1 adenosine receptor knockout male and female mice [182].
Acute and Chronic PTS Assessment After CCI
The studies performed in rodent CCI models revealed the manifestation of acute and chronic behavioral seizures and EEG abnormalities associated with epileptiform activity and hyperexcitability in the brain [173]. The occurrence of acute and chronic PTS after CCI is an important outcome that represents clinical features of human TBI. In addition to behavioral seizures and EEG epileptiform discharges, CCI-induced injury results in several chronic alterations in the brain such as mossy fiber sprouting and delayed hippocampal lesions that are often considered hallmarks of temporal lobe epilepsy.
In wild type control mice following CCI injury, seizures are generally observed within the first hours, and these seizures are characterized by mild-to-moderate severity and short duration (generally within a few seconds), whereas in genetically modified mice lacking the A1 adenosine receptor, the severity of seizures was significantly higher, including the occurrence of tonic–clonic seizures and the development of lethal status epilepsy [182]. Interestingly, the study also showed that A1 adenosine receptor knockout mice exhibited enhanced microglial responses compared to wild type mice, especially in the ipsilateral and contralateral cortices and thalami, and in the ipsilateral CA3 and contralateral CA1 regions of the hippocampus, whereas lesion and cortical volumes were not different between knockout and wild-type mice and Fluoro-Jade staining revealed no neuronal death in CA1 or CA3 regions, suggesting a critical role for microglia in epileptogenic changes after TBI [183]. A study performed in rats has shown that following CCI, 67 % exhibit transient epileptiform discharges that were significantly altered by treatment with lisuride, an agonist of dopamine and a partial agonist of serotonin receptors, including total duration of epileptiform discharges, spectral characteristics such as reduction in delta power; it also resulted in an increase in the mean EEG frequency representing anticonvulsive effects [184]. Although lisuride has significant anticonvulsive effects, it did not have significant effects on secondary brain damage [184].
In the mouse CCI model, spontaneous behavioral seizures occur after several weeks following induction of the injury and the seizure incidence depends on the severity of CCI parameters such as dwell distance [185]. When using 0.5 and 1 mm injury depths, seizures were observed in 20 and 36 % of mice, respectively, and both parameters induced mossy fiber sprouting [185]. A later chronic study performed in mice employing video-EEG monitoring for 16 weeks following CCI has shown that most PTS occur after 10 weeks following TBI [186]. This study was focused on the mammalian target of rapamycin complex 1 (mTORC1) pathway in posttraumatic epileptogenesis and the results showed that inhibition of this pathway with the specific inhibitor (i.e., rapamycin) for 30 days after CCI decreased neuronal degeneration and mossy fiber sprouting, and decreased the frequency of seizures and the rate of PTE [186]. Another study demonstrated that although rats did not develop spontaneous behavioral seizures for up to 8 weeks after CCI, they have increased susceptibility to bicuculline-induced seizures 1 week after injury, suggesting an increased hyperexcitability [187]. A chronic 9-month study performed in C57BL/6S adult male mice that included three 2-week continuous video-EEG monitoring showed that after CCI, 9 % of mice exhibited electro-graphic seizures and 82 % of mice exhibited epileptiform spiking on EEG and increased susceptibility to the PTZ-induced seizure threshold test [188]. These changes developed within 6 months after CCI and did not further progress for up to 9 months [188].
Of interest is a PTE model induced by CCI in the immature rat, which is often called a pediatric PTE model. In this model, CCI is applied to 2- to 3-week-old-rats and epileptogenic changes occurred during maturation [17, 189]. CCI performed in 16- to 18-week-old rats resulted in a sustained reduction of the minimal clonic seizure threshold at 2 months of age [17]. A study using chronic video-EEG monitoring within 4–11 months after CCI showed the occurrence of EEG spiking in 87.5 % of rats who underwent CC and in rare cases even spontaneous recurrent seizures [189].
Electrophysiological In Vitro Assessment of Epileptogenic Changes Induced by CCI
These investigations usually employ patch-clamp, intracellular, or extracellular recording techniques, allowing registration of evoked and spontaneous events at different time points following experimental TBI. The progressive development of neocortical hyperexcitability underlying spontaneous epileptiform firing could be detected 2 weeks following CCI [173]. The mechanisms underlying this hyperexcitability are complex and poorly understood. For example, CCI produces a transient decrease in the expression of the Kv4.2 subunit of the IK(A) channel and a reduction in IK(A) currents in the ipsilateral CA1 pyramidal neurons 1 week after CCI but these changes were no longer significant 8 weeks after injury [187].
Intracellular and extracellular recordings obtained from layer V in neocortical slices from rats who underwent CCI revealed that characteristically evoked epileptiform discharges are evident even in the first week after injury, whereas spontaneous epileptiform discharges are evident after 2 weeks [173]. The data suggest that the hyperexcitability observed following CCI is associated with augmentation of excitatory reduction of inhibitory synaptic activities. Whole-cell patch-clamp recordings in granule cells revealed a reduction in spontaneous and miniature IPSC frequency in GABAergic hilar interneurons after 8–13 weeks of CCI injury, whereas the action potential and EPSC frequencies were increased [190]. The abnormalities in GABAergic transmission in hippocampal dentate granule cells resulted from CCI injury includes differential alterations in the function of synaptic and extrasynaptic GABA A receptors [126]. In addition, CCI results in the altered expression of both GABA A and glutamate receptor subunits, and, remarkably, an increase in NR2B protein and decrease in GluR1 protein expression [191].
2 Materials
2.1 Equipment and Instruments for General Surgical Procedures in Rodents
Stereotaxic apparatus (e.g., David Kopf Instruments, Tujunga, CA, USA) equipped with anesthesia nose cone (if gas anesthesia is used).
Anesthesia machine for gas anesthesia (e.g., tabletop non-rebreathing anesthesia machine, Harvard Apparatus, Holliston, MA, USA or similar).
High speed dental drill or surgical micro drill (e.g., Complete Bone Micro Drill System, Harvard Apparatus; OmniDrill25, World Precision Instruments, Sarasota, FL USA) and 0.5 mm diameter bit for craniotomy procedure (World Precision Instruments).
Closed loop temperature control system for small rodents Homeothermic Monitoring System, Harvard Apparatus). Alternatively, animal’s body temperature could be monitored during surgery using a low-cost electronic thermometer (MicroTherma 2T Hand Held Thermometer, Braintree Scientific Inc., Braintree, MA, USA) equipped with a rodent rectal probe (Braintree Scientific Inc.) and temperature-controlled heating pad (e.g., Fine Science Tools, Vancouver, BC, Canada).
Recovery temperature/humidity-controlled chamber (e.g., Small Animal Recovery Chamber/Warming Cabinet, Harvard Apparatus).
Surgical instruments: forceps, scissors, scalpel.
Surgical retractor (optional) (e.g., small animal retractor system Harvard Apparatus).
Reflex 7 or Reflex 9 wound closure system (CellPoint Scientific, Inc., Gaithersburg, MD, USA) for mice or rats, respectively. Alternatively, surgical sutures can be used.
Isoflurane (or other desired anesthesia gas).
Supplies for aseptic surgical preparation. Although the aseptic technique is critical for the animal surgical studies, specific aseptic techniques might be selected based on the internal guidelines or recommendation of institutional committee (e.g., Betadine, 2 % chlorhexidine, 70 % isopropyl alcohol).
Sterile gauze sponges, cotton tips, sterile surgical spears (e.g., BIO CEL™ and BIO SPEARS™, Biotech Visioncare, India).
Eye ointment.
Triple antibiotic ointment (optional).
Dental cement and/or veterinary acrylic adhesive (e.g., LiquiVet).
2.2 Equipment for Induction of Experimental TBI (Depending on the Experimental Model Used in the Study)
Cortical impactor device [e.g., electromagnetically driven impactor devices: PCI3000 PinPoint Precision Cortical Impactor (Hatteras Instruments, Cary, NC, USA) or Leica Impact One™ Stereotaxic Impactor (Leica), pneumatically driven impactor devices: Pneumatic (Cortical) Impact Device AMS 201 (AmScien Instruments LLC,, Richmond, VA, USA), TBI-0310 Impactor (Precision Systems and Instrumentation, LLC, Fairfax Station, VA].
Fluid Percussion Injury device (Custom Design & Fabrication, Virginia Commonwealth University Medical Center, Richmond, Virginia, USA). FPI assembly includes the base, pendulum mast, graduated protractor, ball bearing-enhanced impact hammer assembly, acrylic fluid cylinder and bracketry, transducer housing, transfer tube, and a digital transducer. The device is completed with a self-calibrating Trauma Inducer Pressure Inducer Amplifier.
2.3 Materials for EEG Monitoring and Seizure/Epilepsy Assessment
2.3.1 Setup for EEG/Video Seizure Monitoring and Analysis
Biopotential amplifier (type and number of channels depend on needs) (e.g., Bio Potential Amplifier or Electroencephalogram Amplifier Modules, Harvard Apparatus).
Analog-to-digital converter for digitizing of EEG (e.g., Model#: PCI-6221, 16-Bit, 250 kS/s, 16 Analog Inputs or Model#: USB-6008, 12-Bit, 10 kS/s Low-Cost Multifunction DAQ with required accessories, National Instruments Corp., Austin, TX, USA).
Tether and commutator to connect the biopotential amplifier to the digitizer (e.g., Cat#: 8204 3-channel mouse commutator/swivel, Pinnacle Technology Inc.).
Preamplifier Mouse Preamplifier for 3-Channel System (optional, depending on the equipment used, e.g., 8202, Pinnacle Technology Inc.).
8-Pin Surface Mount Connector for Mice (8415-SM, Pinnacle Technology Inc.).
EEG headmounts (#8201-EEG headmounts, Pinnacle Technology Inc., Lawrence, KS).
Two-component epoxy for ensuring solid connections at the electrodes (e.g., 8226 Silver Epoxy for use with mouse #8201 or #8201-EEG headmounts).
Mouse or rat stainless EEG screws (e.g., 8209: 0.10″ or 8212: 0.12″, Pinnacle Technology Inc.).
EEG signal acquisition software (e.g., AcqKnowledge BioPac Systems Inc., Santa Barbara, California). Alternatively, a custom specific for the study acquisition/analysis program can be written using Matlab software (Mathworks Co., Natick, MA).
Digital video surveillance system with infrared camera to enable night recording (e.g., Automated Video Systems).
2.3.2 Materials and Equipment: PTZ Challenge Seizure Tests
Pentylenetetrazole (CAS Number 54-95-5) (e.g., Cat#: P6500, Sigma-Aldrich, St. Louis, MO, USA).
~10″ diameter circular mouse cage for performing PTZ challenge test (e.g., Pinnacle Technology Inc., Lawrence, KA, USA) (see Note 1).
Syringe infusion pump for PTZ-induced seizure threshold test (e.g., Standard Infusion Only PHD Ultra Syringe Pump, Harvard Apparatus) (see Note 2).
Syringe (depend on the syringe pump used).
30 G Needles.
Polyethylene tubing (e.g., Cat#: PE10, Braintree Scientific Inc.; Cat#: 51150, Stoelting Co., Wood Dale, IL 60191 USA).
Medical tape to fix a needle to animal’s tail (optional) (e.g., 3M Transpore Transparent Tape).
3 Methods
This section describes common protocols for induction and detection of acute PTS and/or chronic PTE based on the common TBI models: CCI and FPI. The main differences between these experimental models are reflected in the method of TBI induction, whereas most of the general surgical procedures such as craniotomy and electrode implantation, and EEG and behavioral seizure/epilepsy assessments are the same for both these models.
3.1 Animal Selection
Prior experiments, animals should be examined for their general appearance and health condition. The animals showing any unexpected diseases or pathology such as skin lesions, wound from fighting, ophthalmic pathologies should not be used. In addition, the animals should be excluded from the experiments if any phenotypic alternations from group characteristic are present including size and weight, fur condition and/or color, skull and body shape, and behavioral baseline tests (see Note 3). Each animal should be assigned a unique identification and the animals should be randomized into the study groups. It is the best approach to use littermates if available.
3.2 Surgical Procedures
3.2.1 Surgery Preparation
Prior to all procedures, anesthesia has been induced with 4 % isoflurane in a 25 % oxygen-in-air mixture in the anesthesia induction chamber. Exposure to isoflurane for about 1 min is normally enough induce deep anesthesia that allows for removing animal’s hair within surgery site and transferring to the stereotactic apparatus equipped with nose cone for maintaining anesthesia and heating pad.
Animal’s hair is removed using any of the three methods that are currently commonly used: clipping with an electrical rodent hair clipper, using a chemical depilatory agent, or shaving with a razor.
Animal is transferred to the stereotaxic apparatus and connected to nose cone. During all surgical procedures, mice are maintained on 2 % isoflurane anesthesia via nose cone.
Eye ointment is applied on animal’s eyes to prevent drying of cornea.
A lubricated rectal probe is placed in the rectum to monitor animal’s body temperature. The body temperature is maintained with a controlled heating pad.
Surgery site is prepared by appropriate aseptic techniques, e.g., washing with 2 % chlorhexidine and sterile saline for three times, starting from center and moving to edge.
3.3 Electrode Implantation for PTS Detection
For acute recording of acute PTS the electrodes are implanted before TBI induction, whereas for recording of spontaneous seizures for characterization of the experimental PTE, the electrodes are implanted weeks or months after experimental TBI.
3.3.1 General Consideration for Selection of Method of EEG Monitoring and Type of Electrodes
Human PTE is heterogeneous in terms of seizure durations and semiologies and may represent simple or complex partial seizures and secondarily generalized seizures. Generally, PTE may represent simple or complex partial seizures and secondarily generalized seizures. Although initial seizures in PTE most commonly have a focal neocortical origin, both partial neocortical epilepsy and TLE might be developed [2]. For selection of the method for EEG monitoring it is important to take into consideration the characteristic brain pathologies characterized by significant loss of brain tissue and hippocampal degeneration especially at chronic stages (Fig. 1). Both single-channel and multiple-channel array EEG recording systems have been described for the assessment spontaneous seizures following TBI. The methods of EEG acquisition include unipolar, bipolar and average reference, and, in general, the same basic information could be obtained by any of these methods [192]. The choice of a method of EEG acquisition depends to a large extent on the specific problem addressed in the study and on the personal preference of the investigator. The epidural electrodes (e.g., stainless steel, Ag/AgCl) are used for both bipolar and unipolar recordings. Commercially available stainless steel screw electrodes (e.g., Plastics One, Pinnacle Technology) are an example of electrodes suitable for chronic EEG monitoring. However, using unipolar recording techniques allows to determine only overall changes in EEG activity. The EEG waveform parameters strongly depend on the location of the electrodes. Reference electrodes could be pleased on the distal part of skull or other animal’s body part (e.g., tail, cheek). The placing of electrodes at the margins of TBI injury on the ipsilateral side and the corresponding electrodes in the contralateral side allow to measure changes in EEG activity measured as a difference between the two hemispheres versus one reference electrode [193].
Fig. 1.
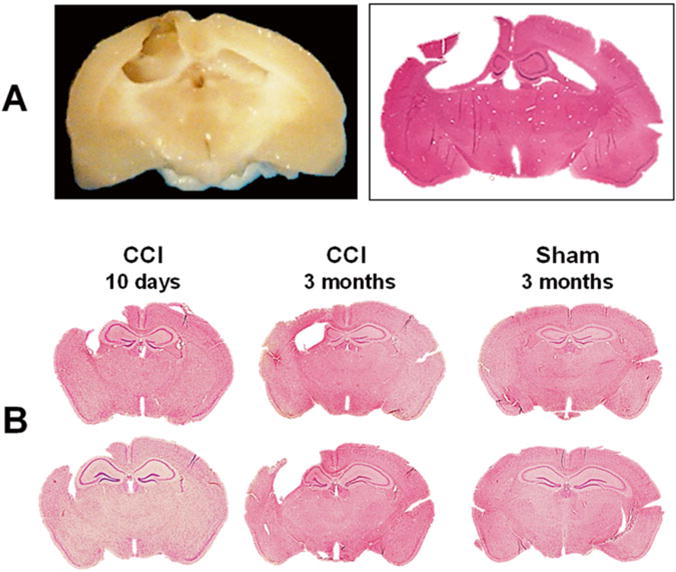
Chronic brain pathology following CCI. Representative photograph of paraformaldehyde-perfused rat brain spacemen collected at 3 months after induction of CCI and corresponding hematoxylin–eosin (H&E)-stained coronal 8-μm paraffin section. The photographs demonstrate marked brain degradation at chronic stages resulted in loss of cortical tissue (cavitation) and significant reduction in hippocampal size ipsilateral to the injury at chronic time points (a). Representative photographs of H&E-stained coronal 8-μm paraffin mouse brain sections demonstrating progression of hippocampal pathology at subacute and chronic time points after CCI. At subacute time point (10 days after CCI) the ipsilateral hippocampus is enlarged due to focal edema that is still evident at this time point, whereas at chronic time point after CCI, the ipsilateral hippocampus is reduced in size due to neurodegenerative processes resulting in neuronal death and loss of brain tissue. Cortical cavitations in CCI-injured mice are presented at both time points. In contrast, no obvious brain pathology is observed in animals at 3 months after sham injury (b)
3.3.2 Selection of Position of EEG Electrode Placement
EEG electrode positions depend on the method of acquisition and available equipment:
Position of the single-channel epidural EEG electrode implantation is at the caudal edge of the TBI lesion.
For the bipolar recording, two stainless steel screws are positioned fronto-lateral and occipito-parasagittal to injury site, respectively.
For multichannel recording, the electrodes are positioned ipsilateral and contralateral to injury hemispheres [194]. In the ipsilateral hemisphere, one frontal and one parietal electrode are positioned rostral and caudal to craniotomy, respectively. In the ipsilateral hemisphere, one frontal and one parietal electrode are inserted in the corresponding positions. An additional ground electrode is positioned into the occipital bone caudal to lambda. The schematic of default EEG multiple electrode placements in rodents is shown in Fig. 2.
Fig. 2.
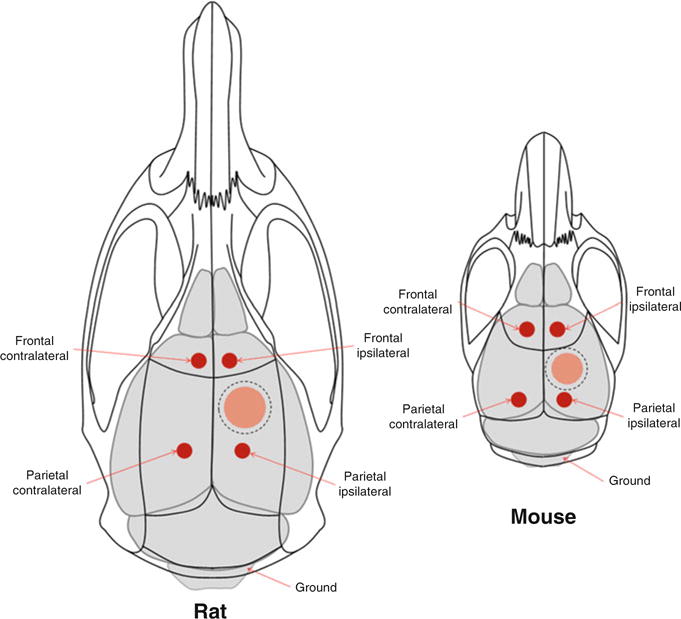
Schematic of the EEG electrode placement in rodents. Position of the EEG leads in rat and mouse TBI models for detection of the PTS and PTE. Scattered line represents positions of craniotomy and red filled areas represent the sites of injury. Ground electrode if required could be positioned over cerebellum
3.3.3 Procedures of EEG Electrode Implantation
Epidural stainless still screw electrodes are inserted prior to CCI induction proximally from the craniotomy boundary and at the same position contralaterally. Alternatively, scalp electrodes could be used [182].
Metal parts are insulated with dental cement.
Baseline EEG recordings are performed for at least 10–30 min under anesthesia downtitrated to about 1 % of isoflurane when animal still has negative pinch toe reflex.
The EEG recording to detect acute PTS will be resumed after induction of experimental TBI CCI (see below).
3.3.4 Craniotomy Procedures
Under 2 % isoflurane anesthesia, central skin incision is made and connecting tissues covering skull are removed with cotton tips. Normally, spreading skin to sides allows for performing all further surgical procedures and other manipulations when gently holding the skin with a cotton tip. Using a surgical restrainer might be helpful.
Craniotomy site according to desired coordinates is marked using appropriate marking tool. Generally in PTE models, circular craniotomy with appropriate diameter to allow freely access for rigid impactor for CCI model or attach connector to FPI is positioned in the center of temporal bone.
After the skull is exposed with a central skin incision and soft tissue is removed with a cotton tip, circular craniotomy, ~4 mm in diameter, is made in the middle of the right parietal bone, about 0.5 mm from sagittal, coronal, and lambdoid sutures.
Circular craniotomy is made using an electric high-speed drill with 0.5 mm diameter bit periodically applying sterile saline to prevent drying of skull bone and overheating. Visual control using a low-power microscope during the whole procedure is highly preferred. It is critical to continue drilling removing a small amount of bone tissue around the edge until the bone cracks. Generally, the craniotomy procedure using electric drill takes less than 5 min. Alternatively, trephine device (preferably manual) might be used. However, when using trephine, it is more difficult to control depth to prevent damage of dura mater.
Bone flap is gently removed with forceps. If craniotomy is performed properly, the bone flap moves freely. If bone flap is still attached to the skull bone at some points, there is a high risk for damaging dura mater (see Note 4).
The exposed brain is washed with warm sterile saline to remove bone dust. Brain tissue should be moist all the time.
3.3.5 CCI Procedures
Excess of saline should be gently removed with cotton tip before position of impactor.
The impactor is positioned on the site of injury under visual control and or using a sound sensor if equipped (see Note 5). Visual control under magnification allows establishing a starting position more precisely.
The impact with desired parameters is executed and the impactor is retracted.
Occasional minor bleeding is stopped with cotton tips from edge of craniotomy not touching brain surface and washed with warm saline.
The example CCI parameters for mice are as follows: impact tip diameter 3–4 mm, velocity 3 m/s, compression time 100–200 ms, and a compression distance of 1–1.5 mm; using these parameters nearly 100 % of WT C57BL/6 mice exhibit behavioral PTS within 1 h after CCI. Characteristic subacute and chronic brain pathologies following CCI with aforementioned parameters are shown in Fig. 1b.
3.3.6 Lateral FPI Procedures
The general craniotomy procedures are the same as those described above.
After performing craniotomy, Luer lock hub fitting is attached to animal’s scull with acrylic adhesive to provide tight seal and filled with sterile saline. Brain tissue should be kept moist with sterile saline.
Animal is removed from the stereotaxic apparatus and allowed to recover for ~1 h in a temperature controlled recovery chamber.
Before induction of injury, the high-pressure tubing of FPI device is connected and filled with sterile water and ensure that it is free of air bubbles and FPI device is settled up to deliver injury pulse free of noise as monitored using oscilloscope.
Prior to TBI procedures, deep anesthesia is induced with 4 % isoflurane in a 25 % oxygen-in-air mixture in the anesthesia induction chamber.
Animal is placed on the platform on its side close to FPI device and connected to the high-pressure tubing of the FPI device.
Experimental TBI is delivered by releasing pendulum to produce a single inquiry pulse. The exact pressure is measured and recorded (the transducer amplifier is calibrated at 10 mV per 1 PSI pressure).
After completion of the experimental TBI, animal is returned to surgery setup and maintained under isoflurane anesthesia to complete surgery.
Luer lock fitting used for connection of the animal to the FPI device is removed from the skull and the incision is closed.
3.3.7 Completion of Surgery
For animals intended for chronic PTS/PTE assessments after surgical procedures, the incision is closed using wound clips (or sutures).
Each animal receives an intraperitoneal injection of warm saline to prevent dehydration and transferred to a temperature-controlled recovery chamber for at least 1 h.
3.3.8 Procedures in Sham Animals
The animals assigned into the sham groups undergo craniotomy procedures only. In animal TBI models, craniotomies with replacing bone flap result in considerable morphological, inflammatory, and behavioral alternations [172], and may cause an increase in allodynia [195]. In the latter case, an increase in allodynia following above-described sham procedures was observed in mice but not in rats [195].
3.4 Acute and Chronic Spontaneous PTS Assessment
3.4.1 General Consideration for EEG/Video Monitoring
The EEG recording to detect acute PTS will be resumed immediately after induction of CCI when animal still under anesthesia of or after waking up after anesthesia using an EEG/video recording setup for recordings in freely moving animals.
For chronic EEG/behavioral seizure activity the EEG and video recordings are continuously performed to assess remote spontaneous seizure activity, which is expected to occur within 2 months after CCI. The approaches for EEG and video monitoring of PTE development require continuous recordings in epoch with duration from weeks-to-months and computerized analysis for EEG episode detection [196–198]. Alternatively, the animals could be monitored for the spontaneous PTS using random 1–2 h intervals within the same time frame [185]. The EEG episodes are confirmed with synchronized video recording for behavioral seizure manifestation which are scored using the Racine’s scale.
3.4.2 EEG Data Analysis
Computer-assisted EEG spectral analysis is performed on selected epochs of fixed duration (e.g., 60 s) of cortical to evaluate changes in EEG power [199]. Compressed spectral arrays, spectral trend graphs, and spectrographic analyses are also used to evaluate EEG changes. The respective power scores across each frequency band are valuated at fixed time points following and compared with baseline and the corresponding controls (e.g., sham-operated animals or vehicle injection). Changes in power are assessed across four following frequency bands:
Delta (0–4 Hz).
Theta (4–8 Hz).
Alpha (8–12 Hz).
Beta (12–30 Hz).
An EEG seizure episode is defined as repetitive spikes or spike-and- wave discharges recurring at frequencies of >1 Hz with amplitude greater than spontaneous activity and duration greater than 2–10 s. The requiring EEG seizures with a short interval between two episodes (e.g., less than 10 s) could be considered as a single event.
Chronic epilepsy is defined as appearance of recurring paroxysmal events detected by EEG and/or behavioral manifestations. Based on these criteria, the following behavioral/EEG seizure parameters are included in the statistical analyses:
Incidence of the chronic PTS is determined as a percentage of animals that have at least one unprovoked seizure.
Incidence of the PTE is determined as a percentage of animals that have at least two unprovoked seizure.
Onset latency of the PTS/PTE is determined as the time between the induction of experimental TBI and the first episode or recurrent EEG seizures.
PTS frequency is determined as the number of EEG episodes observed in individual animals for a fixed period time.
PTS episode duration is determined as an average duration between the onset and offset of each EEG seizure episode.
Total PTS duration is determined as the sum of episode durations.
Time course of PTS frequency, defined as the hourly EEG seizure frequency up to 2 months.
3.4.3 Assessment of Behavioral Seizure Severity
Behavioral acute or chronic seizures are confirmed and scored based on the revised Racine’s seizures severity scale. This scaling system has been adapted to different seizure models including, electrically and chemically induced seizure models [200–203]. The same scaling system is also used for seizure severity assessment for PTZ challenge test. The system is based on the behavioral manifestation with the following hallmarks reflecting the discrete seizure scores: 0, normal response; 1, ear and facial twitching; 2, myoclonic twitches or waves of the whole body; 3, myoclonic jerks and/or rearing; 4, clonic–tonic seizures; 5, generalized clonic–tonic seizures, loss of postural control; 6, death (see Note 6).
In addition, the behavioral seizure parameters such as incidence frequency, seizure episode duration, total duration, onset latency, and time course will be defined and assessed similarly to corresponding parameters of EEG seizures.
3.4.4 PTZ Challenge Seizure Tests
There are two types of PTZ seizures induced by bolus injection or determination of seizure threshold using slow PTZ infusion.
PTZ Challenge with Bolus Injection
Animal is allowed to acclimatize in 10″ d circular mouse cage (Pinnacle Technology Inc., Lawrence, KA) and 10–30 min baseline EEG is recorded.
Animal are removed from the chamber injected intraperitoneally with PTZ (50 mg/kg for rat or 80 mg/kg for mouse).
The seizures are graded using Racine’s scale during 30–60 min with a constant bin intervals selected from 30 s for up to 5 min.
PTZ-Induced Seizure Threshold Test
A PTZ-induced clonic seizure threshold test is widely used as surrogate test for seizure susceptibility [204]. The PTZ-threshold test is performed in all animal groups before terminal time point as a quantitative test of epileptogenesis in PTE [43, 188]. The value is expressed as an amount of PTZ infused via tail vein required to induce clonic seizure in a freely moving animal. However, because of the possible day-to-day variability in the PTZ threshold, appropriate control groups should be included for each testing.
PTZ solution (0.5 % in saline) will be infused into the lateral tail vein at a constant rate of 0.5 mL/min using a and an infusion pump, which is connected by polyethylene tubing to the 30-G dental needle secured to the tail with a narrow piece of adhesive tape (optional).
Infusion is halted when forelimb clonus followed by full clonus of the body is observed. The minimum dose of PTZ (mg/kg) needed to induce a clonic seizure will be measured as an index of clonic seizure threshold.
In addition, the PTZ test allows analysis of different convulsive end points including the following observed seizure phases: general excitation with myoclonic twitches of the whole body, generalized clonic seizures with loss of righting reflex and tonic hind limb extension.
3.4.5 Exclusion Criteria in TBI/PTE Animal Models
The experimental results could be influences by several factors that are not related to the experimental model or treatment such as those results from accidents, human error (e.g., incorrect procedures or treatment) or animal’s health condition resulted from sickness (e.g., infection diseases or wound resulted from fighting). Importantly, because the number of animal experimental groups based on power analysis for each particular study, the presence even a single animal with erroneous outcome measure may lead to marked increase of the standard deviation and loss of statistical significance resulting in wrong experiment interpretation and conclusions. Thus, recognizing and exclusion of the animals with deficiencies or alterations that are not related to the study is critical to ensure quality of the experimental results and reproducibility (see also Subheading 3.1 and Note 7).
Exclusion Criteria Related to Surgical Procedures
Damaged dura mater, meninges, and/or brain tissue during craniotomy.
Subdural hemorrhage after craniotomy
Excessive bleeding during or after craniotomy
Accidental incorrect position of impactor causing brain deformation (e.g., exceeding ~50 μm).
Prolonged surgery or anesthetic exposure (e.g., more than 30 min due to any reasons).
- The following applies for wild type animals, whereas for genetically modified animals they should not be considered as outcomes unless the study confirms that any of these reflects outlier characteristics:
- Excessive bleeding after CCI impact.
- Any alteration in anesthesia response reflected in animal behavior (e.g., waking-up, abnormal breathing pattern) or response to test (e.g., toe pinch reflex) when animal maintained at 2 % isoflurane. In this case, anesthesia should be adjusted to the proper level and the same anesthesia settings should be maintained in all experimental groups.
Exclusion Criteria Post-surgery
Humane end point criteria defined by the animal use protocol or institutional committee.
Removing clips (extremely rare event with proper wound closure technique).
Removing EEG electrode headset.
Any unexpected diseases or pathology in WT mice w/o treatment.
Fighting, cage flooding, mishandling (e.g., accidental falling), etc.
Exclusion Criteria Based on Anatomical Assessment of Brain Pathology
Broken skull or bone pieces (or foreign articles) in brain tissue.
Incorrect implantation of EEG electrodes.
Brain pathology that is not associated with experimental TBI, excessive hematoma and/or cavitation statistically distinct from the same group and injury parameters, excessive brain pathology in sham animals.
4 Notes
Circular chamber is used for grading of the seizures induced with bolus PTZ injection to prevent potential injuries of animal during progression of violent seizures. In contrast, PTZ threshold test is normally performed on the table top.
Low-cost infusion pumps are generally not suitable for the PTZ threshold test because of low precision of the delivery of small volumes of solution.
In genetically modified animals, phenotypic or baseline behavioral characteristics might be different from those observed in their wild type controls.
Damaged dura mater, excessive bleeding, or subdural hemorrhage after craniotomy procedure should be considered as exclusion criteria.
If using sound sensor it is critical to verify if impactor tip is positioned properly and touches the brain surface. Excessive amount of saline may result in a false sound signal.
Generally, seizures with a Racine score of 2 or lower are considered as “non-convulsive” and those with a Racine score of 3 and above are identified as “convulsive.”
The number of animals that are excluded from analyses and condition needs to be reported to determine success rate and reproducibility of the technique used in the study.
References
- 1.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 2.Raymont V, Salazar AM, Lipsky R, Goldman D, Tasick G, Grafman J. Correlates of posttraumatic epilepsy 35 years following combat brain injury. Neurology. 2010;75:224–229. doi: 10.1212/WNL.0b013e3181e8e6d0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–1414. doi: 10.1212/wnl.35.10.1406. [DOI] [PubMed] [Google Scholar]
- 4.Scher AI, Wu H, Tsao JW, Blom HJ, Feit P, Nevin RL, Schwab KA. MTHFR C677T genotype as a risk factor for epilepsy including post-traumatic epilepsy in a representative military cohort. J Neurotrauma. 2011;28:1739–1745. doi: 10.1089/neu.2011.1982. [DOI] [PubMed] [Google Scholar]
- 5.Kazemi H, Hashemi-Fesharaki S, Razaghi S, Najafi M, Kolivand PH, Kovac S, Gorji A. Intractable epilepsy and craniocerebral trauma: analysis of 163 patients with blunt and penetrating head injuries sustained in war. Injury. 2012;43:2132–2135. doi: 10.1016/j.injury.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Bruns J, Jr, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia. 2003;44(Suppl 10):2–10. doi: 10.1046/j.1528-1157.44.s10.3.x. [DOI] [PubMed] [Google Scholar]
- 7.Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- 8.Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J, Jr, Forsgren L, French JA, Glynn M, Hesdorffer DC, Lee BI, Mathern GW, Moshe SL, Perucca E, Scheffer IE, Tomson T, Watanabe M, Wiebe S. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–482. doi: 10.1111/epi.12550. [DOI] [PubMed] [Google Scholar]
- 9.Frey LC. Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia. 2003;44(Suppl 10):11–17. doi: 10.1046/j.1528-1157.44.s10.4.x. [DOI] [PubMed] [Google Scholar]
- 10.Cabral RJ, King TT, Scott DF. Epilepsy after two different neurosurgical approaches to the treatment of ruptured intracranial aneurysm. J Neurol Neurosurg Psychiatry. 1976;39:1052–1056. doi: 10.1136/jnnp.39.11.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 12.Diaz-Arrastia R, Agostini MA, Frol AB, Mickey B, Fleckenstein J, Bigio E, Van Ness PC. Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch Neurol. 2000;57:1611–1616. doi: 10.1001/archneur.57.11.1611. [DOI] [PubMed] [Google Scholar]
- 13.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- 14.Risdall JE, Menon DK. Traumatic brain injury. Philos Trans R Soc Lond B Biol Sci. 2011;366:241–250. doi: 10.1098/rstb.2010.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen LL, Baca CB, Choe J, Chen JW, Ayad ME, Cheng EM. Posttraumatic epilepsy in operation enduring freedom/operation Iraqi freedom veterans. Mil Med. 2014;179:492–496. doi: 10.7205/MILMED-D-13-00413. [DOI] [PubMed] [Google Scholar]
- 16.Temkin NR. Risk factors for posttraumatic seizures in adults. Epilepsia. 2003;44(Suppl 10):18–20. doi: 10.1046/j.1528-1157.44.s10.6.x. [DOI] [PubMed] [Google Scholar]
- 17.Statler KD, Swank S, Abildskov T, Bigler ED, White HS. Traumatic brain injury during development reduces minimal clonic seizure thresholds at maturity. Epilepsy Res. 2008;80:163–170. doi: 10.1016/j.eplepsyres.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer G. Epilepsy in the elderly: some clinical and pharmacotherapeutic aspects. Epilepsia. 2001;42(Suppl 3):55–59. doi: 10.1046/j.1528-1157.2001.042suppl.3055.x. [DOI] [PubMed] [Google Scholar]
- 19.Roberts I, Schierhout G, Alderson P. Absence of evidence for the effectiveness of five interventions routinely used in the intensive care management of severe head injury: a systematic review. J Neurol Neurosurg Psychiatry. 1998;65:729–733. doi: 10.1136/jnnp.65.5.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang YH, Liao CC, Chen WF, Ou CY. Characterization of acute post-craniectomy seizures in traumatically brain-injured patients. Seizure. 2015;25:150–154. doi: 10.1016/j.seizure.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Torbic H, Forni AA, Anger KE, Degrado JR, Greenwood BC. Use of antiepileptics for seizure prophylaxis after traumatic brain injury. Am J Health Syst Pharm. 2013;70:759–766. doi: 10.2146/ajhp120203. [DOI] [PubMed] [Google Scholar]
- 22.Beghi E. Overview of studies to prevent posttraumatic epilepsy. Epilepsia. 2003;44(Suppl 10):21–26. doi: 10.1046/j.1528-1157.44.s10.1.x. [DOI] [PubMed] [Google Scholar]
- 23.Jones KE, Puccio AM, Harshman KJ, Falcione B, Benedict N, Jankowitz BT, Stippler M, Fischer M, Sauber-Schatz EK, Fabio A, Darby JM, Okonkwo DO. Levetiracetam versus phenytoin for seizure prophylaxis in severe traumatic brain injury. Neurosurg Focus. 2008;25:E3. doi: 10.3171/FOC.2008.25.10.E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagni CA, Zenga F. Posttraumatic epilepsy with special emphasis on prophylaxis and prevention. Acta Neurochir Suppl. 2005;93:27–34. doi: 10.1007/3-211-27577-0_3. [DOI] [PubMed] [Google Scholar]
- 25.Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–13. doi: 10.1111/j.1528-1167.2008.02005.x. [DOI] [PubMed] [Google Scholar]
- 26.Kinirons P, McCarthy M, Doherty CP, Delanty N. Predicting drug-resistant patients who respond to add-on therapy with levetiracetam. Seizure. 2006;15:387–392. doi: 10.1016/j.seizure.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Michelucci R. Optimizing therapy of seizures in neurosurgery. Neurology. 2006;67:S14–S18. doi: 10.1212/wnl.67.12_suppl_4.s14. [DOI] [PubMed] [Google Scholar]
- 28.Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, Mamdani MB. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792–798. doi: 10.1056/NEJM199809173391202. [DOI] [PubMed] [Google Scholar]
- 29.Zou H, Brayer SW, Hurwitz M, Niyonkuru C, Fowler LE, Wagner AK. Neuroprotective, neuroplastic, and neurobehavioral effects of daily treatment with levetiracetam in experimental traumatic brain injury. Neurorehabil Neural Repair. 2013;27:878–888. doi: 10.1177/1545968313491007. [DOI] [PubMed] [Google Scholar]
- 30.Cernak I, O’Connor C, Vink R. Inhibition of cyclooxygenase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Exp Brain Res. 2002;147:193–199. doi: 10.1007/s00221-002-1245-z. [DOI] [PubMed] [Google Scholar]
- 31.Velioglu SK, Ozmenoglu M. Migraine-related seizures in an epileptic population. Cephalalgia. 1999;19:797–801. doi: 10.1046/j.1468-2982.1999.1909797.x. discussion 766. [DOI] [PubMed] [Google Scholar]
- 32.Cernak I, O’Connor C, Vink R. Activation of cyclo-oxygenase-2 contributes to motor and cognitive dysfunction following diffuse traumatic brain injury in rats. Clin Exp Pharmacol Physiol. 2001;28:922–925. doi: 10.1046/j.1440-1681.2001.03549.x. [DOI] [PubMed] [Google Scholar]
- 33.Jordan KG. Emergency EEG and continuous EEG monitoring in acute ischemic stroke. J Clin Neurophysiol. 2004;21:341–352. [PubMed] [Google Scholar]
- 34.Menon B, Shorvon SD. Ischaemic stroke in adults and epilepsy. Epilepsy Res. 2009;87:1–11. doi: 10.1016/j.eplepsyres.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Jordan KG. Continuous EEG and evoked potential monitoring in the neuroscience intensive care unit. J Clin Neurophysiol. 1993;10:445–475. doi: 10.1097/00004691-199310000-00006. [DOI] [PubMed] [Google Scholar]
- 36.Privitera MD, Strawsburg RH. Electroencephalographic monitoring in the emergency department. Emerg Med Clin North Am. 1994;12:1089–1100. [PubMed] [Google Scholar]
- 37.Jordan KG. Neurophysiologic monitoring in the neuroscience intensive care unit. Neurol Clin. 1995;13:579–626. [PubMed] [Google Scholar]
- 38.Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, Phelps ME, McArthur DL, Caron MJ, Kraus JF, Becker DP. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. 1997;86:241–251. doi: 10.3171/jns.1997.86.2.0241. [DOI] [PubMed] [Google Scholar]
- 39.Claassen J, Mayer SA, Kowalski RG, Emerson RG, Hirsch LJ. Detection of electrographic seizures with continuous EEG monitoring in critically ill patients. Neurology. 2004;62:1743–1748. doi: 10.1212/01.wnl.0000125184.88797.62. [DOI] [PubMed] [Google Scholar]
- 40.Vespa PM, O’Phelan K, Shah M, Mirabelli J, Starkman S, Kidwell C, Saver J, Nuwer MR, Frazee JG, McArthur DA, Martin NA. Acute seizures after intracerebral hemorrhage: a factor in progressive midline shift and outcome. Neurology. 2003;60:1441–1446. doi: 10.1212/01.wnl.0000063316.47591.b4. [DOI] [PubMed] [Google Scholar]
- 41.Grand’Maison F, Reiher J, Leduc CP. Retrospective inventory of EEG abnormalities in partial status epilepticus. Electroencephalogr Clin Neurophysiol. 1991;79:264–270. doi: 10.1016/0013-4694(91)90121-j. [DOI] [PubMed] [Google Scholar]
- 42.Cavazos JE, Jones SM, Cross DJ. Sprouting and synaptic reorganization in the subiculum and CA1 region of the hippocampus in acute and chronic models of partial-onset epilepsy. Neuroscience. 2004;126:677–688. doi: 10.1016/j.neuroscience.2004.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci. 2001;21:8523–8537. doi: 10.1523/JNEUROSCI.21-21-08523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ariza M, Serra-Grabulosa JM, Junque C, Ramirez B, Mataro M, Poca A, Bargallo N, Sahuquillo J. Hippocampal head atrophy after traumatic brain injury. Neuropsychologia. 2006;44:1956–1961. doi: 10.1016/j.neuropsychologia.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Palacios EM, Sala-Llonch R, Junque C, Fernandez-Espejo D, Roig T, Tormos JM, Bargallo N, Vendrell P. Long-term declarative memory deficits in diffuse TBI: correlations with cortical thickness, white matter integrity and hippocampal volume. Cortex. 2013;49:646–657. doi: 10.1016/j.cortex.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 46.Hickey RW, Adelson PD, Johnnides MJ, Davis DS, Yu Z, Rose ME, Chang YF, Graham SH. Cyclooxygenase-2 activity following traumatic brain injury in the developing rat. Pediatr Res. 2007;62:271–276. doi: 10.1203/PDR.0b013e3180db2902. [DOI] [PubMed] [Google Scholar]
- 47.Miller G. Neuropathology. A battle no soldier wants to fight. Science. 2011;333:517–519. doi: 10.1126/science.333.6042.517. [DOI] [PubMed] [Google Scholar]
- 48.Menzler K, Thiel P, Hermsen A, Chen X, Benes L, Miller D, Sure U, Knake S, Rosenow F. The role of underlying structural cause for epilepsy classification: clinical features and prognosis in mesial temporal lobe epilepsy caused by hippocampal sclerosis versus cavernoma. Epilepsia. 2011;52:707–711. doi: 10.1111/j.1528-1167.2011.02984.x. [DOI] [PubMed] [Google Scholar]
- 49.Glushakov AV, Galvis JM, Solaski SL, Doré S. Hippocampal degeneration after traumatic brain injury: the roles of the PGE2 EP1 receptor. J Trauma Care. 2015;1:1007. [Google Scholar]
- 50.Hellewell SC, Yan EB, Agyapomaa DA, Bye N, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates brain tissue damage: analysis of axonal injury and glial responses. J Neurotrauma. 2010;27:1997–2010. doi: 10.1089/neu.2009.1245. [DOI] [PubMed] [Google Scholar]
- 51.Teasdale GM, Graham DI. Craniocerebral trauma: protection and retrieval of the neuronal population after injury. Neurosurgery. 1998;43:723–737. doi: 10.1097/00006123-199810000-00001. discussion 737–728. [DOI] [PubMed] [Google Scholar]
- 52.Franzblau M, Gonzales-Portillo C, Gonzales-Portillo GS, Diamandis T, Borlongan MC, Tajiri N, Borlongan CV. Vascular damage: a persisting pathology common to Alzheimer’s disease and traumatic brain injury. Med Hypotheses. 2013;81:842–845. doi: 10.1016/j.mehy.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferro JM, Pinto F. Poststroke epilepsy: epidemiology, pathophysiology and management. Drugs Aging. 2004;21:639–653. doi: 10.2165/00002512-200421100-00003. [DOI] [PubMed] [Google Scholar]
- 54.Berges S, Moulin T, Berger E, Tatu L, Sablot D, Challier B, Rumbach L. Seizures and epilepsy following strokes: recurrence factors. Eur Neurol. 2000;43:3–8. doi: 10.1159/000008120. [DOI] [PubMed] [Google Scholar]
- 55.Bladin CF, Alexandrov AV, Bellavance A, Bornstein N, Chambers B, Cote R, Lebrun L, Pirisi A, Norris JW. Seizures after stroke: a prospective multicenter study. Arch Neurol. 2000;57:1617–1622. doi: 10.1001/archneur.57.11.1617. [DOI] [PubMed] [Google Scholar]
- 56.Maganti R, Gerber P, Drees C, Chung S. Nonconvulsive status epilepticus. Epilepsy Behav. 2008;12:572–586. doi: 10.1016/j.yebeh.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 57.Silverman IE, Restrepo L, Mathews GC. Poststroke seizures. Arch Neurol. 2002;59:195–201. doi: 10.1001/archneur.59.2.195. [DOI] [PubMed] [Google Scholar]
- 58.Berger AR, Lipton RB, Lesser ML, Lantos G, Portenoy RK. Early seizures following intracerebral hemorrhage: implications for therapy. Neurology. 1988;38:1363–1365. doi: 10.1212/wnl.38.9.1363. [DOI] [PubMed] [Google Scholar]
- 59.Rhoney DH, Tipps LB, Murry KR, Basham MC, Michael DB, Coplin WM. Anticonvulsant prophylaxis and timing of seizures after aneurysmal subarachnoid hemorrhage. Neurology. 2000;55:258–265. doi: 10.1212/wnl.55.2.258. [DOI] [PubMed] [Google Scholar]
- 60.Pinto AN, Canhao P, Ferro JM. Seizures at the onset of subarachnoid haemorrhage. J Neurol. 1996;243:161–164. doi: 10.1007/BF02444009. [DOI] [PubMed] [Google Scholar]
- 61.Claassen J, Peery S, Kreiter KT, Hirsch LJ, Du EY, Connolly ES, Mayer SA. Predictors and clinical impact of epilepsy after subarachnoid hemorrhage. Neurology. 2003;60:208–214. doi: 10.1212/01.wnl.0000038906.71394.de. [DOI] [PubMed] [Google Scholar]
- 62.Burn J, Dennis M, Bamford J, Sandercock P, Wade D, Warlow C. Epileptic seizures after a first stroke: the Oxfordshire Community Stroke Project. BMJ. 1997;315:1582–1587. doi: 10.1136/bmj.315.7122.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willmore LJ, Sypert GW, Munson JV, Hurd RW. Chronic focal epileptiform discharges induced by injection of iron into rat and cat cortex. Science. 1978;200:1501–1503. doi: 10.1126/science.96527. [DOI] [PubMed] [Google Scholar]
- 64.Willmore LJ, Triggs WJ. Iron-induced lipid peroxidation and brain injury responses. Int J Dev Neurosci. 1991;9:175–180. doi: 10.1016/0736-5748(91)90009-b. [DOI] [PubMed] [Google Scholar]
- 65.Kucukkaya B, Aker R, Yuksel M, Onat F, Yalcin AS. Low dose MK-801 protects against iron-induced oxidative changes in a rat model of focal epilepsy. Brain Res. 1998;788:133–136. doi: 10.1016/s0006-8993(97)01544-8. [DOI] [PubMed] [Google Scholar]
- 66.Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma. 2014;31:1180–1193. doi: 10.1089/neu.2013.3080. [DOI] [PubMed] [Google Scholar]
- 67.Willmore LJ, Rubin JJ. Antiperoxidant pretreatment and iron-induced epileptiform discharges in the rat: EEG and histopathologic studies. Neurology. 1981;31:63–69. doi: 10.1212/wnl.31.1.63. [DOI] [PubMed] [Google Scholar]
- 68.Willmore LJ, Hiramatsu M, Kochi H, Mori A. Formation of superoxide radicals after FeCl3 injection into rat isocortex. Brain Res. 1983;277:393–396. doi: 10.1016/0006-8993(83)90954-x. [DOI] [PubMed] [Google Scholar]
- 69.Glushakov AV, Robbins SW, Bracy CL, Narumiya S, Dore S. Prostaglandin F2 alpha FP receptor antagonist improves outcomes after experimental traumatic brain injury. J Neurotrauma. 2013;30:A163. doi: 10.1186/1742-2094-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47:1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- 71.Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, Gidon M, Cohen A, Zumsteg D, Friedman A. Blood-brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79:774–777. doi: 10.1136/jnnp.2007.126425. [DOI] [PubMed] [Google Scholar]
- 72.Huang XJ, Glushakova O, Mondello S, Van K, Hayes RL, Lyeth BG. Acute temporal profiles of serum levels of UCH-L1 and GFAP and relationships to neuronal and astroglial pathology following traumatic brain injury in rats. J Neurotrauma. 2015;32:1179–1189. doi: 10.1089/neu.2015.3873. [DOI] [PubMed] [Google Scholar]
- 73.Glushakova OY, Jeromin A, Martinez J, Johnson D, Denslow N, Streeter J, Hayes RL, Mondello S. Cerebrospinal fluid protein biomarker panel for assessment of neurotoxicity induced by kainic acid in rats. Toxicol Sci. 2012;130:158–167. doi: 10.1093/toxsci/kfs224. [DOI] [PubMed] [Google Scholar]
- 74.Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, Panikashvili D, Shohami E, Jansen SA, Narayan RK, Strauss KI. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dash PK, Mach SA, Moore AN. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J Neurotrauma. 2000;17:69–81. doi: 10.1089/neu.2000.17.69. [DOI] [PubMed] [Google Scholar]
- 77.Yang T, Zhou D, Stefan H. Why mesial temporal lobe epilepsy with hippocampal sclerosis is progressive: uncontrolled inflammation drives disease progression? J Neurol Sci. 2010;296:1–6. doi: 10.1016/j.jns.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 78.Desjardins P, Sauvageau A, Bouthillier A, Navarro D, Hazell AS, Rose C, Butterworth RF. Induction of astrocytic cyclooxygenase-2 in epileptic patients with hippocampal sclerosis. Neurochem Int. 2003;42:299–303. doi: 10.1016/s0197-0186(02)00101-8. [DOI] [PubMed] [Google Scholar]
- 79.Turrin NP, Rivest S. Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol Dis. 2004;16:321–334. doi: 10.1016/j.nbd.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 80.Takemiya T, Maehara M, Matsumura K, Yasuda S, Sugiura H, Yamagata K. Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci Res. 2006;56:103–110. doi: 10.1016/j.neures.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 81.Yasojima K, Schwab C, McGeer EG, McGeer PL. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999;830:226–236. doi: 10.1016/s0006-8993(99)01389-x. [DOI] [PubMed] [Google Scholar]
- 82.Toti P, DE Felice C, Schürfeld K, Stumpo M, Bartolommei S, Lombardi A, Petraglia E, Buonocore G. Cyclooxygenase-2 immunoreactivity in the ischemic neonatal human brain. An autopsy study. J Submicrosc Cytol Pathol. 2001;33:245–249. [PubMed] [Google Scholar]
- 83.Tomimoto H, Akiguchi I, Wakita H, Lin JX, Budka H. Cyclooxygenase-2 is induced in microglia during chronic cerebral ischemia in humans. Acta Neuropathol (Berl) 2000;99:26–30. doi: 10.1007/pl00007402. [DOI] [PubMed] [Google Scholar]
- 84.Tomimoto H, Shibata M, Ihara M, Akiguchi I, Ohtani R, Budka H. A comparative study on the expression of cyclooxygenase and 5-lipoxygenase during cerebral ischemia in humans. Acta Neuropathol (Berl) 2002;104:601–607. doi: 10.1007/s00401-002-0590-0. [DOI] [PubMed] [Google Scholar]
- 85.Kunz T, Marklund N, Hillered L, Oliw EH. Cyclooxygenase-2, prostaglandin synthases, and prostaglandin H2 metabolism in traumatic brain injury in the rat. J Neurotrauma. 2002;19:1051–1064. doi: 10.1089/089771502760341965. [DOI] [PubMed] [Google Scholar]
- 86.Ahmad M, Rose ME, Vagni V, Griffith RP, Dixon CE, Kochanek PM, Hickey RW, Graham SH. Genetic disruption of cyclooxygenase-2 does not improve histological or behavioral outcome after traumatic brain injury in mice. J Neurosci Res. 2008;86:3605–3612. doi: 10.1002/jnr.21809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Strauss KI. Antiinflammatory and neuroprotective actions of COX2 inhibitors in the injured brain. Brain Behav Immun. 2008;22:285–298. doi: 10.1016/j.bbi.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E 2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- 89.Ahmad AS, Saleem S, Ahmad M, Doré S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–270. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- 90.Abe T, Kunz A, Shimamura M, Zhou P, Anrather J, Iadecola C. The neuroprotective effect of prostaglandin E2 EP1 receptor inhibition has a wide therapeutic window, is sustained in time and is not sexually dimorphic. J Cereb Blood Flow Metab. 2009;29:66–72. doi: 10.1038/jcbfm.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ahmad AS, Kim YT, Ahmad M, Maruyama T, Doré S. Selective blockade of PGE2 EP1 receptor protects brain against experimental ischemia and excitotoxicity, and hippocampal slice cultures against oxygen-glucose deprivation. Neurotox Res. 2008;14:343–351. doi: 10.1007/BF03033858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhen G, Kim YT, Li RC, Yocum J, Kapoor N, Langer J, Dobrowolski P, Maruyama T, Narumiya S, Doré S. PGE(2) EP1 receptor exacerbated neurotoxicity in a mouse model of cerebral ischemia and Alzheimer’s disease. Neurobiol Aging. 2011;33:2215–2219. doi: 10.1016/j.neurobiolaging.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Topol EJ. Failing the public health—rofecoxib, Merck, and the FDA. N Engl J Med. 2004;351:1707–1709. doi: 10.1056/NEJMp048286. [DOI] [PubMed] [Google Scholar]
- 94.Glushakov AV, Robbins SW, Bracy CL, Narumiya S, Doré S. Prostaglandin F2alpha FP receptor antagonist improves outcomes after experimental traumatic brain injury. J Neuroinflammation. 2013;10:132. doi: 10.1186/1742-2094-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Willmore LJ, Ueda Y. Posttraumatic epilepsy: hemorrhage, free radicals and the molecular regulation of glutamate. Neurochem Res. 2009;34:688–697. doi: 10.1007/s11064-008-9841-3. [DOI] [PubMed] [Google Scholar]
- 96.Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Royes LF, Ferreira J, Calixto JB, Mello CF. Modulation of pentylenetetrazol-induced seizures by prostaglandin E2 receptors. Neuroscience. 2008;152:1110–1118. doi: 10.1016/j.neuroscience.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 97.Fischborn SV, Soerensen J, Potschka H. Targeting the prostaglandin E2 EP1 receptor and cyclooxygenase-2 in the amygdala kindling model in mice. Epilepsy Res. 2010;91:57–65. doi: 10.1016/j.eplepsyres.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 98.Khatibi NH, Jadhav V, Matus B, Fathali N, Martin R, Applegate R, Tang J, Zhang JH. Prostaglandin E2 EP1 receptor inhibition fails to provide neuroprotection in surgically induced brain-injured mice. Acta Neurochir Suppl. 2011;111:277–281. doi: 10.1007/978-3-7091-0693-8_46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Glushakov AV, Fazal JA, Narumiya S, Dore S. Role of the prostaglandin E2 EP1 receptor in traumatic brain injury. PLoS One. 2014;9:e113689. doi: 10.1371/journal.pone.0113689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shimamura M, Zhou P, Casolla B, Qian L, Capone C, Kurinami H, Iadecola C, Anrather J. Prostaglandin E2 type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J Cereb Blood Flow Metab. 2013;33:1207–1214. doi: 10.1038/jcbfm.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baik EJ, Kim EJ, Lee SH, Moon C. Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res. 1999;843:118–129. doi: 10.1016/s0006-8993(99)01797-7. [DOI] [PubMed] [Google Scholar]
- 102.Kawaguchi K, Hickey RW, Rose ME, Zhu L, Chen J, Graham SH. Cyclooxygenase-2 expression is induced in rat brain after kainate-induced seizures and promotes neuronal death in CA3 hippocampus. Brain Res. 2005;1050:130–137. doi: 10.1016/j.brainres.2005.05.038. [DOI] [PubMed] [Google Scholar]
- 103.Yoshikawa K, Kita Y, Kishimoto K, Shimizu T. Profiling of eicosanoid production in the rat hippocampus during kainate-induced seizure: dual-phase regulation and differential involvement of COX-1 and COX-2. J Biol Chem. 2006;281:14663–14669. doi: 10.1074/jbc.M511089200. [DOI] [PubMed] [Google Scholar]
- 104.Toscano CD, Kingsley PJ, Marnett LJ, Bosetti F. NMDA-induced seizure intensity is enhanced in COX-2 deficient mice. Neurotoxicology. 2008;29:1114–1120. doi: 10.1016/j.neuro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Berchtold-Kanz E, Anhut H, Heldt R, Neufang B, Hertting G. Regional distribution of arachidonic acid metabolites in rat brain following convulsive stimuli. Prostaglandins. 1981;22:65–79. doi: 10.1016/0090-6980(81)90054-x. [DOI] [PubMed] [Google Scholar]
- 106.Steinhauer HB, Anhut H, Hertting G. The synthesis of prostaglandins and thromboxane in the mouse brain in vivo. Influence of drug induced convulsions, hypoxia and the anticonvulsants trimethadione and diazepam. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:53–58. doi: 10.1007/BF00499874. [DOI] [PubMed] [Google Scholar]
- 107.Steinhauer HB, Hertting G. Lowering of the convulsive threshold by non-steroidal anti-inflammatory drugs. Eur J Pharmacol. 1981;69:199–203. doi: 10.1016/0014-2999(81)90414-3. [DOI] [PubMed] [Google Scholar]
- 108.Forstermann U, Heldt R, Hertting G. Increase in brain prostaglandins during convulsions is due to increased neuronal activity and not to hypoxia. Arch Int Pharmacodyn Ther. 1983;263:180–188. [PubMed] [Google Scholar]
- 109.Forstermann U, Heldt R, Hertting G. Effects of intracerebroventricular administration of prostaglandin D2 on behaviour, blood pressure and body temperature as compared to prostaglandins E2 and F2 alpha. Psychopharmacology (Berl) 1983;80:365–370. doi: 10.1007/BF00432122. [DOI] [PubMed] [Google Scholar]
- 110.Forstermann U, Heldt R, Knappen F, Hertting G. Potential anticonvulsive properties of endogenous prostaglandins formed in mouse brain. Brain Res. 1982;240:303–310. doi: 10.1016/0006-8993(82)90225-6. [DOI] [PubMed] [Google Scholar]
- 111.Naffah-Mazzacoratti MG, Bellissimo MI, Cavalheiro EA. Profile of prostaglandin levels in the rat hippocampus in pilocarpine model of epilepsy. Neurochem Int. 1995;27:461–466. doi: 10.1016/0197-0186(95)00053-b. [DOI] [PubMed] [Google Scholar]
- 112.Okada K, Yuhi T, Tsuji S, Yamashita U. Cyclooxygenase-2 expression in the hippocampus of genetically epilepsy susceptible El mice was increased after seizure. Brain Res. 2001;894:332–335. doi: 10.1016/s0006-8993(01)02019-4. [DOI] [PubMed] [Google Scholar]
- 113.Forstermann U, Seregi A, Hertting G. Anticonvulsive effects of endogenous prostaglandins formed in brain of spontaneously convulsing gerbils. Prostaglandins. 1984;27:913–923. doi: 10.1016/s0090-6980(84)80010-6. [DOI] [PubMed] [Google Scholar]
- 114.Folco GC, Longiave D, Bosisio E. Relations between prostaglandin E2, F2alpha, and cyclic nucleotides levels in rat brain and induction of convulsions. Prostaglandins. 1977;13:893–900. doi: 10.1016/0090-6980(77)90218-0. [DOI] [PubMed] [Google Scholar]
- 115.Kim HJ, Chung JI, Lee SH, Jung YS, Moon CH, Baik EJ. Involvement of endogenous prostaglandin F2alpha on kainic acid-induced seizure activity through FP receptor: the mechanism of proconvulsant effects of COX-2 inhibitors. Brain Res. 2008;1193:153–161. doi: 10.1016/j.brainres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 116.Chung JI, Kim AY, Lee SH, Baik EJ. Seizure susceptibility in immature brain due to lack of COX-2-induced PGF2alpha. Exp Neurol. 2013;249:95–103. doi: 10.1016/j.expneurol.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 117.Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- 118.Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma. 1988;5:1–15. doi: 10.1089/neu.1988.5.1. [DOI] [PubMed] [Google Scholar]
- 119.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 120.Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- 121.Cernak I, Savic J, Malicevic Z, Zunic G, Radosevic P, Ivanovic I, Davidovic L. Involvement of the central nervous system in the general response to pulmonary blast injury. J Trauma. 1996;40:S100–S104. doi: 10.1097/00005373-199603001-00023. [DOI] [PubMed] [Google Scholar]
- 122.Diaz-Arrastia R, Agostini MA, Madden CJ, Van Ness PC. Posttraumatic epilepsy: the endophenotypes of a human model of epileptogenesis. Epilepsia. 2009;50(Suppl 2):14–20. doi: 10.1111/j.1528-1167.2008.02006.x. [DOI] [PubMed] [Google Scholar]
- 123.Kovacs SK, Leonessa F, Ling GS. Blast TBI models, neuropathology, and implications for seizure risk. Front Neurol. 2014;5:47. doi: 10.3389/fneur.2014.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li BC, Li Y, Xu C, Wang J, Chen Z, Li G, Zhang J, Hu S, Wang L, Feng H. Blast-induced traumatic brain injury of goats in confined space. Neurol Res. 2014;36:974–982. doi: 10.1179/1743132813Y.0000000314. [DOI] [PubMed] [Google Scholar]
- 125.Cernak I. The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front Neurol. 2010;1:151. doi: 10.3389/fneur.2010.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma. J Neuropathol Exp Neurol. 2000;59:641–651. doi: 10.1093/jnen/59.8.641. [DOI] [PubMed] [Google Scholar]
- 127.Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- 128.Kabadi SV, Hilton GD, Stoica BA, Zapple DN, Faden AI. Fluid-percussioninduced traumatic brain injury model in rats. Nat Protoc. 2010;5:1552–1563. doi: 10.1038/nprot.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hayes RL, Stalhammar D, Povlishock JT, Allen AM, Galinat BJ, Becker DP, Stonnington HH. A new model of concussive brain injury in the cat produced by extradural fluid volume loading: II. Physiological and neuropathological observations. Brain Inj. 1987;1:93–112. doi: 10.3109/02699058709034449. [DOI] [PubMed] [Google Scholar]
- 130.Hartl R, Medary M, Ruge M, Arfors KE, Ghajar J. Blood-brain barrier breakdown occurs early after traumatic brain injury and is not related to white blood cell adherence. Acta Neurochir Suppl. 1997;70:240–242. doi: 10.1007/978-3-7091-6837-0_74. [DOI] [PubMed] [Google Scholar]
- 131.Stalhammar D, Galinat BJ, Allen AM, Becker DP, Stonnington HH, Hayes RL. A new model of concussive brain injury in the cat produced by extradural fluid volume loading: I. Biomechanical properties. Brain Inj. 1987;1:73–91. doi: 10.3109/02699058709034448. [DOI] [PubMed] [Google Scholar]
- 132.Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- 133.McIntosh TK, Noble L, Andrews B, Faden AI. Traumatic brain injury in the rat: characterization of a midline fluid-percussion model. Cent Nerv Syst Trauma. 1987;4:119–134. doi: 10.1089/cns.1987.4.119. [DOI] [PubMed] [Google Scholar]
- 134.Carbonell WS, Maris DO, McCall T, Grady MS. Adaptation of the fluid percussion injury model to the mouse. J Neurotrauma. 1998;15:217–229. doi: 10.1089/neu.1998.15.217. [DOI] [PubMed] [Google Scholar]
- 135.Sullivan HG, Martinez J, Becker DP, Miller JD, Griffith R, Wist AO. Fluid-percussion model of mechanical brain injury in the cat. J Neurosurg. 1976;45:521–534. [PubMed] [Google Scholar]
- 136.Millen JE, Glauser FL, Fairman RP. A comparison of physiological responses to percussive brain trauma in dogs and sheep. J Neurosurg. 1985;62:587–591. doi: 10.3171/jns.1985.62.4.0587. [DOI] [PubMed] [Google Scholar]
- 137.Dixon CE, Lighthall JW, Anderson TE. Physiologic, histopathologic, and cineradiographic characterization of a new fluid-percussion model of experimental brain injury in the rat. J Neurotrauma. 1988;5:91–104. doi: 10.1089/neu.1988.5.91. [DOI] [PubMed] [Google Scholar]
- 138.Alder J, Fujioka W, Lifshitz J, Crockett DP, Thakker-Varia S. Lateral fluid percussion: model of traumatic brain injury in mice. J Vis Exp. 2011;54 doi: 10.3791/3063. pii 3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pfenninger EG, Reith A, Breitig D, Grunert A, Ahnefeld FW. Early changes of intracranial pressure, perfusion pressure, and blood flow after acute head injury. Part 1: an experimental study of the underlying pathophysiology. J Neurosurg. 1989;70:774–779. doi: 10.3171/jns.1989.70.5.0774. [DOI] [PubMed] [Google Scholar]
- 140.Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- 141.Zink BJ, Walsh RF, Feustel PJ. Effects of ethanol in traumatic brain injury. J Neurotrauma. 1993;10:275–286. doi: 10.1089/neu.1993.10.275. [DOI] [PubMed] [Google Scholar]
- 142.Santhakumar V, Ratzliff AD, Jeng J, Toth Z, Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol. 2001;50:708–717. doi: 10.1002/ana.1230. [DOI] [PubMed] [Google Scholar]
- 143.Neuberger EJ, Abdul Wahab R, Jayakumar A, Pfister BJ, Santhakumar V. Distinct effect of impact rise times on immediate and early neuropathology after brain injury in juvenile rats. J Neurosci Res. 2014;92:1350–1361. doi: 10.1002/jnr.23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang Y, Hameed MQ, Rakhade SN, Iglesias AH, Muller PA, Mou DL, Rotenberg A. Hippocampal immediate early gene transcription in the rat fluid percussion traumatic brain injury model. Neuroreport. 2014;25:954–959. doi: 10.1097/WNR.0000000000000219. [DOI] [PubMed] [Google Scholar]
- 146.D’Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL, Miller JW. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127:304–314. doi: 10.1093/brain/awh038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Impaired K(+) homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci. 1999;19:8152–8162. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cao R, Hasuo H, Ooba S, Akasu T, Zhang X. Facilitation of glutamatergic synaptic transmission in hippocampal CA1 area of rats with traumatic brain injury. Neurosci Lett. 2006;401:136–141. doi: 10.1016/j.neulet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 149.Toth Z, Hollrigel GS, Gorcs T, Soltesz I. Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci. 1997;17:8106–8117. doi: 10.1523/JNEUROSCI.17-21-08106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Coulter DA, Rafiq A, Shumate M, Gong QZ, DeLorenzo RJ, Lyeth BG. Brain injury-induced enhanced limbic epileptogenesis: anatomical and physiological parallels to an animal model of temporal lobe epilepsy. Epilepsy Res. 1996;26:81–91. doi: 10.1016/s0920-1211(96)00044-7. [DOI] [PubMed] [Google Scholar]
- 151.Dewitt DS, Kong DL, Lyeth BG, Jenkins LW, Hayes RL, Wooten ED, Prough DS. Experimental traumatic brain injury elevates brain prostaglandin E2 and thromboxane B2 levels in rats. J Neurotrauma. 1988;5:303–313. doi: 10.1089/neu.1988.5.303. [DOI] [PubMed] [Google Scholar]
- 152.D’Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, Miller JW. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain. 2005;128:174–188. doi: 10.1093/brain/awh337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Curia G, Levitt M, Fender JS, Miller JW, Ojemann J, D’Ambrosio R. Impact of injury location and severity on posttraumatic epilepsy in the rat: role of frontal neocortex. Cereb Cortex. 2011;21:1574–1592. doi: 10.1093/cercor/bhq218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Campbell JN, Gandhi A, Singh B, Churn SB. Traumatic brain injury causes a tacrolimus-sensitive increase in non-convulsive seizures in a rat model of post-traumatic epilepsy. Int J Neurol Brain Disord. 2014;1:1–11. doi: 10.15436/2377-1348.14.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kharatishvili I, Nissinen JP, McIntosh TK, Pitkanen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140:685–697. doi: 10.1016/j.neuroscience.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 156.Bao YH, Bramlett HM, Atkins CM, Truettner JS, Lotocki G, Alonso OF, Dietrich WD. Post-traumatic seizures exacerbate histopathological damage after fluid-percussion brain injury. J Neurotrauma. 2011;28:35–42. doi: 10.1089/neu.2010.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huusko N, Pitkanen A. Parvalbumin immunoreactivity and expression of GABAA receptor subunits in the thalamus after experimental TBI. Neuroscience. 2014;267:30–45. doi: 10.1016/j.neuroscience.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 158.Atkins CM, Truettner JS, Lotocki G, Sanchez-Molano J, Kang Y, Alonso OF, Sick TJ, Dietrich WD, Bramlett HM. Post-traumatic seizure susceptibility is attenuated by hypothermia therapy. Eur J Neurosci. 2010;32:1912–1920. doi: 10.1111/j.1460-9568.2010.07467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mishra AM, Bai X, Sanganahalli BG, Waxman SG, Shatillo O, Grohn O, Hyder F, Pitkanen A, Blumenfeld H. Decreased resting functional connectivity after traumatic brain injury in the rat. PLoS One. 2014;9:e95280. doi: 10.1371/journal.pone.0095280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wallis RA, Panizzon KL. Felbamate neuroprotection against CA1 traumatic neuronal injury. Eur J Pharmacol. 1995;294:475–482. doi: 10.1016/0014-2999(95)00568-4. [DOI] [PubMed] [Google Scholar]
- 161.Pitkanen A, Immonen R, Ndode-Ekane X, Grohn O, Stohr T, Nissinen J. Effect of lacosamide on structural damage and functional recovery after traumatic brain injury in rats. Epilepsy Res. 2014;108:653–665. doi: 10.1016/j.eplepsyres.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 162.Lighthall JW, Goshgarian HG, Pinderski CR. Characterization of axonal injury produced by controlled cortical impact. J Neurotrauma. 1990;7:65–76. doi: 10.1089/neu.1990.7.65. [DOI] [PubMed] [Google Scholar]
- 163.Hamm RJ, Dixon CE, Gbadebo DM, Singha AK, Jenkins LW, Lyeth BG, Hayes RL. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma. 1992;9:11–20. doi: 10.1089/neu.1992.9.11. [DOI] [PubMed] [Google Scholar]
- 164.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 165.Brody DL, MacDonald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E, Schwetye KE, Holtzman DM, Bayly PV. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma. 2007;24:657–673. doi: 10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lighthall JW, Dixon CE, Anderson TE. Experimental models of brain injury. J Neurotrauma. 1989;6:83–97. doi: 10.1089/neu.1989.6.83. [DOI] [PubMed] [Google Scholar]
- 167.Sanders MJ, Dietrich WD, Green EJ. Cognitive function following traumatic brain injury: effects of injury severity and recovery period in a parasagittal fluid-percussive injury model. J Neurotrauma. 1999;16:915–925. doi: 10.1089/neu.1999.16.915. [DOI] [PubMed] [Google Scholar]
- 168.Alessandri B, Heimann A, Filippi R, Kopacz L, Kempski O. Moderate controlled cortical contusion in pigs: effects on multi-parametric neuromonitoring and clinical relevance. J Neurotrauma. 2003;20:1293–1305. doi: 10.1089/089771503322686094. [DOI] [PubMed] [Google Scholar]
- 169.Manley GT, Rosenthal G, Lam M, Morabito D, Yan D, Derugin N, Bollen A, Knudson MM, Panter SS. Controlled cortical impact in swine: pathophysiology and biomechanics. J Neurotrauma. 2006;23:128–139. doi: 10.1089/neu.2006.23.128. [DOI] [PubMed] [Google Scholar]
- 170.King C, Robinson T, Dixon CE, Rao GR, Larnard D, Nemoto CE. Brain temperature profiles during epidural cooling with the ChillerPad in a monkey model of traumatic brain injury. J Neurotrauma. 2010;27:1895–1903. doi: 10.1089/neu.2009.1178. [DOI] [PubMed] [Google Scholar]
- 171.Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma. 2005;22:252–265. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- 172.Goodman JC, Cherian L, Bryan RM, Jr, Robertson CS. Lateral cortical impact injury in rats: pathologic effects of varying cortical compression and impact velocity. J Neurotrauma. 1994;11:587–597. doi: 10.1089/neu.1994.11.587. [DOI] [PubMed] [Google Scholar]
- 173.Yang L, Afroz S, Michelson HB, Goodman JH, Valsamis HA, Ling DS. Spontaneous epileptiform activity in rat neocortex after controlled cortical impact injury. J Neurotrauma. 2010;27:1541–1548. doi: 10.1089/neu.2009.1244. [DOI] [PubMed] [Google Scholar]
- 174.Sutton RL, Lescaudron L, Stein DG. Unilateral cortical contusion injury in the rat: vascular disruption and temporal development of cortical necrosis. J Neurotrauma. 1993;10:135–149. doi: 10.1089/neu.1993.10.135. [DOI] [PubMed] [Google Scholar]
- 175.Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- 176.Dixon CE, Kraus MF, Kline AE, Ma X, Yan HQ, Griffith RG, Wolfson BM, Marion DW. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restor Neurol Neurosci. 1999;14:285–294. [PubMed] [Google Scholar]
- 177.Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, DeKosky ST. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- 178.Vink R, Mullins PG, Temple MD, Bao W, Faden AI. Small shifts in craniotomy position in the lateral fluid percussion injury model are associated with differential lesion development. J Neurotrauma. 2001;18:839–847. doi: 10.1089/089771501316919201. [DOI] [PubMed] [Google Scholar]
- 179.Fox GB, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- 180.Kochanek PM, Hendrich KS, Dixon CE, Schiding JK, Williams DS, Ho C. Cerebral blood flow at one year after controlled cortical impact in rats: assessment by magnetic resonance imaging. J Neurotrauma. 2002;19:1029–1037. doi: 10.1089/089771502760341947. [DOI] [PubMed] [Google Scholar]
- 181.Hunt RF, Haselhorst LA, Schoch KM, Bach EC, Rios-Pilier J, Scheff SW, Saatman KE, Smith BN. Posttraumatic mossy fiber sprouting is related to the degree of cortical damage in three mouse strains. Epilepsy Res. 2012;99:167–170. doi: 10.1016/j.eplepsyres.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, Jenkins LW, Clark RS, Homanics GE, Dixon CE, Schnermann J, Jackson EK. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26:565–575. doi: 10.1038/sj.jcbfm.9600218. [DOI] [PubMed] [Google Scholar]
- 183.Haselkorn ML, Shellington DK, Jackson EK, Vagni VA, Janesko-Feldman K, Dubey RK, Gillespie DG, Cheng D, Bell MJ, Jenkins LW, Homanics GE, Schnermann J, Kochanek PM. Adenosine A1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice. J Neurotrauma. 2010;27:901–910. doi: 10.1089/neu.2009.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zweckberger K, Simunovic F, Kiening KL, Unterberg AW, Sakowitz OW. Anticonvulsive effects of the dopamine agonist lisuride maleate after experimental traumatic brain injury. Neurosci Lett. 2010;470:150–154. doi: 10.1016/j.neulet.2009.12.075. [DOI] [PubMed] [Google Scholar]
- 185.Hunt RF, Scheff SW, Smith BN. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol. 2009;215:243–252. doi: 10.1016/j.expneurol.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One. 2013;8:e64078. doi: 10.1371/journal.pone.0064078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lei Z, Deng P, Li J, Xu ZC. Alterations of A-type potassium channels in hippocampal neurons after traumatic brain injury. J Neurotrauma. 2012;29:235–245. doi: 10.1089/neu.2010.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bolkvadze T, Pitkanen A. Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma. 2012;29:789–812. doi: 10.1089/neu.2011.1954. [DOI] [PubMed] [Google Scholar]
- 189.Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE. A potential model of pediatric posttraumatic epilepsy. Epilepsy Res. 2009;86:221–223. doi: 10.1016/j.eplepsyres.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hunt RF, Scheff SW, Smith BN. Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J Neurosci. 2011;31:6880–6890. doi: 10.1523/JNEUROSCI.0032-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kharlamov EA, Lepsveridze E, Meparishvili M, Solomonia RO, Lu B, Miller ER, Kelly KM, Mtchedlishvili Z. Alterations of GABA(A) and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res. 2011;95:20–34. doi: 10.1016/j.eplepsyres.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 192.Osselton JW. Acquisition of EEG data by bipolar, unipolar and average reference methods: a theoretical comparison. Electroencephalogr Clin Neurophysiol. 1965;19:527–528. doi: 10.1016/0013-4694(65)90195-1. [DOI] [PubMed] [Google Scholar]
- 193.Stover JF, Sakowitz OW, Beyer TF, Dohse NK, Kroppenstedt SN, Thomale UW, Schaser KD, Unterberg AW. Effects of LY379268, a selective group II metabotropic glutamate receptor agonist on EEG activity, cortical perfusion, tissue damage, and cortical glutamate, glucose, and lactate levels in brain-injured rats. J Neurotrauma. 2003;20:315–326. doi: 10.1089/089771503765172273. [DOI] [PubMed] [Google Scholar]
- 194.Frey L, Lepkin A, Schickedanz A, Huber K, Brown MS, Serkova N. ADC mapping and T1-weighted signal changes on post-injury MRI predict seizure susceptibility after experimental traumatic brain injury. Neurol Res. 2014;36:26–37. doi: 10.1179/1743132813Y.0000000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Macolino CM, Daiutolo BV, Albertson BK, Elliott MB. Mechanical alloydnia induced by traumatic brain injury is independent of restraint stress. J Neurosci Methods. 2014;226:139–146. doi: 10.1016/j.jneumeth.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 196.Sanchez JC, Alba N, Nishida T, Batich C, Carney PR. Structural modifications in chronic microwire electrodes for cortical neuroprosthetics: a case study. IEEE Trans Neural Syst Rehabil Eng. 2006;14:217–221. doi: 10.1109/TNSRE.2006.875581. [DOI] [PubMed] [Google Scholar]
- 197.Nandan M, Talathi SS, Myers S, Ditto WL, Khargonekar PP, Carney PR. Support vector machines for seizure detection in an animal model of chronic epilepsy. J Neural Eng. 2010;7:036001. doi: 10.1088/1741-2560/7/3/036001. [DOI] [PubMed] [Google Scholar]
- 198.MacLennan AJ, Carney PR, Zhu WJ, Chaves AH, Garcia J, Grimes JR, Anderson KJ, Roper SN, Lee N. An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. Eur J Neurosci. 2001;14:203–209. doi: 10.1046/j.0953-816x.2001.01634.x. [DOI] [PubMed] [Google Scholar]
- 199.Lu XC, Williams AJ, Tortella FC. Quantitative electroencephalography spectral analysis and topographic mapping in a rat model of middle cerebral artery occlusion. Neuropathol Appl Neurobiol. 2001;27:481–495. doi: 10.1046/j.1365-2990.2001.00357.x. [DOI] [PubMed] [Google Scholar]
- 200.Cao W, Glushakov A, Shah HP, Mecca AP, Sumners C, Shi P, Seubert CN, Martynyuk AE. Halogenated aromatic amino acid 3,5-dibromo-D: -tyrosine produces beneficial effects in experimental stroke and seizures. Amino Acids. 2011;40:1151–1158. doi: 10.1007/s00726-010-0739-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cao W, Shah HP, Glushakov AV, Mecca AP, Shi P, Sumners C, Seubert CN, Martynyuk AE. Efficacy of 3,5-dibromo-L-phenylalanine in rat models of stroke, seizures and sensorimotor gating deficit. Br J Pharmacol. 2009;158:2005–2013. doi: 10.1111/j.1476-5381.2009.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lopez PH, Ahmad AS, Mehta NR, Toner M, Rowland EA, Zhang J, Dore S, Schnaar RL. Myelin-associated glycoprotein protects neurons from excitotoxicity. J Neurochem. 2011;116:900–908. doi: 10.1111/j.1471-4159.2010.07069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Martynyuk AE, Seubert CN, Yarotskyy V, Glushakov AV, Gravenstein N, Sumners C, Dennis DM. Halogenated derivatives of aromatic amino acids exhibit balanced anti-glutamatergic actions: potential applications for the treatment of neurological and neuropsychiatric disorders. Recent Pat CNS Drug Discov. 2006;1:261–270. doi: 10.2174/157488906778773706. [DOI] [PubMed] [Google Scholar]
- 204.Moezi L, Shafaroodi H, Hojati A, Dehpour AR. The interaction of melatonin and agmatine on pentylenetetrazole-induced seizure threshold in mice. Epilepsy Behav. 2011;22:200–206. doi: 10.1016/j.yebeh.2011.07.002. [DOI] [PubMed] [Google Scholar]