

Abstract
Matrix-assisted laser desorption ionization (MALDI) imaging mass spectrometry (IMS) is a powerful tool for the generation of multidimensional spatial expression maps of biomolecules directly from a tissue section. From a clinical proteomics perspective, this method correlates molecular detail to histopathological changes found in patient-derived tissues, enhancing the ability to identify candidates for disease biomarkers. The unbiased analysis and spatial mapping of a variety of molecules directly from clinical tissue sections can be achieved through this method. Conversely, targeted IMS, by the incorporation of laser-reactive molecular tags onto antibodies, aptamers, and other affinity molecules, enables analysis of specific molecules or a class of molecules. In addition to exploring tissue during biomarker discovery, the integration of MALDI-IMS methods into existing clinical pathology laboratory practices could prove beneficial to diagnostics. Querying tissue for the expression of specific biomarkers in a biopsy is a critical component in clinical decision-making and such markers are a major goal of translational research. An important challenge in cancer diagnostics will be to assay multiple parameters in a single slide when tissue quantities are limited. The development of multiplexed assays that maximize the yield of information from a small biopsy will help meet a critical challenge to current biomarker research. This review focuses on the use of MALDI-IMS in biomarker discovery and its potential as a clinical diagnostic tool with specific reference to our application of this technology to prostate cancer.
Keywords: Matrix-assisted laser desorption ionization tissue imaging, Biomarker discovery, Cancer
Introduction
Direct analysis of clinical tissues using matrix-assisted laser desorption ionization (MALDI) imaging mass spectrometry (IMS) represents a unique approach to assessing spatial expression of molecules linked with histopathology and compiled clinical information. In this regard, MALDI-IMS can be utilized as a biomarker discovery tool as it facilitates a pathology-directed, unbiased approach to identifying the cellular origins and relative concentrations of biomarker candidates across an entire tissue section. MALDI ion sources are well suited to this application since they enable the ionization of diverse biomolecules, including peptides, proteins, oligonucleotides, sugars, and lipids [1, 2]. Once a marker of interest has been identified, properties related to its localization, abundance, regulation, and function can be assessed across multiple tissues to better understand disease progression at the molecular level. Our group has applied MALDI-IMS to the analysis of over 200 individual prostate-tumor-associated tissue samples [3], as well as over 100 tissues related to kidney and bladder cancers. From these studies and others, it is clear that issues related to sample manipulation, pathology-directed interrogation, improved and class-specific ionization, and across-class multiplexing present significant challenges to and opportunities for progress in this emerging field.
The value of using tumor tissues as a primary source for biomarker discovery is self-evident, but there are many issues associated with obtaining quality tissue samples. For many types of cancers, the most common tissue samples are obtained via biopsy and are destined for primary use by pathologists. The tumors themselves are frequently highly heterogeneous, and the quality of the sample can be greatly affected by the amount of adipose, connective tissue and cell types present within each biopsy. Obtaining enough cancer versus nontumor/uninvolved epithelial cell tissue can thus be challenging, and requires concerted input from pathologists to select the most appropriate regions of tissue relevant to disease. Also, fresh frozen tissues, the optimal sample for MALDI IMS, are expensive to obtain and store, and require institutionally approved standard operating procedures for informed consent and collection at the time of surgery. Compared with the collection of serum, tissues require significantly greater clinical resources and more complex follow-up evaluations.
In addition, critical questions remain regarding the integration of MALDI-IMS with clinical decision-making. The information obtained from MALDI-IMS should significantly augment and enhance existing clinical pathology practices. Obtaining biomarkers that are specific for a disease or different stages of a disease would greatly empower physicians, surgeons, and pathologists to provide patients with a “personalized” diagnosis and prognosis. Current strategies and new approaches for use of MALDI-IMS to achieve these personalized diagnostic medicine goals will be summarized in this review. Prior reviews have summarized the basic method and optimization of analysis parameters for MALDI-IMS [4–8], as well as overviews of different applications of the technology to biological systems and disease states [9–13]. In this review, specific examples of the application of MALDI-IMS will be shown for prostate and renal tissues from our own studies, as will examples from other published studies that have advanced the use of this emerging technique toward clinical proteomics applications. Emphasis will be placed on MALDI-IMS as applied to clinical applications, as well as comment on specific areas that need to be improved or optimized to better facilitate diagnostic use of the technology.
Pathology-directed MALDI-IMS
The potential application of MALDI-IMS in clinical workflows was first suggested by Caprioli and colleagues. Protein expression profiles obtained using MALDI-IMS were reported to discriminate lung cancer subtype [14]. Similarly, tumor histology findings, therapeutic response, and patient survival were shown to correlate with the protein expression patterns obtained from direct tissue analysis in breast tumors [10, 15], and protein expression patterns and images were also found to correlate with brain tumor histology findings and patient survival [16]. Following these earlier studies, our own laboratory reported the application of MALDI-IMS to the diagnosis of prostate cancer [3], in which 75 prostate tissues were analyzed. A study design of 21 tissues was used in the initial discovery set, and 54 tissues were used in a subsequent verification study, to determine differential expression between tumor and normal prostate tissues. The overexpression of a single peptide at m/z 4,355 was detected and could be used to accurately define cancer tissue from adjacent normal tissue. This peptide was subsequently identified as a fragment of MEKK2, a member of the mitogen-activated protein kinase signaling pathway. The workflow for preparation and analysis of these tissues is provided in Fig. 1. A key step for this study and any study using clinical specimens was the initial evaluation of each tissue by a pathologist for selection of regions of interest (ROIs) for feature content (e. g., tumor, stroma, immune infiltrate) and assessment of tissue quality and cellular morphology. In Fig. 1, these ROIs are highlighted by circles on the corresponding hematoxylin and eosin (H&E)-stained slide and overlaid with the related MALDI-IMS and immunohistochemistry (IHC) slide images. An example of the spatial expression of m/z 4,355 MEKK2 peptide in prostate tissue is shown in Fig. 1, panel c. The immunohistochemical staining for MEKK2 expression is shown in Fig. 1, panel d, the levels of which could be used to discriminate tumor from adjacent and distal uninvolved tissues. Cumulatively, this study illustrates the types of clinical and experimental data that can be obtained with a MALDI-IMS workflow integrated with standard pathology practices.
Fig. 1.
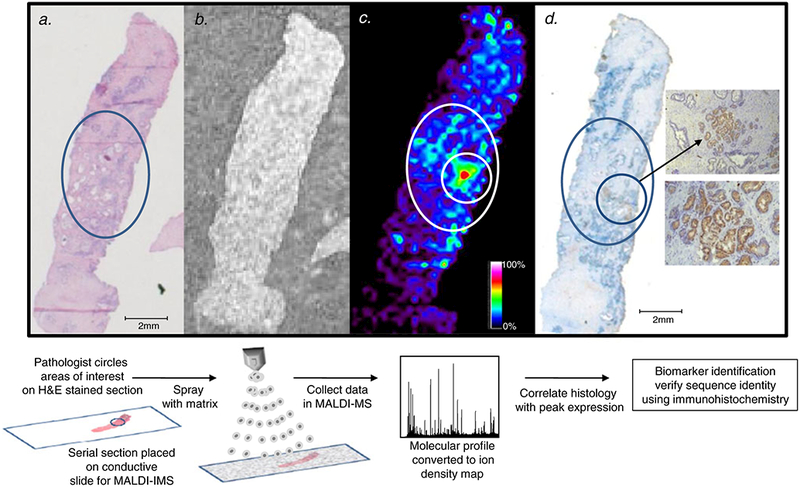
Workflow of matrix-assisted laser desorption ionization (MALDI) imaging mass spectrometry (IMS) for biomarker discovery. a a pathologist reviews the hematoxylin and eosin (H&E)-stained tissue and circles a region of prostate cancer. b a serial section is placed on a conductive slide and sprayed with matrix. c the spatial expression of the m/z 4,355 MEKK2 peptide in the prostate tissue collected using MALDI-IMS with a raster width of 200 μm correlates to the circled prostate cancer region. The relative expression of m/z 4,355 is color-coded according to the inset scale. d immunohisto-chemistry (IHC) staining of MEKK2 expression, showing strong staining in the circled areas that correspond to regions of high m/z 4,355 expression. Magnified views of the stained prostate cancer cells are shown in the insets: top ×10 and bottom ×40
From the biomarker characterization standpoint, MALDI-IMS can be applied to multiple tissues reflective of the spectrum of disease that is being targeted. Using the MEKK2 peptide as an example, we first normalized the peak intensity values for m/z 4,355 to the total ion current, then we determined expression levels correlating to localization within the prostate tissue ROIs (cancer, benign adjacent, and benign distal). As previously reported [3], an average intensity cutoff value of 23.8 was selected for maximal sensitivity of 96.8% and a specificity of 81.8% when comparing tumor versus benign tissue. This cutoff value was further used to compare the expression of the m/z 4,355 MEKK2 peptide across Gleason grade and pathological stage. Particularly for prostate tumors with a Gleason score of 8 or more and pathological stage pT3b or higher, there was a notable reduction in expression of the m/z 4,355 peak. Using IHC for whole protein, we confirmed this reduction in MEKK2 expression in more advanced Gleason grade/pathological stage prostate cancers relative to lower Gleason grade/pathological stage cancers (data not shown). Currently, we are conducting a large-scale validation study using this combination of MALD-IMS and IHC to better define the clinical utility of detecting MEKK2 as a potential biomarker of prostate cancer aggressiveness at the time of biopsy.
A major challenge in cancer diagnosis and treatment is related to the fact that the histopathological features of a tumor do not provide complete guidance and accuracy in patient risk assessment and treatment response [17]. Furthermore, histopathological analysis often fails to detect neoplastic cells in surgical margins, since these cells often exhibit normal histological features but have already undergone genomic and proteomic changes. Recent studies have shown that normal-appearing cells at tumor margins interrogated with MALDI-IMS produce protein and peptide patterns which resemble those found in tumor profiles [18, 19]. In one study, 34 tissue sections containing both normal cells and clear cell renal carcinoma (ccRCC) found the reduced expression of peptide peaks indicated a compromised histological margin [19]. The proteins giving rise to the diagnostic peaks were identified as cytochrome c, cytochrome c oxidase, and NADH ubiquinone reductase MLRQ subunit. Interestingly, each of these biomarkers were previously shown to be decreased in ccRCC using 2D electrophoresis, enzyme activity determination, and Western blotting [20–22] and purported to indicate dysfunctional mitochondrial enzymes that may foreshadow tumor development.
It is clear that morphological analysis has benefitted from concurrent biochemical, immunohistochemical, and nucleic acid examination of tissue samples [23]. Pathology reports containing molecular data of clinical relevance are now being generated in many tumor types. For example, the reporting of breast carcinomas includes, in most instances, the levels of expression of the estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2/Neu) [24, 25]. Overexpression of HER2/ Neu oncoprotein is an important factor associated with a poor prognosis in breast cancer. Thus, an obvious challenge in biomarker discovery is the ability to meet and exceed the performance of these emerging clinical tools. A recent MALDI-IMS biomarker discovery study used HER2 status to stratify tissue samples. A peptide ion (m/z 8,403) overexpressed in HER2-positive breast cancer tissue was identified as cysteine-rich intestinal protein 1 (CRIP1) in a study reported last year by Rauser et al. [26], confirming previous findings of the coexpression of CRIP1 and ERBB2 messenger RNAs [27]. Additionally, in this study, HER2 status of breast cancer tissues was accurately determined on the basis of a MALDI-IMS proteomic algorithm, which resulted in a sensitivity of 83% and a specificity of 92%. Thus, even without sequence identification of all differentially expressed peaks, the spectral features found via MALDI-IMS can be used in combinatorial algorithms for tissue classification [3, 28, 29].
MALDI-IMS also has utility in examining the distribution of small biomolecules in tissue sections [30, 31]. Many compounds that are biologically or pharmacologically active are less than 1 kDa in size and thus fall into the broad category of small molecules. Levels of drugs such as chemotherapeutic agents or hormonal therapies can be measured directly from tissue specimens to assess the level of delivery to a particular organ site [32], opening new possibilities for the measurement of concomitant protein changes in specific tissues after systemic drug administration. MALDI-IMS can be performed with Fourier transform mass spectrometers, which allows the measurement of mass signals with very high resolution and precision [33]. The spatial distributions of molecules differing in mass by less than 0.1 Da can be resolved, representing a mass resolution currently not attainable with other mass spectrometry (MS) imaging platforms. This makes Fourier transform MS imaging an ideal technique for the analysis of small metabolites, drugs, and lipid molecules. Additionally, a new generation of instrumentation boasts greatly improved scan speeds, which dramatically reduces the previously lengthy acquisition process of tissue imaging using Fourier transform MS.
Thus, MALDI-IMS can detect native biomolecules that correlate with histological features, morphology, and pathological aggressiveness. Except for standard tissue processing protocols of fresh frozen tissues, visualization of the biomolecules by MALDI-IMS does not require use of chemical affinity or modification reagents, or treatment with degradative enzymes. However, this does not preclude use of MALDI-IMS with tissues processed in different fixatives, or combining the technique with any available molecular characterization agent, particularly for application to biomarker discovery projects. In the following sections, application of MALDI-IMS with different tissue fixation approaches, protein modification and identification strategies, and targeted affinity methods will be summarized.
MALDI-IMS and tissue fixation
Most methods developed for peptide and protein analysis by MALDI-IMS have been optimized for freshly frozen tissue specimens. Snap-freezing of tissue samples is ideal because, when performed rapidly, it preserves the maximum content. In general, however, tissue samples analyzed in pathology laboratories undergo formalin fixation, and paraffin embedding (FFPE). Although the morphology of formalin-fixed tissues is superior to that of fresh frozen samples, the molecular cross-linking by formalin and subsequent paraffin embedding significantly impairs subsequent proteomic analysis. However, FFPE tissues have been analyzed by MALDI-IMS. Heat-induced antigen retrieval coupled with in situ tryptic digestion of proteins followed by interrogation of the resulting peptides has yielded successful MALDI-IMS [34–37].
There also exist many tissue fixation methods as an alternative to formalin fixation that promise better utility in protein analysis. Various alcohol-based fixation methods followed by paraffin embedding have been shown to be compatible with MALDI-IMS without trypsin digestion, including PAXGene and RCL2/CS100 (both of which are proprietary mixes of alcohol and acid) [38, 39]. Universal molecular fixative (UMFix), which is methanol-based with a small percentage of polyethylene glycol, is one of the most widely used and commercially available alcohol-based histological fixatives. Currently, UMFix processing of tissue is in use at our clinical sites for surgery and biopsy specimens. It preserves macromolecules (DNA, RNA, and proteins) in tissue at ambient temperature [40, 41], and we have confirmed the findings of previous studies [42] which found morphology and IHC results from UMFix-treated tissue to be equal to or superior to those of FFPE tissues (data not shown). We have also assessed the quality of the spectral data for MALDI-IMS by comparing profiles and images obtained from the UMFix-processed samples with those obtained from optimal cutting temperature embedded frozen tissue. The UMFix specimens, after sectioning, paraffin removal, and partial rehydration, were sprayed with a sinapinic acid matrix using our established protocol on an ImagePrep workstation (Bruker Daltonics). The matched frozen tissue was cryosectioned and processed as previously described [3]. As shown in Fig. 2, the area of overexpression and peak pattern of an ion at m/z 4,355 (fragment of MEKK2 [3]) corresponds well in both the UMFix-treated tissue and the frozen tissue. The m/z 4,355 peak is not detected in formalin-fixed tissues (data not shown). As more pathology laboratories move away from formalin fixation, the stocks of tissues fixed with protein-friendly fixatives will increase, although the effects of long-term storage on these samples needs to be evaluated.
Fig. 2.
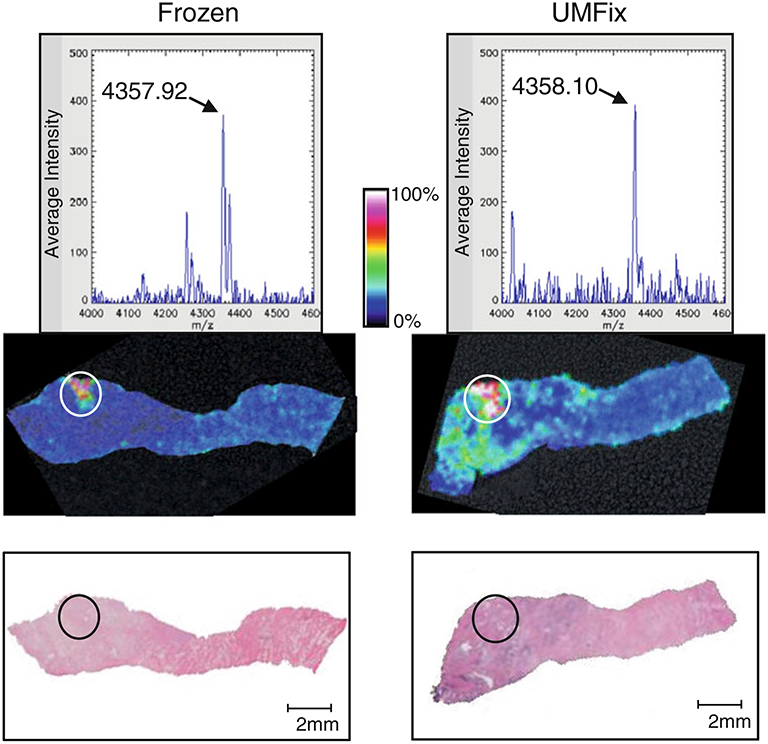
Comparison of MALDI-IMS data in frozen and universal molecular fixative (UMFix)-processed tissue in a pair of matched prostate tissue samples. A tissue sample was harvested from a prostate surgery specimen and bisected. One half was snap-frozen (left) and the other was treated with UMFix (right). Top panels: Representative spectra of the m/z 4,355 ion. Middle panels: MALDI-IMS map showing high expression of m/z 4,355 in specific areas where tumor cells are located (circled) in the H&E staining image (bottom panel)
As we are evaluating detection of MEKK2 as a potential biomarker of prostate cancer aggressiveness at the time of biopsy, preserving the biopsy samples in UMFix would facilitate integration into current clinical collection and pathology assessment workflows. An example of a MALDI-IMS image of two 18-guage-needle biopsy cores preserved in UMFix is shown in Fig. 3. The m/z 4,355 MEKK2 peak is easily detected and high expression corresponds to the indicated ROIs determined by a pathologist. In addition, there is enough tissue from the biopsy cores to allow effective standard pathological H&E staining and IHC determinations, as well as MALDI-IMS of an adjacent tissue slice. Our cumulative results indicate that the UMFix-processed tissue is compatible with MALDI-IMS and preserves detail in the subsequent images, producing results comparable to those obtain with frozen tissue. Combined with results from other studies using alcohol-based fixative formulations, use of these types of fixatives with MALDI-IMS and standard pathology workflows is feasible and could be ideal for large cohort studies involving multiple institutions. As with the other fixatives, use of UMFix tissue preservation in this context will require continued development and optimization of sample acquisition, sample processing, storage, and data collection protocols. We are currently evaluating this type of workflow using the prostate cancer biopsy and standard tissue specimens described in Figs. 2 and 3.
Fig. 3.
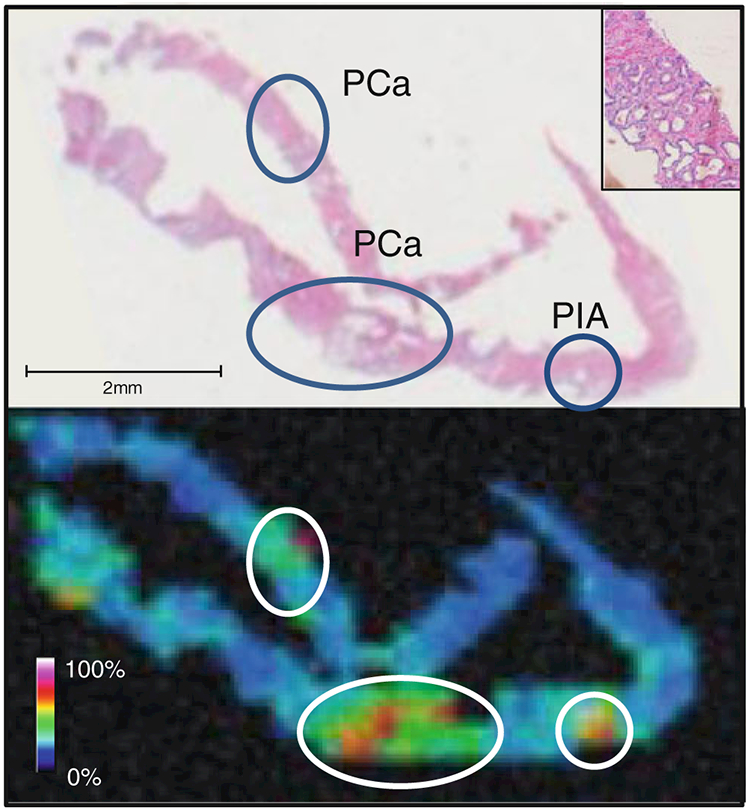
MALDI-IMS of prostate biopsy cores. Top panel·. Two 18-guage-needle biopsy cores preserved in UMFix with tumor regions of interest determined by a pathologist circled in the H&E-stained serial section. The inset shows the area of prostate cancer at ×10 magnification. Bottom panel: An example of a MALDI-IMS map showing detection of the m/z 4,355 MEKK2 in a serial section of the biopsy cores. PCa prostrate cancer, P1A Prostatic Inflammatory Atrophy
Biomarker discovery and clinical applications of MALDI-IMS
Cancer development demands tissue remodeling, a process attributed to proteolytic degradation of the extracellular matrix. Degradation of extracellular matrix proteins by proteases, secreted by different cell types, is a proteolytic balancing act governing tumor cell invasion [43], making them attractive targets if they are present in high abundance. Proteins and endogenous peptides of mass less than 15 kDa are the predominant analytes detected by MALDI-IMS. Demonstrating whether the detected analyte is a native low molecular weight protein or a proteolytic fragment of a larger protein by sequence identification has been one of the more difficult hurdles to overcome for MALDI-IMS. Methods for endogenous peptide and protein sequence identity in MALDI-IMS discovery efforts are shown in Fig. 4a, and c. The identities of small endogenous peptides and lipids have been determined through MS/MS analysis directly on tissue sections [44–46]. Unfortunately, most endogenous peptide analytes that compose a typical MALDI-IMS spectrum fall below the level of sensitivity currently available with MALDI time-of-flight (TOF) MS/MS instruments for direct tissue sequencing analysis. Thus, it is routine to employ scale-up methods using tissue lysates. In such approaches, tissue is harvested from the ROI and is subjected to fractionation by high-performance liquid chromatography. In spite of these limitations, it appears that upregulation of proteolytic activity in malignant cells preferentially generates analytes well suited to MALDI-IMS. This was demonstrated in a MALDI-IMS study of ovarian cancer tissue where the level of an 84 amino acid peptide fragment of the 11S proteasome activator complex (Reg-alpha) was found increased in tumor samples [47]. Alteration of this protein by cleavage was hypothesized to be a mechanism of immune evasion in ovarian carcinoma. The sequence identification of the 38 amino acid MEKK2 peptide fragment in prostate tissues by our group is another example [3].
Fig. 4.
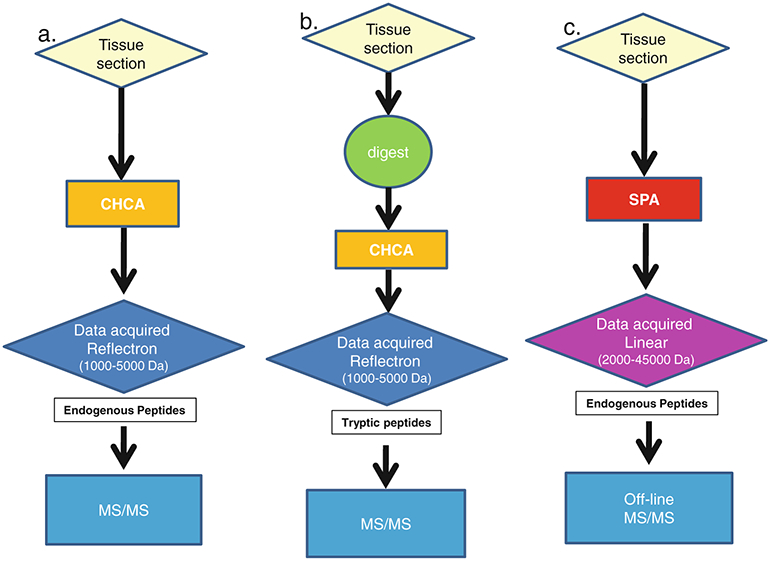
Schemes for data collection and sequence identification after MALDI-IMS methods for endogenous peptides and proteins. a. Scheme for the identification of abundant endogenous peptides (1–5 kDa) directly from a tissue section. b. Scheme for the identification tryptic peptides using “in situ” protease digestion followed by data acquisition in reflectron mode and MS/MS analysis. c. Scheme for the identification of endogenous proteins and large peptides using linear mode acquisition followed by off-line MS/MS. CHCA α-cyano-4-hydroxycinnamic acid, SPA sinapinic acid
Although MALDI-TOF-MS is highly flexible with respect to the detection of low and high mass compounds [48, 49], most signals detected are well below m/z 20,000. Higher molecular weight molecules tend to be underrepresented because of poor detector efficiency in the high mass range and/or detector saturation. Furthermore, most matrix preparations used in MALDI-IMS contain at least 50% organic solvents, which may not effectively solubilize higher mass proteins. The enhancement of higher molecular weight signals has been achieved through the addition of chaotropic agents or detergents to the matrix as well as by the incorporation of improved mass detectors that are less prone to saturation [50, 51]. The detection of larger proteins may also be facilitated by the use of in situ digestion in a tissue section, while preserving the spatial distribution of the intact proteins. The smaller peptide fragments generated by such enzymatic cleavage are easier to detect by standard MALDI-IMS techniques, and will generally reflect the abundance of the parent protein. In fact, we and other groups have shown that the spatial distribution of the parent protein can be inferred from the localization of the fragment [3, 35, 52]. Thus, the resulting peptides from in situ trypsinization can be visualized to provide spectra for profiling, but they can also be sequence-identified via MALDI-TOF MS/MS directly on tissue provided the masses are 4 kDa of less. Direct tissue sequencing was used to identify several proteins specifically overexpressed in the coronal sections of a rat brain [52]. This study clearly demonstrated the feasibility of an automated on-tissue peptide identification strategy that should be applicable to human tissues. Our laboratory has employed a similar approach for the sequence identification of peptides directly from both frozen and UMFix-treated prostate cancer tissue. As expected, the yield is extremely modest, such that in our studies we identified 68 peptides derived from 50 unique proteins by performing MS/MS directly on prostate cancer tissue after in situ trypsinization. One example is shown in Fig. 5, where a tissue section containing a region of prostate cancer was subjected to trypsin digestion in situ. A peptide at m/z 1,408.75 can be seen in the MALDI-IMS ion intensity map in a region designated as tumor (Fig. 5, panel a). This peptide was selected for MS/MS performed directly from the tissue section and was identified as a fragment of transgelin (Fig. 5, panel b), an actin-binding protein with tumor suppressor function [53]. From the list of proteins identified using this method, 13 of the 50 identified peptides (26%) originate from proteins present in high abundance, such as actin, myosin, and collagen. However, the remaining 37 proteins (74%) represent lower-abundance and prostate-specific molecules such as neuroendocrine convertase, 17β-hydroxysteroid dehydrogenase, and testes-specific protein kinase 1. These results underscore the ability of this technique to identify proteins directly from tissue.
Fig. 5.
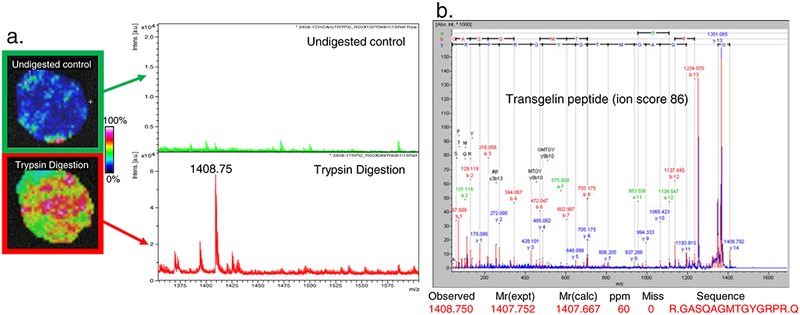
In situ trypsin digestion of prostate tissue. A tissue core was harvested from a prostate surgery specimen and serial sections were made. a. One section was sprayed with trypsin, and one was used as a control. Tissues were sprayed with CHCA and spectra were collected in reflectron mode. b. one peptide at m/z 1,408.75 was selected for in situ MS/MS and produced a fragmentation spectrum which was identified as corresponding to a peptide of transgelin with a MASCOT ion score of 86
In situ trypsin digestion is also used commonly for the MALDI-IMS investigation of FFPE tissues after heat-induced antigen retrieval techniques [34]. A large number of FFPE tissue samples arrayed on a single slide (tumor microarrays, TMAs) with known outcome are extremely useful for the discovery and validation of diagnostic and prognostic biomarkers [54]. This approach was used in a study of non-small-cell lung cancer and biopsy tissues in a TMA containing 112 needle core biopsies [52]. Trypsin was spotted onto the tissues using an automated spotting device. The trypsin deposition was performed a total of 30 times with a 5-min interval between applications, which allowed for adequate digestion of the tissue proteins. MALDI MS/MS measurements of selected peptides were acquired directly from the digested TMA sections. A support vector machine algorithm incorporating 73 peaks classified the spectra from regions marked by the pathologist as adenocarcinoma with an accuracy of 97.9% and as squamous cell carcinoma with an accuracy of 98.6%. The authors also sequence-identified a peptide from heat shock protein beta-1, which showed selective expression in the squamous cell carcinoma subtype. A more recent study using a TMA of pancreatic tissues used principal component analysis-discriminant analysis to generate molecular classification models based on MALDI-IMS tryptic peaks, which separated the resulting spectra into various tumor classes [55].
Targeted MALDI-IMS for the analysis of selected biomolecules
IHC with the targeted detection of protein biomarkers in tissues is the backbone of diagnostic clinical pathology. Clearly, IHC is limited by the availability of biomarker antibodies, but in fact is often further restricted by the lack of standardization in essential steps such as tissue pretreatment, reagents, detection methods, and interpretation [56]. The current state of the art in IHC also limits the number of different antibodies that can be placed onto one section even with highly sensitive fluorescent probes such as quantum dots [57]. A recently developed variant of MALDI-IMS, termed “targeted imaging mass spectrometry” (TIMS) or “targeted multiplex mass spectrometry imaging,” first described by Theiry et al. [58], seeks to overcome some of the limitations of standard IHC. TIMS allows the targeted analysis and spatial visualization of a molecule of interest directly from tissue sections by the use of laser-reactive photocleavable molecular tags attached to antibodies and potentially other affinity molecules [59, 60]. The bond conjugating the mass tag to the affinity molecule is photocleavable, so exposure to the UV laser in a MALDI mass spectrometer releases the tag, which is readily detected without the use of a matrix. Because no matrix is used, tissue preparation is streamlined, and few endogenous molecules are desorbed, which provides excellent signal-to-noise ratios in resulting spectra. Changing the mass of the tags also allows multiplexed detection of different molecules simultaneously within the same tissue, and in contrast to fluorescent tags, mass tags do not exhibit neighbor quenching. Tissues and sections prepared by standard methods (fixed or frozen) can be used for TIMS, so the pathology workflows remain unchanged. The mass tag employed is essentially a histochemical detection reagent and images are created for the mass of each tag. Previous studies have successfully shown simultaneous detection of three mass tags at femtomole levels using TIMS [58–60]. Furthermore, TIMS has the potential to enable across-class analysis of multiple molecular types not readily available to standard MALDI-MS such as oligonucleotides and sugars, thus enabling expanded multiplexing within limited tissue samples.
Our laboratory is currently examining the ability of TIMS to detect and quantitate a variety of biomarker reagents. The photocleavable tags our laboratory uses are triarylmethyl (trityl)-based compounds with a typical molecular mass of 650–750 Da [61]. As shown in Fig. 6, the conjugation of the N-hydroxysuccinimide group of the trityl compound with an antibody occurs via ε-amino acid groups on lysine and the primary amine of the N-terminal amino acids. Cleavage of this trityl compound from the antibody when subjected to the MALDI laser results in the generation of a mass tag at m/z 519.5 In a proof of principal experiment, we linked a trityl molecule to an antibody for prostate-specific antigen (PSA), which is expressed in normal prostate tissue. Conjugation was performed and the probe was purified as previously described [58]. The trityl-linked antibody was then incubated directly on frozen prostate tissue sections. The tissues were washed, air-dried, and desiccated before being subjected to MALDI-MS. As shown in Fig. 7, targeted labeling by the trityl-anti-PSA is compared with standard IHC staining with the same anti-PSA in serial sections. A specific signal at m/z 519.5 was detected in the locations of the tissue where the antibody was bound (glandular epithelium) and is shown in the resulting image. This same probe was equally effective in alcohol and formalin fixed prostate tissues (data not shown).
Fig. 6.
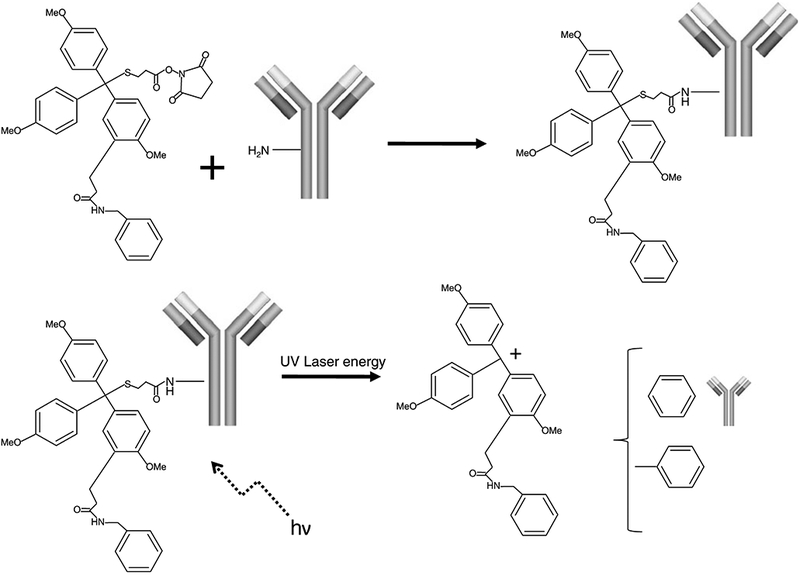
Conjugation of a triarylmethyl (trityl) mass tag to an antibody molecule. Covalent attachment of the trityl mass tag to the primary amine of an antibody is facilitated by the N-hydroxysuccinimide ester group on the trityl molecule. In the MALDI mass spectrometer the trityl group absorbs the energy from the laser, resulting in cleavage of the C–S bond, creation of a stable carbocation, and release of the trityl mass tag
Fig. 7.
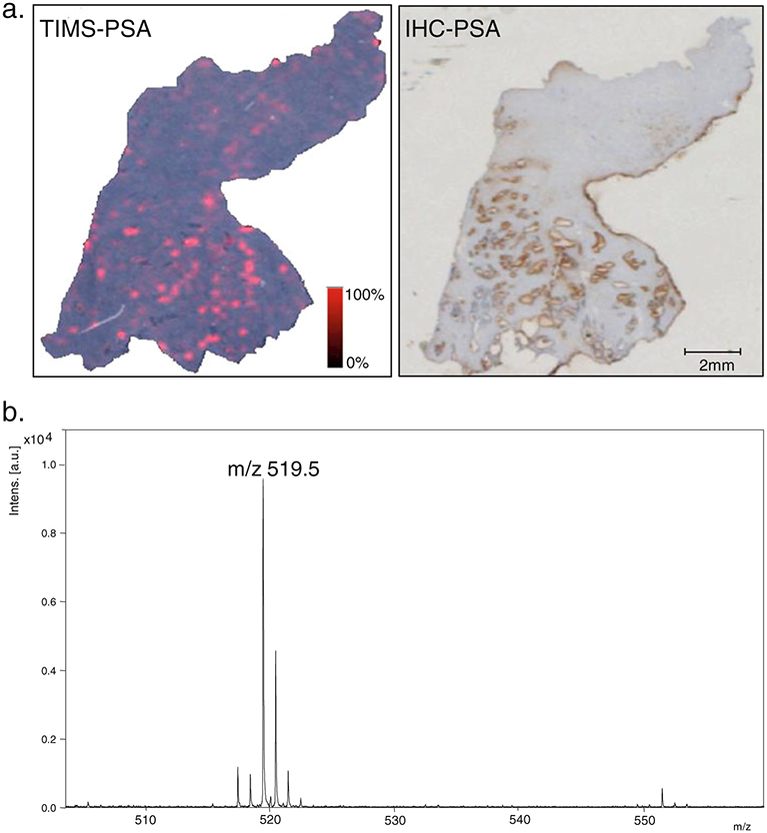
Targeted MALDI-IMS for prostate-specific antigen (PSA). a. Resulting image from targeted IMS (TIMS) of prostate tissue showing the spatial distribution of the trityl tag (m/z 519.5) (left panel) which was bound to PSA in areas which also stained for PSA in traditional IHC for PSA (right panel) in a serial section. b. Resulting reflectron mode spectrum after release of the mass tag from the antibody by the MALDI laser
Obviously the spatial resolution of microscopic techniques such as IHC is superior to that currently attainable with MALDI-IMS. However, all current methods for biomarker testing in tissue to date generate a continuous optical signal, and quantitation is done by estimating the signal intensity either with the human eye or using image analysis software, which converts the optical signal into a digital signal. The quantum nature of the TIMS signal offers inherent advantages to signal quantitation. As biopsies for cancer diagnosis become smaller, combined with more diagnostic demands on tissue, the ability to quantitate multiple biomarkers on a single histological section will be an important tool for future clinical applications. Methods that test for biomarkers in tissue and preserve morphology but enable multiple simultaneous tests (multiplexing) will be much in demand. Therefore, the combination of the inherent ease of quantitation of MALDI-MS and its theoretical multiplexing capabilities make TIMS attractive for the investigation of protein biomarkers and a promising technology in clinical diagnostics.
Perspective
Although MALDI-IMS is a preferred approach for determining spatially resolved analyte expression profiles, it suffers from poor overall analyte volume (numbers of ions resolved). Thus, the utility of this technique will depend upon the development of new strategies for obtaining an increased volume of analysis. Some of these advances will be in the area of tissue preparation, washing protocols, fixation, and novel/selective solubilization approaches. In addition, the application of molecular class-specific chemistries for targeted tissue mining and ionization is sure to expand the analyte horizon. Practitioners in the field should also anticipate the added value of applying specific chemistries for the covalent addition of mass tags to targeted side groups, chemical modifications, and enzymatic intermediates, as well as metabolic incorporation of affinity and/or mass tags into live cellular and animal models.
One class of biomarkers for imaging and histology studies which has been underexplored is carbohydrates, which are known to be correlated with the initiation, progression, and aggressiveness of cancer [62–64]. Carbohydrate biomarkers have the advantage of being resistant to proteolytic digestion and not being affected by cross-linking chemistry at the tissue fixation stage. Intense efforts in the last decade have yielded compounds capable of recognizing certain carbohydrate molecules for biomarker applications [65–69]. In an effort to create a carbohydrate-specific affinity TIMS-like approach, we have combined a sialyl Lewis X-specific compound for conjugation with a trityl MS tag. Using a section of human renal tissue containing a region of ccRCC that is known to express sialyl Lewis X antigens, we found this TIMS conjugate was effective at labeling the tumor-specific regions of the section (see Fig. 8). The continued development and future use of such class-specific expression-profiling reagents will expand the context of applications for MALDI-IMS, and will allow improved detection and quantification of the expression of normal/altered glycans.
Fig. 8.
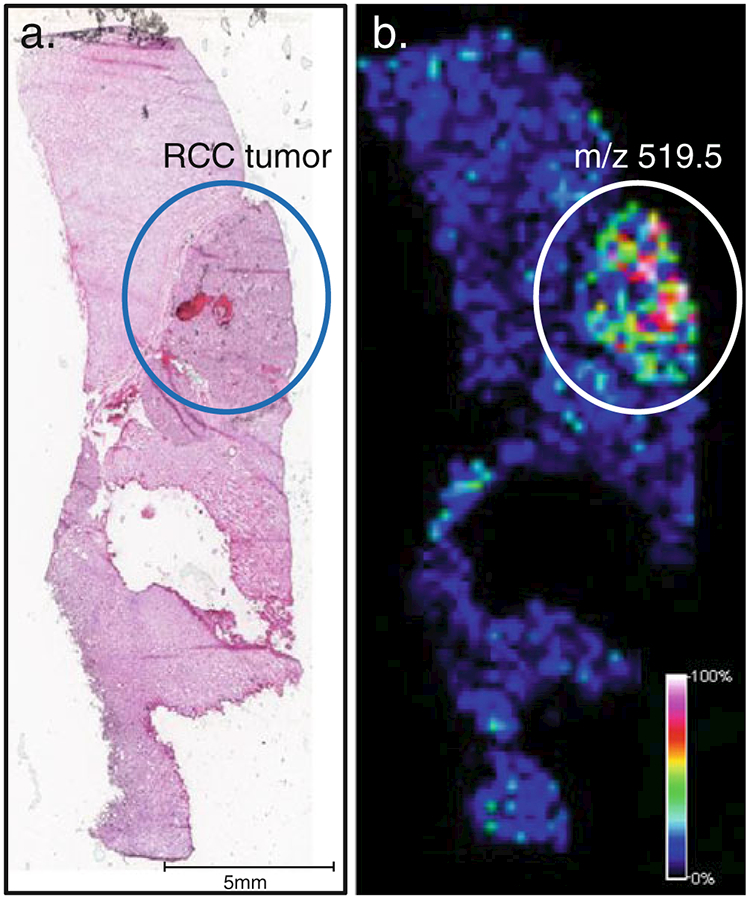
Targeted carbohydrate affinity labeling of sialyl Lewis X antigen expression in renal tissue. a. An H&E image of a clear cell renal carcinoma (RCC) tissue slice. The area of tumor is circled in blue. b. A serial slice of the same tissue incubated with a sialyl Lewis antigen targeting affinity molecule linked to a trityl reporter tag. Shown is the image of the spatial distribution of the trityl tag (m/z 519.5), highlighting the most intense signal in the circled tumor area
The use of TMAs can greatly reduce the time periods between initial discovery, verification, and validation of cancer biomarkers. Well-annotated arrays can populate a study with a comprehensive series of tissues derived from various disease states. In our laboratory we have constructed high-density TMAs onto MALDI-IMS-compatible glass slides with adjacent sections fixed and prepared for histology. In this way the expression of newly discovered biomarkers can be compared with the complete clinical data (disease state, pathology record, and outcome) associated with each tissue. An assessment of the relative expression of whole proteins (when antibodies are available) to the putative diagnostic ion can be achieved by IHC of the adjacent TMA. Although TMAs can streamline biomarker discovery, the use of these tools demands methods to control for tissue variability. In addition to the need for standardization in tissue processing, sectioning, and storage required for typical TMA utilization, the specific sensitivity of IMS to surface topology poses an as yet unsolved problem area. Thus, there is a need for both software and automation that can ensure uniformity in tissue preparation as high-throughput systems are implemented.
There has also been much discussion regarding the entry of IMS into a clinical pathology workflow. Experience clearly suggests that the easiest route to biomarker utility would relegate IMS to a discovery platform from which protein identification is applied toward development of reagents that would facilitate commonly used analytical platforms such as IHC and ELISA. However, MS platforms may discover structures for which specific antibodies cannot easily be developed. In addition, quantitative microscopy for IHC in a clinical pathology setting is far from routine. Consequently, the performance of an antibody-based assay in IHC is always degraded to easily observed changes in intensity or localization. Thus, future rendition of IMS may provide a more robust and quantitative assay. As with any technology, utility in the clinic will ultimately depend upon whether the approach can reveal novel information of clinical relevance to a large enough number of patients. This may be in the form of greatly improved quantitation of existing biomarkers, the ability to multiplex this quantitation, or as a new biomarker for which MS is best suited for detection. The demands on this technology as a diagnostic will be very different from those imposed by the analytical chemist. Speed, sensitivity, resolution, and reproducibility will dominate in the clinical laboratory. In addition, the information and data pipeline from patient to laboratory will need to be seamless. This includes the digitization of pathology records/slides, MS data, and optical images and correlation with each patient from whom the elements were derived. Even at the level of discovery it is critical to integrate all patient data with all experimental data. The laboratories that are able to establish solutions to the bottlenecks described above will derive the most benefit from this rapidly evolving technology.
Contributor Information
Lisa H. Cazares, Department of Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, Norfolk, VA 23507, USA The Leroy T. Canoles Jr. Cancer Research Center, Eastern Virginia Medical School, Norfolk, VA 23507, USA.
Dean A. Troyer, The Leroy T. Canoles Jr. Cancer Research Center, Eastern Virginia Medical School, Norfolk, VA 23507, USA
Binghe Wang, Department of Chemistry, Georgia State University, Atlanta, GA 30303, USA.
Richard R. Drake, Department of Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, Norfolk, VA 23507, USA The Leroy T. Canoles Jr. Cancer Research Center, Eastern Virginia Medical School, Norfolk, VA 23507, USA.
O. John Semmes, Department of Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, Norfolk, VA 23507, USA; The Leroy T. Canoles Jr. Cancer Research Center, Eastern Virginia Medical School, Norfolk, VA 23507, USA.
References
- 1.Hillenkamp F et al. (1991) Anal Chem 63(24):1193A–1203A [DOI] [PubMed] [Google Scholar]
- 2.Vestal ML (2009) J Mass Spectrom 44(3):303–317 [DOI] [PubMed] [Google Scholar]
- 3.Cazares LH et al. (2009) Clin Cancer Res 15(17):5541–5551 [DOI] [PubMed] [Google Scholar]
- 4.Agar NY et al. (2010) Methods Mol Biol 656:415–431 [DOI] [PubMed] [Google Scholar]
- 5.Caldwell RL, Caprioli RM (2005) Mol Cell Proteomics 4(4):394–401 [DOI] [PubMed] [Google Scholar]
- 6.Chaurand P et al. (2005) Toxicol Pathol 33(1):92–101 [DOI] [PubMed] [Google Scholar]
- 7.Franck J et al. (2009) Anal Chem 81(19):8193–8202 [DOI] [PubMed] [Google Scholar]
- 8.Vegvari A et al. (2010) J Proteomics 73(6):1270–1278 [DOI] [PubMed] [Google Scholar]
- 9.Chaurand P et al. (2006) J Proteome Res 5(11):2889–2900 [DOI] [PubMed] [Google Scholar]
- 10.Cornett DS et al. (2006) Mol Cell Proteomics 5(10):1975–1983 [DOI] [PubMed] [Google Scholar]
- 11.Fournier I, Wisztorski M, Salzet M (2008) Expert Rev Proteomics 5(3):413–424 [DOI] [PubMed] [Google Scholar]
- 12.Franck J et al. (2009) Mol Cell Proteomics 8(9):2023–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwamborn K, Caprioli RM (2010) Nat Rev Cancer 10(9):639–646 [DOI] [PubMed] [Google Scholar]
- 14.Yanagisawa K et al. (2003) Lancet 362(9382):433–439 [DOI] [PubMed] [Google Scholar]
- 15.Reyzer ML et al. (2004) Cancer Res 64(24):9093–9100 [DOI] [PubMed] [Google Scholar]
- 16.Schwartz SA et al. (2005) Cancer Res 65(17):7674–7681 [DOI] [PubMed] [Google Scholar]
- 17.Gofrit ON et al. (2007) J Urol 178(5):1925–1928 [DOI] [PubMed] [Google Scholar]
- 18.Herring KD, Oppenheimer SR, Caprioli RM (2007) Semin Nephrol 27(6):597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oppenheimer SR et al. (2010) J Proteome Res 9(5):2182–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hervouet E et al. (2005) Carcinogenesis 26(3):531–539 [DOI] [PubMed] [Google Scholar]
- 21.Simonnet H et al. (2002) Carcinogenesis 23(5):759–768 [DOI] [PubMed] [Google Scholar]
- 22.Simonnet H et al. (2003) Carcinogenesis 24(9):1461–1466 [DOI] [PubMed] [Google Scholar]
- 23.Finn WG (2007) J Mol Diagn 9(4):431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allemani C et al. (2004) Br J Cancer 91(7):1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hadjisavvas A et al. (2002) Ultrastruct Pathol 26(4):237–244 [DOI] [PubMed] [Google Scholar]
- 26.Rauser S et al. (2010) J Proteome Res 9(4):1854–1863 [DOI] [PubMed] [Google Scholar]
- 27.Wilson KS et al. (2002) Am J Pathol 161(4):1171–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaurand P et al. (2004) Am J Pathol 165(4):1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cornett DS et al. (2007) Nat Methods 4(10):828–833 [DOI] [PubMed] [Google Scholar]
- 30.Hsieh Y, Chen J, Korfmacher WA (2007) J Pharmacol Toxicol Methods 55(2):193–200 [DOI] [PubMed] [Google Scholar]
- 31.Reyzer ML, Caprioli RM (2007) Curr Opin Chem Biol 11(1):29–35 [DOI] [PubMed] [Google Scholar]
- 32.Hsieh Y et al. (2006) Rapid Commun Mass Spectrom 20(6):965–972 [DOI] [PubMed] [Google Scholar]
- 33.Cornett DS, Frappier SL, Caprioli RM (2008) Anal Chem 80 (14):5648–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groseclose MR et al. (2008) Proteomics 8(18):3715–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemaire R et al. (2007) J Proteome Res 6(4):1295–1305 [DOI] [PubMed] [Google Scholar]
- 36.Morita Y et al. (2009) Cancer Sci 101(1):267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronci M et al. (2008) Proteomics 8(18):3702–3714 [DOI] [PubMed] [Google Scholar]
- 38.Chaurand P et al. (2008) J Proteome Res 7(8):3543–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ergin B et al. (2010) J Proteome Res 9(10):5188–5196 [DOI] [PubMed] [Google Scholar]
- 40.Morales AR et al. (2004) Am J Clin Pathol 121(4):528–536 [DOI] [PubMed] [Google Scholar]
- 41.Vincek V et al. (2003) Lab Invest 83(10):1427–1435 [DOI] [PubMed] [Google Scholar]
- 42.Nadji M et al. (2005) Appl Immunohistochem Mol Morphol 13(3):277–282 [DOI] [PubMed] [Google Scholar]
- 43.Skrzydlewska E et al. (2005) World J Gastroenterol 11(9):1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaurand P et al. (2001) Proteomics 1(10):1320–1326 [DOI] [PubMed] [Google Scholar]
- 45.Shimma S et al. (2007) J Chromatogr B Anal Technol Biomed Life Sci 855:98–103 [DOI] [PubMed] [Google Scholar]
- 46.Shimma S et al. (2008) Anal Chem 80(3):878–885 [DOI] [PubMed] [Google Scholar]
- 47.Lemaire R et al. (2007) J Proteome Res 6(11):4127–4134 [DOI] [PubMed] [Google Scholar]
- 48.Bahr U et al. (1997) J Mass Spectrom 32(10):1111–1116 [DOI] [PubMed] [Google Scholar]
- 49.Westmacott G et al. (2000) Rapid Commun Mass Spectrom 14 (7):600–607 [DOI] [PubMed] [Google Scholar]
- 50.van Remoortere A et al. (2010) J Am Soc Mass Spectrom 21 (11):1922–1929 [DOI] [PubMed] [Google Scholar]
- 51.Leinweber BD et al. (2009) J Am Soc Mass Spectrom 20(1):89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Groseclose MR et al. (2007) J Mass Spectrom 42(2):254–262 [DOI] [PubMed] [Google Scholar]
- 53.Yang Z et al. (2007) Mol Endocrinol 21(2):343–358 [DOI] [PubMed] [Google Scholar]
- 54.Simon R (2010) Methods Mol Biol 664:1–16 [DOI] [PubMed] [Google Scholar]
- 55.Djidja MC et al. (2010) Anal Bioanal Chem 397(2):587–601 [DOI] [PubMed] [Google Scholar]
- 56.Rojo MG, Bueno G, Slodkowska J (2009) Folia Histochem Cytobiol 47(3):349–354 [DOI] [PubMed] [Google Scholar]
- 57.Sweeney E et al. (2008) Biochem Biophys Res Commun 374 (2):181–186 [DOI] [PubMed] [Google Scholar]
- 58.Thiery G et al. (2007) Rapid Commun Mass Spectrom 21(6):823–829 [DOI] [PubMed] [Google Scholar]
- 59.Thiery G et al. (2008) Proteomics 8(18):3725–3734 [DOI] [PubMed] [Google Scholar]
- 60.Lemaire R et al. (2007) J Proteome Res 6(6):2057–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ustinov AV et al. (2008) Org Biomol Chem 6(24):4593–4608 [DOI] [PubMed] [Google Scholar]
- 62.Cheng Y et al. (2010) Sci China Ser B Chem 53:3–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fukuda M, Hindsgaul O (eds) (1994) Molecular glycobiology. Oxford University Press, New York, pp 1–52 [Google Scholar]
- 64.Wang B, Boons G-J (eds) (2011) Carbohydrate recognition: biological problems, methods, and potential applications. Wiley, Hoboken [Google Scholar]
- 65.Burroughs S, Wang B (2010) ChemBioChem 11(16):2245–2246. doi: 10.1002/cbic.201000462 [DOI] [PubMed] [Google Scholar]
- 66.Jin S et al. (2010) Med Res Rev 30(2):171–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pal A, Bérubé M, Hall DG (2010) Angew Chem Int Ed 49:1492–1495 [DOI] [PubMed] [Google Scholar]
- 68.Yang W et al. (2004) Chem Biol 11:439–448 [DOI] [PubMed] [Google Scholar]
- 69.Yang W et al. (2002) Bioorg Med Chem Lett 12:2175–2177 [DOI] [PubMed] [Google Scholar]