

Abstract
This study evaluated the short-term effects of tofacitinib treatment on peripheral blood leukocyte phenotype and function, and the reversibility of any such effects following treatment withdrawal in healthy volunteers. Cytomegalovirus (CMV)-seropositive subjects received oral tofacitinib 10 mg twice daily for 4 weeks and were followed for 4 weeks after drug withdrawal. There were slight increases in total lymphocyte and total T-cell counts during tofacitinib treatment, and B-cell counts increased by up to 26%. There were no significant changes in granulocyte or monocyte counts, or granulocyte function. Naïve and central memory T-cell counts increased during treatment, while all subsets of activated T cells were decreased by up to 69%. T-cell subsets other than effector memory cluster of differentiation CD4 +, activated naïve CD4 + and effector CD8 + T-cell counts and B-cell counts, normalized 4 weeks after withdrawal. Following ex vivo activation, measures of CMV-specific T-cell responses, and antigen non-specific T-cell-mediated cytotoxicity and interferon (IFN)-γ production, decreased slightly. These T-cell functional changes were most pronounced at Day 15, partially normalized while still on tofacitinib and returned to baseline after drug withdrawal. Total natural killer (NK)-cell counts decreased by 33%, returning towards baseline after drug withdrawal. NK-cell function decreased during tofacitinib treatment, but without a consistent time course across measured parameters. However, markers of NK-cell-mediated cytotoxicity, antibody-dependent cellular cytotoxicity and IFN-γ production were decreased up to 42% 1 month after drug withdrawal. CMV DNA was not detectable in whole blood, and there were no cases of herpes zoster reactivation. No new safety concerns arose. In conclusion, the effect of short-term tofacitinib treatment on leukocyte composition and function in healthy CMV+ volunteers is modest and largely reversible 4weeks after withdrawal.
Keywords: Tofacitinib, Healthy volunteers, T cells, Natural killer cells, Interferon-γ, Cytotoxicity
1. Introduction
Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of rheumatoid arthritis (RA). The efficacy and safety of tofacitinib 5 and 10mg twice daily (BID), administered as monotherapy or in combination with conventional synthetic disease-modifying antirheumatic drugs - mainly methotrexate – in patients with moderately to severely active RA, have been demonstrated in Phase 2 [1–5] and Phase 3 [6–11] studies of up to 24months duration, and in long-term extension (LTE) studies with up to 105months of observation [12–14].
Tofacitinib preferentially inhibits signaling by receptors associated with JAK1 and JAK3, with functional selectivity over receptors that signal via JAK2 [15,16]. JAK-dependent cytokines are important mediators of lymphocyte homeostasis, differentiation and function, and also modulate aspects of innate immunity [17].
The effect of tofacitinib on lymphocyte numbers has been characterized in patients with psoriasis [18] and for up to 8years in patients with RA [19]. Treatment up to 6months resulted in a reduction in natural killer (NK) cells, with estimated maximum reduction occurring at 8 to 10 weeks. These changes resolved within 6 weeks of discontinuation. Treatment was associated with increases in B cells and a transient increase in T cells, with small and inconsistent changes at intermediate times (up to 12months). With long-term treatment, total absolute lymphocyte count (ALC) decreased gradually by ~25% over 4years, and then plateaued. Over long-term treatment, cluster of differentiation (CD)4+ and CD8 + T-cell numbers correlated with ALC, whereas NK cells increased by a median of 73%. There was no evidence of a relationship between serious or opportunistic infections or herpes zoster (HZ) infection and lymphocyte subset counts.
The effects of tofacitinib on vaccine responses have been evaluated in patients with RA. Patients with RA immunized after 4weeks of treatment with tofacitinib monotherapy appeared to have a nominally diminished antibody response to pneumococcal polysaccharide (T-cell independent), but not to influenza (T-cell dependent) vaccines [20].
However, the antibody response to both vaccines appeared relatively unaffected when administered after longer-term treatment with tofacitinib, either as monotherapy or with background methotrexate, supporting the absence of a major effect of long-term tofacitinib treatment on antigen presentation, B-cell, and T-cell functions required for humoral immune responses to vaccines [20].
Despite the data on vaccination and immune cell numbers, the effect of in vivo administration of tofacitinib on immune cell function in healthy populations free of confounding variables, such as the RA disease state and concomitant immunosuppressant use, has not been specifically measured. In this study, the effect of 28days of tofacitinib treatment on functional parameters, including monocyte and granulocyte phagocytosis and oxidative burst capacity, direct and antibody-directed NK-cell-mediated cytolytic activity, measures of CD8 + T-cell cytotoxicity, and the number of T cells that respond to anti-CD3 cross-linking or cytomegalovirus (CMV) antigen presentation, were measured. In addition to the functional assays, a more detailed analysis of lymphocyte subsets was performed.
The objectives of the present study were to evaluate the effects of tofacitinib administered to healthy CMV+ volunteers at a dose of 10mg BID for 4 weeks on the number and function of T cells, NK cells, granulocytes and monocytes, and on the number of B cells, together with the potential reversibility of any such effects over the 4 weeks following drug withdrawal.
2. Material and methods
2.1. Study design
Study A3921252 was an exploratory, Phase 1, open-label, single-center, healthy-volunteer study of the effects of tofacitinib on the immune system, using ex vivo blood samples. Following a 4-week screening period, subjects received oral tofacitinib 10mg BID for 4weeks and were followed for an additional 4 weeks after drug withdrawal. The effect of tofacitinib 10mg BID on the number and function of T cells, NK cells, granulocytes and monocytes, and on the number of B cells, was assessed.
2.2. Subjects
Subjects were healthy males or females aged between 18 and 70 years. No clinically relevant abnormalities were identified from a detailed medical history, full physical examination, 12-lead electrocardiogram and clinical laboratory tests. Female subjects were of non-childbearing potential. Subjects were CMV seropositive, with no evidence of active, latent or inadequately treated infection with Mycobacterium tuberculosis. Testing for CMV seropositivity was performed, since it was a requirement for a recall response to CMV, and only 50% of US inhabitants are CMV seropositive [21]. Subjects with any infection requiring treatment within 2weeks prior to Day 1, or requiring hospitalization, parenteral antimicrobial therapy or judged to be opportunistic or clinically significant by the investigator within the past 6 months, were excluded from the study. Malignancy or a history of malignancy, with the exception of adequately treated or excised non-metastatic basal cell or squamous cell cancer of the skin or cervical carcinoma in situ, was also an exclusion criterion.
Healthy volunteers were chosen to allow for a washout period of tofacitinib to assess the reversibility of any alterations in immune function. The choice of healthy volunteers also removed the potential effects of disease state and concomitant immunosuppressive medications. A dose of 10mg BID, which is higher than the approved 5 mg BID dose in RA, was used to adjust for the lower serum drug levels that are observed in healthy volunteers compared to patients with RA.
2.3. Study procedures
Blood samples were collected from consenting subjects 4weeks before dosing commenced, to confirm CMV+ status. Blood samples were then collected from confirmed CMV+ subjects at various times; leukocyte counts and granulocyte and monocyte functional assays were performed using fresh whole blood. Lymphocyte phenotyping and functional assays were performed on viably cryopreserved peripheral blood mononuclear cells (PBMCs) to allow batch testing of longitudinal samples and minimize assay variation. Duplicate baseline samples were collected (each separated by a minimum of 2days) through Day -2, and then again at pre-dose on Day 1 (a total of three baseline samples were collected per subject). Subjects received oral tofacitinib 10mg BID from Day 1 through Day 29 (±3 days). On Days 15 and 29, pre-dose blood samples were collected for the performance of flow cytometry and immune function assays. Subjects entered a 4-week post-treatment phase on Day 29, with blood samples collected on Days 14 and 28 post-treatment during this phase.
To minimize variability in the functional and flow cytometry assessments, most assays were performed using PBMCs isolated at the clinical site and cryopreserved for later evaluation at a single site; batch testing of this type is standard practice in longitudinal studies as it eliminates assay-to-assay variation [22,23]. Where possible, all samples from an individual subject were evaluated together. Since the process of isolating PBMCs removes tofacitinib from the cells, changes in cell function are most likely due to altered gene expression or epigenetic changes that occurred in vivo. The results from this study are, therefore, distinct from those of in vitro functional assays where cells are incubated with a fixed concentration of drug for a finite period of time, since in vitro exposure to tofacitinib does not mimic the variation in tofacitinib exposure over a dosing period or the possible alteration in the immune response that can occur over days in vivo.
2.4. Flow cytometry
The cell subtypes identified via flow cytometry are shown in Fig. 1.
Fig. 1.
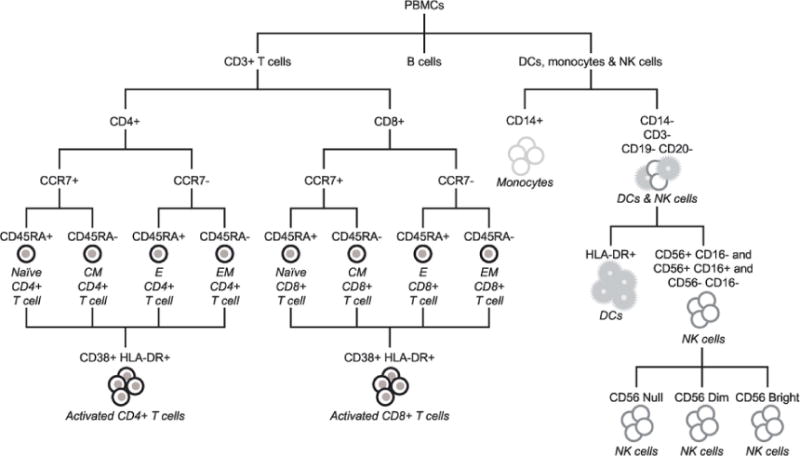
Cell subtypes identified via flow cytometry. CCR7 is also designated as CD197. CCR, CC-chemokine receptor; CD, cluster of differentiation; CM, central memory; DCs, dendritic cells; E, effector; EM, effector memory; HLA-DR, human leukocyte antigen - antigen D related; NK, natural killer; PBMC, peripheral blood mononuclear cell.
Cryopreserved PBMCs were thawed according to previously published procedures [24], and 2 × 106 viable PBMCs were stained for 45 min using five lyophilized, standardized, eight-color antibody panels (BD Biosciences, Franklin Lakes, NJ, USA) [25]. Cells were washed, fixed and acquired within 6h of staining using a calibrated 5 laser, 20-color BD SORP LSRII analyzer. Gating and analysis regions were established using appropriate fluorescence minus one controls. Flow Cytometry Standard files were analyzed using a template with uniform gating. Absolute numbers and percentages of circulating T cells (total and naïve, central memory [CM], effector memory [EM], effector [E] and activated CD4 + and CD8 +), NK cells, B cells, monocytes and dendritic cells (plasmacytoid and myeloid) were measured. Supplementary Fig. 1. shows the gating strategy for each polychromatic flow cytometry panel.
2.5. Functional assays
2.5.1. T-cell function assays
2.5.1.1. Anti-CD3/CD28 stimulated CD8 T-cell assay (CD107a and IFN- γ)
CD8 T-cell function was assessed by monitoring modulation to the cell surface of the degranulation marker CD107a, and intracellular accumulation of interferon (IFN)-γ upon anti-CD3/anti-CD28 cross-linking.
Unless otherwise noted, all reagents were obtained from BD Biosciences. Cryopreserved PBMCs were thawed at 37 °C and transferred immediately to 10mL of warm complete medium (Roswell Park Memorial Institute [RPMI]-1640 containing 10% fetal bovine serum [FBS]). Cells were centrifuged at 250 ×g for 5min and resuspended in fresh complete medium for cell count and viability assessment. PBMCs were centrifuged again and resuspended into a cocktail of Brefeldin A (GolgiPlug), Monensin (GolgiStop) and CD107a-APC, prior to addition of the appropriate stimulus. Approximately 3 × 105 PBMCs were incubated with an equal number of anti-CD3/CD28 Dynabeads (Life Technologies/Thermo Fisher, Waltham, MA, USA) or medium alone, and incubated overnight at 37°C/5% CO2. Cells were collected and washed by centrifugation and then incubated for 30 min at ambient temperature with LIVE/DEAD™ Aqua Fixable Stain (Life Technologies/Thermo Fisher). Cells were washed, and then incubated on ice in a cocktail of labeled antibodies consisting of CD16-PE, CD56-PE, human leukocyte antigen - antigen D related (HLA-DR)-APC-H7, CD8-PerCP-Cy5.5 and CD3-BV421, for 30 min. Cells were then washed and incubated for 20 min in BD Cytofix™ Fixation Buffer, before being washed and incubated for a further 20 min in BD fluorescence activated cell sorting (FACS™) Permeabilizing Solution 2. Cells were again washed and incubated for 30 min at ambient temperature with anti-IFN-γ-fluorescein isothiocyanate. Cells were then washed and resuspended in a 1% solution of paraformaldehyde, and stored at 4 °C until ready for flow cytometry. Data were reported as the percentage of CD8 T cells (live CD3 + CD8 +) that were CD107a + or IFN-γ + after CD3/CD28 cross-linking. Specific T-cell stimulation was calculated by subtracting background stimulation (medium alone) from anti-CD3/CD28 stimulated values.
2.5.1.2. CMV-specific T-cell function (IFN-γELISPOT)
T-cell function was assessed by monitoring the number of IFN-γ-secreting cells/106 PBMCs in response to either CMV peptide antigen or activation by CD3 cross-linking.
Cryopreserved PBMCs were thawed at 37 °C and added slowly to AIM-V medium supplemented with 20 U/mL of Benzonase Stain (Life Technologies/Thermo Fisher). PBMCs were centrifuged at 325 ×g for 10 min at 25 °C. Cell pellets were resuspended in AIM-V medium Stain (Life Technologies/Thermo Fisher) and counted, then centrifuged and resuspended in 5% FBS/AIM-V medium at a concentration of 2× 106cells/mL. PBMCs were incubated at 37°C/5% CO2 overnight (18–20 h). The following day, PBMCs were centrifuged, counted and adjusted to 8× 106 PBMCs/mL in AIM-V medium (without FBS).
CMV-induced IFN-γ production was measured using a human IFN-γ ELISPOT PLUS kit (MABTECH Inc., Stockholm, Sweden), and was conducted by the Pfizer Vaccine Research Unit (Pearl River, NY, USA). In 96-well plates coated with anti-IFN-γ in a 100 μL volume, 4 × 105 PBMCs were incubated with 50 ng (1 μg/mL) of CMV pp65 peptide pool (65 kDa phosphoprotein of human CMV strain human herpesvirus 5 [HHV-5)], JPT, Acton, MA, USA) for 18 ± 2 h at 37°C/5% CO2. PepMix™ HCMVA is a peptide pool of 138 peptides derived from a peptide scan (15 mers with 11 aa overlap) through 65kDa phosphoprotein (pp65) (Swiss-Prot ID: P06725) of human CMV (HHV-5). Alternatively, PBMCs were stimulated with anti-CD3 (MABTECH) diluted 1:3000 in medium. Plates were washed and incubated with biotinylated-detection antibody for 2 h at room temperature. Plates were washed, incubated with Streptavidin-HRP for 1 h at room temperature, and then incubated with colorimetric substrate for 10 min. After stopping the reaction and drying the plates, the number of spots was quantified using an ELISPOT plate reader (Cellular Technologies Ltd. Immunospot Micro-Analyzer, Shaker Heights, OH, USA) using Immunospot v5.1 software. The average number of background spots (no stimulation) was subtracted from the average stimulated spot count for each control or test antigen. Data were reported as the number of spot-forming cells (SFCs)/106 PBMCs, after stimulation.
2.5.2. NK-cell function assays
2.5.2.1. NK-cell functional assays (CD107a and IFN-γ)
Cryopreseved PBMCs were thawed, as described for the anti-CD3/CD28 stimulated CD8-T cell assay, and resuspended into complete medium containing Brefeldin A (GolgiPlug), Monensin (GolgiStop) and CD107a-APC, prior to activation. 1 × 105 K562 human erythroleukemia cells (ATCC, Manassas, VA, USA) were added to 3 × 105 PBMCs and incubated overnight at 37 °C/5% CO2. Cells were collected and washed by centrifugation, and then stained as per the anti-CD3/CD28-stimulated CD8 T-cell assay described above.
Data were reported as the percentage of NK cells (live CD3-HLA-DR-CD16 + CD56 + /dim) that were CD107a + or IFN-γ + after incubation with K562 target cells. Specific NK-cell stimulation was calculated by subtracting background stimulation (medium alone) from K562-stimulated values.
2.5.2.2. NK-cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) assay
In this modified ADCC assay, NK cells - via Fc-receptors - bind anti-CD20-bound B cells present in the PBMCs, and subsequently upregulate the degranulation marker CD107a. 3 × 105 PBMCs were resuspended in medium containing Brefeldin A (GolgiPlug), Monensin (GolgiStop) and CD107a-APC, and ADCC was initiated by addition of 10 μg/mL rituximab (Genentech) or control human immunoglobulin G (IgG; Life Technologies/Thermo Fisher). Cells were incubated overnight at 37°C/5% CO2 and then collected and washed by centrifugation. Cells were incubated for 30 min at ambient temperature with LIVE/DEAD Aqua fixable stain (Life Technologies/Thermo Fisher), washed and then incubated on ice in a cocktail of labeled antibodies consisting of CD16-PE, CD56-PE, HLA-DR-APC-H7 and CD3-BV421, for 30 min. Cells were washed and incubated for 2 min with 1% paraformaldehyde, and stored at 4 °C until ready for flow cytometry.
Data were reported as the percentage of NK cells (live CD3-HLA-DR-CD16 + CD56 + /dim) that were CD107a + after incubation with rituximab.
Specific NK-cell stimulation was calculated by subtracting background stimulation (human IgG) from rituximab-stimulated values.
2.5.3. Granulocyte and monocyte function
The granulocyte and monocyte oxidative burst and phagocytosis assays were performed within 4 h of blood draw on fresh whole blood. Oxidative burst assay was performed as previously described [26]. Briefly, resting and PMA-stimulated (1 μM, 25 min at 37 °C) whole blood was incubated with 5 μg/mL dihydrorhodamine (DHR123; Sigma-Aldrich, St. Louis, MO, USA) for 5min at 37°C. Red blood cells were lysed with ammonium-chloride-potassium (ACK) lysing buffer (Thermo Fisher Scientific, Waltham, MA, USA), rinsed with 1% FBS in phosphate-buffered saline (PBS) and then fixed with 1% formaldehyde in PBS. Granulocytes and monocytes were analyzed by FACS, gating on forward scattered light/side scattered light (FSC/SSC) and measuring DHR123 fluorescence on FL1. Oxidative burst was quantified as the median fluorescence intensity ratio of stimulated and resting granulocytes and monocytes (FS/FR).
Granulocyte and monocyte phagocytosis assay was performed using pHrodo Red E. coli BioParticles (Thermo Fisher Scientific) per the supplied protocol. Briefly, following incubation of heparanized whole blood with the BioParticles for 30 min at 37 °C, the red blood cells were lysed with ACK lysing buffer, rinsed with 1% FBS in PBS and then fixed with 1% formaldehyde in PBS. Negative control was whole blood incubated with BioParticles on ice for 30 min. Phagocytic activity was determined from the percentage positive and the median fluorescence intensity (FL2) ratio of experiment to control granulocytes and monocytes, as gated by FSC and SSC.
2.6. Statistical methods
Sample size calculations were made based on longitudinal absolute NK-cell counts from a dose-ranging study in patients with RA. No transformation of the data was made. The tofacitinib 5 mg BID and placebo arms from this study were combined and an estimate of the within-patient longitudinal variability (or standard deviation) was obtained as 0.23. A one-sample t-test was used with the (two-sided) Type-I error rate at 5%. Based on these calculations, as well as logistical feasibility considerations, a sample size of approximately 30 subjects was proposed, which would provide 80% power to detect mean decreases (relative to pre-treatment baseline) of 13% or larger in absolute NK-cell counts, for comparing baseline to Day 29 (drug effect) as well as comparing Day 29 to Day 28 post-treatment (reversibility effect). This sample size also accounted for an estimated 20% rate in non-compliance and drop-out.
All statistical analyses were descriptive in nature and estimation methods were used, providing point estimates and 95% confidence intervals (CIs). Calculation of CIs did not exclude outliers; CIs were calculated based on a logarithmic scale and then back-transformed to the original units to mitigate the effect of outliers.
Single baseline values for all parameters were derived based on the mean of the three pre-dose blood samples. Descriptive statistics of observed values, change from baseline and percentage change from baseline were obtained for each of the endpoints. In general, medians and ranges are presented in Tables 1–4 and in Sections 3.2–3.6. Geometric means are shown using connecting lines in the box and whisker plots included in the accompanying supplementary materials. For the comparisons between study days, all immune function endpoints were analyzed after natural logarithmic transformation to provide test statistics that have robust distribution characteristics, since many of the observed immune function endpoints were not normally distributed. Longitudinal repeated-measures models for the transformed data were used to estimate the change over time from baseline through Day 29 (treatment) and, separately, from Day 29 through Day 28 post-treatment. The model included visit and baseline as fixed effects; subjects were repeated within visit. The baseline, as defined above, was used for both the treatment and post-treatment phases. Exploratory hypothesis tests with nominal p-values are presented for the evaluation of whether changes from baseline in immune function were statistically detectable.
Table 1.
T-cell counts during and following treatment with tofacitinib.
| Cell count, cells/μL, median (range) | |||||
|---|---|---|---|---|---|
|
| |||||
| Baseline | Day 15 | Day 29 | Day 14 post-treatment | Day 28 post-treatment | |
| Total CD4 +a | 831 (452–1743) | 903* (337–2191) | 1054* (523–3034) | 833*††† (477–1531) | 812†† (453–1884) |
| Naïve CD4 +b | 439 (106–1397) | 522* (109–1805) | 625* (94–2501) | 428*†††(60–1402) | 464†† (57–1076) |
| Activated naïve CD4 +c | 1.69 (0.65–6.34) | 1.42* (0.45–7.02) | 1.43* (0.24–4.03) | 1.43 (0.44–13.33) | 1.37* (0.31–4.38) |
| Central memory CD4 +d | 265 (7–791) | 297* (14–766) | 302* (12–1089) | 231††† (9–894) | 248††† (8–1125) |
| Activated central memory CD4 +e | 4.54 (0.30–9.53) | 3.10*** (0.24–8.69) | 4.08* (0.28–6.97) | 5.43*†† (0.32–14.76) | 5.55*††† (0.29–9.94) |
| Effector memory CD4 +f | 52.2 (1.6–429.9) | 45.3 (3.2–480.0) | 47.1* (2.4–382.0) | 44.5 (2.7–285.0) | 42.3 (2.1–349.6) |
| Activated effector memory CD4 +g | 2.30 (0.13–6.69) | 1.72*** (0.14–5.72) | 2.21 (0.17–7.18) | 3.04**††† (0.32–29.33) | 2.68*†† (0.15–23.22) |
| Effector CD4 +h | 15.0 (1.7–679.8) | 15.8* (1.1–697.5) | 13.6** (1.7–562.0) | 16.8* (1.8–483.0) | 18.1† (1.8–562.4) |
| Activated effector CD4 +I | 0.22 (0.06–4.12) | 0.11*** (0.02–1.06) | 0.15** (0.02–0.88) | 0.36*††† (0.06–10.60) | 0.45*††† (0.06–16.65) |
| Total CD8 +j | 359 (209–789) | 372 (212–755) | 382 (190–805) | 329*† (180–673) | 330 (191–942) |
| Naïve CD8 +k | 164 (19–519) | 170* (22–570) | 175* (20–553) | 138*††† (10–456) | 153†† (11–633) |
| Activated naïve CD8 +l | 0.73 (0.28–2.47) | 0.42*** (0.13–1.88) | 0.45*** (0.18–1.86) | 0.631† (0.17–4.46) | 0.73† (0.23–4.96) |
| Central memory CD8 +m | 32.4 (2.7–85.4) | 40.6* (6.7–103.9) | 41.2* (5.0–117.3) | 28.6† (3.5–71.8) | 32.8†† (3.7–99.3) |
| Activated central memory CD8 +n | 0.87 (0.26–3.60) | 0.46*** (0.11–2.41) | 0.57*** (0.13–2.00) | 1.17††† (0.44–6.28) | 1.32††† (0.18–3.05) |
| Effector memory CD8 +o | 23.6 (0.2–125.5) | 22.4* (0.2–153.0) | 21.6* (0.2–149.9) | 20.9 (0.2–133.8) | 22.3 (0.1–100.4) |
| Activated effector memory CD8 +p | 1.31 (0.02–13.20) | 0.75*** (0.03–15.07) | 1.01* (0.11–9.56) | 2.41*††† (0.08–23.75) | 2.14*††† (0.64–19.07) |
| Effector CD8 +q | 124 (40–474) | 93** (31–305) | 97*** (31–295) | 92** (34–378) | 93†(40–508) |
| Activated effector CD8 +r | 2.70 (0.50–54.74) | 0.84*** (0.20–8.85) | 1.46*** (0.24–5.78) | 4.16*†††(0.74–74.71) | 4.05*††† (0.83–101.37) |
CCR, CC-chemokine receptor; CD, cluster of differentiation; HLA-DR, human leukocyte antigen - antigen D related.
Test statistics are based on log-transformed data.
p < 0.05,
p < 0.001,
p < 0.0001 compared with baseline,
p < 0.05,
p < 0.001,
p < 0.0001 compared with Day 29.
CD3 + CD4 +.
CD3+/CD4 + CD8−/CCR7 + CD45RA +.
CD3+/CD4 + CD8−/CCR7 + CD45RA+/CD38 + HLA−DR+.
CD3+/CD4 + CD8−/CCR7 + CD45RA−.
CD3+/CD4 + CD8−/CCR7 + CD45RA−/CD38 + HLA−DR+.
CD3+/CD4 + CD8−/CCR7−CD45RA−.
CD3+/CD4 + CD8−/CCR7−CD45RA−/CD38 + HLA−DR +.
CD3+/CD4 + CD8−/CCR7−CD45RA+.
CD3+/CD4 + CD8−/CCR7−CD45RA + /CD38 + HLA−DR +.
CD3 + CD8 +.
CD3+/CD4−CD8 +/CCR7 + CD45RA +.
CD3+/CD4−CD8 +/CCR7 + CD45RA+/CD38 + HLA−DR +.
CD3+/CD4−CD8 +/CCR7 + CD45RA−.
CD3+/CD4−CD8 +/CCR7 + CD45RA−/CD38 + HLA−DR +.
CD3+/CD4−CD8 +/CCR7−CD45RA−.
CD3+/CD4−CD8 +/CCR7−CD45RA−/CD38 + HLA−DR+.
CD3+/CD4−CD8 +/CCR7−CD45RA +.
CD3+/CD4−CD8 +/CCR7−CD45RA + /CD38 + HLA−DR+.
Table 4.
NK-cell function during and following treatment with tofacitinib.
| Median (range) | |||||
|---|---|---|---|---|---|
|
| |||||
| Baseline | Day 15 | Day 29 | Day 14 post-treatment |
Day 28 post-treatment |
|
| CD107a + NK cells following stimulation by K562 cells (%) | 12.70 (5.56–26.05) |
13.81 (4.45–27.96) |
10.80 (4.37–29.90) |
13.16 (3.84–32.80) |
10.44*** (4.02–23.62) |
| IFN-γ + NK cells following stimulation by K562 cells (%) | 8.48 (2.79–19.71) |
8.17 (3.07–18.86) |
7.58* (1.78–14.35) |
7.66 (0.56–18.14) |
6.15** (1.49–18.18) |
| CD107a + NK cells following rituximab-induced ADCC (%) | 5.32 (1.08–27.69) |
4.23* (0.18–16.50) |
4.82 (0.52–19.23) |
4.77 (0.97–26.50) |
3.07**†† (0.08–25.27) |
Test statistics are based on log-transformed data.
ADCC, antibody-dependent cell-mediated cytotoxicity; CD, cluster of differentiation; IFN-γ, interferon-gamma; NK, natural killer.
p < 0.05,
p < 0.001,
p < 0.0001 compared with baseline,
p < 0.001 compared with Day 29.
For safety laboratory test endpoints that were continuous, statistical methods for non-transformed data were used. Safety analyses considered data separately for the treatment and post-treatment phases.
2.7. Ethics
This study was conducted in compliance with the ethical principles originating in, or derived from, the Declaration of Helsinki, and in compliance with all international Conference on Harmonization Good Clinical Practice guidelines. The study and all relevant documentation were reviewed and approved by the Duke University Institutional Review Board. All subjects gave written informed consent prior to screening.
3. Results
3.1. Subjects
A total of 32 subjects were enrolled. Of these, 27 (84%) were male and 17 (53%) were Black. The mean age of subjects was 40.2years (standard deviation 13.3; range: 20–69years) and the mean body mass index was 25.8kg/m2 (range: 19.9–30.4kg/m2).
Twenty-seven subjects completed the study. Five other subjects discontinued the study, three during the treatment phase and two in the post-treatment phase. All discontinuations were due to non-serious adverse events (AEs), except for one discontinuation during the post-treatment phase, which was due to the subject being unable to attend study visits.
3.2. Total lymphocyte counts
Total lymphocyte count increased from a median of 1.80 × 103/mm3 at baseline to 2.00 × 103/mm3 after 4weeks of tofacitinib, decreasing during the post-treatment phase to 1.60 × 103/mm3 (p = 0.0024 vs baseline, based on log-transformed data) after both 2 and 4 weeks off treatment.
3.3. B-cell counts
Increases in absolute B-cell counts from a median of 173 cells/μL at baseline were observed during the treatment phase, with counts statistically significantly elevated at Day 15 (26% increase to 218 cells/μL; p < 0.0001) and Day 29 (193 cells/μL; p = 0. 0038). During the post-treatment phase, the B-cell count decreased to a level below (although not statistically significantly below) that at baseline; counts were significantly lower than on Day 29 at both Day 14 post-treatment (149cells/μL) and Day 28 post-treatment (165 cells/μL) (p<0.0001 both days).
3.4. T-cell counts and function
3.4.1. T-cell counts
Counts for total and subpopulations of CD4 + and CD8 + T cells are shown in Table 1, with box and whisker plots of absolute values for each population shown in Supplementary Fig. 2.
3.4.2. CD4 + T-cell counts
During tofacitinib treatment, total CD4 +, naïve CD4 + and CM CD4 + T-cell counts increased to a peak after 4weeks of treatment. Following treatment withdrawal, counts for all three subtypes returned to near baseline levels.
EM and E CD4 + T-cell counts decreased during treatment, EM CD4 + T cells to a trough at Day 15 and E CD4 + T cells to below baseline at Day 29. After drug withdrawal, EM CD4 + T-cell counts further decreased, while E CD4 + T-cell counts recovered to a level above baseline, as shown in Fig. 2, depicting representative data for activated EM CD4 + and CD8 + cell subsets throughout the treatment and post-treatment phases.
Fig. 2.
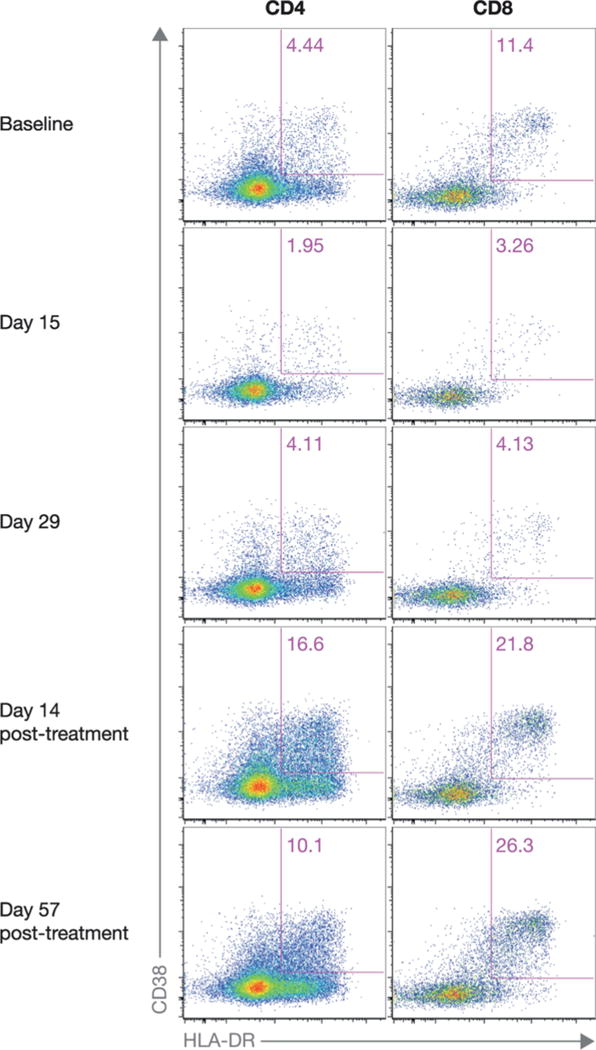
Frequencies of activated EM CD4 + and CD8 + cell subsets from a single, representative participant from baseline through Day 57 post-treatment. The value in the upper right quadrant of each plot represents the percentage of CD4 + or CD8 + cells that co-expressed CD38 and HLA-DR. CD, cluster of differentiation; EM, effector memory; HLA-DR, human leukocyte antigen - antigen D related antigen.
All activated CD4 + T-cell subsets decreased by up to 50% during the 4-week tofacitinib treatment period. Activated naïve, CM, EM and E CD4 + T-cell counts reached their lowest levels at Day 15. In the post-treatment phase, activated CM, EM and E CD4 + T-cell counts all increased up above baseline. Activated naïve CD4 + T cells dropped to their lowest level at Day 28 post-treatment.
3.4.3. CD8 + T-cell counts
Total CD8 + T-cell count remained fairly constant during tofacitinib treatment. Naïve CD8 + and CM CD8 + T-cell counts showed increases above baseline, peaking after 4 weeks of treatment, then decreasing to levels below baseline 2 weeks after treatment withdrawal.
During tofacitinib treatment, CM CD8 + T-cell count increased above baseline, while EM and E CD8 + T-cell counts decreased below baseline. Following drug withdrawal, CM CD8 + T-cell count returned to baseline level, EM CD8 + T cells recovered to a level close to baseline and E CD8 + T-cell counts remained below baseline.
All activated CD8 + T-cell subsets decreased by up to 69% by Day 15 of tofacitinib treatment, without further decrease at Day 29, while still on drug. Activated naïve CD8 + T cells returned to baseline after drug withdrawal, while all other activated CD8 + T-cell subsets increased above baseline.
3.4.4. T-cell function
The results of the T-cell function assays are shown in Table 2, with box and whisker plots of values shown in Supplementary Fig. 3.
Table 2.
T-cell function during and following treatment with tofacitinib.
| Median (range) | |||||
|---|---|---|---|---|---|
|
| |||||
| Baseline | Day 15 | Day 29 | Day 14(BR/)post-treatment | Day 28(BR/)post-treatment | |
| CD107a + CD8 T cells following stimulation with anti-CD3/CD28 (%) | 4.42 (0.87–20.80) | 2.77*** (0.50–10.78) | 3.72 (0.59–15.20) | 5.01† (0.70–18.24) | 4.43 (0.44–18.21) |
| IFN-γ + CD8 T cells following stimulation with anti-CD3/CD28 (%) | 8.54 (3.29–23.11) | 5.73*** (2.17–19.11) | 7.45* (1.89–22.11) | 7.76 (1.42–24.18) | 7.47* (1.97–23.16) |
| IFN-γ-producing cells following stimulation with anti-CD3 (SFCs/106 PBMCs) | 1784 (1118–1920) | 1785 (967–2000) | 1741 (757–2000) | 1745 (1332–2000) | 1780 (966–2000) |
| IFN-γ-producing cells following stimulation with CMV antigen (SFCs/106 PBMCs) | 834 (199–1869) | 777** (181–1928) | 848* (166–1884) | 909 (138–2000) | 960 (99–2000) |
Test statistics are based on log-transformed data.
CD, cluster of differentiation; CMV, cytomegalovirus; IFN-γ, interferon-gamma; PBMC, peripheral blood mononuclear cell; SFC, spot-forming cell.
p < 0.05,
p < 0.001,
p < 0.0001 compared with baseline,
p < 0.05 compared with Day 29.
During treatment with tofacitinib, the percentage of CD8 + T cells that were CD107a + following stimulation with anti-CD3/CD28 decreased by Day 15, partially normalized by Day 29 while still on treatment, and then returned to baseline levels. Similar results were observed with the percentage of CD8 + T cells expressing IFN-γ post stimulation.
The number of IFN-γ producing cells responding to CMV antigen as assessed by IFN-γ ELISPOT formation also decreased by Day 15 of treatment, returning to baseline levels by Day 29 while still on treatment (Table 2). CMV DNA was undetectable in whole blood. Levels then increased above baseline levels, reaching a peak by Day 28 post-treatment. Similar trends in the ELISPOT results were observed with CD3 stimulation, although the assay was near the upper limit of detection. Overall, tofacitinib transiently decreased both antigen-specific and non-specific T-cell-mediated responses (Table 2).
3.5. NK-cell counts and function
3.5.1. NK-cell counts
Counts for total and subpopulations of NK cells are shown in Table 3, with box and whisker plots of absolute values for each population shown in Supplementary Fig. 4.
Table 3.
NK-cell counts during and following treatment with tofacitinib.
| Cell count, cells/μL, median (range) |
|||||
|---|---|---|---|---|---|
|
| |||||
| Baseline | Day 15 | Day 29 | Day 14 post-treatment |
Day 28 post-treatment |
|
| Total NK | 213 (65–510) | 144*** (71–400) | 142*** (49–567) | 151*** (58–577) | 161† (74–561) |
| CD56 null NK | 7.14 (2.76–20.86) | 5.51 (1.74–30.42) | 7.94 (2.37–29.50) | 7.19 (2.54–19.62) | 8.17* (2.43–41.50) |
| CD56 dim NK | 146 (36–405) | 99** (35–340) | 94** (27–410) | 107* (34–391) | 127†(45–422) |
| CD56 bright NK | 51.0 (20.4–123.1) | 36.3** (13.1–88.0) | 29.8*** (13.9–148.1) | 30.9*** (12.7–167.4) | 35.7***† (16.6–120.3) |
Test statistics are based on log-transformed data.
CD, cluster of differentiation; NK, natural killer.
p < 0.05,
p < 0.001,
p< 0.0001 compared with baseline,
p < 0.05 compared with Day 29.
During tofacitinib treatment, total NK, CD56 null NK, CD56 dim NK and CD56 bright NK-cell counts all decreased below baseline, with total NK-cell count 33% below baseline at Day 29. Total NK, CD56 dim NK and CD56 bright NK-cell counts increased following drug withdrawal, but remained below baseline. CD56 null NK-cell counts initially decreased, then increased above baseline after 4 weeks of treatment, reaching a peak at the end of the post-treatment phase.
3.5.2. NK-cell function
The results of the NK-cell functional assays are shown in Table 4, with box and whisker plots of values shown in Supplementary Fig. 5.
Changes in the percentage of functionally activated NK cells were modest and without clear directionality until Day 28 post-treatment. Decreased NK functional activity was observed on Day 28 post-treatment following stimulation with K562 target cells (NK functional assay; 18% and 27% vs baseline monitoring CD107a and IFN-γ, respectively) or rituximab (ADCC assay; 42% vs baseline).
3.6. Granulocyte, monocyte and dendritic cell counts and function
Tofacitinib treatment had no significant effect on granulocyte, monocyte or dendritic cell counts, with the exception of a decrease in myeloid dendritic cells as a percentage of PBMCs from a median of 0.73 at baseline to 0.61 at Day 15 (p = 0.0293). There were also no statistically significant changes seen in monocyte or granulocyte function, based on oxidative burst or phagocytosis analysis during or following treatment with tofacitinib, except for an increase in monocyte phagocytosis from a median of 3.96 (ratio of mean fluorescence intensity experimental samples to negative control samples) at baseline to 5.90 at Day 14 post-treatment (p = 0.0238).
3.7. Safety
A total of 29 AEs were reported for 18 (56%) subjects. There were no serious AEs. The most frequently reported AEs during the treatment phase were fatigue and headache, each reported by four subjects. Three mild infections occurred during treatment (herpes simplex, oral herpes, and upper respiratory tract infection). There were no cases of HZ.
Four subjects discontinued the study due to AEs. AEs leading to discontinuation were fatigue, headache, depression and sexually transmitted disease (not a treatment-emergent AE); the AEs of fatigue and depression were considered to be related to study drug. There were no discontinuations for leukopenia, lymphopenia, neutropenia, thrombocytopenia or anemia. No patients had total lymphocyte count <500/mm3 or CD4 + lymphocyte count <200/mm3 at any time during the study (i.e., there were no cases meeting Common Terminology Criteria for Adverse Events [CTCAE] Grade 3 or worse).
Supplementary Table 1 shows the incidence of treatment-emergent AEs.
4. Discussion
In this study, the effect of 10 mg BID tofacitinib treatment on a number of immune cell parameters was monitored in healthy volunteers both during and after a 4-week treatment period. The 10 mg dose in healthy volunteers results in similar serum drug level concentrations to the 5 mg dose in patients with RA. The current study aimed to determine if JAK inhibition at clinically relevant levels in vivo leads to alterations in immune cell function in ex vivo assays.
There were slight increases in total lymphocyte count during tofacitinib treatment that normalized at the end of the study, as was previously observed in a dose-ranging Phase 2 study in psoriasis [18], and as has been observed over the longer-term in the tofacitinib RA clinical development program [12–14]. There were no changes in granulocyte or monocyte counts.
The observed decrease in activated T cells after tofacitinib treatment is consistent with inherent mechanisms that reduce the number of activated T cells after antigenic challenge, in order to avoid excessive accumulation that could otherwise lead to autoimmunity or neoplasia [27]. Cytokines dependent on JAK1 and/or JAK3 including interleukin (IL)-2, IL-7, IL-15 and IL-6 have been shown to partially prevent apoptosis of in vivo activated T cells [27]. Reduction in the signaling of these cytokines by tofacitinib would be expected to enhance apoptosis, at least temporarily, and may be beneficial in RA where the population of CD38 + HLA−DR+ CD4 + T cells are elevated relative to healthy individuals [28]. Additionally, in vitro studies have shown that tofacitinib reduces surface expression of co-stimulatory molecules on dendritic cells [29], which may lead to a more tolerogenic state including an increase in the ratio of regulatory T cells to Th1 and Th17 cells when dendritic cells are pulsed with a specific antigen [30]. We did not detect a change in regulatory T cells, but we did not monitor antigen-specific responses following in vivo antigen challenge. The reduction in activated T cells either plateaued or began to normalize by Day 29, while tofacitinib treatment was ongoing, indicating a compensatory mechanism. Upon discontinuation of tofacitinib, activated effector and memory T-cell counts rebounded to levels up to two times higher than baseline, which may be due to the return of normal IL-2, IL-7, IL-15 and IL-6 signaling before the compensatory mechanism could reequilibrate. In vitro studies at the relatively high concentrations of 10 and 100 μM demonstrated a decrease in proliferation of PHA-activated T cells, but after removal of tofacitinib a higher percentage of treated cells proliferated [31]. No prior studies have reported the impact of tofacitinib treatment on numbers of activated T cells in patients with RA. However, since activated T cells contribute to RA pathogenesis [32], it is likely that the therapeutic effects of tofacitinib are at least in part attributable to decreases in activated T cells.
Tofacitinib had little or no effect on overall CD4 + regulatory T-cell subset numbers (data not shown).
CD8 T-cell function was assessed by monitoring the percentage of CD8 T cells on which the degranulation marker CD107a was modulated on the cell surface, and the percentage of CD8 + T cells that produced IFN-γ upon anti-CD3/anti-CD28 cross-linking. T-cell function was also assessed by monitoring the number of cells/106 PBMCs that produced IFN-γ in response to either CMV antigen, or activation with anti-CD3. The number of CMV-specific IFN-γ-producing cells, as assessed by ELISPOT, and the percentage of CD107a + and IFN-γ + CD8 + T cells following anti-CD3/CD28 activation, were modestly decreased by Day 15 of tofacitinib treatment, largely normalized by Day 29 while still on treatment, and then returned to baseline levels. Similar results were observed in the ELISPOT assay with CD3 stimulation, where the decrease did not reach statistical significance. Overall, the data suggest that tofacitinib mediated a modest, reversible effect on both antigen-specific and non-specific T-cell function.
Modest decreases in NK-cell counts were observed with tofacitinib treatment, as in previous studies [18]. NK-cell function, as measured by the percentage of CD107a + or IFN-γ + cells in response to stimulation with K562 tumor cells, and the percentage of CD107a + cells in the ADCC assay, varied in an inconsistent manner with time, but all parameters were reduced 1 month after treatment discontinuation (Day 28 post-treatment). Multiple JAK1/3-dependent cytokines - including IL-2, IL-15 and IL-21 - may support the cytolytic activity of NK cells [33,34], and IL-12 signaling – which is modestly inhibited by tofacitinib – supports NK cell cytotoxicity and IFN-γ production [34,35]. However, it is difficult to rationalize why the effect would be more pronounced after discontinuation of treatment. The reason for this persistent decrease in NK-cell function is unknown, but may result from excess IL-2 consumption by activated T cells [36], which recovered to levels considerably higher than baseline after drug withdrawal in this study. However, these short-term findings may be of only temporary consequence, since NK-cell levels were actually increased from baseline after 8 years of tofacitinib treatment in patients with RA [19].
The clinical consequences of these findings are unknown at present, but the decreases in T-cell and NK-cell function did not result in reactivation of latent CMV in this CMV-seropositive population, as CMV DNA remained undetectable in whole blood.
In contrast to decreases in granulocytes observed in patients with RA treated with tofacitinib [16], short-term tofacitinib treatment in healthy volunteers was not associated with decreased granulocyte counts, and granulocyte function was unaffected in the present study. This difference may be explained by an excess of activated granulocytes in patients with RA [37]. Tofacitinib targets JAK1 pathways associated with activation of T cells that secrete granulocyte-activating cytokines such as IL-6. Since granulocytes contribute to inflammation in RA, tofacitinib-mediated decreases in activated granulocytes may contribute at least in part to its therapeutic effect. The present study, together with studies of immune function in RA, suggests that tofacitinib preferentially targets activated lymphocytes such as CD38 + HLA−DR+ T cells and activated granulocytes, while leaving recall functions of memory T cells relatively intact.
Consistent with results observed in short-term RA studies, B-cell counts had increased by 26% by the end of the treatment phase in this study. B-cell function was not tested in this short-term healthy volunteer study, but in patients with RA treated with short-term tofacitinib there was a mild decrease in humoral responses to T-independent, but not T-dependent, antigens [20]. Previous in vitro studies monitoring B-cell function including class switching [38] and differentiation into plasmablasts [39] observed a marked effect of tofacitinib when the process was driven with JAK-dependent cytokines such as IL-4 or IL-21, but not when the cytokine was replaced by toll-like receptor (TLR) signaling. Therefore, following the humoral response to vaccination may be a more accurate assessment of the impact on the integrated immune response.
A limitation of this study, as it relates to RA, is that the demographic profile of the healthy CMV+ volunteers differed from that of an RA patient population treated with tofacitinib in a number of aspects. Demographic data have been reported for 4102 patients with RA in the ongoing tofacitinib LTE study, and show the volunteers in the current study to be younger (mean age: 40.2years vs 53.2years in the LTE study), with a higher proportion of males (84% vs 17% in the LTE study) and a higher proportion of Black subjects (53% vs 3% in the LTE study) [12]. It is possible that the effects of tofacitinib on the number and function of various cell types might vary depending on age, gender, race and pre-existing immune status. Another limitation is the lack of a placebo-controlled population for comparison. However, since there were no disease activity measurements in this healthy volunteer population, and all assays were performed blinded to time and in coded batches, the likelihood of placebo effect bias was limited.
In summary, tofacitinib treatment in healthy CMV+ volunteers had modest and largely reversible effects on B-cell numbers, T-cell numbers and function, and on NK-cell numbers. Modest decreases in NK-cell function were more pronounced a month after drug withdrawal. There was little or no effect on monocyte and granulocyte function. Tofacitinib appears to target activated leukocytes while mostly sparing immune recall functions, making it a suitable choice for treatment of inflammatory disease.
Supplementary Material
Acknowledgments
The authors would like to thank the volunteers who were involved in this study, Francine Sproul and Lucian Cappoli for their study conduct expertise, and Samuel Zwillich and Jack Cook for critical review of the manuscript.
Editorial support was provided, under the guidance of the authors, by Carole Evans, PhD, on behalf of Complete Medical Communications, and was funded by Pfizer Inc.
Funding
This study was funded by Pfizer Inc.
Abbreviations list
- ACK
ammonium-chloride-potassium
- ADCC
antibody-dependent cell-mediated cytotoxicity
- AE
adverse event
- ALC
absolute lymphocyte count
- BID
twice daily
- CCR
CC-chemokine receptor
- CD
cluster of differentiation
- CI
confidence interval
- CM
central memory
- CMV
cytomegalovirus
- CTCAE
Common Terminology Criteria for Adverse Events
- DC
dendritic cell
- E
effector
- EM
effector memory
- FACS
fluorescence activated cell sorting
- FBS
fetal bovine serum
- FSC
forward scattered light
- SSC
side scattered light
- HHV-5
human herpesvirus 5
- HLA-DR
human leukocyte antigen - antigen D related
- HZ
herpes zoster
- IFN
interferon
- IgG
immunoglobulin G
- IL
interleukin
- JAK
Janus kinase
- LTE
long-term extension
- PBS
phosphate-buffered saline
- NK
natural killer
- PBMC
peripheral blood mononuclear cells
- RA
rheumatoid arthritis
- RPMI
Roswell Park Memorial Institute
- SFC
spot-forming cells
- TLR
toll-like receptor
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.clim.2018.03.002.
Footnotes
Conflicts of interest
KJW, TVB, RJN, JSS, LL and LS have nothing to disclose.
JFB, CH, AW, CFM, JC, MC, MIJ, AH, PB, SL, JDC and JAH are employees and shareholders of Pfizer Inc.
References
- 1.Fleischmann R, Cutolo M, Genovese MC, Lee EB, Kanik KS, Sadis S, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64:617–629. doi: 10.1002/art.33383. [DOI] [PubMed] [Google Scholar]
- 2.Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60:1895–1905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 3.Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez-Reino J, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64:970–981. doi: 10.1002/art.33419. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka Y, Suzuki M, Nakamura H, Toyoizumi S, Zwillich SH. Tofacitinib Study Investigators, Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res. 2011;63:1150–1158. doi: 10.1002/acr.20494. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka Y, Takeuchi T, Yamanaka H, Nakamura H, Toyoizumi S, Zwillich S. Efficacy and safety of tofacitinib as monotherapy in Japanese patients with active rheumatoid arthritis: a 12-week, randomized, phase 2 study. Mod Rheumatol. 2015;25:514–521. doi: 10.3109/14397595.2014.995875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–460. doi: 10.1016/S0140-6736(12)61424-X. [DOI] [PubMed] [Google Scholar]
- 7.Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507. doi: 10.1056/NEJMoa1109071. [DOI] [PubMed] [Google Scholar]
- 8.Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin-Mola E, et al. To- facitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159:253–261. doi: 10.7326/0003-4819-159-4-201308200-00006. [DOI] [PubMed] [Google Scholar]
- 9.Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley J, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377–2386. doi: 10.1056/NEJMoa1310476. [DOI] [PubMed] [Google Scholar]
- 10.van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65:559–570. doi: 10.1002/art.37816. [DOI] [PubMed] [Google Scholar]
- 11.van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–519. doi: 10.1056/NEJMoa1112072. [DOI] [PubMed] [Google Scholar]
- 12.Wollenhaupt J, Silverfield J, Lee EB, Curtis JR, Wood SP, Soma K, et al. Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J Rheumatol. 2014;41:837–852. doi: 10.3899/jrheum.130683. [DOI] [PubMed] [Google Scholar]
- 13.Wollenhaupt J, Silverfield J, Lee EB, Terry K, Kwok K, Abramsky S, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: safety and efficacy in open-label, long-term extension studies over 8 years. Arthritis Rheum. 2016;68(Suppl 10):1647. [Google Scholar]
- 14.Yamanaka H, Tanaka Y, Takeuchi T, Sugiyama N, Yuasa H, Toyoizumi S, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open-label, long-term extension study. Arthritis Res Ther. 2016;18:34. doi: 10.1186/s13075-016-0932-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186:4234–4243. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016;34:318–328. [PubMed] [Google Scholar]
- 17.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 18.Strober B, Buonanno M, Clark JD, Kawabata T, Tan H, Wolk R, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. 2013;169:992–999. doi: 10.1111/bjd.12517. [DOI] [PubMed] [Google Scholar]
- 19.van Vollenhoven R, Choy E, Lee EB, Hazra A, Anisfeld A, Lazariciu I, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: changes in lymphocytes and lymphocyte subset counts and reversibility after up to 8 years of tofacitinib treatment. Ann Rheum Dis. 2016;75:258. [Google Scholar]
- 20.Winthrop KL, Silverfield J, Racewicz A, Neal J, Lee EB, Hrycaj P, et al. The effect of tofacitinib on pneumococcal and influenza vaccine responses in rheumatoid arthritis. Ann Rheum Dis. 2016;75:687–695. doi: 10.1136/annrheumdis-2014-207191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988-2004. Clin Infect Dis. 2010;50:1439–1447. doi: 10.1086/652438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lauer FT, Denson JL, Burchiel SW. Isolation, Cryopreservation, and Immunophenotyping of human peripheral blood mononuclear cells. Curr Protoc Toxicol. 2017;74:18. doi: 10.1002/cptx.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberg A, Song LY, Wilkening C, Sevin A, Blais B, Louzao R, et al. Optimization and limitations of use of cryopreserved peripheral blood mononuclear cells for functional and phenotypic T-cell characterization. Clin Vaccine Immunol. 2009;16:1176–1186. doi: 10.1128/CVI.00342-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murdoch DM, Suchard MS, Venter WD, Mhlangu P, Ottinger JS, Feldman C, et al. Polychromatic immunophenotypic characterization of T cell profiles among HIV-infected patients experiencing immune reconstitution inflammatory syndrome (IRIS) AIDS Res Ther. 2009;6:16. doi: 10.1186/1742-6405-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the human immunology project. Nat Rev Immunol. 2012;12:191–200. doi: 10.1038/nri3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson MP, Ayliffe MJ, Helbert M, Davies EG. A simple flow cytometry assay using dihydrorhodamine for the measurement of the neutrophil respiratory burst in whole blood: comparison with the quantitative nitrobluetetrazolium test. J Immunol Methods. 1998;219:187–193. doi: 10.1016/s0022-1759(98)00136-7. [DOI] [PubMed] [Google Scholar]
- 27.Pajusto M, Ihalainen N, Pelkonen J, Tarkkanen J, Mattila PS. Human in vivo-activated CD45R0(+) CD4(+) T cells are susceptible to spontaneous apoptosis that can be inhibited by the chemokine CXCL12 and IL-2, -6, -7, and -15. Eur J Immunol. 2004;34:2771–2780. doi: 10.1002/eji.200324761. [DOI] [PubMed] [Google Scholar]
- 28.Lurati A, Laria A, Gatti A, Brando B, Scarpellini M. Different T cells’ distribution and activation degree of Th17 CD4 + cells in peripheral blood in patients with osteoarthritis, rheumatoid arthritis, and healthy donors: preliminary results of the MAGENTA CLICAO study. Open Access Rheumatol. 2015;7:63–68. doi: 10.2147/OARRR.S81905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheum Dis. 2014;73:2192–2198. doi: 10.1136/annrheumdis-2013-203756. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y, Leng X, Luo S, Su Z, Luo X, Guo H, et al. Tolerogenic dendritic cells generated with tofacitinib ameliorate experimental autoimmune encephalomyelitis through modulation of Th17/Treg balance. J Immunol Res. 2016;2016:5021537. doi: 10.1155/2016/5021537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piscianz E, Valencic E, Cuzzoni E, De Iudicibus S, De Lorenzo E, Decorti G, et al. Fate of lymphocytes after withdrawal of tofacitinib treatment. PLoS One. 2014;9:e85463. doi: 10.1371/journal.pone.0085463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cope AP. T cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10(Suppl 1):S1. doi: 10.1186/ar2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parrish-Novak J, Foster DC, Holly RD, Clegg CH. Interleukin-21 and the IL-21 receptor: novel effectors of NK and T cell responses. J Leukoc Biol. 2002;72:856–863. [PubMed] [Google Scholar]
- 34.Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. 2013;132:515–525. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm (Lond) 2010;7:41. doi: 10.1186/1476-9255-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humblet-Baron S, Franckaert D, Dooley J, Bornschein S, Cauwe B, Schönefeldt S, et al. IL-2 consumption by highly activated CD8 T cells induces regulatory T-cell dysfunction in patients with hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 2016;138:200–209. doi: 10.1016/j.jaci.2015.12.1314. [DOI] [PubMed] [Google Scholar]
- 37.Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 2010;49:1618–1631. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- 38.Wang SP, Iwata S, Nakayamada S, Sakata K, Yamaoka K, Tanaka Y. Tofacitinib, a JAK inhibitor, inhibits human B cell activation in vitro. Ann Rheum Dis. 2014;73:2213–2215. doi: 10.1136/annrheumdis-2014-205615. [DOI] [PubMed] [Google Scholar]
- 39.Rizzi M, Lorenzetti R, Fischer K, Staniek J, Janowska I, Troilo A, et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J Autoimmun. 2017;77:55–66. doi: 10.1016/j.jaut.2016.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.