

Abstract
Protein kinase signaling pathways, such as Janus kinase 2-Signal transducer and activator of transcription 5A/B (JAK2-STAT5A/B), are of significant interest in the search for new therapeutic strategies in both breast and prostate cancers. In prostate cancer, the components of the JAK2-STAT5A/B signaling pathway provide molecular targets for small-molecule inhibition of survival and growth signals of the cells. At the same time, new evidence suggests that the STAT5A/B signaling pathway is involved in the transition of organ-confined prostate cancer to hormone-refractory disease. This implies that the active JAK2-STAT5A/B signaling pathway potentially provides the means for pharmacological intervention of clinical prostate cancer progression. In addition, active STAT5A/B may serve as a prognostic marker for identification of those primary prostate cancers that are likely to progress to aggressive disease. In breast cancer, the role of STAT5A/B is more complex. STAT5A/B may have a dual role in the regulation of malignant mammary epithelium. Data accumulated from mouse models of breast cancer suggest that in early stages of breast cancer STAT5A/B may promote malignant transformation and enhance growth of the tumor. This is in contrast to established breast cancer, where STAT5A/B may mediate the critical cues for maintaining the differentiation of mammary epithelium. In addition, present data suggest that activation of STAT5A/B in breast cancer predicts favorable clinical outcome. The dual nature of STAT5A/B action in breast cancer makes the therapeutic use of STAT5 A/B more complex.
Introduction
Signal transducer and activator of transcription 5 (STAT5) was originally identified in the mouse mammary gland (Schmitt-Ney et al. 1991) and it was further characterized in the mouse system (Schmitt-Ney et al. 1992a,b, Wakao et al. 1992) before a ‘mammary gland factor’ (MGF) was cloned from sheep mammary gland. MGF was identified as the signaling protein that mediates the effects of prolactin (PRL) (Gouilleux et al. 1994, Wakao et al. 1994). It soon became clear that MGF had a significant sequence homology with the members of the STAT transcription factor family, and MGF was renamed STAT5 (Gouilleux et al. 1994, 1995a,b, Wakao et al. 1994). Later, another isoform of STAT5 was discovered in the mammary gland (Liu et al. 1995, 1996a, Lin et al. 1996), which was encoded by a separate gene. This STAT5 homolog was named STAT5B, whereas the original MGF carried the name STAT5A. Further studies revealed that STAT5B was a crucial signaling protein mediating the biological effects of growth hormone (GH), while the key function of STAT5A was to transduce the signals initiated by PRL receptors. In parallel with these significant findings, it was realized that STAT5A/B becomes activated by phosphorylation on a tyrosine residue (Gouilleux et al. 1994, Liu et al. 1995, 1996a). Concurrently, the PRL receptor-associated tyrosine kinase, Janus kinase 2 (JAK2), was discovered (Rui et al. 1994).
At the same time as the components of the PRL receptor-JAK2-STAT5A/B signaling pathway were identified, understanding of PRL as a classical peptide hormone underwent significant changes. Specifically, it was realized that PRL, which is known as a pituitary hormone secreted by the cells in the anterior lobe of the hypophysis, is actually produced as a local growth factor both in normal and malignant prostate (Nevalainen et al. 1997a,b, Li et al. 2004, Dagvadorj et al. 2007) and mammary glands (Ginsburg & Vonderhaar 1995). It was known that PRL has both mitogenic and secretory effects on breast epithelial cells. Unexpectedly, PRL was found to be a significant mitogen and survival factor for prostate epithelial cells as well (Nevalainen et al. 1991, 1997b, Wennbo et al. 1997, Kindblom et al. 2002, 2003). Autocrine PRL in prostate and breast tissue became important findings, since local production of PRL provided an explanation as to why the results of studies trying to link breast or prostate cancer incidence or progression with the circulating PRL levels yielded controversial results. Potential molecular mechanisms and factors, in addition to pituitary and autocrine GH and PRL underlying the constitutive activation of STAT5A/B signaling pathway in prostate and breast cancers will be discussed. Furthermore, involvement of STAT5A/B signaling pathway in the regulation of growth and progression of prostate and breast cancers will be reviewed.
Structure and function of STAT5A/B proteins
The STAT family of transcription factors has seven members (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) that are all encoded by separate genes (Darnell et al. 1994, Zhong et al. 1994b). The STAT proteins are likely to have diverged from a single gene through several consecutive duplications into three genetic loci. Specifically, in humans, the STAT genes map to three chromosomal regions (Copeland et al. 1995): STAT3, STAT5A, and STAT5B map to chromosome 17 (bands q11-1 to q22); STAT1 and STAT4 map to chromosome 2 (bands q12 to q33) (Yamamoto et al. 1997, Haddad et al. 1998); and STAT2 and STAT6 map to chromosome 12 (bands q13 to q14-1) (Leek et al. 1997, Goureau et al. 2001; Table 1). Differential splicing and proteolytic processing further increase the diversity of STAT function in cells (Haddad et al. 1998, Ihle 2001). In the mouse, Stat1 and Stat4 are located on chromosome 1 (band 1 C1.1) (Schindler et al. 1992, Yamamoto et al. 1994); Stat2 and Stat6 are on chromosome 10 (band 10 D3) (Fu et al. 1992, Quelle et al. 1995); and Stat3, Stat5a, and Stat5b map to chromosome 11 (band 11 D) (Zhong et al. 1994a, Copeland et al. 1995, Shi et al. 1996, Levy et al. 1998; Table 1).
Table 1.
Chromosomal mapping and sequence identity between human and mouse signal transducer and activator of transcriptions (STATs)
| STAT | Human | Mouse | % Identity | References | ||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Amino acid | Chromosome | Amino acid | Chromosome | |||
| STAT1 | 750 | 2 | 749 | 1 | 93 | Schindler et al. (1992) and Haddad et al. (1998) |
| STAT2 | 846 | 12 | 915 | 10 | 64 | Fu et al. (1992) and Goureau et al. (2001) |
| STAT3 | 769 | 17 | 769 | 11 | 99 | Zhong et al. (1994a) and Choi et al. (1996) |
| STAT4 | 748 | 2 | 748 | 1 | 94 | Yamamoto et al. (1994, 1997) |
| STAT5A | 794 | 17 | 793 | 11 | 96 | Mui et al. (1995) and Lin et al. (1996) |
| STAT5B | 787 | 17 | 786 | 11 | 96 | Mui et al. (1995) and Lin et al. (1996) |
| STAT6 | 847 | 12 | 837 | 10 | 85 | Quelle et al. (1995) and Leek et al. (1997) |
STATs are proteins of 750–900 amino acids (90–115 kDa) with five structurally and functionally conserved domains that allow the transduction of ligand-specific signals (Schindler & Darnell 1995). The 94 kDa STAT5A (human, 794 amino acids; mouse, 793 amino acids) and 92 kDa STAT5B (human, 787 amino acids; mouse 786 aa) are distinct, but highly homologous isoforms (Mui et al. 1994, Liu et al. 1995, 1996a) (Table 1). Both carry five domains (Table 2) that are structurally and functionally conserved with other STAT proteins and allow the transduction of ligand-specific signals (Schindler & Darnell 1995) (Fig. 1). There is a unique stretch of 20 and 8 amino acids at the C-terminus of STAT5A and STAT5B respectively. The domain of STAT5A/B that is most highly conserved with other STAT proteins is the SH2 domain (aa 593–670, Table 1) that mediates both receptor-specific recruitment and Stat dimerization (Stocklin et al. 1996) through the phosphorylated tyrosine residue of one STAT5 to the SH2 domain of another (Wakao et al. 1994, Welte et al. 1994). The central DNA-binding domain (DBD, aa 332–583) of STAT5A/B is another conserved domain that allows binding of STAT5A/B to consensus GAS (γ-interferon activation sequence) sites (TTC(C/T)N(G/A)GAA) within gene regulatory elements (Decker et al. 1991, Horvath et al. 1995, Soldaini et al. 2000). The linker domain (aa 475–592) of STATs, named based on crystallographic studies, in fact overlaps in function with the DBD (Yang et al. 2002). Site-directed mutagenesis of the STAT1 linker domain resulted in a mutant dimer that binds and dissociates from DNA more rapidly than the wild-type protein (Yang et al. 2002). The N-terminal domain of STAT5A/B (aa 1–126) stabilizes interactions between two STAT dimers to form tetramers. STAT5–DNA interaction, particularly at adjacent non-consensus STAT5-binding sites, is reinforced by tetramerization that enhances transcriptional activation of weak promoters (Horvath et al. 1995, Meyer et al. 1997, John et al. 1999, Soldaini et al. 2000). Homodimers of STAT5A and STAT5B were found to share similar binding specificities for half palindromes that are spaced 3 bp apart (Soldaini et al. 2000, Ehret et al. 2001). The non-redundant functions of STAT5A and STAT5B during development are likely due to their cell type-specific expression or due to the interactions of their divergent C-terminus with different co-regulators rather than a result of differences in DNA-binding specificity (Ehret et al. 2001). Tetrameric binding is made possible by tandemly linked GAS (gamma - interferon activation sequence) motifs with non-consensus motifs that are optimally spaced 6 bp apart (Soldaini et al. 2000).
Table 2.
Location of conserved functional domains of human and mouse signal transducer and activator of transcription 5A/B (STAT5A/B)
| Human | Mouse | |||
|---|---|---|---|---|
|
|
|
|||
| STAT5A | STAT5B | STAT5a | STAT5b | |
| N-domain | 1–126 | 1–126 | 1–126 | 1–126 |
| Coiled-coil domain (STAT protein, all-α domain) | 138–330 | 138–330 | 138–330 | 138–330 |
| DNA-binding domain | 332–583 | 332–583 | 332–583 | 332–583 |
| Linker domain | 475–592 | 475–592 | 475–592 | 475–592 |
| SH2 domain | 593–670 | 593–670 | 593–670 | 593–670 |
| Phosphotyrosyl segment | 686–701 | 686–706 | 686–701 | 686–706 |
| Transactivation domain | 722–794 | 727–787 | 721–793 | 726–786 |
Figure 1.
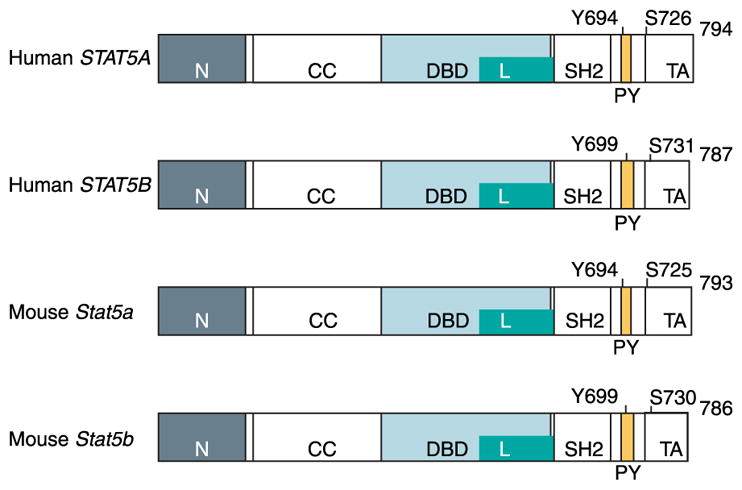
The major structural and functional domains and the phosphorylation sites of human and mouse STAT5A and STAT5B proteins. STAT5 proteins share an overall general structure that is organized into functional modular domains: N-terminal domain (N), coiled-coil domain (CC), DNA-binding domain (DBD), linker domain (L), SH2 domain (SH2), and transactivation domain (TA). All STAT molecules have a highly conserved tyrosine phosphorylation site (Y) at or around residue 700, labeled as the phosphotyrosyl segment (PY). Tyrosine phosphorylation follows ligand-induced activation and is required for dimerization and nuclear translocation.
The glycosylation on threonine 92 of STAT5 is found to enhance interaction with the co-activator of transcription CREB-binding protein (CBP; Gewinner et al. 2004). Adjacent to the N-terminus of STAT5A/B is the coiled-coil domain (aa 138–330), consisting of a four-helix bundle (Soldaini et al. 2000) (Fig. 1; Table 2), which facilitates multiple protein–protein interactions crucial for transcriptional regulation by interacting with chaperones (Xu et al. 2004), with Nmi, which helps the recruitment of co-activators (Zhu et al. 1999), and with co-repressors (Nakajima et al. 2001, Maurer et al. 2002). The transcriptional activation domain in the C-terminus of STAT5A/B (STAT5A, aa 722–794; STAT5B, aa 727–787) is the most variable region and interacts with critical co-activators, including the p300/CBP-associated co-activator NcoA-1 (Litterst et al. 2003), centrosomal P4.1-associated protein (Peng et al. 2002), P100 (Paukku et al. 2003) and Oct-1 (Magne et al. 2003, Litterst et al. 2005).
In contrast to other STAT transcription factors that have a narrow activation profile, STAT5A and STAT5B transduce signals triggered by multiple ligands (Table 3). In addition to being activated by PRL (Kazansky et al. 1999), STAT5A/B are activated by interleukin-2 (IL-2; Hou et al. 1995), IL-3 (Mui et al. 1995), IL-5 (Mui et al. 1995)), and IL-7 (Foxwell et al. 1995), granulocyte–macrophage colony-stimulating factor (Barahmand-pour et al. 1995, Mui et al. 1995), insulin (Wartmann et al. 1996), erythropoietin (Wakao et al. 1994, Gouilleux et al. 1995b, Pallard et al. 1995b), thrombopoietin (Pallard et al. 1995a), and GH (Gouilleux et al. 1995b, Galsgaard et al. 1996) (Table 3).
Table 3.
Ligands activating signal transducer and activator of transcription 5A/B (STAT5A/B)
| Ligand | Reference |
|---|---|
| Prolactin | Gouilleaux et al. (1994) and Wakao et al. (1994) |
| IL-2 | Hou et al. (1995) |
| IL-3 | Mui et al. (1995) |
| IL-5 | Mui et al. (1995) |
| IL7 | Foxwell et al. (1995) |
| Granulocyte–macrophage colony-stimulating factor (GM-CSF) | Barahmand-pour et al. (1995), Gouilleux et al. (1995a), Mui et al. (1995), Pallard et al. (1995a) and Rosen et al. (1996) |
| Insulin | Wartmann et al. (1996) and Chen et al. (1997) |
| Erythropoietin (EPO) | Gouilleux et al. (1995b), Pallard et al. (1995b) |
| Thrombopoietin (TPO) | Pallard et al. (1995a) |
| GH | Gouilleux et al. (1995b) and Galsgaard et al. (1996) |
Initially, insight into the distinct roles of STAT5A/B in mediating biological responses was gleaned mainly from gene targeting studies, which revealed that the phenotypes of Stat5a/b knockout (KO) mice are not fully functionally redundant. Stat5a-null female mice are defective in PRL-dependent mammary gland development (Liu et al. 1997), whereas Stat5a-null male mice exhibit defective prostate epithelium (Nevalainen et al. 2000). By contrast, Stat5b-null mice fail to respond effectively to GH (Udy et al. 1997, Teglund et al. 1998) and are severely anemic, indicating defective hematopoiesis due to impaired response to hematopoietins (Socolovsky et al. 1999). In Stat5a/b double KO mice, lymphoid development and differentiation were impaired (Yao et al. 2006). In addition, T-cell receptor γ-rearrangement and peripheral CD8+T-cell survival were abrogated in the absence of Stat5a/b (Yao et al. 2006).
The Jak-STAT5A/B signaling cascade
STAT5A/B-activating cytokine receptors do not typically posses tyrosine kinase activity that is provided by receptor-associated cytoplasmic proteins from the JAK family (Darnell 1997, Ihle 2001, Schindler 2002). In mammalian cells, there are four JAK proteins (120–130 kDa), JAK1, JAK2, JAK3, and TYK2 (tyrosine kinase 2) (Darnell 1997, Ihle 2001, Aaronson & Horvath 2002, Levy & Darnell 2002), which are, except for JAK3, ubiquitously expressed (Leonard & O’Shea 1998). JAK proteins have seven highly homologous domains. Located at the carboxyl terminus, the JAK homology domain 1 (JH1) has the kinase activity. Directly upstream of JH1 is the JH2 pseudokinase domain that resembles JH1 but has a negative regulatory function. The domains that mediate association with cytokine receptors are JH3–JH7 at the amino-terminus of JAKs, which constitute a four-point-one, ezrin, radixin, moesin domain (Schindler & Darnell 1995, Darnell 1997, Ihle 2001, Aaronson & Horvath 2002, Levy & Darnell 2002, Schindler 2002). The primary JAK protein that activates STAT5A/B is JAK2 (Gouilleux et al. 1994).
Ligand-induced receptor dimerization brings two JAK2 molecules into close proximity allowing them to phosphorylate specific tyrosine motifs of the receptor and activate each other (Fig. 2). STAT5A/B and other signaling molecules that recognize these tyrosine motifs, typically through their SH2 domains, are recruited to the docking sites. This is followed by rapid phosphorylation of a conserved tyrosine residue in the C-terminus of STAT5A/B by JAK2. Phosphorylation of the tyrosine residues Y694 and Y699 (identical in human and mouse) activates STAT5A and STAT5B respectively, leading them to homo- or heterodimerize through a phosphotyrosine–SH2 domain (Becker et al. 1998, Chen et al. 1998). A variety of protein kinases phosphorylate STATs on serine residues, allowing additional signaling pathways to potentiate the primary STAT-activating stimulus (Decker & Kovarik 2000). By contrast, phosphorylation of the serine residues, S726 (human)/S725 (mouse) on STAT5A (Beuvink et al. 2000) and S731 (human)/S730 (mouse) on STAT5B, may inhibit the transcriptional activity of STAT5 (Kirken et al. 1997a,b, Yamashita et al. 2001).
Figure 2.

Functional interaction between JAK2-STAT5A/B and androgen receptor (AR) signaling in prostate cancer cells. Binding of prolactin (PRL) to prolactin receptor (PRLR) results in receptor dimerization and activates the receptor associated kinase, Janus kinase 2 (JAK2). JAK2 phosphorylates the PRLR on tyrosine residues, thereby creating docking sites for the SH2 domain of STAT5A/B monomers. STAT5A/B molecules are phosphorylated on unique tyrosine residues by JAK2, dissociate from the receptors, dimerize, and translocate to the nucleus where they bind to specific STAT5A/B-binding GAS (γ-interferon activation sequence) sites within the promoters of target genes. Binding of androgens (DHT) to the androgen receptor (AR) induces the dissociation from heat shock proteins (HSPs), receptor phosphorylation, and dimerization. Dimerized AR is translocated into the nucleus and bind to androgen response elements at the promoter of target genes. In prostate cancer cells, liganded AR interacts with activated STAT5A/B and enhances the nuclear translocation STAT5A/B. STAT5A/B, in turn, increases the nuclear translocation of AR. STAT5A/B and AR are both growth promoting signaling pathways in prostate cancer cells.
Phosphorylated STAT5 dimers translocate from the cytoplasm into the nucleus, where they bind to the 8–10 bp inverted repeat consensus, TTC(C/T)N(G/A)-GAA, referred to as the GAS element (γ-interferon activation sequence) (Decker et al. 1991, Horvath et al. 1995, Soldaini et al. 2000). Recent reports support the notion that nuclear and cytoplasmic pools of unphosphorylated STAT proteins, including STAT5A/B, shuttle freely at high exchange rates in the absence of cytokine activation (Meyer et al. 2002, Zeng et al. 2002, Marg et al. 2004, Vinkemeier 2004, Reich & Liu 2006). While non-phosphorylated STATs cycle between the cytoplasm and nucleus, the translocation of dimerized STAT proteins has been suggested to be an active, energy-dependent process (Vinkemeier 2004, Reich & Liu 2006) and utilize components of Ran-dependent nuclear import machinery (Sekimoto et al. 1996, 1997). The karyopherin importin-b (p97) has been identified as the carrier that transports importin complexed with STATs into the nuclear compartment. Intriguing recent evidence suggests that unphosphorylated STATs may be able to bind to DNA in association with other transcription factors (Chatterjee-Kishore et al. 2000, Yang et al. 2005, 2007b).
Negative regulators of STAT5 signaling
A number of different mechanisms regulate the duration and magnitude of STAT5 activation at the cytoplasmic and nuclear levels. First, both cytoplasmic and/or nuclear phosphatases inactivate STAT5 proteins. The protein tyrosine phosphatase (PTP), SHP-2, directly interacts with STATs in the cytoplasm and translocates as a complex into the nucleus (Chughtai et al. 2002, Chen et al. 2003). Other phophatases known to inactivate STATs are cytosolic PTP, PTP1B, and a nuclear phosphatase, TCPTP (T-cell protein tyrosine phosphatase) (Aoki & Matsuda 2000, 2002). The second mechanism includes protein inhibitors of activated STAT proteins (PIAS), which inhibit STAT protein activation by direct association to STATs. The mammalian PIAS family members include PIAS1, PIAS3, PIASx, PIASy, and alternative splicing variants of PIASx (Chung et al. 1997, Schmidt & Muller 2003). DNA binding of STAT1 and STAT3 is selectively inhibited by PIAS1 and PIAS3 respectively (Chung et al. 1997, Rogers et al. 2004). Moreover, PIAS proteins are E3 ligases for the small ubiquitin-like modifier (SUMO; Jackson 2001, Kahyo et al. 2001, Muller et al. 2001, Sachdev et al. 2001). SUMO is a family of four proteins of about 100 amino acids, SUMO-1, -2, -3, and -4, which is conjugated to the consensus site (Ψ(Psi)KXE) on protein substrates. Similar to ubiquitin conjugation, SUMO proteins are activated by E1 enzymes (Aos1 and Uba2) and conjugated by an E2 enzyme (Ubc9) (Muller et al. 2001, Seeler et al. 2007). Diverse SUMO ligases, including PIAS proteins (Sharrocks 2006, Shuai 2006), define the specificity of SUMOylation. SUMO modification of a protein may alter its function, localization, or extent of ubiquitination (Muller et al. 2001, Seeler et al. 2007).
A third mechanism for the down-regulation of signaling by STAT5 proteins involves cytokine-inducible suppressors of cytokine signaling (SOCS) proteins (Alexander & Hilton 2004). There are eight members of the SOCS family, including cytokine-inducible SH2 (CIS domain protein) and SOCS-1 to -7. These proteins are all structurally related and they possess a central SH2 domain and a conserved C-terminal motif, termed the SOCS box (Alexander & Hilton 2004). The SOCS proteins also appear to target signal transducers for proteasomal destruction (Zhang et al. 1999, Kamizono et al. 2001, Ungureanu et al. 2002). SOCS are rapidly induced by activated STATs and act to block the cytokine signal by direct inhibition of JAKs (SOCS-1) and by competitive binding to tyrosine phosphorylated receptors so as to exclude further binding of signaling proteins such as STAT5, or by both mechanisms (Ram & Waxman 1999, Alexander & Hilton 2004).
The caveolin-1 (CAV-1) gene maps to chromosome 7q31.1 and encodes a 21–24 kDa integral membrane protein of caveolae, plasma membrane invaginations that are involved in vesicular transport, cholesterol homeostasis, signal transduction, and cell transformation (Williams & Lisanti 2005). The CAV-1 scaffolding domain shares homology with the SOCS pseudosubstrate domain, suggesting a negative regulatory function in the JAK/STAT5 signaling pathway. Indeed, CAV-1 was found to inhibit the kinase activity of JAK2 and suppress JAK2/STAT5A signaling (Park et al. 2001, Jasmin et al. 2006).
STAT5A/B in prostate cancer
Transcription factor STAT5A/B regulates the viability of prostate cancer cells
The understanding of the importance and function of transcription factor STAT5A/B in normal and malignant prostate tissues has been obtained from the analysis of Stat5a KO mice, studies in human prostate cancer cells in vitro and in vivo, and from the TRAMP (transgenic adrenocarcinoma of mouse prostate) mouse model of prostate cancer. Originally, STAT5A and STAT5B were identified as the key signaling proteins activated by PRL in both normal and malignant prostate tissues (Ahonen et al. 2002, Li et al. 2004). PRL, in turn, had been shown to be an autocrine mitogen and survival factor for androgen-deprived prostate cells (Ahonen et al. 1999). In determining the role of STAT5A/B in the growth of normal prostate epithelium, the first step was the analysis of the phenotype of Stat5a KO mice (Nevalainen et al. 2000). These studies revealed that the prostate epithelium of Stat5a-null mice was defective. Specifically, the prostate epithelium in Stat5a−/− mice was characterized by acinar cyst formation, local disorganization, and shedding of the epithelial cells to the glandular lumini (Nevalainen et al. 2000). The deformed prostate acini were filled with desquamated, granular epithelial cells embedded in dense, coagulated secretory material inside the broken acini (Nevalainen et al. 2000). The defective prostate tissue architecture in Stat5a−/− mice did not show increased prostate size or morphological hallmarks of epithelial hyperplasia. These results implied that STAT5A is not likely to mediate growth inhibition of prostate epithelium.
Presently, no reports of prostate phenotypes of Stat5b or Stat5a/b double KO mice exist. Analysis of the prostate phenotype of Stat5b or Stat5a/b double KO mice would be essential for more complete understanding of the importance of STAT5A/B for the integrity and maintenance of normal prostate epithelium. This is because the prostate phenotype of Stat5a-null mice may have been undermined by redundant functions of other STAT family proteins, particularly STAT5B (Liu et al. 1998b, Nevalainen et al. 2002). Specifically, in the mammary glands of Stat5a−/− mice, STAT5B compensated for the lack of Stat5a after multiple pregnancies (Liu et al. 1998b, Nevalainen et al. 2002). Likewise, in the prostates of Stat5a-deficient mice, STAT5B may have compensated for the lack of STAT5A. Furthermore, the loss of Stat5a stemmed from a germ line mutation of the Stat5a gene and, therefore, allowed sufficient time for STAT5B to functionally offset the loss of STAT5A throughout the development of the mice. Based on these facts, more specific results on the significance of STAT5A/B in growth regulation of normal prostate epithelium would be obtained from the studies that utilize conditional prostate-specific targeting of Stat5a or Stat5b or Stat5a/b.
Transcription factor STAT5A/B is highly critical for the viability of human prostate cancer cells in culture. Moreover, STAT5A/B is crucial for the regulation of prostate tumor growth in vivo. The novel concept of STAT5A/B being a prostate cell growth-controlling protein is supported by the reports from several different groups. First, Ahonen et al. (2003) showed that inhibition of STAT5A/B in STAT5-positive human prostate cancer cells by adenoviral expression of a dominant-negative mutant of STAT5A/B induced massive apoptotic death of the cells as determined by cell morphology, cell viability assays, DNA fragmentation, and activation of caspase-3 and caspase-9. The demonstration of the vital contribution of STAT5A/B to the viability of prostate cancer cells was later confirmed by the studies in the TRAMP mouse prostate cancer model (Kazansky et al. 2003). In the TRAMP mouse prostate tumor cell lines, inhibition of STAT5A/B by inducible expression of a carboxy terminal-truncated STAT5B mutant decreased the growth of the cells in soft agar and tumor formation in nude mice. For further validation of STAT5A/B as a therapeutic target protein for prostate cancer, recent work from the Nevalainen Laboratory (Dagvadorj et al. 2008) established the critical role of STAT5A/B for human prostate xenograft tumor growth in nude mice. The study also demonstrated that, regardless of the methodological approach, STAT5 inhibition in all STAT5-positive human prostate cancer cells resulted in massive cell death. In addition, CYCLIN-D1 and BCL-XL were identified as target genes of STAT5A/B in human prostate cancer cells (Dagvadorj et al. 2008). What remains unclear so far is the individual role of STAT5A versus STAT5B in the maintenance of growth regulation of prostate cancer cells. Moreover, identification of the molecular mechanisms underlying rapid apoptosis of prostate cancer cells upon STAT5A/B inhibition should be investigated, as it may reveal additional therapeutic target proteins for prostate cancer.
Active STAT5A/B in clinical progression of prostate cancer
STAT5A/B is constitutively activated in human prostate cancer cells, but not in the epithelium of adjacent normal prostate glands (Ahonen et al. 2003). Moreover, activation of STAT5A/B is associated with high-grade prostate cancer (Li et al. 2004). The distribution of active STAT5A/B in clinical prostate cancers of different histological grades was first demonstrated in a study that analyzed STAT5A/B activation in 114 paraffin-embedded prostate cancer samples (Li et al. 2004). Examination of tissue microarrays of an independent set of 357 prostate cancer patients further confirmed that activation of STAT5A/B associated with high Gleason grades of prostate cancer (Li et al. 2005). Active STAT5A/B in primary prostate tumors predicted an early recurrence of prostate cancer after the initial treatment of prostate cancer in the patient (Li et al. 2005). This finding further supported the concept of involvement of STAT5A/B in clinical progression of prostate cancer. Most importantly, active STAT5A/B remained an independent prognostic marker of early disease recurrence even if only prostate cancers of intermediate Gleason grades were analyzed (Li et al. 2005). It is therefore possible that the presence of active STAT5A/B in primary prostate cancers of intermediate histological grade is associated with progressive disease and would serve as a prognostic marker for the identification of prostate cancer patients who would benefit from a more aggressive therapeutic intervention. Future studies should determine the distribution of active STAT5A versus STAT5B in prostate cancers of different histological grades and assess the individual prognostic value of STAT5A versus STAT5B in prostate cancer. The ideal material for such studies would be primary prostate cancer specimens from patients who have undergone radical prostatectomy but have not received adjuvant therapies, since they may affect the activation of STAT5A/B.
Because active STAT5A/B promotes the growth of prostate cancer cells and active STAT5A/B in primary prostate cancer predicted early disease recurrence, which is often hormone-refractory cancer, the contribution of STAT5A/B to androgen-independent growth of prostate cancer is an important open question. Hormone-refractory prostate cancer is characterized by continued expression of the androgen receptor (AR) and androgen-regulated genes, suggesting that the AR signaling pathway remains active despite low levels of circulatory androgens (Isaacs & Isaacs 2004). Recently, the Nevalainen Laboratory (Tan et al. 2008) demonstrated that the active STAT5A/B signaling pathway increased transcriptional activity of AR in prostate cancer cells. Ligand-bound AR, in turn, increases transcriptional activity of STAT5A/B. AR expression is known to persist in hormone-refractory prostate cancer. Similar to AR, STAT5A/B was shown to be in the active state in 95% of hormone-refractory clinical human prostate cancers (Tan et al. 2008). The functional synergism between STAT5A/B and AR in prostate cancer cells was demonstrated to involve direct physical interaction between the two. Intriguingly, liganded AR was shown to enhance nuclear localization of STAT5A/B and active STAT5 promoted nuclear translocation of AR (Tan et al. 2008). These findings are important because STAT5A/B and AR are both transcription factors that inhibit apoptosis and promote the growth of prostate cancer cells (Fig. 2). Specifically, promotion of AR transcriptional activity by STAT5A/B in the presence of low levels of androgens may contribute to androgen-independent growth of prostate cancer. AR, in turn, by promoting transcriptional activity of STAT5A/B, may critically support the viability of prostate cancer cells in growth conditions where prostate cancer cells would normally undergo apoptosis. Among outstanding questions are the molecular mechanisms underlying the co-action between STAT5A/B and AR, the effect of STAT5A/B–AR synergy on prostate tumor growth in vivo and the STAT5 interaction with mutated liganded AR by non-testicular androgens.
Pathways leading to constitutive activation of STAT5A/B in prostate cancer cells
The molecular mechanisms underlying constitutive activation of STAT5A/B in primary and hormone-refractory human prostate cancer are presently unclear. Such mechanisms may involve autocrine PRL (Nevalainen et al. 1997a,b, Li et al. 2004; Fig. 2). Specifically, PRL is one of the predominant peptide factors presently known to activate JAK2-STAT5A/B in normal and malignant prostate epithelia (Ahonen et al. 2002, Li et al. 2004, Dagvadorj et al. 2007). PRL promotes proliferation and survival of prostate cells, and PRL is produced locally by normal prostate epithelium and prostate cancer (Nevalainen et al. 1991, 1996, 1997a,b, Wennbo et al. 1997, Ahonen et al. 1999, Kindblom et al. 2002, 2003, Dagvadorj et al. 2007). Analysis of clinical human prostate cancer specimens showed that PRL protein expression is associated with a high histological grade of human prostate cancer (Li et al. 2004). Autocrine PRL in prostate cancer may be one of the factors responsible for the constitutive activation of STAT5A/B in human prostate cancer. It has been shown that JAK2 is the predominant kinase that activates STAT5A/B in prostate cancer cells (Li et al. 2004). Activating mutations of JAK2 have been recently described in hematopoietic malignancies resulting in constitutive activation of STAT5 (Baxter et al. 2005). Such JAK2 mutations may also occur in advanced prostate cancer. A third potential mechanism for the high abundance of STAT5A/B in prostate cancer is the amplification of STAT5A/B genes. This is particularly important since the STAT5A/B genes are located on chromosome 17 (Clark et al. 2003) that is frequently altered in both incidental and hereditary prostate cancers (Gillanders et al. 2004). Chromosome 17q showed allelic imbalance in prostate cancer (Latil et al. 1994, Bova & Isaacs 1996, Alers et al. 2000, Kasahara et al. 2002, Wolter et al. 2002a,b, Verhage et al. 2003, von Knobloch et al. 2004), and gains in chromosome 17q were detected in five studies (Bova & Isaacs 1996, Alers et al. 2000, Kasahara et al. 2002, Wolter et al. 2002a,b). Moreover, three large studies linked a prostate cancer susceptibility gene to chromosome 17q (17q22) (Lange et al. 2003, Gillanders et al. 2004, Zuhlke et al. 2004), suggesting involvement of genes in this region in an inherited form of prostate cancer.
STAT5A/B might also be activated by tyrosine kinases such as Src (Silva 2004, Yu & Jove 2004), Bcr-Abl (de Groot et al. 1999), or Tel-JAK (Schwaller et al. 2000). Although GH, a principal activator of STAT5B in a number of tissues, might be involved in activating STAT5A/B in malignant prostate epithelium (Chopin et al. 2002, Halmos et al. 2002, Letsch et al. 2003, Weiss-Messer et al. 2004, Stangelberger et al. 2005, Wang et al. 2005), there is presently no evidence of direct effects of GH on the stimulation of prostate cancer cell growth.
Negative regulators of STAT5A/B signaling in prostate cancer
Constitutive activation of STAT5A/B in malignant prostate epithelium may result from the loss of STAT5A/B phosphatases or inhibitory proteins of STAT5A/B (PIAS, CAV-1). STAT proteins are inactivated by both cytoplasmic and nuclear PTPs, such as SHP-1, SHP-2, CD45, cytosolic PTP, PTP1B, and a nuclear phosphatase, TCPTP (Aoki & Matsuda 2000, 2002, Shuai & Liu 2003). However, studies on direct regulation of STAT5A/B activation by tyrosine phosphatases in prostate cancer cells have not been reported. Nevertheless, expression of the tyrosine phosphatase SHP-1 has been detected in both PC-3 and LNCaP prostate cancer cells (Zapata et al. 2002). SHP-1 is also expressed in normal human prostate, benign prostate hyperplasia, and well-differentiated prostate cancer, but is undetectable in poorly differentiated advanced prostate cancer (Zapata et al. 2002).
The PIAS family of proteins are localized within the nucleus and function as constitutive repressors of STAT activity (Chung et al. 1997, Shuai 2000, Schmidt & Muller 2003). In addition, PIAS1, PIAS3 (Junicho et al. 2000, Gross et al. 2001, Wang & Banerjee 2004), and PIAS-like proteins Zimp7 (Huang et al. 2005) and Zimp10 (Sharma et al. 2003) have been shown to function as co-activators to AR-mediated transcription in human prostate epithelial cells, while PIASy acts as a co-repressor of AR (Junicho et al. 2000, Gross et al. 2001). Importantly, the only member of the PIAS family that has been shown to interact with STAT5A/B is PIAS3. Specifically, the repressive action of PIAS3 on STAT5-mediated transcription was shown in CHO and lymphoid cells (Rycyzyn & Clevenger 2002), but not in prostate cells. PIAS3 is expressed in prostate cancer tissues and cell lines (Gross et al. 2001, Wang & Banerjee 2004). Moreover, PIAS3 acts as a co-regulator of AR-regulated transcription in LNCaP cells, and its expression is enhanced in response to DHT (dihydro-testosterone) treatment (Junicho et al. 2000, Gross et al. 2001). PIAS1 expression, in turn, has been shown to be 33% higher in primary prostate cancers compared with normal prostates, but this overexpression did not correlate with the Gleason score as determined by in situ hybridization of PIAS1mRNA (Li et al. 2002). Another study showed that PIAS1 expression is significantly lower in hormone-refractory prostate tumors than in untreated prostate tumors (Linja et al. 2004). However, interaction and the effects of PIAS1 on STAT5A/B activity in prostate cancer cells remain unclear. PIAS1 has been shown to act as an E3 ligase for AR (Nishida & Yasuda 2002), enhance the transcriptional activity of AR in LNCaP cells (Gross et al. 2001), and inhibit STAT1-mediated transcription (Liu et al. 1998a). In contrast to PIAS1 and PIAS3, PIASy has been shown to interact with AR and act as an inhibitor of AR in prostate cancer cell without interfering with the DNA binding of AR (Gross et al. 2001). Additional studies on the interaction of PIAS proteins with STAT5A/B- and STAT5A/B-regulated gene transcription in prostate cancer cells would give more insight into how this interaction contributes to prostate cancer progression.
A comparison of SOCS expression in three prostate cancer cell lines (LNCaP, PC-3, and DU145) with a normal prostate cell line (RWPE-1) showed that the expression levels of SOCS-1, SOCS-3, SOCS-5, and CIS genes in PC-3 and DU145 cells are significantly lower than in the normal RWPE prostate cells. On the other hand, in LNCaP cells the expression of SOCS-1, SOCS-3, SOCS-5, and CIS genes were at levels comparable with RWPE cells (Evans et al. 2007). These results indicate that SOCS genes are not silenced in all prostate cancer cells. It remains to be determined whether the SOCS proteins directly regulate the JAK2-STAT5A/B pathway in human prostate cancer cells (Neuwirt et al. 2007) and, therefore, would contribute to constitutive activation of STAT5A/B in clinical prostate cancer.
In mammary epithelial cells, CAV-1 has been shown to repress JAK2-STAT5A/B signaling pathway (Park et al. 2002, Jasmin et al. 2006). In prostate cancer, over-expression of CAV-1 has been associated with higher Gleason score, positive surgical margins, metastasis to lymph nodes, aggressive PSA (prostate specific antigen) recurrence, and a higher likelihood of disease recurrence in patients treated with radical prostatectomy (Yang et al. 1999, Tahir et al. 2006, Karam et al. 2007). Moreover, it has been suggested recently that CAV-1 mediates angiogenesis during prostate cancer progression (Yang et al. 2007a). In the CAV-1 deficient human prostate cancer cell line, LNCaP, the overexpression of CAV-1 stimulated cell proliferation and promoted tumor growth in nude mice (Bartz et al. 2008). To date, there are no studies on the interaction of CAV-1 expression and JAK2/STAT5 signaling pathway in human prostate cancer cells. In summary, little is presently known about how changes in the expression patterns of negative regulatory proteins of STAT5A/B signaling contribute to the constitutive activation of STAT5A/B in advanced prostate cancer.
STAT5A/B and breast cancer
STAT5A/B regulation of normal mammary gland
The present understanding of the role of STAT5A/B in the regulation of normal mammary gland has been obtained for the most part from studies using murine mammary gland as a model system. Before and after the onset of puberty and during the reproductive cycle, the mouse mammary gland undergoes major morphological changes. Signals from steroid hormones, lactogenic hormones, and peptide growth factors all coordinate the expansion and differentiation of the alveolar compartment of the mouse mammary gland during the reproductive cycle (Hennighausen & Robinson 2005). Throughout all the stages of mammary gland development, the expression of STAT5A/B proteins is detected in the breast epithelium with a minor increase during the final stages of pregnancy and the onset of lactation (Kazansky et al. 1995, Liu et al. 1995). Of the two homologs of STAT5, STAT5A is more abundant in mammary epithelial cells than STAT5B (Liu et al. 1997). In contrast to the relatively steady expression pattern of STAT5A/B proteins, the kinetics of STAT5 tyrosine phosphorylation correlates with the developmental profile of the mammary gland and the activation of milk protein genes (Liu et al. 1995, 1996b, Nevalainen et al. 2002). In late pregnancy and early lactation, marked stimulation in STAT5 phosphorylation occurs, followed by a decrease during the involution phase of the post-lactational mammary gland (Liu et al. 1995, 1996b, Nevalainen et al. 2002). In addition to PRL, STAT5A/B is also activated in mouse mammary gland by GH or epidermal growth factor (EGF). However, while PRL activates STAT5A/B uniformly in the mammary epithelium, GH, and EGF activate STAT5A/B only in a subset of epithelial cells, and the main targets of GH and EGF in mouse mammary gland are mammary stromal cells (Gallego et al. 2001, Nevalainen et al. 2002).
Creation of Stat5a-null mice revealed a specific role for STAT5A in the mouse mammary gland during the reproductive cycle (Liu et al. 1997). The inactivation of Stat5a gene in mice by homologous recombination showed disrupted mammary gland development and a failure to lactate. Specifically, the postpartum lobulo-alveolar outgrowth was reduced and the alveolar lumina in the mammary gland failed to terminally differentiate (Liu et al. 1997). The mice developed otherwise normally and were indistinguishable from their wild-type litter mates in size, weight, and fertility (Liu et al. 1997). Interestingly, in the non-pregnant murine mammary gland, STAT5A/B is basally active within the ductal compartment (Nevalainen et al. 2002). However, the key functions of PRL-STAT5 signaling during pregnancy and lactation are located to the alveolar compartment of the gland (Horseman et al. 1997, Ormandy et al. 1997). These findings are in line with the results of the analysis of mice lacking two alleles of the PRL receptor in mammary epithelial cells that exhibited normal morphogenesis of the ducts of the mammary glands but lacked milk-producing alveoli (Brisken et al. 1999, Miyoshi et al. 2001). Further studies using transgenic mice overexpressing the native ovine STAT5A (94% identical to mouse STAT5A) in the mammary glands showed that STAT5A promoted lobulo-alveolar development and delayed the onset of apoptosis (Iavnilovitch et al. 2002). This suggested that STAT5A is a survival factor for terminally differentiated non-malignant mammary epithelial cells. Collectively, these studies demonstrated that STAT5A is the principal mediator of lobulo-alveolar differentiation and expansion in mice during pregnancy, as well as the key mediator of lactogenic signaling. Moreover, transcriptionally active STAT5A critically promotes the viability and survival of terminally differentiated alveolar mammary epithelial cells.
Stat5b-null mice demonstrated a decreased lobulo-alveolar development, albeit a less severe phenotype than that of the Stat5a-null mice, with lactation unimpeded. The key function of STAT5B is to mediate the effects of GH, as Stat5b-null mice failed to respond effectively to GH (Udy et al. 1997, Teglund et al. 1998) and had severe anemia suggesting defective hematopoiesis due to impaired response to hematopoietins (Socolovsky et al. 1999). KO of both Stat5a and Stat5b, in turn, is perinatally lethal and the mice had severely impaired lymphoid development and differentiation. Absence of STAT5A/B also abrogated T-cell receptor γ-rearrangement and peripheral CD8+ T-cell survival (Yao et al. 2006). In addition, Stat5a/b KO mice were found to have a defect in the development of functional corpora lutea in the ovary, resultingin female infertility. Interestingly, while STAT5A primarily mediates biological effects distinct from those of STAT5B, it has been shown that STAT5B can compensate for the lack of STAT5A in the mammary gland function (Liu et al. 1998b, Nevalainen et al. 2002). After multiple pregnancies, lactation was partially established by activation of STAT5B in the mammary gland epithelium of the STAT5A-null mice. These results imply functional redundancy between STAT5A and STAT5B when sufficient time is provided, which is inherent to germ line KO studies.
STAT5A/B in tumorigenesis of rodent mammary gland
Present evidence suggests that STAT5A/B promotes tumorigenesis of rodent mammary gland. In a study using the rat mammary gland as a model, Shan et al. (2004) examined the immunohistochemical expression and activation of STAT5A in a panel of rat mammary gland carcinomas induced by the chemical carcinogens, 7,12-dimethylbenz[a]anthracene and 2-amino-1-methyl-6 phenylimidazo[4,5-b]pyridine, to assess the percentage of cells in carcinomas and preneoplastic lesions showing tyrosine phosphorylated and nuclear localized STAT5A. Nuclear immunostaining of STAT5A was detected in 65% of carcinomas, while STAT5A in control normal mammary gland tissue was cytosolic. STAT5A immunostaining patterns (cytoplasmic or nuclear) were compared with proliferating cell nuclear antigen immunostaining, tumor differentiation, nuclear grade, mitotic activity, and tumor size. The results indicated that intense nuclear immunostaining for STAT5A was associated with high-grade rat mammary gland carcinomas and STAT5A immunostaining was detected in intraductal proliferating cells and in ductal carcinomas in situ (Shan et al. 2004).
The effect of STAT5A on mouse mammary gland carcinogenesis has been tested in several different mouse breast cancer models. First, the effect of hemizygous loss of Stat5a was determined in a SV40-T antigen transgenic mouse model of mammary cancer. The breeding of WAP-TAg mice to mice carrying germ line hemizygous deletions of the Stat5a allele generated mice with reduced levels of STAT5A without altering mammary gland development or transgene expression levels. In comparison with mice carrying two wild-type Stat5a alleles, hemizygous loss of the Stat5a allele demonstrated a modest decrease in the number of tumor-bearing mice. Moreover, the tumors were smaller and the tumor formation was delayed (Ren et al. 2002). In another mouse breast cancer model system, Stat5a-null females exhibited delayed transforming growth factor-α-induced tumorigenesis (Humphreys & Hennighausen 2000). Third, a recent study by Oakes et al. (2007) suggested that PRL receptor deficiency may prevent early progression of mammary gland neoplasia to invasive carcinoma. However, it is unclear so far whether this inhibition of mammary gland cancer progression would be due to the lack of JAK2/STAT5 signaling or inhibition of other PRL receptor-activated signal transduction pathways. Cav-1, in turn, which inhibits JAK2-STAT5 signaling (Park et al. 2002), has been shown to affect mammary tumor formation in mice (Sotgia et al. 2006b). The mammary glands of Cav-1 KO mice expressed a hyperactive JAK2-STAT5 pathway and developed hyperplastic, well-differentiated mammary tumors (Sotgia et al. 2006b). An interesting recent study reported that overexpression of wild-type or constitutively active Stat5a in a transgenic mouse model promoted the occurrence of sporadic mammary cancers. Specifically, mammary-directed expression of wild-type Stat5a or constitutively active Stat5a developed well-differentiated micropapillary and papillary adenocarcinomas after latency periods of 8–12 months. Unexpectedly, mammary-directed overexpression of a carboxyl terminal-truncated STAT5 was able to promote mammary gland adenocarcinoma development in mice (Iavnilovitch et al. 2004). This result was surprising because C-terminally truncated STAT5A is transcriptionally inactive, and thus acts as a dominant-negative variant of STAT5A/B inhibiting endogenous STAT5A/B. Of note, the mammary gland adenocarcinomas in these mice were less well differentiated than carcinomas that occurred in wild-type or constitutively active STAT5 expressing transgenic mice (Iavnilovitch et al. 2004). Further observation showed that the expression of the carboxy terminally truncated Stat5 in the mammary glands of transgenic mice inhibits cell proliferation during pregnancy, delays onset of milk secretion, and induces apoptosis upon involution (Iavnilovitch et al. 2006). In another study, Eilon et al. (2007) reported that in transgenic mice overexpressing Stat5 or a constitutively activated Stat5 variant, mammary tumorigenesis occurred more frequently in multiparous females compared with their age-matched virgin counterparts. The fact that mammary tumorigenesis was more frequent in multiparous females may be due to the use β-lactoglobulin gene promoter, which is a PRL-inducible promoter, to drive the expression of Stat5. The role of STAT5A/B in differentiation of malignant mammary epithelial cells is supported by the studies conducted in HC11 cells that are well-differentiated mouse mammary cancer cells. Specifically, inhibition of PRL-JAK2 signaling blocked cell differentiation and induced a hyperproliferative phenotype characterized by increased mitotic rate, reduced apoptosis, and reduced contact inhibition (Xie et al. 2002). In conclusion, several lines of evidence suggest that STAT5A/B may promote mammary gland tumorigenesis in rat and mice. The results of several independent studies suggest that STAT5A/B may also affect differentiation of mouse mammary gland carcinomas. However, it is unclear at this point to what extent the observed phenotypes in these studies with somewhat conflicting results were affected by crossing different mouse strains with one another.
STAT5A/B signaling pathway in human breast cancer
Activation of STAT5A/B in human breast cancer has been shown to positively correlate with the differentiation status of the tumor. In one study, STAT5A/B was found to be constitutively active in ~76% of human breast tumors (n=83) and the authors observed a positive correlation between tumor differentiation and active STAT5A (Cotarla et al. 2004). No association of active STAT5 was found with recognized prognostic indicators such as lymph node metastases, tumor size, ploidy, percentage of cells in S-phase, estrogen receptor (ER), ErbB2, or nuclear localized p21. In another study, STAT5A/B activation in primary breast cancer (n=1100) was found to predict favorable clinical outcome (Nevalainen et al. 2004). This study describes a gradual inactivation of STAT5A/B during metastatic progression of human breast cancer, with less than 20% of metastases exhibiting active STAT5. Importantly, loss of tyrosine phosphorylated STAT5A/B in the primary tumor of patients with lymph node-negative breast cancer was associated with an eightfold increased risk of death from breast cancer (Nevalainen et al. 2004). In multivariate analyses, STAT5A/B was an independent prognostic factor in node-negative breast cancer when other markers, including age, ER, progesterone receptor, HER2, (human epidermal growth factor receptor-2) tumor size, and grade were included. Subsequent studies focusing on histological differentiation of breast cancer and STAT5A/B activation confirmed that STAT5A/B levels positively correlated with breast cancer differentiation in the material of 517 cases (Yamashita et al. 2006). Moreover, high STAT5A/B levels predicted better response to endocrine therapy and longer survival after relapse than tumors that were STAT5A/B negative (Yamashita et al. 2006). Interestingly, examination of secretory carcinomas for the presence of STAT5A by immunohistochemistry showed that STAT5A was expressed at a high level in secretory carcinomas (11 invasive and 7 in situ, including 4 cases with both), but was absent in in situ or invasive ductal carcinomas and apocrine breast metaplasias (Strauss et al. 2006). Moreover, STAT5A was not expressed in other specialized histological types of breast carcinomas such as mutinous or clear cell carcinomas (Strauss et al. 2006). The authors suggested that STAT5A/B expression in the secretory breast carcinomas may be due to the secretory changes occurring in the cells. In addition, the levels of STAT5A were reported to be reduced in breast cancers in general compared with STAT5A levels in normal luminal breast epithelial cells (Bratthauer et al. 2006).
The favorable prognosis associated with active STAT5A/B may be a result of reduced metastatic dispersal of breast cancer cells from the primary tumor caused by STAT5A/B. Specifically, STAT5A/B had an invasion-suppressive role in human breast cancer cells (Sultan et al. 2005) as determined by cell clustering assays, E-cadherin expression, matrix metalloproteinase secretion, cell migration, and invasion assays (Sultan et al. 2005). On the other hand, hypoxia, a common consequence of solid tumor growth in breast cancer or other cancers, was reported to activate STAT5A/B and increase its transcriptional activity and binding to the GAS element in breast cancer cells (Joung et al. 2005, Lee et al. 2006). In addition, inhibition of STAT5A/B by adenoviral expression of a dominant-negative STAT5 mutant in T47D cells was described to cause apoptosis of the cells (Yamashita et al. 2003, 2004). These studies, however, did not include critical controls for the cytopathic effects of adenovirus, and therefore it is difficult to interpret the results. In conclusion, further studies are needed to determine whether STAT5A/B is not only a promoter of differentiation of breast cancer, but also a survival factor for established human breast tumors.
Negative regulation of STAT5 signaling in breast cancer
The molecular mechanisms underlying constitutive activation of STAT5A/B in clinical human breast cancer are presently unclear. First, the down-regulation of STAT5A/B phosphorylation during breast cancer progression to metastatic disease could be due to decreased STAT5A/B protein expression caused by genetic or epigenetic changes occurring in breast cancer cells. Second, STAT5A/B phosphorylation may be reduced because of increased expression or activation of phosphatases that dephosphorylate STAT5A/B. Both TCPTP and PTP1B were shown to form a complex with and dephosphorylate STAT5A/B, resulting in inactivation of STAT5A/B-mediated gene expression in mammary epithelial cells (Aoki & Matsuda 2000, 2002). Analysis of SHP-1 expression in 72 primary breast cancers showed a 2- to 12-fold increase in 58% of the samples compared with normal breast epithelial cells (Yip et al. 2000). No reports on the distribution of TCPTP or PTP1B protein expression in human breast cancers of different histological grades exist to date. Third, changes in the expression levels of PIAS proteins may result in reduced transcriptional activity of STAT5A/B in high-grade breast cancer. PIAS3 has been shown to inhibit PRL-induced STAT5A/B-mediated transactivation of the β-casein promoter (Rycyzyn et al. 2000). This inhibitory interaction between PIAS3 and STAT5A/B activity was disrupted by increasing amounts of a complex formed by intra-nuclear PRL and the peptidyl prolyl isomerase cyclophilin B, suggesting an indirect regulatory role of nuclear PRL on STAT5A/B transcriptional activity (Rycyzyn & Clevenger 2002).
Elevated expression levels of SOCS-1, SOCS-2, SOCS-3, and CIS were found in breast carcinomas and breast cancer cell lines compared with normal breast tissue and cell lines (Raccurt et al. 2003). In another study, the mRNA expression levels of SOCS-1, SOCS-2, SOCS-3, CIS, and the STAT5-induced growth factor, insulin-like growth factor-I, were examined by RT-PCR followed by immunohistochemical analysis of SOCS-2 protein expression (Haffner et al. 2007). SOCS-2 expression level was inversely correlated with histopathological grade of breast cancer. Moreover, high SOCS-2 expression were found in ER-positive tumors and correlated with higher survival rates.
The mammary glands of Cav-1 null mice showed premature lactation, with accelerated development of the lobulo-alveolar compartment and hyperactivation of the JAK2/STAT5A signaling compared with the mammary gland of normal mice (Park et al. 2002). Cav-1−/− mammary epithelia were hyperproliferative in vivo, with dramatic increases in terminal end bud areas and mammary ductal thickness as well as increases in bromodeoxyuridine incorporation, extra-cellular signal-regulated kinase 1/2 hyperactivation, and up-regulation of Stat5a and Cyclin-D1. Consistent with these findings, loss of Cav-1 dramatically exacerbated mammary lobulo-alveolar hyperplasia in Cyclin-D1 transgenic mice, whereas overexpression of Cav-1 caused reversal of this phenotype (Williams et al. 2006). However, the role of CAV-1 human cancers has been controversial (Lee et al. 1998, Hurlstone et al. 1999, Chen et al. 2004, Sagara et al. 2004, Williams & Lisanti 2005). CAV-1 is believed to be a tumor suppressor gene (Williams & Lisanti 2005, Sotgia et al. 2006a) based on the high frequency of deletions of 7q31 in human cancers (Lee et al. 2002), the presence of CAV-1 gene promoter methylation (Engelman et al. 1999), and inactivating gene mutation (Hayashi et al. 2001). By contrast, other reports suggest that CAV-1 may behave as an oncogene in breast (Hurlstone et al. 1999, Van den Eynden et al. 2006, Pinilla et al. 2006) and prostate (Thompson et al. 1999, Timme et al. 2000) cancers.
Reduced CAV-1 expression in breast carcinomas has been reported in several independent studies (Chen et al. 2004, Park et al. 2005). In another study, mRNA levels of CAV-1 were analyzed in breast cancer tissues by RT-PCR and showed that the down-regulation of CAV-1 mRNA levels in breast cancer tissues compared with normal tissues and correlated positively with tumor size and negative ER status (Sagara et al. 2004) A comparison of CAV-1 expression in normal breast tissues, benign lesions, breast cancer precursors, metastatic, and invasive breast cancers using a combination of immunofluorescence, ultrastructural analysis, and immunohistochemistry showed preferential CAV-1 expression in myoepithelial cells (MECs), fibroblasts, and endothelial cells in normal breast tissue (Savage et al. 2007). CAV-1 was found to be expressed in 90% metaplastic breast carcinomas and in 9.4% invasive breast carcinomas, and, importantly, the expression of CAV-1 in the latter group was associated with basal-like immunophenotype, shorter disease free, and lower survival based on univariate analysis.
Targeting STAT5A/B activation
There are multiple levels at which STAT5A/B function could be targeted in cells. These include STAT5A/B gene expression, STAT5A/B recruitment to a receptor, STAT5A/B tyrosine phosphorylation, STAT5A/B dimerization, STAT5A/B nuclear translocation, STAT5A/B DNA binding, and its interaction with other transcriptional co-regulators. Down-regulation of STAT5A/B expression by RNA interference in both mammary epithelial cells and prostate cancer cells (Dagvadorj et al. 2008) has been performed successfully. SH2 domain-binding phosphotyrosyl peptides or peptidomimetics that have been used to selectively inhibit STAT3 dimerization (Turkson et al. 2001, 2004) could be applied to inhibit STAT5A/B receptor recruitment and dimerization. In addition, decoy oligonucleotides that mimic STAT5A/B-binding sites could theoretically sequester STAT5 from its target genes and inhibit DNA binding as shown by the inhibition of CYCLIN-D1 protein expression in NRK-49F kidney cells (Guh et al. 2001). Furthermore, peptide aptamers selected from combinatorial peptide libraries could potentially be used to block STAT5 dimerization or DNA binding (Nagel-Wolfrum et al. 2004). Another approach to inhibit STAT5A/B would be a disruption of transcriptional activation of STAT5 by the expression of a truncated dominant negative variant that lacks the C-terminal transactivation domain (Ahonen et al. 2003, Dagvadorj et al. 2008). The involvement of human autocrine PRL in activating the JAK2/STAT5A/B signaling pathway in breast and prostate epithelial cells inspired the development of hPRL antagonists such as G129R-PRL (Llovera et al. 2000) that may have potential for therapeutic use (Dagvadorj et al. 2007). Finally, identification of pharmacological small-molecule lead compounds that would disrupt STAT5A/B dimerization or DNA binding would be critical for the development of conventional pharmaceuticals for the inhibition of STAT5A/B.
Conclusions
Transcription factor STAT5A/B is constitutively active in human prostate and breast cancers. In prostate cancer, activation of STAT5A/B is associated with high histological grade of clinical prostate cancer, and activation of STAT5A/B in primary prostate cancer may predict early disease recurrence. Moreover, STAT5A/B functionally interacts with AR in prostate cancer cells. Importantly, STAT5A/B is highly critical for the viability of human prostate cancer cells and therefore presents a potential therapeutic target protein for high-grade prostate cancer (Table 4).
Table 4.
Findings on the role of signal transducer and activator of transcription 5A/B (STAT5A/B) in mammary gland and prostate in mouse models versus human
| Mouse | Human (cell lines) | Human | |
|---|---|---|---|
| Breast |
|
|
|
| Prostate |
|
|
|
In breast cancer, the role of STAT5A/B is more complex (Table 4). In mouse models of breast cancer, STAT5A/B promotes tumor formation. By contrast, activation of STAT5A/B in human breast cancer is a robust predictor of favorable clinical outcome, and STAT5A/B inhibits metastatic behavior of human breast cancer cells. Moreover, expression of active STAT5A/B is lost during histological dedifferentiation of human breast cancer. It is therefore possible that STAT5A/B has a dual role in breast cancer. STAT5A/B may promote the growth of the early malignant lesions, whereas it may maintain differentiation of established breast cancer. It is also possible that the role of STAT5A/B in mouse mammary gland regulation is different from that in humans. The specific and possibly different roles of STAT5A versus STAT5B in the regulation of breast cancer cells are not yet clear and some of the conflicting data may have resulted from the analysis of STAT5A and STAT5B collectively in the majority of the studies carried out to date. Finally, little is known about STAT5A/B activation and regulation in human breast cancer stem cells and how STAT5A/B activation in different cell types of the human breast translates into the effect of STAT5A/B on growth promotion versus differentiation of breast cancer.
Acknowledgments
We would like to thank Ms Jacqueline Lutz for her help in designing the graphs. This work was supported by the American Cancer Society (RSG-04-196-01-MGO), DOD Department of Defense Prostate Cancer Grants (W81XWH-05-01-0062, W81XWH-06-01-0076, W81XWH-07-01-0411), and NIH NCI grant (1RO1CA113580-01A1). The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
References
- Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Ahonen TJ, Harkonen PL, Laine J, Rui H, Martikainen PM, Nevalainen MT. Prolactin is a survival factor for androgen-deprived rat dorsal and lateral prostate epithelium in organ culture. Endocrinology. 1999;140:5412–5421. doi: 10.1210/endo.140.11.7090. [DOI] [PubMed] [Google Scholar]
- Ahonen TJ, Harkonen PL, Rui H, Nevalainen MT. PRL signal transduction in the epithelial compartment of rat prostate maintained as long-term organ cultures in vitro. Endocrinology. 2002;143:228–238. doi: 10.1210/endo.143.1.8576. [DOI] [PubMed] [Google Scholar]
- Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, Rui H, Nevalainen MT. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. Journal of Biological Chemistry. 2003;278:27287–27292. doi: 10.1074/jbc.M304307200. [DOI] [PubMed] [Google Scholar]
- Alers JC, Rochat J, Krijtenburg PJ, Hop WC, Kranse R, Rosenberg C, Tanke HJ, Schroder FH, van Dekken H. Identification of genetic markers for prostatic cancer progression. Laboratory Investigation. 2000;80:931–942. doi: 10.1038/labinvest.3780096. [DOI] [PubMed] [Google Scholar]
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annual Review of Immunology. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Aoki N, Matsuda T. A cytosolic protein-tyrosine phosphatase PTP1B specifically dephosphorylates and deactivates prolactin-activated STAT5a and STAT5b. Journal of Biological Chemistry. 2000;275:39718–39726. doi: 10.1074/jbc.M005615200. [DOI] [PubMed] [Google Scholar]
- Aoki N, Matsuda T. A nuclear protein tyrosine phosphatase TC-PTP is a potential negative regulator of the PRL-mediated signaling pathway: dephosphorylation and deactivation of signal transducer and activator of transcription 5a and 5b by TC-PTP in nucleus. Molecular Endocrinology. 2002;16:58–69. doi: 10.1210/mend.16.1.0761. [DOI] [PubMed] [Google Scholar]
- Barahmand-pour F, Meinke A, Eilers A, Gouilleux F, Groner B, Decker T. Colony-stimulating factors and interferon-gamma activate a protein related to MGF-Stat 5 to cause formation of the differentiation-induced factor in myeloid cells. FEBS Letters. 1995;360:29–33. doi: 10.1016/0014-5793(95)00072-h. [DOI] [PubMed] [Google Scholar]
- Bartz R, Zhou J, Hsieh JT, Ying Y, Li W, Liu P. Caveolin-1 secreting LNCaP cells induce tumor growth of caveolin-1 negative LNCaP cells in vivo. International Journal of Cancer. 2008;122:520–525. doi: 10.1002/ijc.23142. [DOI] [PubMed] [Google Scholar]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- Beuvink I, Hess D, Flotow H, Hofsteenge J, Groner B, Hynes NE. Stat5a serine phosphorylation. Serine 779 is constitutively phosphorylated in the mammary gland, and serine 725 phosphorylation influences prolactin-stimulated in vitro DNA binding activity. Journal of Biological Chemistry. 2000;275:10247–10255. doi: 10.1074/jbc.275.14.10247. [DOI] [PubMed] [Google Scholar]
- Bova GS, Isaacs WB. Review of allelic loss and gain in prostate cancer. World Journal of Urology. 1996;14:338–346. doi: 10.1007/BF00184607. [DOI] [PubMed] [Google Scholar]
- Bratthauer GL, Strauss BL, Tavassoli FA. STAT 5a expression in various lesions of the breast. Virchows Archiv. 2006;448:165–171. doi: 10.1007/s00428-005-0056-6. [DOI] [PubMed] [Google Scholar]
- Brisken C, Kaur S, Chavarria TE, Binart N, Sutherland RL, Weinberg RA, Kelly PA, Ormandy CJ. Prolactin controls mammary gland development via direct and indirect mechanisms. Developmental Biology. 1999;210:96–106. doi: 10.1006/dbio.1999.9271. [DOI] [PubMed] [Google Scholar]
- Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO Journal. 2000;19:4111–4122. doi: 10.1093/emboj/19.15.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Sadowski HB, Kohanski RA, Wang LH. Stat5 is a physiological substrate of the insulin receptor. Proc Natl Acad Sci U S A. 1997;94:2295–2300. doi: 10.1073/pnas.94.6.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao H, Harada N, Kitamura T, Mui AL, Miyajima A. Interleukin 2 and erythropoietin activate STAT5/MGF via distinct pathways. EMBO J. 1995;14:2527–2535. doi: 10.1002/j.1460-2075.1995.tb07250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wen R, Yang S, Schuman J, Zhang EE, Yi T, Feng GS, Wang D. Identification of Shp-2 as a Stat5A phosphatase. Journal of Biological Chemistry. 2003;278:16520–16527. doi: 10.1074/jbc.M210572200. [DOI] [PubMed] [Google Scholar]
- Chen ST, Lin SY, Yeh KT, Kuo SJ, Chan WL, Chu YP, Chang JG. Mutational, epigenetic and expressional analyses of caveolin-1 gene in breast cancers. International Journal of Molecular Medicine. 2004;14:577–582. [PubMed] [Google Scholar]
- Choi JY, Li WL, Kouri RE, Yu J, Kao FT, Ruano G. Assignment of the acute phase response factor (APRF) gene to 17q21 by microdissection clone sequencing and fluorescence in situ hybridization of a P1 clone. Genomics. 1996;37:264–265. doi: 10.1006/geno.1996.0556. [DOI] [PubMed] [Google Scholar]
- Chopin LK, Veveris-Lowe TL, Philipps AF, Herington AC. Co-expression of GH and GHR isoforms in prostate cancer cell lines. Growth Hormone and IGF Research. 2002;12:126–136. doi: 10.1054/ghir.2002.0271. [DOI] [PubMed] [Google Scholar]
- Chughtai N, Schimchowitsch S, Lebrun JJ, Ali S. Prolactin induces SHP-2 association with Stat5, nuclear translocation, and binding to the beta-casein gene promoter in mammary cells. Journal of Biological Chemistry. 2002;277:31107–31114. doi: 10.1074/jbc.M200156200. [DOI] [PubMed] [Google Scholar]
- Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- Clark J, Edwards S, Feber A, Flohr P, John M, Giddings I, Crossland S, Stratton MR, Wooster R, Campbell C, et al. Genome-wide screening for complete genetic loss in prostate cancer by comparative hybridization onto cDNA microarrays. Oncogene. 2003;22:1247–1252. doi: 10.1038/sj.onc.1206247. [DOI] [PubMed] [Google Scholar]
- Copeland NG, Gilbert DJ, Schindler C, Zhong Z, Wen Z, Darnell JE, Jr, Mui AL, Miyajima A, Quelle FW, Ihle JN, et al. Distribution of the mammalian Stat gene family in mouse chromosomes. Genomics. 1995;29:225–228. doi: 10.1006/geno.1995.1235. [DOI] [PubMed] [Google Scholar]
- Cotarla I, Ren S, Zhang Y, Gehan E, Singh B, Furth PA. Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers. International Journal of Cancer. 2004;108:665–671. doi: 10.1002/ijc.11619. [DOI] [PubMed] [Google Scholar]
- Dagvadorj A, Collins S, Jomain JB, Abdulghani J, Karras J, Zellweger T, Li H, Nurmi M, Alanen K, Mirtti T, et al. Autocrine prolactin promotes prostate cancer cell growth via Janus kinase-2-signal transducer and activator of transcription-5a/b signaling pathway. Endocrinology. 2007;148:3089–3101. doi: 10.1210/en.2006-1761. [DOI] [PubMed] [Google Scholar]
- Dagvadorj A, Kirken RA, Leiby B, Karras J, Nevalainen MT. Transcription factor Stat5 promotes growth of human prostate cancer cells in vivo. Clinical Cancer Research. 2008;14:1317–1324. doi: 10.1158/1078-0432.CCR-07-2024. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- Decker T, Lew DJ, Mirkovitch J, Darnell JE., Jr Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. EMBO Journal. 1991;10:927–932. doi: 10.1002/j.1460-2075.1991.tb08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M, Bucher P. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. Journal of Biological Chemistry. 2001;276:6675–6688. doi: 10.1074/jbc.M001748200. [DOI] [PubMed] [Google Scholar]
- Eilon T, Groner B, Barash I. Tumors caused by overexpression and forced activation of Stat5 in mammary epithelial cells of transgenic mice are parity-dependent and developed in aged, postestropausal females. International Journal of Cancer. 2007;121:1892–1902. doi: 10.1002/ijc.22954. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zhang XL, Lisanti MP. Sequence and detailed organization of the human caveolin-1 and -2 genes located near the D7S522 locus (7q31.1). Methylation of a CpG island in the 5′ promoter region of the caveolin-1 gene in human breast cancer cell lines. FEBS Letters. 1999;448:221–230. doi: 10.1016/s0014-5793(99)00365-8. [DOI] [PubMed] [Google Scholar]
- Evans MK, Yu CR, Lohani A, Mahdi RM, Liu X, Trzeciak AR, Egwuagu CE. Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene. 2007;26:1941–1948. doi: 10.1038/sj.onc.1209993. [DOI] [PubMed] [Google Scholar]
- Van den Eynden GG, Van Laere SJ, Van der Auwera I, Merajver SD, Van Marck EA, van Dam P, Vermeulen PB, Dirix LY, van Golen KL. Overexpression of caveolin-1 and -2 in cell lines and in human samples of inflammatory breast cancer. Breast Cancer Research and Treatment. 2006;95:219–228. doi: 10.1007/s10549-005-9002-1. [DOI] [PubMed] [Google Scholar]
- Foxwell BM, Beadling C, Guschin D, Kerr I, Cantrell D. Interleukin-7 can induce the activation of Jak 1, Jak 3 and STAT 5 proteins in murine T cells. European Journal of Immunology. 1995;25:3041–3046. doi: 10.1002/eji.1830251109. [DOI] [PubMed] [Google Scholar]
- Fu XY, Schindler C, Improta T, Aebersold R, Darnell JE., Jr The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. PNAS. 1992;89:7840–7843. doi: 10.1073/pnas.89.16.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego MI, Binart N, Robinson GW, Okagaki R, Coschigano KT, Perry J, Kopchick JJ, Oka T, Kelly PA, Hennighausen L. Prolactin, growth hormone, and epidermal growth factor activate Stat5 in different compartments of mammary tissue and exert different and overlapping developmental effects. Developmental Biology. 2001;229:163–175. doi: 10.1006/dbio.2000.9961. [DOI] [PubMed] [Google Scholar]
- Galsgaard ED, Gouilleux F, Groner B, Serup P, Nielsen JH, Billestrup N. Identification of a growth hormone-responsive STAT5-binding element in the rat insulin 1 gene. Molecular Endocrinology. 1996;10:652–660. doi: 10.1210/mend.10.6.8776725. [DOI] [PubMed] [Google Scholar]
- Gewinner C, Hart G, Zachara N, Cole R, Beisenherz-Huss C, Groner B. The coactivator of transcription CREB-binding protein interacts preferentially with the glycosylated form of Stat5. Journal of Biological Chemistry. 2004;279:3563–3572. doi: 10.1074/jbc.M306449200. [DOI] [PubMed] [Google Scholar]
- Gillanders EM, Xu J, Chang BL, Lange EM, Wiklund F, Bailey-Wilson JE, Baffoe-Bonnie A, Jones M, Gildea D, Riedesel E, et al. Combined genome-wide scan for prostate cancer susceptibility genes. Journal of National Cancer Institute. 2004;96:1240–1247. doi: 10.1093/jnci/djh228. [DOI] [PubMed] [Google Scholar]
- Ginsburg E, Vonderhaar BK. Prolactin synthesis and secretion by human breast cancer cells. Cancer Research. 1995;55:2591–2595. [PubMed] [Google Scholar]
- Gouilleux F, Wakao H, Mundt M, Groner B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO Journal. 1994;13:4361–4369. doi: 10.1002/j.1460-2075.1994.tb06756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouilleux F, Moritz D, Humar M, Moriggl R, Berchtold S, Groner B. Prolactin and interleukin-2 receptors in T lymphocytes signal through a MGF-STAT5-like transcription factor. Endocrinology. 1995a;136:5700–5708. doi: 10.1210/endo.136.12.7588326. [DOI] [PubMed] [Google Scholar]
- Gouilleux F, Pallard C, Dusanter-Fourt I, Wakao H, Haldosen LA, Norstedt G, Levy D, Groner B. Prolactin, growth hormone, erythropoietin and granulocyte-macrophage colony stimulating factor induce MGF-Stat5 DNA binding activity. EMBO Journal. 1995b;14:2005–2013. doi: 10.1002/j.1460-2075.1995.tb07192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goureau A, Garrigues A, Tosser-Klopp G, Lahbib-Mansais Y, Chardon P, Yerle M. Conserved synteny and gene order difference between human chromosome 12 and pig chromosome 5. Cytogenetics and Cell Genetics. 2001;94:49–54. doi: 10.1159/000048782. [DOI] [PubMed] [Google Scholar]
- de Groot RP, Raaijmakers JA, Lammers JW, Jove R, Koenderman L. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood. 1999;94:1108–1112. [PubMed] [Google Scholar]
- Gross M, Liu B, Tan J, French FS, Carey M, Shuai K. Distinct effects of PIAS proteins on androgen-mediated gene activation in prostate cancer cells. Oncogene. 2001;20:3880–3887. doi: 10.1038/sj.onc.1204489. [DOI] [PubMed] [Google Scholar]
- Guh JY, Huang JS, Chen HC, Hung WC, Lai YH, Chuang LY. Advanced glycation end product-induced proliferation in NRK-49F cells is dependent on the JAK2/STAT5 pathway and cyclin D1. American Journal of Kidney Diseases. 2001;38:1096–1104. doi: 10.1053/ajkd.2001.28616. [DOI] [PubMed] [Google Scholar]
- Haddad B, Pabon-Pena CR, Young H, Sun WH. Assignment1 of STAT1 to human chromosome 2q32 by FISH and radiation hybrids. Cytogenetics and Cell Genetics. 1998;83:58–59. doi: 10.1159/000015126. [DOI] [PubMed] [Google Scholar]
- Haffner MC, Petridou B, Peyrat JP, Revillion F, Muller-Holzner E, Daxenbichler G, Marth C, Doppler W. Favorable prognostic value of SOCS2 and IGF-I in breast cancer. BMC Cancer. 2007;7:136. doi: 10.1186/1471-2407-7-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halmos G, Schally AV, Czompoly T, Krupa M, Varga JL, Rekasi Z. Expression of growth hormone-releasing hormone and its receptor splice variants in human prostate cancer. Journal of Clinical Endocrinology and Metabolism. 2002;87:4707–4714. doi: 10.1210/jc.2002-020347. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Matsuda S, Machida K, Yamamoto T, Fukuda Y, Nimura Y, Hayakawa T, Hamaguchi M. Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Research. 2001;61:2361–2364. [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Information networks in the mammary gland. Nature Reviews. Molecular and Cellular Biology. 2005;6:715–725. doi: 10.1038/nrm1714. [DOI] [PubMed] [Google Scholar]
- Horseman ND, Zhao W, Montecino-Rodriguez E, Tanaka M, Nakashima K, Engle SJ, Smith F, Markoff E, Dorshkind K. Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO Journal. 1997;16:6926–6935. doi: 10.1093/emboj/16.23.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath CM, Wen Z, Darnell JE., Jr A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes and Development. 1995;9:984–994. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- Hou J, Schindler U, Henzel WJ, Wong SC, McKnight SL. Identification and purification of human Stat proteins activated in response to interleukin-2. Immunity. 1995;2:321–329. doi: 10.1016/1074-7613(95)90140-x. [DOI] [PubMed] [Google Scholar]
- Huang CY, Beliakoff J, Li X, Lee J, Sharma M, Lim B, Sun Z. hZimp7, a novel PIAS-like protein, enhances androgen receptor-mediated transcription and interacts with SWI/SNF-like BAF complexes. Molecular Endocrinology. 2005;19:2915–2929. doi: 10.1210/me.2005-0097. [DOI] [PubMed] [Google Scholar]
- Humphreys RC, Hennighausen L. Transforming growth factor alpha and mouse models of human breast cancer. Oncogene. 2000;19:1085–1091. doi: 10.1038/sj.onc.1203278. [DOI] [PubMed] [Google Scholar]
- Hurlstone AF, Reid G, Reeves JR, Fraser J, Strathdee G, Rahilly M, Parkinson EK, Black DM. Analysis of the CAVEOLIN-1 gene at human chromosome 7q31.1 in primary tumours and tumour-derived cell lines. Oncogene. 1999;18:1881–1890. doi: 10.1038/sj.onc.1202491. [DOI] [PubMed] [Google Scholar]
- Iavnilovitch E, Groner B, Barash I. Overexpression and forced activation of stat5 in mammary gland of transgenic mice promotes cellular proliferation, enhances differentiation, and delays postlactational apoptosis. Molecular Cancer Research. 2002;1:32–47. [PubMed] [Google Scholar]
- Iavnilovitch E, Cardiff RD, Groner B, Barash I. Deregulation of Stat5 expression and activation causes mammary tumors in transgenic mice. International Journal of Cancer. 2004;112:607–619. doi: 10.1002/ijc.20484. [DOI] [PubMed] [Google Scholar]
- Iavnilovitch E, Eilon T, Groner B, Barash I. Expression of a carboxy terminally truncated Stat5 with no transactivation domain in the mammary glands of transgenic mice inhibits cell proliferation during pregnancy, delays onset of milk secretion, and induces apoptosis upon involution. Molecular Reproduction and Development. 2006;73:841–849. doi: 10.1002/mrd.20479. [DOI] [PubMed] [Google Scholar]
- Ihle JN. The Stat family in cytokine signaling. Current Opinion in Cell Biology. 2001;13:211–217. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- Isaacs JT, Isaacs WB. Androgen receptor outwits prostate cancer drugs. Nature Medicine. 2004;10:26–27. doi: 10.1038/nm0104-26. [DOI] [PubMed] [Google Scholar]
- Jackson PK. A new RING for SUMO: wrestling transcriptional responses into nuclear bodies with PIAS family E3 SUMO ligases. Genes and Development. 2001;15:3053–3058. doi: 10.1101/gad.955501. [DOI] [PubMed] [Google Scholar]
- Jasmin JF, Mercier I, Sotgia F, Lisanti MP. SOCS proteins and caveolin-1 as negative regulators of endocrine signaling. Trends in Endocrinology and Metabolism. 2006;17:150–158. doi: 10.1016/j.tem.2006.03.007. [DOI] [PubMed] [Google Scholar]
- John S, Vinkemeier U, Soldaini E, Darnell JE, Jr, Leonard WJ. The significance of tetramerization in promoter recruitment by Stat5. Molecular Cell Biology. 1999;19:1910–1918. doi: 10.1128/mcb.19.3.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung YH, Lim EJ, Lee MY, Park JH, Ye SK, Park EU, Kim SY, Zhang Z, Lee KJ, Park DK, et al. Hypoxia activates the cyclin D1 promoter via the Jak2/STAT5b pathway in breast cancer cells. Experimental and Molecular Medicine. 2005;37:353–364. doi: 10.1038/emm.2005.45. [DOI] [PubMed] [Google Scholar]
- Junicho A, Matsuda T, Yamamoto T, Kishi H, Korkmaz K, Saatcioglu F, Fuse H, Muraguchi A. Protein inhibitor of activated STAT3 regulates androgen receptor signaling in prostate carcinoma cells. Biochemical and Biophysical Research Communications. 2000;278:9–13. doi: 10.1006/bbrc.2000.3753. [DOI] [PubMed] [Google Scholar]
- Kahyo T, Nishida T, Yasuda H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Molecular Cell. 2001;8:713–718. doi: 10.1016/s1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- Kamizono S, Hanada T, Yasukawa H, Minoguchi S, Kato R, Minoguchi M, Hattori K, Hatakeyama S, Yada M, Morita S, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. Journal of Biological Chemistry. 2001;276:12530–12538. doi: 10.1074/jbc.M010074200. [DOI] [PubMed] [Google Scholar]
- Karam JA, Lotan Y, Roehrborn CG, Ashfaq R, Karakiewicz PI, Shariat SF. Caveolin-1 overexpression is associated with aggressive prostate cancer recurrence. Prostate. 2007;67:614–622. doi: 10.1002/pros.20557. [DOI] [PubMed] [Google Scholar]
- Kasahara K, Taguchi T, Yamasaki I, Kamada M, Yuri K, Shuin T. Detection of genetic alterations in advanced prostate cancer by comparative genomic hybridization. Cancer Genetics and Cytogenetic. 2002;137:59–63. doi: 10.1016/s0165-4608(02)00552-6. [DOI] [PubMed] [Google Scholar]
- Kazansky AV, Raught B, Lindsey SM, Wang YF, Rosen JM. Regulation of mammary gland factor/Stat5a during mammary gland development. Molecular Endocrinology. 1995;9:1598–1609. doi: 10.1210/mend.9.11.8584036. [DOI] [PubMed] [Google Scholar]
- Kazansky AV, Kabotyanski EB, Wyszomierski SL, Mancini MA, Rosen JM. Differential effects of prolactin and src/abl kinases on the nuclear translocation of STAT5B and STAT5A. Journal of Biological Chemistry. 1999;274:22484–22492. doi: 10.1074/jbc.274.32.22484. [DOI] [PubMed] [Google Scholar]
- Kazansky AV, Spencer DM, Greenberg NM. Activation of signal transducer and activator of transcription 5 is required for progression of autochthonous prostate cancer: evidence from the transgenic adenocarcinoma of the mouse prostate system. Cancer Research. 2003;63:8757–8762. [PubMed] [Google Scholar]
- Kindblom J, Dillner K, Ling C, Tornell J, Wennbo H. Progressive prostate hyperplasia in adult prolactin transgenic mice is not dependent on elevated serum androgen levels. Prostate. 2002;53:24–33. doi: 10.1002/pros.10113. [DOI] [PubMed] [Google Scholar]
- Kindblom J, Dillner K, Sahlin L, Robertson F, Ormandy C, Tornell J, Wennbo H. Prostate hyperplasia in a transgenic mouse with prostate-specific expression of prolactin. Endocrinology. 2003;144:2269–2278. doi: 10.1210/en.2002-0187. [DOI] [PubMed] [Google Scholar]
- Kirken RA, Malabarba MG, Xu J, DaSilva L, Erwin RA, Liu X, Hennighausen L, Rui H, Farrar WL. Two discrete regions of interleukin-2 (IL2) receptor beta independently mediate IL2 activation of a PD98059/rapamycin/wortmannin-insensitive Stat5a/b serine kinase. Journal of Biological Chemistry. 1997a;272:15459–15465. doi: 10.1074/jbc.272.24.15459. [DOI] [PubMed] [Google Scholar]
- Kirken RA, Malabarba MG, Xu J, Liu X, Farrar WL, Hennighausen L, Larner AC, Grimley PM, Rui H. Prolactin stimulates serine/tyrosine phosphorylation and formation of heterocomplexes of multiple Stat5 isoforms in Nb2 lymphocytes. Journal of Biological Chemistry. 1997b;272:14098–14103. doi: 10.1074/jbc.272.22.14098. [DOI] [PubMed] [Google Scholar]
- von Knobloch R, Konrad L, Barth PJ, Brandt H, Wille S, Heidenreich A, Moll R, Hofmann R. Genetic pathways and new progression markers for prostate cancer suggested by microsatellite allelotyping. Clinical Cancer Research. 2004;10:1064–1073. doi: 10.1158/1078-0432.ccr-03-0070. [DOI] [PubMed] [Google Scholar]
- Lange EM, Gillanders EM, Davis CC, Brown WM, Campbell JK, Jones M, Gildea D, Riedesel E, Albertus J, Freas-Lutz D, et al. Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan prostate cancer genetics project finds evidence for linkage on chromosome 17 near BRCA1. Prostate. 2003;57:326–334. doi: 10.1002/pros.10307. [DOI] [PubMed] [Google Scholar]
- Latil A, Baron JC, Cussenot O, Fournier G, Boccon-Gibod L, Le Duc A, Lidereau R. Oncogene amplifications in early-stage human prostate carcinomas. International Journal of Cancer. 1994;59:637–638. doi: 10.1002/ijc.2910590510. [DOI] [PubMed] [Google Scholar]
- Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene. 1998;16:1391–1397. doi: 10.1038/sj.onc.1201661. [DOI] [PubMed] [Google Scholar]
- Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP. Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (−/−) null mice show mammary epithelial cell hyperplasia. American Journal of Pathology. 2002;161:1357–1369. doi: 10.1016/S0002-9440(10)64412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Joung YH, Lim EJ, Park JH, Ye SK, Park T, Zhang Z, Park DK, Lee KJ, Yang YM. Phosphorylation and activation of STAT proteins by hypoxia in breast cancer cells. Breast. 2006;15:187–195. doi: 10.1016/j.breast.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Leek JP, Hamlin PJ, Bell SM, Lench NJ. Assignment of the STAT6 gene (STAT6) to human chromosome band 12q13 by in situ hybridization. Cytogenetics and Cell Genetics. 1997;79:208–209. doi: 10.1159/000134723. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annual Review of Immunology. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Letsch M, Schally AV, Busto R, Bajo AM, Varga JL. Growth hormone-releasing hormone (GHRH) antagonists inhibit the proliferation of androgen-dependent and -independent prostate cancers. PNAS. 2003;100:1250–1255. doi: 10.1073/pnas.0337496100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nature Reviews. Molecular and Cellular Biology. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Levy N, Boettger-Tong H, Dohmae K, Agoulnik AI, Ty TI, Nishimune Y, Bishop CE. Physical and genetic linkage of glutaminase (Gls), signal transducer and activator of transcription 1 (Stat1), and xeroderma pigmentosum complementation group G (Xpg) on mouse proximal chromosome 1. Genomics. 1998;54:355–356. doi: 10.1006/geno.1998.5532. [DOI] [PubMed] [Google Scholar]
- Li P, Yu X, Ge K, Melamed J, Roeder RG, Wang Z. Heterogenous expression and functions of androgen receptor co-factors in primary prostate cancer. American Journal of Pathology. 2002;161:1467–1474. doi: 10.1016/S0002-9440(10)64422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ahonen TJ, Alanen K, Xie J, LeBaron MJ, Pretlow TG, Ealley EL, Zhang Y, Nurmi M, Singh B, et al. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Research. 2004;64:4774–4782. doi: 10.1158/0008-5472.CAN-03-3499. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang Y, Glass A, Zellweger T, Gehan E, Bubendorf L, Gelmann EP, Nevalainen MT. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clinical Cancer Research. 2005;11:5863–5868. doi: 10.1158/1078-0432.CCR-05-0562. [DOI] [PubMed] [Google Scholar]
- Lin JX, Mietz J, Modi WS, John S, Leonard WJ. Cloning of human Stat5B. Reconstitution of interleukin-2-induced Stat5A and Stat5B DNA binding activity in COS-7 cells. Journal of Biological Chemistry. 1996;271:10738–10744. [PubMed] [Google Scholar]
- Linja MJ, Porkka KP, Kang Z, Savinainen KJ, Janne OA, Tammela TL, Vessella RL, Palvimo JJ, Visakorpi T. Expression of androgen receptor coregulators in prostate cancer. Clinical Cancer Research. 2004;10:1032–1040. doi: 10.1158/1078-0432.ccr-0990-3. [DOI] [PubMed] [Google Scholar]
- Litterst CM, Kliem S, Marilley D, Pfitzner E. NCoA-1/SRC-1 is an essential coactivator of STAT5 that binds to the FDL motif in the alpha-helical region of the STAT5 transactivation domain. Journal of Biological Chemistry. 2003;278:45340–45351. doi: 10.1074/jbc.M303644200. [DOI] [PubMed] [Google Scholar]
- Litterst CM, Kliem S, Lodrini M, Pfitzner E. Coactivators in gene regulation by STAT5. Vitamins and Hormones. 2005;70:359–386. doi: 10.1016/S0083-6729(05)70012-1. [DOI] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. PNAS. 1995;92:8831–8835. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Mietz J, Modi WS, John S, Leonard WJ. Cloning of human Stat5B. Reconstitution of interleukin-2-induced Stat5A and Stat5B DNA binding activity in COS-7 cells. Journal of Biological Chemistry. 1996a;271:10738–10744. [PubMed] [Google Scholar]
- Liu X, Robinson GW, Hennighausen L. Activation of Stat5a and Stat5b by tyrosine phosphorylation is tightly linked to mammary gland differentiation. Molecular Endocrinology. 1996b;10:1496–1506. doi: 10.1210/mend.10.12.8961260. [DOI] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes and Development. 1997;11:179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K. Inhibition of Stat1-mediated gene activation by PIAS1. PNAS. 1998a;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gallego MI, Smith GH, Robinson GW, Hennighausen L. Functional release of Stat5a-null mammary tissue through the activation of compensating signals including Stat5b. Cell Growth and Differentiation. 1998b;9:795–803. [PubMed] [Google Scholar]
- Llovera M, Pichard C, Bernichtein S, Jeay S, Touraine P, Kelly PA, Goffin V. Human prolactin (hPRL) antagonists inhibit hPRL-activated signaling pathways involved in breast cancer cell proliferation. Oncogene. 2000;19:4695–4705. doi: 10.1038/sj.onc.1203846. [DOI] [PubMed] [Google Scholar]
- Magne S, Caron S, Charon M, Rouyez MC, Dusanter-Fourt I. STAT5 and Oct-1 form a stable complex that modulates cyclin D1 expression. Molecular Cell Biology. 2003;23:8934–8945. doi: 10.1128/MCB.23.24.8934-8945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. Journal of Cell Biology. 2004;165:823–833. doi: 10.1083/jcb.200403057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer AB, Wichmann C, Gross A, Kunkel H, Heinzel T, Ruthardt M, Groner B, Grez M. The Stat5-RARalpha fusion protein represses transcription and differentiation through interaction with a corepressor complex. Blood. 2002;99:2647–2652. doi: 10.1182/blood.v99.8.2647. [DOI] [PubMed] [Google Scholar]
- Meyer WK, Reichenbach P, Schindler U, Soldaini E, Nabholz M. Interaction of STAT5 dimers on two low affinity binding sites mediates interleukin 2 (IL-2) stimulation of IL-2 receptor alpha gene transcription. Journal of Biological Chemistry. 1997;272:31821–31828. doi: 10.1074/jbc.272.50.31821. [DOI] [PubMed] [Google Scholar]
- Meyer T, Begitt A, Lodige I, van Rossum M, Vinkemeier U. Constitutive and IFN-gamma-induced nuclear import of STAT1 proceed through independent pathways. EMBO Journal. 2002;21:344–354. doi: 10.1093/emboj/21.3.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Shillingford JM, Smith GH, Grimm SL, Wagner KU, Oka T, Rosen JM, Robinson GW, Hennighausen L. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. Journal of Cell Biology. 2001;155:531–542. doi: 10.1083/jcb.200107065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui A, Muto A, Sakamaki K, Sato N, Kinoshita T, Watanabe S, Yokota T, Arai K, Miyajima A. Function of the common beta subunit of the GM-CSF/IL-3/IL-5 receptors. Advances in Experimental Medicine and Biology. 1994;365:217–223. doi: 10.1007/978-1-4899-0987-9_22. [DOI] [PubMed] [Google Scholar]
- Mui AL, Wakao H, O’Farrell AM, Harada N, Miyajima A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO Journal. 1995;14:1166–1175. doi: 10.1002/j.1460-2075.1995.tb07100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nature Reviews. Molecular and Cellular Biology. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- Nagel-Wolfrum K, Buerger C, Wittig I, Butz K, Hoppe-Seyler F, Groner B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Molecular Cancer Research. 2004;2:170–182. [PubMed] [Google Scholar]
- Nakajima H, Brindle PK, Handa M, Ihle JN. Functional interaction of STAT5 and nuclear receptor co-repressor SMRT: implications in negative regulation of STAT5-dependent transcription. EMBO Journal. 2001;20:6836–6844. doi: 10.1093/emboj/20.23.6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwirt H, Puhr M, Cavarretta IT, Mitterberger M, Hobisch A, Culig Z. Suppressor of cytokine signalling-3 is upregulated by androgen in prostate cancer cell lines and inhibits androgen-mediated proliferation and secretion. Endocrine-Related Cancer. 2007;14:1007–1019. doi: 10.1677/ERC-07-0172. [DOI] [PubMed] [Google Scholar]
- Nevalainen MT, Valve EM, Makela SI, Blauer M, Tuohimaa PJ, Harkonen PL. Estrogen and prolactin regulation of rat dorsal and lateral prostate in organ culture. Endocrinology. 1991;129:612–622. doi: 10.1210/endo-129-2-612. [DOI] [PubMed] [Google Scholar]
- Nevalainen MT, Valve EM, Ingleton PM, Harkonen PL. Expression and hormone regulation of prolactin receptors in rat dorsal and lateral prostate. Endocrinology. 1996;137:3078–3088. doi: 10.1210/endo.137.7.8770934. [DOI] [PubMed] [Google Scholar]
- Nevalainen MT, Valve EM, Ahonen T, Yagi A, Paranko J, Harkonen PL. Androgen-dependent expression of prolactin in rat prostate epithelium in vivo and in organ culture. FASEB Journal. 1997a;11:1297–1307. doi: 10.1096/fasebj.11.14.9409549. [DOI] [PubMed] [Google Scholar]
- Nevalainen MT, Valve EM, Ingleton PM, Nurmi M, Martikainen PM, Harkonen PL. Prolactin and prolactin receptors are expressed and functioning in human prostate. Journal of Clinical Investigation. 1997b;99:618–627. doi: 10.1172/JCI119204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevalainen MT, Ahonen TJ, Yamashita H, Chandrashekar V, Bartke A, Grimley PM, Robinson GW, Hennighausen L, Rui H. Epithelial defect in prostates of Stat5a-null mice. Laboratory Investigation. 2000;80:993–1006. doi: 10.1038/labinvest.3780105. [DOI] [PubMed] [Google Scholar]
- Nevalainen MT, Xie J, Bubendorf L, Wagner KU, Rui H. Basal activation of transcription factor signal transducer and activator of transcription (Stat5) in nonpregnant mouse and human breast epithelium. Molecular Endocrinology. 2002;16:1108–1124. doi: 10.1210/mend.16.5.0839. [DOI] [PubMed] [Google Scholar]
- Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, Sauter G, Rui H. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. Journal of Clinical Oncology. 2004;22:2053–2060. doi: 10.1200/JCO.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Nishida T, Yasuda H. PIAS1 and PIASxalpha function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. Journal of Biological Chemistry. 2002;277:41311–41317. doi: 10.1074/jbc.M206741200. [DOI] [PubMed] [Google Scholar]
- Oakes SR, Robertson FG, Kench JG, Gardiner-Garden M, Wand MP, Green JE, Ormandy CJ. Loss of mammary epithelial prolactin receptor delays tumor formation by reducing cell proliferation in low-grade preinvasive lesions. Oncogene. 2007;26:543–553. doi: 10.1038/sj.onc.1209838. [DOI] [PubMed] [Google Scholar]
- Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, Edery M, Brousse N, Babinet C, Binart N, et al. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes and Development. 1997;11:167–178. doi: 10.1101/gad.11.2.167. [DOI] [PubMed] [Google Scholar]
- Pallard C, Gouilleux F, Benit L, Cocault L, Souyri M, Levy D, Groner B, Gisselbrecht S, Dusanter-Fourt I. Thrombopoietin activates a STAT5-like factor in hematopoietic cells. EMBO Journal. 1995a;14:2847–2856. doi: 10.1002/j.1460-2075.1995.tb07284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallard C, Gouilleux F, Charon M, Groner B, Gisselbrecht S, Dusanter-Fourt I. Interleukin-3, erythropoietin, and prolactin activate a STAT5-like factor in lymphoid cells. Journal of Biological Chemistry. 1995b;270:15942–15945. doi: 10.1074/jbc.270.27.15942. [DOI] [PubMed] [Google Scholar]
- Park C, Lee I, Kang WK. Lovastatin-induced E2F-1 modulation and its effect on prostate cancer cell death. Carcinogenesis. 2001;22:1727–1731. doi: 10.1093/carcin/22.10.1727. [DOI] [PubMed] [Google Scholar]
- Park DS, Lee H, Frank PG, Razani B, Nguyen AV, Parlow AF, Russell RG, Hulit J, Pestell RG, Lisanti MP. Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation, and hyperactivation of the Jak-2/STAT5a signaling cascade. Molecular Biology of the Cell. 2002;13:3416–3430. doi: 10.1091/mbc.02-05-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SS, Kim JE, Kim YA, Kim YC, Kim SW. Caveolin-1 is down-regulated and inversely correlated with HER2 and EGFR expression status in invasive ductal carcinoma of the breast. Histopathology. 2005;47:625–630. doi: 10.1111/j.1365-2559.2005.02303.x. [DOI] [PubMed] [Google Scholar]
- Paukku K, Yang J, Silvennoinen O. Tudor and nuclease-like domains containing protein p100 function as coactivators for signal transducer and activator of transcription 5. Molecular Endocrinology. 2003;17:1805–1814. doi: 10.1210/me.2002-0256. [DOI] [PubMed] [Google Scholar]
- Peng B, Sutherland KD, Sum EY, Olayioye M, Wittlin S, Tang TK, Lindeman GJ, Visvader JE. CPAP is a novel stat5-interacting cofactor that augments stat5-mediated transcriptional activity. Molecular Endocrinology. 2002;16:2019–2033. doi: 10.1210/me.2002-0108. [DOI] [PubMed] [Google Scholar]
- Pinilla SM, Honrado E, Hardisson D, Benitez J, Palacios J. Caveolin-1 expression is associated with a basal-like phenotype in sporadic and hereditary breast cancer. Breast Cancer Research and Treatment. 2006;99:85–90. doi: 10.1007/s10549-006-9184-1. [DOI] [PubMed] [Google Scholar]
- Quelle FW, Thierfelder W, Witthuhn BA, Tang B, Cohen S, Ihle JN. Phosphorylation and activation of the DNA binding activity of purified Stat1 by the Janus protein-tyrosine kinases and the epidermal growth factor receptor. Journal of Biological Chemistry. 1995;270:20775–20780. doi: 10.1074/jbc.270.35.20775. [DOI] [PubMed] [Google Scholar]
- Raccurt M, Tam SP, Lau P, Mertani HC, Lambert A, Garcia-Caballero T, Li H, Brown RJ, McGuckin MA, Morel G, et al. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. British Journal of Cancer. 2003;89:524–532. doi: 10.1038/sj.bjc.6601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. Journal of Biological Chemistry. 1999;274:35553–35561. doi: 10.1074/jbc.274.50.35553. [DOI] [PubMed] [Google Scholar]
- Reich NC, Liu L. Tracking STAT nuclear traffic. Nature Reviews of Immunology. 2006;6:602–612. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- Ren S, Cai HR, Li M, Furth PA. Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene. 2002;21:4335–4339. doi: 10.1038/sj.onc.1205484. [DOI] [PubMed] [Google Scholar]
- Rogers RS, Inselman A, Handel MA, Matunis MJ. SUMO modified proteins localize to the XY body of pachytene spermatocytes. Chromosoma. 2004;113:233–243. doi: 10.1007/s00412-004-0311-7. [DOI] [PubMed] [Google Scholar]
- Rosen RL, Winestock KD, Chen G, Liu X, Hennighausen L, Finbloom DS. Granulocyte-macrophage colony-stimulating factor preferentially activates the 94-kD STAT5A and an 80-kD STAT5A isoform in human peripheral blood monocytes. Blood. 1996;88:1206–1214. [PubMed] [Google Scholar]
- Rui H, Kirken RA, Farrar WL. Activation of receptor-associated tyrosine kinase JAK2 by prolactin. Journal of Biological Chemistry. 1994;269:5364–5368. [PubMed] [Google Scholar]
- Rycyzyn MA, Clevenger CV. The intranuclear prolactin/cyclophilin B complex as a transcriptional inducer. PNAS. 2002;99:6790–6795. doi: 10.1073/pnas.092160699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycyzyn MA, Reilly SC, O’Malley K, Clevenger CV. Role of cyclophilin B in prolactin signal transduction and nuclear retrotranslocation. Molecular Endocrinology. 2000;14:1175–1186. doi: 10.1210/mend.14.8.0508. [DOI] [PubMed] [Google Scholar]
- Sachdev S, Bruhn L, Sieber H, Pichler A, Melchior F, Grosschedl R. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes and Development. 2001;15:3088–3103. doi: 10.1101/gad.944801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara Y, Mimori K, Yoshinaga K, Tanaka F, Nishida K, Ohno S, Inoue H, Mori M. Clinical significance of Caveolin-1, Caveolin-2 and HER2/neu mRNA expression in human breast cancer. British Journal of Cancer. 2004;91:959–965. doi: 10.1038/sj.bjc.6602029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage K, Lambros MB, Robertson D, Jones RL, Jones C, Mackay A, James M, Hornick JL, Pereira EM, Milanezi F, et al. Caveolin 1 is overexpressed and amplified in a subset of basal-like and metaplastic breast carcinomas: a morphologic, ultrastructural, immunohistochemical, and in situ hybridization analysis. Clinical Cancer Research. 2007;13:90–101. doi: 10.1158/1078-0432.CCR-06-1371. [DOI] [PubMed] [Google Scholar]
- Schindler CW. Series introduction. JAK-STAT signaling in human disease. Journal of Clinical Investigation. 2002;109:1133–1137. doi: 10.1172/JCI15644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annual Review of Biochemistry. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- Schindler C, Fu XY, Improta T, Aebersold R, Darnell JE., Jr Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon alpha. PNAS. 1992;89:7836–7839. doi: 10.1073/pnas.89.16.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Muller S. PIAS/SUMO: new partners in transcriptional regulation. Cellular and Molecular Life Sciences. 2003;60:2561–2574. doi: 10.1007/s00018-003-3129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-Ney M, Doppler W, Ball RK, Groner B. Beta-casein gene promoter activity is regulated by the hormone-mediated relief of transcriptional repression and a mammary-gland-specific nuclear factor. Molecular Cell Biology. 1991;11:3745–3755. doi: 10.1128/mcb.11.7.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-Ney M, Happ B, Ball RK, Groner B. Developmental and environmental regulation of a mammary gland-specific nuclear factor essential for transcription of the gene encoding beta-casein. PNAS. 1992a;89:3130–3134. doi: 10.1073/pnas.89.7.3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-Ney M, Happ B, Hofer P, Hynes NE, Groner B. Mammary gland-specific nuclear factor activity is positively regulated by lactogenic hormones and negatively by milk stasis. Molecular Endocrinology. 1992b;6:1988–1997. doi: 10.1210/mend.6.12.1491685. [DOI] [PubMed] [Google Scholar]
- Schwaller J, Parganas E, Wang D, Cain D, Aster JC, Williams IR, Lee CK, Gerthner R, Kitamura T, Frantsve J, et al. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Molecular Cell. 2000;6:693–704. doi: 10.1016/s1097-2765(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Seeler JS, Bischof O, Nacerddine K, Dejean A. SUMO, the three Rs and cancer. Current Topics in Microbiology and Immunology. 2007;313:49–71. doi: 10.1007/978-3-540-34594-7_4. [DOI] [PubMed] [Google Scholar]
- Sekimoto T, Nakajima K, Tachibana T, Hirano T, Yoneda Y. Interferon-gamma-dependent nuclear import of Stat1 is mediated by the GTPase activity of Ran/TC4. Journal of Biological Chemistry. 1996;271:31017–31020. doi: 10.1074/jbc.271.49.31017. [DOI] [PubMed] [Google Scholar]
- Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO Journal. 1997;16:7067–7077. doi: 10.1093/emboj/16.23.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Yu M, Clark BD, Snyderwine EG. Possible role of Stat5a in rat mammary gland carcinogenesis. Breast Cancer Research and Treatment. 2004;88:263–272. doi: 10.1007/s10549-004-0805-2. [DOI] [PubMed] [Google Scholar]
- Sharma M, Li X, Wang Y, Zarnegar M, Huang CY, Palvimo JJ, Lim B, Sun Z. hZimp10 is an androgen receptor co-activator and forms a complex with SUMO-1 at replication foci. EMBO Journal. 2003;22:6101–6114. doi: 10.1093/emboj/cdg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks AD. PIAS proteins and transcriptional regulation – more than just SUMO E3 ligases? Genes and Development. 2006;20:754–758. doi: 10.1101/gad.1421006. [DOI] [PubMed] [Google Scholar]
- Shi W, Inoue M, Minami M, Takeda K, Matsumoto M, Matsuda Y, Kishimoto T, Akira S. The genomic structure and chromosomal localization of the mouse STAT3 gene. International Immunology. 1996;8:1205–1211. doi: 10.1093/intimm/8.8.1205. [DOI] [PubMed] [Google Scholar]
- Shuai K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 2000;19:2638–2644. doi: 10.1038/sj.onc.1203522. [DOI] [PubMed] [Google Scholar]
- Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Research. 2006;16:196–202. doi: 10.1038/sj.cr.7310027. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nature Reviews of Immunology. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004;23:8017–8023. doi: 10.1038/sj.onc.1208159. [DOI] [PubMed] [Google Scholar]
- Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Soldaini E, John S, Moro S, Bollenbacher J, Schindler U, Leonard WJ. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Molecular Cell Biology. 2000;20:389–401. doi: 10.1128/mcb.20.1.389-401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgia F, Rui H, Bonuccelli G, Mercier I, Pestell RG, Lisanti MP. Caveolin-1, mammary stem cells, and estrogen-dependent breast cancers. Cancer Research. 2006a;66:10647–10651. doi: 10.1158/0008-5472.CAN-06-2805. [DOI] [PubMed] [Google Scholar]
- Sotgia F, Williams TM, Schubert W, Medina F, Minetti C, Pestell RG, Lisanti MP. Caveolin-1 deficiency (−/−) conveys premalignant alterations in mammary epithelia, with abnormal lumen formation, growth factor independence, and cell invasiveness. American Journal of Pathology. 2006b;168:292–309. doi: 10.2353/ajpath.2006.050429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangelberger A, Schally AV, Varga JL, Zarandi M, Szepeshazi K, Armatis P, Halmos G. Inhibitory effect of antagonists of bombesin and growth hormone-releasing hormone on orthotopic and intraosseous growth and invasiveness of PC-3 human prostate cancer in nude mice. Clinical Cancer Research. 2005;11:49–57. [PubMed] [Google Scholar]
- Stocklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- Strauss BL, Bratthauer GL, Tavassoli FA. STAT 5a expression in the breast is maintained in secretory carcinoma, in contrast to other histologic types. Human Pathology. 2006;37:586–592. doi: 10.1016/j.humpath.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, Rui H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene. 2005;24:746–760. doi: 10.1038/sj.onc.1208203. [DOI] [PubMed] [Google Scholar]
- Tahir SA, Frolov A, Hayes TG, Mims MP, Miles BJ, Lerner SP, Wheeler TM, Ayala G, Thompson TC, Kadmon D. Preoperative serum caveolin-1 as a prognostic marker for recurrence in a radical prostatectomy cohort. Clinical Cancer Research. 2006;12:4872–4875. doi: 10.1158/1078-0432.CCR-06-0417. [DOI] [PubMed] [Google Scholar]
- Tan SH, Dagvadorj A, Shen F, Gu L, Liao Z, Abdulghani J, Zhang Y, Gelmann EP, Zellweger T, Culig Z, et al. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Research. 2008;68:236–248. doi: 10.1158/0008-5472.CAN-07-2972. [DOI] [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Thompson TC, Timme TL, Li L, Goltsov A. Caveolin-1, a metastasis-related gene that promotes cell survival in prostate cancer. Apoptosis. 1999;4:233–237. doi: 10.1023/a:1009612708099. [DOI] [PubMed] [Google Scholar]
- Timme TL, Goltsov A, Tahir S, Li L, Wang J, Ren C, Johnston RN, Thompson TC. Caveolin-1 is regulated by c-myc and suppresses c-myc-induced apoptosis. Oncogene. 2000;19:3256–3265. doi: 10.1038/sj.onc.1203654. [DOI] [PubMed] [Google Scholar]
- Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. Journal of Biological Chemistry. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Molecular Cancer Therapeutics. 2004;3:261–269. [PubMed] [Google Scholar]
- Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. PNAS. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungureanu D, Saharinen P, Junttila I, Hilton DJ, Silvennoinen O. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Molecular Cell Biology. 2002;22:3316–3326. doi: 10.1128/MCB.22.10.3316-3326.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage BA, van Houwelingen K, Ruijter TE, Kiemeney LA, Schalken JA. Allelic imbalance in hereditary and sporadic prostate cancer. Prostate. 2003;54:50–57. doi: 10.1002/pros.10148. [DOI] [PubMed] [Google Scholar]
- Vinkemeier U. Getting the message across, STAT! Design principles of a molecular signaling circuit. Journal of Cell Biology. 2004;167:197–201. doi: 10.1083/jcb.200407163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao H, Schmitt-Ney M, Groner B. Mammary gland-specific nuclear factor is present in lactating rodent and bovine mammary tissue and composed of a single polypeptide of 89 kDa. Journal of Biological Chemistry. 1992;267:16365–16370. [PubMed] [Google Scholar]
- Wakao H, Gouilleux F, Groner B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO Journal. 1994;13:2182–2191. doi: 10.1002/j.1460-2075.1994.tb06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Banerjee S. Differential PIAS3 expression in human malignancy. Oncology Reports. 2004;11:1319–1324. [PubMed] [Google Scholar]
- Wang Z, Prins GS, Coschigano KT, Kopchick JJ, Green JE, Ray VH, Hedayat S, Christov KT, Unterman TG, Swanson SM. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/Tag mouse. Endocrinology. 2005;146:5188–5196. doi: 10.1210/en.2005-0607. [DOI] [PubMed] [Google Scholar]
- Wartmann M, Cella N, Hofer P, Groner B, Liu X, Hennighausen L, Hynes NE. Lactogenic hormone activation of Stat5 and transcription of the beta-casein gene in mammary epithelial cells is independent of p42 ERK2 mitogen-activated protein kinase activity. Journal of Biological Chemistry. 1996;271:31863–31868. doi: 10.1074/jbc.271.50.31863. [DOI] [PubMed] [Google Scholar]
- Weiss-Messer E, Merom O, Adi A, Karry R, Bidosee M, Ber R, Kaploun A, Stein A, Barkey RJ. Growth hormone (GH) receptors in prostate cancer: gene expression in human tissues and cell lines and characterization, GH signaling and androgen receptor regulation in LNCaP cells. Molecular Cell Endocrinology. 2004;220:109–123. doi: 10.1016/j.mce.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Welte T, Garimorth K, Philipp S, Doppler W. Prolactin-dependent activation of a tyrosine phosphorylated DNA binding factor in mouse mammary epithelial cells. Molecular Endocrinology. 1994;8:1091–1102. doi: 10.1210/mend.8.8.7527899. [DOI] [PubMed] [Google Scholar]
- Wennbo H, Kindblom J, Isaksson OG, Tornell J. Transgenic mice overexpressing the prolactin gene develop dramatic enlargement of the prostate gland. Endocrinology. 1997;138:4410–4415. doi: 10.1210/endo.138.10.5461. [DOI] [PubMed] [Google Scholar]
- Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. American Journal of Physiology Cell Physiology. 2005;288:C494–C506. doi: 10.1152/ajpcell.00458.2004. [DOI] [PubMed] [Google Scholar]
- Williams TM, Sotgia F, Lee H, Hassan G, Di Vizio D, Bonuccelli G, Capozza F, Mercier I, Rui H, Pestell RG, et al. Stromal and epithelial caveolin-1 both confer a protective effect against mammary hyperplasia and tumorigenesis: Caveolin-1 antagonizes cyclin D1 function in mammary epithelial cells. American Journal of Pathology. 2006;169:1784–1801. doi: 10.2353/ajpath.2006.060590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter H, Gottfried HW, Mattfeldt T. Genetic changes in stage pT2N0 prostate cancer studied by comparative genomic hybridization. BJU International. 2002a;89:310–316. doi: 10.1046/j.1464-4096.2001.01722.x. [DOI] [PubMed] [Google Scholar]
- Wolter H, Trijic D, Gottfried HW, Mattfeldt T. Chromosomal changes in incidental prostatic carcinomas detected by comparative genomic hybridization. European Urology. 2002b;41:328–334. doi: 10.1016/s0302-2838(02)00035-0. [DOI] [PubMed] [Google Scholar]
- Xie J, LeBaron MJ, Nevalainen MT, Rui H. Role of tyrosine kinase Jak2 in prolactin-induced differentiation and growth of mammary epithelial cells. Journal of Biological Chemistry. 2002;277:14020–14030. doi: 10.1074/jbc.M112399200. [DOI] [PubMed] [Google Scholar]
- Xu W, Yu F, Yan M, Lu L, Zou W, Sun L, Zheng Z, Liu X. Geldanamycin, a heat shock protein 90-binding agent, disrupts Stat5 activation in IL-2-stimulated cells. Journal of Cell Physiology. 2004;198:188–196. doi: 10.1002/jcp.10403. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Quelle FW, Thierfelder WE, Kreider BL, Gilbert DJ, Jenkins NA, Copeland NG, Silvennoinen O, Ihle JN. Stat4, a novel gamma interferon activation site-binding protein expressed in early myeloid differentiation. Molecular Cell Biology. 1994;14:4342–4349. doi: 10.1128/mcb.14.7.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Kobayashi H, Arai A, Miura O, Hirosawa S, Miyasaka N. cDNA cloning, expression and chromosome mapping of the human STAT4 gene: both STAT4 and STAT1 genes are mapped to 2q32.2→q32.3. Cytogenetics and Cell Genetics. 1997;77:207–210. doi: 10.1159/000134578. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Nevalainen MT, Xu J, LeBaron MJ, Wagner KU, Erwin RA, Harmon JM, Hennighausen L, Kirken RA, Rui H. Role of serine phosphorylation of Stat5a in prolactin-stimulated beta-casein gene expression. Molecular Cell Endocrinology. 2001;183:151–163. doi: 10.1016/s0303-7207(01)00546-9. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Iwase H, Toyama T, Fujii Y. Naturally occurring dominant-negative Stat5 suppresses transcriptional activity of estrogen receptors and induces apoptosis in T47D breast cancer cells. Oncogene. 2003;22:1638–1652. doi: 10.1038/sj.onc.1206277. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Nishio M, Fujii Y, Iwase H. Dominant-negative Stat5 inhibits growth and induces apoptosis in T47D-derived tumors in nude mice. Cancer Science. 2004;95:662–665. doi: 10.1111/j.1349-7006.2004.tb03326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita H, Nishio M, Ando Y, Zhang Z, Hamaguchi M, Mita K, Kobayashi S, Fujii Y, Iwase H. Stat5 expression predicts response to endocrine therapy and improves survival in estrogen receptor-positive breast cancer. Endocrine-Related Cancer. 2006;13:885–893. doi: 10.1677/erc.1.01095. [DOI] [PubMed] [Google Scholar]
- Yang G, Truong LD, Wheeler TM, Thompson TC. Caveolin-1 expression in clinically confined human prostate cancer: a novel prognostic marker. Cancer Research. 1999;59:5719–5723. [PubMed] [Google Scholar]
- Yang E, Henriksen MA, Schaefer O, Zakharova N, Darnell JE., Jr Dissociation time from DNA determines transcriptional function in a STAT1 linker mutant. Journal of Biological Chemistry. 2002;277:13455–13462. doi: 10.1074/jbc.M112038200. [DOI] [PubMed] [Google Scholar]
- Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Research. 2005;65:939–947. [PubMed] [Google Scholar]
- Yang G, Addai J, Wheeler TM, Frolov A, Miles BJ, Kadmon D, Thompson TC. Correlative evidence that prostate cancer cell-derived caveolin-1 mediates angiogenesis. Human Pathology. 2007a;38:1688–1695. doi: 10.1016/j.humpath.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes and Development. 2007b;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. Stat5a/b are essential for normal lymphoid development and differentiation. PNAS. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip SS, Crew AJ, Gee JM, Hui R, Blamey RW, Robertson JF, Nicholson RI, Sutherland RL, Daly RJ. Up-regulation of the protein tyrosine phosphatase SHP-1 in human breast cancer and correlation with GRB2 expression. International Journal of Cancer. 2000;88:363–368. [PubMed] [Google Scholar]
- Yu H, Jove R. The STATs of cancer – new molecular targets come of age. Nature Review Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- Zapata PD, Ropero RM, Valencia AM, Buscail L, Lopez JI, Martin-Orozco RM, Prieto JC, Angulo J, Susini C, Lopez-Ruiz P, et al. Autocrine regulation of human prostate carcinoma cell proliferation by somatostatin through the modulation of the SH2 domain containing protein tyrosine phosphatase (SHP)-1. Journal of Clinical Endocrinology and Metabolism. 2002;87:915–926. doi: 10.1210/jcem.87.2.8194. [DOI] [PubMed] [Google Scholar]
- Zeng R, Aoki Y, Yoshida M, Arai K, Watanabe S. Stat5B shuttles between cytoplasm and nucleus in a cytokine-dependent and -independent manner. Journal of Immunology. 2002;168:4567–4575. doi: 10.4049/jimmunol.168.9.4567. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. PNAS. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JE., Jr Stat3 and Stat4: members of the family of signal transducers and activators of transcription. PNAS. 1994a;91:4806–4810. doi: 10.1073/pnas.91.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JEJ. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994b;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- Zhu M, John S, Berg M, Leonard WJ. Functional association of Nmi with Stat5 and Stat1 in IL-2- and IFNgamma-mediated signaling. Cell. 1999;96:121–130. doi: 10.1016/s0092-8674(00)80965-4. [DOI] [PubMed] [Google Scholar]
- Zuhlke KA, Madeoy JJ, Beebe-Dimmer J, White KA, Griffin A, Lange EM, Gruber SB, Ostrander EA, Cooney KA. Truncating BRCA1 mutations are uncommon in a cohort of hereditary prostate cancer families with evidence of linkage to 17q markers. Clinical Cancer Research. 2004;10:5975–5980. doi: 10.1158/1078-0432.CCR-04-0554. [DOI] [PubMed] [Google Scholar]