

Abstract
Liver is the major organ that regulates whole body cholesterol metabolism. Disrupted hepatic cholesterol homeostasis contributes to the pathogenesis of nonalcoholic steatohepatitis, dyslipidemia, atherosclerosis, and cardiovascular diseases. Hepatic bile acid synthesis is the major catabolic mechanism for cholesterol elimination from the body. Furthermore, bile acids are signaling molecules that regulate liver metabolism and inflammation. Autophagy is a highly-conserved lysosomal degradation mechanism, which plays an essential role in maintaining cellular integrity and energy homeostasis. In this review, we discuss emerging evidence linking hepatic cholesterol and bile acid metabolism to cellular autophagy activity in hepatocytes and macrophages, and how these interactions may be implicated in the pathogenesis and treatment of fatty liver disease and atherosclerosis.
Keywords: nutrient signaling, nuclear receptor, liver injury, hyperlipidemia, enterohepatic circulation, hepatocyte, macrophage
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease in the Western countries, and it is more common in the obese and type-2 diabetic population (1). Simple steatosis usually does not require treatment, while some NAFLD patients may progress to nonalcoholic steatohepatitis (NASH). NASH is a debilitating form of NAFLD characterized by hepatocellular injury, chronic inflammation, and a higher risk of end stage liver diseases such as cirrhosis and liver cancer (1). The mechanisms underlying NASH pathogenesis are still incompletely understood. The role of free fatty acids in causing lipotoxic liver injury in NASH has been extensively studied (2). Emerging evidence supports that cellular non-esterified free cholesterol (FC) accumulation contributes significantly to hepatocyte injury and inflammation in NASH (3,4). Indeed, FC accumulated at significantly higher levels in NASH livers than in normal livers and simple steatotic liver in humans (5). FC accumulation in hepatocytes causes mitochondrial dysfunction, which sensitizes hepatocytes to cytokine and stress-induced cell death (3). In addition, cholesterol-laden foamy Kupffer cells were found at early stage of NASH and showed pro-inflammatory phenotypes (6), suggesting cholesterol accumulation triggers macrophage activation in NASH. Disrupted hepatic cholesterol homeostasis is also a major cause of hyperlipidemia. Intracellular lipid accumulation is a driver of hepatic secretion of very low density lipoprotein (VLDL), which is the precursor of cholesterol-rich low density lipoprotein (LDL). Elevated circulating VLDL and LDL concentration is a major contributor to the development of atherosclerosis and higher risk of cardiovascular disease (CVD) (7). Indeed, CVD is the leading cause of mortality in patients with NASH and type-2 diabetes (7). Therapeutic interventions that improve hepatic cholesterol homeostasis are expected to ameliorate both hepatic and cardiovascular-related complications in NASH and type-2 diabetes.
Autophagy is a highly-conserved lysosomal degradation mechanism, which plays an essential role in maintaining cellular integrity by eliminating protein aggregates and damaged or excessive organelles in mammalian cells (8). Autophagy is also a catabolic process used by cells to generate nutrients and energy by degrading macromolecules in response to nutrient deprivation (8). The process of autophagy initiates from the formation of a double membrane vesicle called autophagosome, which may sequester protein aggregates, intracellular organelles, lipids, etc. Autophagosomes then fuse with lysosomes to form autolysosomes where the autophagy cargos are degraded by lysosomal enzymes and released into the cytosol for various utilizations. Autophagy is a highly complex cellular process regulated by diverse cellular signaling pathways, among which the mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) reciprocally regulate autophagy activity in response to changes of cellular nutrient abundance, stress, growth factor stimuli, etc. (8). Lipophagy is a type of autophagy that transports intracellular triglycerides (TG) and cholesterol esters (CE) stored in the lipid droplets to the lysosomes to be hydrolyzed, and thus controls cellular lipid mobilization and energy homeostasis (9). Furthermore, the complex interactions among over-nutrition, defective autophagy and inflammation are thought to play important roles in the pathogenesis of fatty liver diseases (9–13) and atherosclerosis (14–16). In this concise review, we will mainly focus on recent findings on cholesterol and bile acid-mediated regulation of autophagy in hepatocytes and macrophages and the relevant implications in the pathogenesis and treatment of fatty liver disease and atherosclerosis.
1. REGULATION OF HEPATIC CHOLESTEROL AND BILE ACID HOMEOSTASIS
The liver plays a central role in regulating whole body cholesterol homeostasis. Hepatocytes maintain cellular cholesterol homeostasis by coordinately controlling several cholesterol input and elimination pathways (Figure 1). Hepatocytes acquire cholesterol primarily via de novo synthesis and receptor-mediated lipoprotein uptake from the circulation. These input pathways are mainly regulated by the sterol regulatory element-binding protein-2 (SREBP-2)-mediated cholesterol sensing mechanism (17). SREBP-2 is synthesized as a precursor protein retained in the endoplasmic reticulum (ER) membrane and associated with the sterol-sensing SREBP cleavage-activating protein (SCAP). When cellular cholesterol level is high, cholesterol binding to SCAP causes a conformational change that induces SCAP interaction with the ER membrane protein insulin-induced genes (Insigs), which retains the SREBP-2-SCAP complex in the ER. A decrease of cellular cholesterol promotes SREBP-2-SCAP complex translocation to the Golgi where proteolytic cleavage of SREBP-2 occurs. The released truncated and mature SREBP-2 enters the nucleus to induce a large set of cholesterol synthesis and transport genes including 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and low density lipoprotein receptor (LDLR). Activation of these genes raises intracellular cholesterol levels, which in turn inhibits SREBP-2 cleavage activation via a negative feedback loop (17).
Figure 1. Bile acid synthesis and enterohepatic circulation.
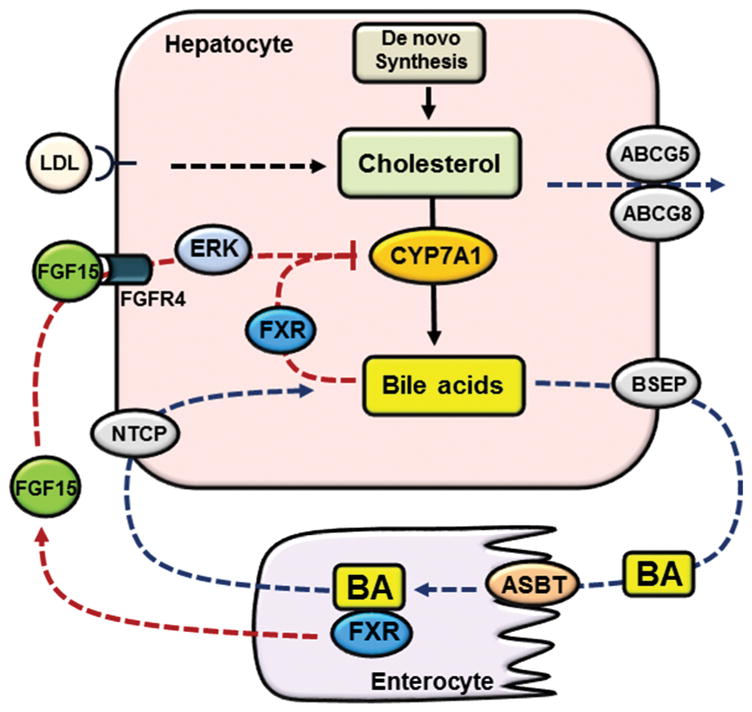
Hepatocytes acquire cholesterol via de novo synthesis and receptor-mediated endocytosis of cholesterol-rich lipoproteins. Hepatocytes eliminate cholesterol via bile acid synthesis and biliary secretion of cholesterol via ABCG5/ABCG8. Bile acids are synthesized from cholesterol in hepatocytes. CYP7A1 catalyzes the first and rate-limiting step in cholesterol conversion into bile acids. Bile acids are secreted into the bile via BSEP and subsequently released into the small intestine. The majority of bile acids is re-absorbed into the enterocytes via ASBT and transported back to the liver via portal circulation. Basolateral NTCP transports conjugated bile acids into the hepatocytes. Bile acids in the hepatocytes activate FXR to inhibit CYP7A1. Bile acids in the small intestine activate FXR to induce FGF15, which binds and activates FGFR4 to inhibit CYP7A1 partially via ERK signaling.
Hepatocytes eliminate the majority of cholesterol from the body by converting cholesterol into bile acids and secreting cholesterol into the bile (Figure 1) (18). Bile acid synthesis occurs exclusively in hepatocytes and is the only quantitatively significant cholesterol catabolic mechanism. The ER resident cytochrome p-450 enzyme cholesterol 7α-hydroxylase (CYP7A1) catalyzes the first and rate-limiting step in the conversion of cholesterol into bile acids (18). Bile acids are secreted into the bile via the bile salt export pump (BSEP) (19), while cholesterol is secreted into the bile by the ATP binding cassette transporter G5 (ABCG5) and ABCG8 functional heterodimer on the canalicular side of the hepatocytes (20). Biliary bile acid secretion generates bile flow and helps solubilize cholesterol in the bile by forming micelles. Once released into the small intestine after a meal, bile acids help emulsify dietary fat and facilitate intestine absorption of lipid and fat-soluble vitamins. It is estimated that over 95% of the bile acids is re-absorbed via the apical sodium-dependent bile acid transporter (ASBT) mainly in the terminal ileum and transported back to the liver for re-secretion into the bile. The transport of bile acids between the liver and the intestine is a process termed the enterohepatic circulation of bile acids. The significant intestinal bile acid conservation means that the liver needs to synthesize a small fraction of the total bile acid pool to compensate for the daily fecal loss of bile acids in order to maintain a constant bile acid pool over time.
Under normal physiology, bile acid homeostasis is mainly achieved through bile acid-mediated feedback inhibition of hepatic bile acid synthesis (Figure 1) (18). Bile acids are endogenous ligands for the nuclear receptor farnesoid x receptor (FXR) (21,22). FXR is highly expressed in the hepatocytes and enterocytes that are routinely exposed to high concentrations of bile acids. The bile acid-sensing FXR exerts a tight control of the hepatic bile acid synthesis rate by inhibiting the transcription of the CYP7A1 gene via several redundant mechanisms (23). In hepatocytes, FXR induces a repressor small heterodimer partner (SHP) to inhibit the CYP7A1 gene transcription (24). More recently, a posttranscriptional mechanism has been reported whereby FXR induces a RNA-binding protein ZFP36L1 to decrease the stability of CYP7A1 mRNA (25). The small intestine is a major reservoir of the bile acid pool. Intestinal FXR senses elevated bile acids to transcriptionally induce mouse fibroblast growth factor 15 (FGF15), which acts as an endocrine hormone to inhibit the hepatic CYP7A1 gene transcription (26). In the enterohepatic system, FGF15 is highly expressed in the terminal ileum but not expressed in mouse hepatocytes (26). Fibroblast growth factor 19 (FGF19) is the human ortholog of the mouse FGF15. FGF19 is expressed in both the hepatocytes and the enterocytes and its transcription is induced by bile acids and FXR in both types of cells (27). FGF15 and FGF19 bind the cell surface receptor FGF receptor 4 (FGFR4) in hepatocytes to inhibit the CYP7A1 gene via ERK1/2-dependent mechanisms. The downstream targets mediating this inhibition remain to be determined.
2. AUTOPHAGY AND HEPATIC CHOLESTEROL HOMEOSTASIS
Given the important roles of autophagy in maintaining cellular organelle function, lipid metabolism, and energy homeostasis, defective hepatic autophagy is considered to be directly involved in the pathogenesis of fatty liver disease (28). A better understanding of the underlying causes of impaired hepatic autophagic flux in steatotic livers may provide molecular basis needed to develop therapeutic approaches. Hepatic steatosis is an over-nutrition state with increased mTOR activity (29), which is expected to inhibit autophagy initiation. In addition, hepatic lipid accumulation has also been shown to impair autophagic flux (30). Recent studies showed that autophagy is highly sensitive to altered cellular cholesterol homeostasis, and hepatic cholesterol accumulation may significantly contribute to defective autophagy in fatty liver disease.
2.1 CHOLESTEROL-MEDIATED REGULATION OF HEPATIC AUTOPHAGY
Evidence linking intracellular cholesterol homeostasis with autophagy was initially reported by studies showing that experimental depletion of cholesterol by cyclodextrin in human fibroblasts induced autophagosome formation, which was associated with reduced mTOR phosphorylation in cholesterol-depleted cells (31). More recent studies further supported the existence of this link by showing that cholesterol lowering agents including statin, an HMGCR inhibitor, cholestyramine, a bile acid sequestrant, and ezetimibe, a Niemann-Pick-type C1-like1 (NPC1L1) inhibitor, decreased AKT and mTOR activity and increased autophagy in hepatocytes (32–34). The molecular links between these cholesterol lowering agents and cellular mTOR activity are likely complex. One possible mechanism underlying the reduced mTOR activation may be altered membrane cholesterol content and lipid raft composition (35). A more recent study shows that lysosomal FC derived from endocytosed LDL directly activates mTOR via a lysosomal membrane protein SLC38A9-mediated cholesterol sensing mechanism (36). The mTOR signaling is a major pathway that stimulates cellular anabolic metabolism in the presence of sufficient nutrients and growth stimuli. As cholesterol is an essential molecule required for cell survival and growth, cholesterol-depleted cells may decrease their anabolic metabolism by reducing mTOR activity. In addition, activation of autophagy may be a way to replenish cellular FC pool by promoting acidic CE hydrolysis in cells.
In mammalian cells, FC predominantly resides in the plasma membrane (37,38), and a plasma membrane cholesterol: phospholipid ratio of ~0.4 or less in isolated hepatocytes has been reported by various studies (39–42). In contrast, FC content in the intracellular organelles such as the ER and mitochondria is kept at a much lower level. Cells need to produce FC via hydrolysis for membrane synthesis, cholesterol efflux, and other cholesterol-demanding cellular pathways. On the other hand, cholesterol esterification prevents excessive FC accumulation in cholesterol-poor intracellular organelles. Hepatocytes efficiently convert excessive FC to CE in the ER by acyl-CoA: cholesterol acyltransferase (ACAT) and CE is subsequently stored in lipid droplets. It should be noted that ACAT1 and ACAT2, encoded by different genes, can catalyze cholesterol esterification in mammalian cells and these two enzymes show both species-dependent and cell type-dependent expression pattern (43). Liver expresses both ACAT1 and ACAT2, while macrophages mainly express ACAT1. Intracellular cholesterol constantly undergoes active hydrolysis and re-esterification cycles even without significant extracellular stimuli. When this cycle is overwhelmed by excessive cholesterol accumulation, FC content may increase in the ER and mitochondria, causing organelle dysfunction, oxidative stress, and hepatocellular injury.
A few recent studies conducted in several experimental models have provided new evidence that hepatic accumulation of cholesterol impairs autophagy. The role and regulation of autophagy in hepatic cholesterol metabolism is illustrated in Figure 2. Insights supporting that cholesterol-mediated lysosomal dysfunction can cause autophagy impairment come from studies of Niemann-Pick disease, type C (NPC), a fatal progressive neurological disorder associated with defective lysosomal cholesterol export due to mutations in NPC1 or NPC2 genes. Although autophagy was initially reported to be induced in NPC cells (44), new evidence suggested that NPC cells also had delayed clearance of autophagosomes, leading to accumulation of autophagy vacuoles (45,46). Acid sphingomyelinase (ASMase) converts sphingomyelins to ceramides in the lysosomes. Lack of ASMase increases lysosomal sphingomyelin and secondary increase of lysosomal cholesterol (47). ASMase deficiency in humans causes lysosomal storage disease Niemann-Pick disease type A/B (48). A more recent study showed that hepatocytes of ASMase knockout mice also showed decreased autophagosome/lysosome fusion (49). In fatty livers, FC accumulation impairs mitochondrial function and causes oxidative stress (3). We recently found that FC loading in cultured liver cells resulted in a marked elevation of microtubule-associated protein 1 light chain 3 (LC3) protein (34). Further results suggested that this was primarily due to accumulation of enlarged autolysosomes with impaired autolysosome cargo clearance, although autophagosome formation or autophagosome/lysosome fusion could also be altered in the cholesterol-laden cells. FC loading increased intracellular CE content by a few folds but caused much milder elevation of cellular FC by ~10–20% (34). However, treating cells with an ACAT inhibitor, which prevented FC loading-induced intracellular CE accumulation but further increased cellular FC content, significantly worsened autolysosome clearance, suggesting that excessive FC, but not CE, is more likely the underlying cause of autophagy impairment. Cholesterol loading in the presence of ACAT inhibitor significantly reduced autolysosome and lysosome hydrolytic activity and increased autolysosome/lysosome membrane permeability, suggesting that lysosomal dysfunction contributed to the impaired autolysosome cargo clearance. In contrast, cells exposed to high concentrations of human LDL had mildly elevated CE but did not show elevated intracellular FC or impaired autophagy flux (34). These cells also showed increased number of late endosomes/lysosomes and higher cathepsin activity. These results suggest that controlled lipoprotein uptake via the endocytic route does not significantly disrupt intracellular cholesterol homeostasis or autophagy function in hepatocytes. These recent studies in cholesterol-laden hepatocytes and lysosomal storage disease models suggest that increased lysosomal cholesterol accumulation impaired autolysosome clearance in the final step of autophagy flux, and meanwhile may result in compensatory induction of autophagy initiation. Over time this vicious cycle may cause detrimental consequences in cells. Plenty of studies have shown that targeting lysosomal cholesterol accumulation by cyclodextrin treatments was feasible and effective in restoring autophagy flux and attenuating autophagy vacuole accumulation in cells with defective lysosomal cholesterol export (50,51). Another genetic condition associated with lysosomal cholesterol accumulation is cholesterol ester storage disease (CESD) resulting from lysosomal acid lipase (LAL) deficiency (52). LAL hydrolyzes CE and TG in lysosomes and thus its deficiency leads to excessive CE accumulation in lysosomes. LAL deficiency causes hepatic steatosis and dyslipidemia in human patients. Recently, NASH was associated with reduced LAL activity in humans (53), which implied a potential link between lysosomal dysfunction and NASH pathogenesis. Currently, the causative relationship between hepatic steatosis, lysosomal dysfunction, and LAL deficiency in NASH is still not clear. How LAL deficiency affects autophagy in hepatocytes remains to be determined.
Figure 2. Role of autophagy in cholesterol metabolism in hepatocytes.
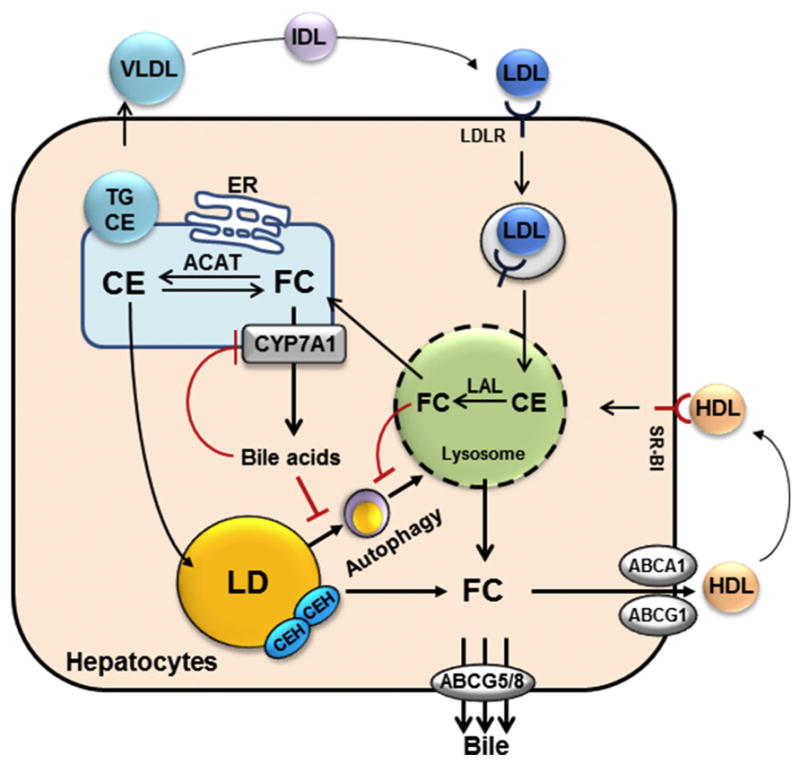
Lysosome-derived free cholesterol (FC) can be transported to the plasma membrane, effluxed via ABCA1 and ABCG1 to HDL, or converted to cholesterol ester (CE) or enter the bile acid synthesis pathway in the ER. CE can be incorporated into VLDL for secretion into the blood, or stored in the lipid droplets (LD). Neutral cholesterol ester hydrolases (CEH) hydrolyze CE in the lipid droplets and the resulting FC can re-enter the cellular FC pool. In addition, autophagy delivers CE from the lipid droplets to the lysosomes where lysosomal acid lipase (LAL) converts CE to FC. Excessive cholesterol accumulation disrupts intrahepatic cholesterol homeostasis, resulting in elevated FC content in the “cholesterol poor” intracellular organelles such as the mitochondria and the ER. In addition, FC accumulation in the lysosomes impairs lysosomal function, resulting in delayed autophagosome clearance. Increased bile acids also inhibit autophagy gene expression. Blocking intestine bile acid re-uptake such as by bile acid sequestrants can decrease hepatic bile acid signaling. As a result, induction of CYP7A1 causes increased ER cholesterol catabolism, relative ER cholesterol depletion and SREBP-2 activation, leading to induction of LDLR and HMGCR expression to replenish cellular cholesterol. Removal of bile acids and stimulation of cholesterol catabolism induce autophagic flux, which promotes acidic CE hydrolysis to help replenish cellular FC pool.
2.2 AUTOPHAGY FUNCTION IN HEPATOCELLULAR CHOLESTEROL METABOLISM
Current understanding of how autophagy regulates hepatic cholesterol metabolism is still limited. It was shown that total cholesterol content increased in the liver of autophagy defective Atg7−/− mice (9). A simple explanation would be decreased lipophagy-mediated acidic CE hydrolysis. However, other potential alterations in cellular cholesterol synthesis, catabolism, trafficking, and secretion in autophagy detective livers are still largely unknown. In addition, this line of inquiry may be further complicated by the profoundly altered hepatocellular function in mice lacking hepatic autophagy (54). Therefore, studies conducted in other experimental models may be needed to obtain new insights into the physiological role of hepatic autophagy in regulating cholesterol metabolism. Both neutral lipase-mediated hydrolysis of CE in lipid droplets and acidic hydrolysis of CE in the lysosomes are quantitatively significant in mediating intracellular CE hydrolysis. Hepatocytes are highly active in receptor-mediated uptake of circulating lipoproteins and a large portion of the late endosomal/lysosomal CE is originated from circulating lipoproteins. Therefore, to what extent hepatic autophagy contributes to the overall CE pool in the acidic compartments remains to be determined. It may depend on the metabolic status of the hepatocytes. In addition, whether autophagy-mediated CE mobilization affects downstream sterol secretion is not known. In comparison, previous studies have shown that the hepatic neutral CE hydrolyzing enzymes enhance bile acid synthesis and biliary sterol secretion (55). Inhibition of neutral CE hydrolases was also associated with increased hepatic apolipoprotein B100 (ApoB100) lipoprotein secretion and hypercholesterolemia, suggesting that the liver adapts to decreased intracellular TG and CE hydrolysis by secreting excessive lipids into the circulation (56,57). Hepatic VLDL secretion rate is controlled by both hepatic fat content and ApoB100 availability (58). In the process of VLDL assembly, an ApoB100 molecule is first lipidated in the ER lumen by the microsomal triglyceride transfer protein (MTTP), and bulk TG was later added. The co-translational and post-translational ApoB100 degradation is a significant regulatory mechanism controlling hepatic ApoB100 secretion. Hepatocytes can degrade ApoB100 via the ubiquitin-proteasome system and lysosomes. Current evidence suggests that both the basal ApoB100 turnover and n-3 polyunsaturated fatty acids (n-3 PUFA) and ER stress inducer-stimulated ApoB100 degradation involve autophagy-mediated lysosomal targeting of ApoB100 (59–61). Insulin signaling also promotes ApoB100 degradation, and impaired insulin signaling is thought to be a cause of hepatic VLDL overproduction in type-2 diabetes and fatty liver disease (62,63). Recently, the Golgi-to-late endosomal/lysosomal trafficking receptor Sortilin 1 has been shown to mediate ApoB100 secretion and lysosomal ApoB100 transport (64). Hepatic Sortilin 1 was positively regulated by insulin (65). As a result, hepatic Sortilin 1 protein was markedly reduced in fatty livers in diet-induced obese mice, which was restored by dietary n-3 PUFA supplementation (66,67). Interestingly, it has been shown recently that Sortilin 1-mediated ApoB100 lysosomal targeting also converges on the autophagy pathway (68). Autophagy induction by n-3 PUFA and ER stress inducers has been reported (69,70), which is consistent with the effects of these compounds in promoting ApoB100 degradation. On the other hand, insulin-activated growth factor signaling inhibits autophagy (8), suggesting that insulin-stimulated ApoB100 degradation may not be a direct result of un-regulated autophagic flux in hepatocytes. Given that autophagy mediates both intracellular lipid mobilization and ApoB100 degradation, defective hepatic autophagy is expected to play a causative role in promoting hepatic ApoB100 secretion and hypercholesterolemia in fatty liver diseases. However, this hypothesis remains to be further substantiated experimentally.
2.3 BILE ACID REGULATION OF HEPATIC AUTOPHAGY
Recent studies indicate that bile acids may regulate hepatic autophagy by two different mechanisms. Firstly, bile acids may activate cellular signaling pathways to regulate autophagy activity (71–73); secondly, changes in bile acid synthesis may modulate hepatic autophagy by altering intracellular cholesterol metabolism (34). A meal intake stimulates gallbladder release of bile acids into the small intestine and subsequently increases trans-flux of bile acids across the enterocytes. As a result, both bile acid and FGF15/19 increases in the portal circulation (74,75). Studies have shown that bile acids and FGF15/19 can act as nutrient-sensing hormones to regulate postprandial hepatic transition from catabolic to anabolic state, such as promoting protein synthesis and inhibiting gluconeogenesis (76–78). Autophagy is considered a catabolic process. In this regard, several recent studies have shown that bile acids inhibit hepatic autophagic activity. Whole genome chromatin binding assays identified FXR binding to many autophagy genes, and pharmacological activation of FXR resulted in transcriptional repression of autophagy genes and decreased hepatic autophagy (71,72). These studies suggest that in concert with postprandial activation of nutrient and growth factor signal transduction pathways, bile acids transcriptionally inhibit hepatic autophagy gene expression via an FXR-mediated mechanism. It was shown that hepatic LC3 protein increased in mice lacking FXR (73). However, it appeared that this was not a result of autophagy induction but impaired autophagosome-lysosome fusion due to increased circulating bile acid concentration, because treating cultured hepatocytes with high concentrations of bile acids also reduced autophagosome-lysosome fusion. Hepatic autophasosome clearance may also be impaired in bile duct ligation mouse models of obstructive cholestasis (79). In contrast, it has been reported that ursodeoxycholic acid, a hydrophilic bile acid used to treat cholestasis, may promote autophagy in the liver (80). How high concentration of hydrophobic bile acids disrupt autophagic flux is not clear. Bile acids at high concentrations may alter membrane structure to inhibit autophagic flux, but this possible mechanism has not been demonstrated.
Recently, studies showed that pharmacological inhibition of intestine bile acid re-uptake such as by bile acid sequestrants or ASBT inhibitors may represent a potential therapeutic approach to treat fatty liver disease and type-2 diabetes (81,82). Blocking intestine bile acid reabsorption by bile acid sequestrants induces hepatic CYP7A1 expression and bile acid synthesis, and has been used for treating hypercholesterolemia in humans (83). Emerging new evidence showed that this therapeutic strategy led to additional beneficial effects notably improved insulin sensitivity and glycaemia and reduced hepatic steatosis and inflammation in experimental animal models and/or human diabetic patients (81,82,84). In a recent study, we reported that treating mice with cholestyramine induced hepatic autophagy (34). Although this effect may be partially explained by reduced hepatic bile acids and FXR activity, we unexpectedly found that hepatic mTOR and AKT signaling was markedly reduced in cholestyramine-treated mice. CYP7A1 induction has a major impact on ER cholesterol catabolism as evidenced by markedly increased SREBP-2 transcriptional network in Cyp7a1 transgenic mice (85). Cholesterol in the membrane rafts is known to be essential in PI3K/AKT activation (35). Since a significant amount of plasma membrane cholesterol traffics to the ER (37), increased ER cholesterol catabolism may possibly cause dynamic changes in plasma membrane cholesterol and lipid composition. As mentioned earlier, experimental approaches that reduced cellular cholesterol have been shown to decrease mTOR activation in mammalian cells (31,32,86). These results support that blocking intestine bile acid re-uptake probably induced hepatic autophagy via the combined effects of reduced bile acid signaling and altered cellular cholesterol content. Future studies employing ASBT inhibitor treatment or genetic Asbt or Ost genetic knockout models could further investigate this molecular link.
3. AUTOPHAGY AND MACROPHAGY CHOLESTEROL METABOLISM IN ATHEROSCLEROSIS
Atherosclerosis is a major underlying cause of cardiovascular diseases worldwide (87). Hyperlipidemia, in the presence of oxidative stress and endothelial dysfunction, is one of the major risk factors for atherosclerosis. In the early stage of atherosclerotic lesion formation, deposition of LDL and the subsequent formation of oxidized LDL (oxLDL) in the sub-endothelial space promote circulating monocyte recruitment and maturation into macrophages. Once in the intima, macrophages take up oxLDL primarily via the Scavenger Receptors (87). Although it is seemingly protective against lipid deposition in the arterial wall, uncontrolled uptake of modified lipoproteins and poorly processed oxLDL in the lysosomes disrupts intracellular lipid homeostasis in macrophages, which eventually leads to the formation of foam cells with accumulation of CE abundant lipid droplets. Activated macrophages contribute to the progression of atherosclerotic lesion by secreting chemotactic and pro-inflammatory mediators. In addition, lipid-laden macrophages undergo apoptosis and release intracellular contents, which further propagates lipid deposition, inflammation, plaque instability and thrombosis (87).
Unlike hepatocytes, macrophages do not possess a catabolic machinery to eliminate cholesterol, and FC efflux to extracellular acceptors apolipoprotein A-I (ApoA-I) or high density lipoprotein (HDL) is the major mechanism by which macrophages ameliorate intracellular cholesterol accumulation (88). For this reason, stimulating macrophage reverse cholesterol transport (RCT), a process of transporting cholesterol from peripheral macrophages by circulating HDL to the liver for fecal excretion, has been considered a therapeutic strategy to prevent foam cell formation in atherosclerotic plaques (88). Cholesterol efflux in macrophages is primarily mediated by the cell surface transporters ATP binding cassette transporter A1 (ABCA1) and ATP-binding cassette transport G1 (ABCG1) (89,90). These cholesterol transporters are transcriptionally induced by the oxysterol-activated nuclear receptor liver X receptor (LXR) (91,92), which provides a feedforward mechanism for macrophages to eliminate excessive intracellular cholesterol. Intracellular CE hydrolysis is an important upstream process regulating cellular cholesterol efflux. In macrophages, hydrolysis of lipid droplet-associated CE by neutral lipases is a significant mechanism in regulating intracellular lipid accumulation and foam cell formation. Neutral lipases have been implicated in mediating the neutral CE hydrolysis by in vitro and in vivo studies (93). However, recent studies showed that blocking neutral hydrolases decreased CE hydrolysis by ~60%, while simultaneous blockage of the acidic hydrolysis by the lysosomotropic agent chloroquine together with neutral hydrolysis inhibition caused a close to complete inhibition of CE hydrolysis in cholesterol-laden bone marrow derived macrophages, suggesting that lysosomes account for a significant portion of the CE hydrolysis in macrophages (16). In addition, inhibition of lysosomal function by chloroquine also significantly reduced cholesterol efflux to ApoA-I in lipid-laden macrophages, suggesting that acidic CE hydrolysis is linked to subsequent cholesterol efflux (16). Cholesterol efflux was significantly reduced in lipid-laden autophagy defective Atg5−/− macrophages, and macrophage-to-feces RCT was also decreased in Atg5−/− macrophages in mice (16). These findings suggest that defective autophagy reduces cholesterol efflux via reduced acidic CE hydrolysis. The role of autophagy in macrophage cholesterol metabolism and transport is illustrated in Figure 3. Inhibition of ACAT1 in macrophages is expected to reduce CE formation and intracellular accumulation and promote FC efflux from macrophages. In one study, deletion of ACAT1 in myeloid cells attenuated lesion formation in Apoe knockout mice, suggesting that macrophage ACAT1 inhibition may be anti-atherogenic (94). However, transfer of macrophages from ACAT1 null mice to Ldlr knockout mice promoted atherosclerotic lesion formation, suggesting that deletion of ACAT1 in macrophages may be pro-atherogenic (95). There are apparent differences in the model systems used in the two studies, which is beyond the scope of this review and will not be further discussed. What should be noted here is that the effects of macrophage ACAT1 deletion (either complete or partial deletion) on lesion formation may not be simply explained by the altered intracellular cholesterol metabolism, while other functional changes in macrophages should be considered. Knockout of ACAT1 in microglia has been shown to increase cellular autophagic activity without causing cellular CE and FC elevation (96). The significance of this molecular link in macrophages may be further investigated in the setting of experimental atherosclerosis.
Figure 3. Regulation of macrophage cholesterol metabolism.

Macrophages acquire cholesterol via LDLR-mediated uptake of LDL and scavenger receptor (CD36)-mediated uptake of oxLDL. Endocytosed cholesterol-rich particles are delivered to the lysosomes where lysosomal acid lipase (LAL) converts CE to FC. FC can be delivered to the plasma membrane or to the ER where FC is converted to CE by ACAT. Excessive CE is stored in the lipid droplets (LD). Neutral cholesterol ester hydrolases (CEH) can convert lipid droplet CE back to FC for either efflux via the ABCA1 and ABCG1 to HDL or for re-esterification by ACAT in the ER. Autophagy delivers lipids from lipid droplets to the lysosomes and thus contributes to acidic CE hydrolysis. FC generated in the lysosomes can also be effluxed to HDL via ABCA1 and ABCG1. Lipid overloading can induce lysosomal stress and dysfunction. This may impair autophagy by causing autophagosome/lysosome fusion defects or delayed autophagosome cargo clearance. Lysosomal permeabilization and content release can also activate inflammasomes and cytokine secretion. As cellular compensatory mechanisms, intracellular cholesterol accumulation and subsequently elevated oxysterols activate LXR to transcriptionally induce ABCA1 and ABCG1. Furthermore, lysosomal stress can cause TFEB to translocate to the nucleus where TFEB induces genes to promote lysosomal biogenesis and autophagy.
Modified atherogenic lipoproteins are not ideal substrates for lysosomal acid lipase and are poorly processed once engulfed by macrophages. Uptake of these particles is associated with lysosomal impairment and lysosomal membrane permeability. Phagocytosed cholesterol crystal in the atherosclerotic lesion can also result in phagolysosomal damage in macrophages (97). Permeable or ruptured lysosomes can release cholesterol crystals and oxLDL to activate NLRP3 inflammasomes, which leads to increased cytokine secretion. In autophagy defective Atg5−/− macrophages, cholesterol crystal appeared to be increased in the atherosclerotic plaque, which was associated with the activation of inflammasomes and pro-inflammatory phenotype in macrophages (14). Extracellular oxLDL or cholesterol crystal uptake and delivery to the lysosomal compartment presumably do not require autophagy process, and how defective autophagy eventually exacerbates intracellular cholesterol crystal deposition is likely complex and remains to be determined. It should also be kept in mind that lack of functional autophagy can cause significant mitochondrial dysfunction, oxidative stress, and hypersensitivity to apoptosis (54). In the setting of lipid overloading and a pro-inflammatory environment, complete inactivation of autophagy may sensitize the macrophage to extracellular insults and apoptosis via mechanisms in addition to reduced autophagy-mediated cholesterol hydrolysis and efflux.
In vitro studies have shown that exposing macrophages to oxLDL or oxysterol 7-ketocholesterol induces LC3 puncta accumulation in macrophages (16), suggesting that induction of autophagy flux may be an adaptive response to intracellular lipid accumulation. However, macrophages in atherosclerotic plaques appeared to have dysfunctional lysosomes and impaired autophagy (14,98). Currently, the mechanisms by which lipid loading affects autophagy in macrophages remain elusive. Recent studies suggest that the transcriptional factor EB (TFEB) may be implicated in linking intracellular lipid accumulation to autophagy activation in macrophages. TFEB belongs to the basic helix-loop-helix leucine zipper family of transcriptional factors that recognize E-box sequences in the target genes (99). TFEB was recently identified as a nutrient and stress-sensing master regulator of lysosomal biogenesis and autophagy in various cell types and organ systems (100,101), which has led to a paradigm shift in our understanding of how lysosomal pathways can be dynamically regulated in response to various nutrient and stress signals to maintain cellular homeostasis. Under fed or over-nutrition conditions, nutrient signaling phosphorylates TFEB to cause its cytoplasmic retention. Under starvation or lysosomal stress, TFEB is de-phosphorylated and enters the nucleus to induce a network of genes involved in lysosomal biogenesis and autophagy. In cultured macrophages, both oxLDL and cholesterol crystal have been shown to cause lysosomal stress and TFEB nuclear translocation, resulting in TFEB-dependent transcriptional activation of lysosomal and autophagy genes (98). Direct overexpression of TFEB in cultured macrophages enhanced cholesterol efflux and decreased inflammasome activation (98). Consistently, TFEB activation has also been shown to reduce atherosclerosis in experimental models in vivo (102,103). Therefore, it is possible that lipid-laden macrophages are under a state of compensatory autophagy activation and lysosomal biogenesis secondary to lysosomal dysfunction. A typical autophagic flux assay using LC3 as a marker could suggest overall increased autophagic flux, while significantly impaired autophagosome/lysosome fusion and autolysosome clearance may also exist in lipid-laden macrophages.
4. CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Recent studies have reported important links between cholesterol and bile acid metabolism and cellular autophagy activity. Disrupted intracellular cholesterol homeostasis in hepatocytes and macrophages contributes to autophagy defect, which is implicated in fatty liver disease and atherosclerosis. Currently, the underlying mechanisms linking these cellular pathways are only partially understood. Hepatic cholesterol metabolism, transport, and organelle trafficking are highly dynamic and interconnected, and their relationship with autophagy remains to be further elucidated under different physiological and pathophysiological conditions. Bile acids can activate various intracellular signal transduction pathways including ERK1/2, AKT, and mTOR via inducing FGF15/19 and/or activating the sphingosine-1-phosphate receptor 2 (S1PR2) (26,104,105). Although these signaling pathways are known to regulate autophagy in hepatocytes, whether they mediate bile acid control of hepatic autophagy is not clear. Lastly, bile acid-based therapies have great promise in treating metabolic and inflammatory liver diseases (18), and future studies may determine if targeting the enterohepatic bile acid signaling to induce hepatic autophagy has therapeutic implications in other forms of genetic and acquired liver diseases.
Highlight.
Disrupted cholesterol homeostasis is critically implicated in the pathogenesis of fatty liver disease and atherosclerosis
Autophagy regulates organelle homeostasis and cellular integrity
New studies show that cholesterol and bile acids modulate hepatic autophagy
Autophagy regulates macrophage cholesterol homeostasis in atherosclerosis
Acknowledgments
This work was supported in part by the National Institutes of Health grants 1R01DK102487-01 (TL), U01 AA024733, R01 AA020518 and R01 DK102142 (WXD), and P20GM103549 & P30GM118247.
ABBREVIATIONS
- NAFLD
Nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- FC
free cholesterol
- VLDL
very low density lipoprotein
- LDL
low density lipoprotein
- mTOR
mechanistic target of rapamycin signaling
- AMPK
AMP-activated protein kinase
- TG
triglycerides
- CE
cholesterol ester
- SREBP-2
sterol regulatory element-binding protein-2
- ER
endoplasmic reticulum
- SCAP
sterol-sensing SREBP cleavage-activating protein
- Insig
insulin-induced genes
- HMGCR
3-hydroxy-3-methylglutaryl coenzyme A reductase
- LDLR
low density lipoprotein receptor
- CYP7A1
cholesterol 7α-hydroxylase
- BSEP
bile salt export pump
- ABCG5
ATP binding cassette transporter G5
- ASBT
apical sodium-dependent bile acid transporter
- FXR
farnesoid x receptor
- SHP
small heterodimer partner
- FGF15
fibroblast growth factor 15
- FGF19
Fibroblast growth factor 19
- FGFR4
FGF receptor 4
- NPC1L1
Niemann-Pick-type C1-like1
- ACAT
acyl-coA cholesterol acyltransferase
- NPC
Niemann-Pick disease, type C,
- ASMase
Acid sphingomyelinase
- LC3
microtubule-associated protein 1 light chain 3
- CESD
cholesterol ester storage disease
- ApoB100
apolipoprotein B100
- LAL
lysosomal acid lipase
- n-3 PUFA
n-3 polyunsaturated fatty acids
- oxLDL
oxidized LDL
- ApoA-I
apolipoprotein A-I
- HDL
high density lipoprotein
- RCT
reverse cholesterol transport
- ABCA1
ATP binding cassette transporter A1
- ABCG1
ATP-binding cassette transport G1
- LXR
liver X receptor
- TFEB
transcriptional factor EB
- S1PR2
sphingosine-1-phosphate receptor 2
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Byrne CD, Targher G. NAFLD: A multisystem disease. J Hepatol. 2015;62:S47–S64. doi: 10.1016/j.jhep.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs M, Sanyal AJ. Lipotoxicity in NASH. J Hepatol. 2012;56:291–293. doi: 10.1016/j.jhep.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 3.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, van Bilsen M, Shiri-Sverdlov R, Hofker MH. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–486. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]
- 5.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 6.Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, Naveau S, Prevot S, Makhzami S, Perlemuter G, Cassard-Doulcier AM. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57:141–149. doi: 10.1016/j.jhep.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 7.Lonardo A, Sookoian S, Chonchol M, Loria P, Targher G. Cardiovascular and systemic risk in nonalcoholic fatty liver disease - atherosclerosis as a major player in the natural course of NAFLD. Curr Pharm Des. 2013;19:5177–5192. [PubMed] [Google Scholar]
- 8.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell research. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284:31484–31492. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 18.Li T, Chiang JY. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol Rev. 2014;66:948–983. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, Hofmann AF, Meier PJ. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273:10046–10050. doi: 10.1074/jbc.273.16.10046. [DOI] [PubMed] [Google Scholar]
- 20.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 21.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 23.Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–1043. doi: 10.1002/hep.25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 25.Tarling EJ, Clifford BL, Cheng J, Morand P, Cheng A, Lester E, Sallam T, Turner M, de Aguiar Vallim TQ. RNA-binding protein ZFP36L1 maintains posttranscriptional regulation of bile acid metabolism. J Clin Invest. 2017;127:3741–3754. doi: 10.1172/JCI94029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49:297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czaja MJ, Ding WX, Donohue TM, Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, Yin XM. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Current biology: CB. 2009;19:R1046–1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng J, Ohsaki Y, Tauchi-Sato K, Fujita A, Fujimoto T. Cholesterol depletion induces autophagy. Biochem Biophys Res Commun. 2006;351:246–252. doi: 10.1016/j.bbrc.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 32.Yamamura T, Ohsaki Y, Suzuki M, Shinohara Y, Tatematsu T, Cheng J, Okada M, Ohmiya N, Hirooka Y, Goto H, Fujimoto T. Inhibition of Niemann-Pick-type C1-like1 by ezetimibe activates autophagy in human hepatocytes and reduces mutant alpha1-antitrypsin Z deposition. Hepatology. 2014;59:1591–1599. doi: 10.1002/hep.26930. [DOI] [PubMed] [Google Scholar]
- 33.Wang HJ, Park JY, Kwon O, Choe EY, Kim CH, Hur KY, Lee MS, Yun M, Cha BS, Kim YB, Lee H, Kang ES. Chronic HMGCR/HMG-CoA reductase inhibitor treatment contributes to dysglycemia by upregulating hepatic gluconeogenesis through autophagy induction. Autophagy. 2015;11:2089–2101. doi: 10.1080/15548627.2015.1091139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Ding Y, Li J, Chavan H, Matye D, Ni HM, Chiang JY, Krishnamurthy P, Ding WX, Li T. Targeting the Enterohepatic Bile Acid Signaling Induces Hepatic Autophagy via a CYP7A1-AKT-mTOR Axis in Mice. Cell Mol Gastroenterol Hepatol. 2017;3:245–260. doi: 10.1016/j.jcmgh.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lasserre R, Guo XJ, Conchonaud F, Hamon Y, Hawchar O, Bernard AM, Soudja SM, Lenne PF, Rigneault H, Olive D, Bismuth G, Nunes JA, Payrastre B, Marguet D, He HT. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nature chemical biology. 2008;4:538–547. doi: 10.1038/nchembio.103. [DOI] [PubMed] [Google Scholar]
- 36.Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, Jiang X, van Eijkeren RJ, Davis OB, Louie SM, Perera RM, Covey DF, Nomura DK, Ory DS, Zoncu R. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. doi: 10.1126/science.aag1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soccio RE, Breslow JL. Intracellular cholesterol transport. Arterioscler Thromb Vasc Biol. 2004;24:1150–1160. doi: 10.1161/01.ATV.0000131264.66417.d5. [DOI] [PubMed] [Google Scholar]
- 38.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kremmer T, Wisher MH, Evans WH. The lipid composition of plasma membrane subfractions originating from the three major functional domains of the rat hepatocyte cell surface. Biochim Biophys Acta. 1976;455:655–664. doi: 10.1016/0005-2736(76)90039-0. [DOI] [PubMed] [Google Scholar]
- 40.Meier PJ, Sztul ES, Reuben A, Boyer JL. Structural and functional polarity of canalicular and basolateral plasma membrane vesicles isolated in high yield from rat liver. The Journal of cell biology. 1984;98:991–1000. doi: 10.1083/jcb.98.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yano M, Marinelli RA, Roberts SK, Balan V, Pham L, Tarara JE, de Groen PC, LaRusso NF. Rat hepatocytes transport water mainly via a non-channel-mediated pathway. J Biol Chem. 1996;271:6702–6707. doi: 10.1074/jbc.271.12.6702. [DOI] [PubMed] [Google Scholar]
- 42.Burger HM, Abel S, Snijman PW, Swanevelder S, Gelderblom WC. Altered lipid parameters in hepatic subcellular membrane fractions induced by fumonisin B1. Lipids. 2007;42:249–261. doi: 10.1007/s11745-007-3025-9. [DOI] [PubMed] [Google Scholar]
- 43.Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab. 2009;297:E1–9. doi: 10.1152/ajpendo.90926.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16:1495–1503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- 45.Elrick MJ, Yu T, Chung C, Lieberman AP. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet. 2012;21:4876–4887. doi: 10.1093/hmg/dds324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao G, Yao Y, Liu J, Yu Z, Cheung S, Xie A, Liang X, Bi X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 −/− mouse brain. Am J Pathol. 2007;171:962–975. doi: 10.2353/ajpath.2007.070052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ridgway ND. Interactions between metabolism and intracellular distribution of cholesterol and sphingomyelin. Biochim Biophys Acta. 2000;1484:129–141. doi: 10.1016/s1388-1981(00)00006-8. [DOI] [PubMed] [Google Scholar]
- 48.Schuchman EH, Desnick RJ. Types A and B Niemann-Pick disease. Molecular genetics and metabolism. 2017;120:27–33. doi: 10.1016/j.ymgme.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fucho R, Martinez L, Baulies A, Torres S, Tarrats N, Fernandez A, Ribas V, Astudillo AM, Balsinde J, Garcia-Roves P, Elena M, Bergheim I, Lotersztajn S, Trautwein C, Appelqvist H, Paton AW, Paton JC, Czaja MJ, Kaplowitz N, Fernandez-Checa JC, Garcia-Ruiz C. ASMase regulates autophagy and lysosomal membrane permeabilization and its inhibition prevents early stage non-alcoholic steatohepatitis. J Hepatol. 2014;61:1126–1134. doi: 10.1016/j.jhep.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, Cohen MA, Chakraborty S, Wang H, Spooner E, Ploegh H, Gsponer J, Korolchuk VI, Jaenisch R. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell reports. 2013;5:1302–1315. doi: 10.1016/j.celrep.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meske V, Priesnitz T, Albert F, Ohm TG. How to reduce the accumulation of autophagic vacuoles in NPC1-deficient neurons: a comparison of two pharmacological strategies. Neuropharmacology. 2015;89:282–289. doi: 10.1016/j.neuropharm.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 52.Reiner Z, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, Jones S, Coric M, Calandra S, Hamilton J, Eagleton T, Ros E. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235:21–30. doi: 10.1016/j.atherosclerosis.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 53.Baratta F, Pastori D, Del Ben M, Polimeni L, Labbadia G, Di Santo S, Piemonte F, Tozzi G, Violi F, Angelico F. Reduced Lysosomal Acid Lipase Activity in Adult Patients With Non-alcoholic Fatty Liver Disease. EBioMedicine. 2015;2:750–754. doi: 10.1016/j.ebiom.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 55.Zhao B, Song J, Ghosh S. Hepatic overexpression of cholesteryl ester hydrolase enhances cholesterol elimination and in vivo reverse cholesterol transport. J Lipid Res. 2008;49:2212–2217. doi: 10.1194/jlr.M800277-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Quiroga AD, Li L, Trotzmuller M, Nelson R, Proctor SD, Kofeler H, Lehner R. Deficiency of carboxylesterase 1/esterase-x results in obesity, hepatic steatosis, and hyperlipidemia. Hepatology. 2012;56:2188–2198. doi: 10.1002/hep.25961. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Wang Y, Matye DJ, Chavan H, Krishnamurthy P, Li F, Li T. Sortilin 1 Modulates Hepatic Cholesterol Lipotoxicity in Mice via Functional Interaction with Liver Carboxylesterase 1. J Biol Chem. 2017;292:146–160. doi: 10.1074/jbc.M116.762005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nielsen S, Karpe F. Determinants of VLDL-triglycerides production. Curr Opin Lipidol. 2012;23:321–326. doi: 10.1097/MOL.0b013e3283544956. [DOI] [PubMed] [Google Scholar]
- 59.Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell. 2006;17:2674–2683. doi: 10.1091/mbc.E05-07-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan M, Maitin V, Parathath S, Andreo U, Lin SX, St Germain C, Yao Z, Maxfield FR, Williams KJ, Fisher EA. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc Natl Acad Sci U S A. 2008;105:5862–5867. doi: 10.1073/pnas.0707460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiu W, Zhang J, Dekker MJ, Wang H, Huang J, Brumell JH, Adeli K. Hepatic autophagy mediates endoplasmic reticulum stress-induced degradation of misfolded apolipoprotein B. Hepatology. 2011;53:1515–1525. doi: 10.1002/hep.24269. [DOI] [PubMed] [Google Scholar]
- 62.Sparks JD, Sparks CE. Insulin modulation of hepatic synthesis and secretion of apolipoprotein B by rat hepatocytes. J Biol Chem. 1990;265:8854–8862. [PubMed] [Google Scholar]
- 63.Wiegman CH, Bandsma RH, Ouwens M, van der Sluijs FH, Havinga R, Boer T, Reijngoud DJ, Romijn JA, Kuipers F. Hepatic VLDL production in ob/ob mice is not stimulated by massive de novo lipogenesis but is less sensitive to the suppressive effects of insulin. Diabetes. 2003;52:1081–1089. doi: 10.2337/diabetes.52.5.1081. [DOI] [PubMed] [Google Scholar]
- 64.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, Rader DJ. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li J, Matye DJ, Li T. Insulin resistance induces posttranslational hepatic sortilin 1 degradation in mice. J Biol Chem. 2015;290:11526–11536. doi: 10.1074/jbc.M115.641225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bi L, Chiang JY, Ding WX, Dunn W, Roberts B, Li T. Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. J Lipid Res. 2013;54:2754–2762. doi: 10.1194/jlr.M039347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li J, Bi L, Hulke M, Li T. Fish oil and fenofibrate prevented phosphorylation-dependent hepatic sortilin 1 degradation in Western diet-fed mice. J Biol Chem. 2014;289:22437–22449. doi: 10.1074/jbc.M114.548933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amengual J, Guo L, Strong A, Madrigal-Matute J, Wang H, Kaushik S, Brodsky JL, Rader DJ, Cuervo AM, Fisher EA. Autophagy Is Required for Sortilin-Mediated Degradation of Apolipoprotein B100. Circ Res. 2018 doi: 10.1161/CIRCRESAHA.117.311240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Y, Xu C, Yan T, Yu C, Li Y. omega-3 Fatty acids reverse lipotoxity through induction of autophagy in nonalcoholic fatty liver disease. Nutrition. 2015;31:1423–1429. e1422. doi: 10.1016/j.nut.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 70.Zhang J, Morris MW, Jr, Dorsett-Martin WA, Drake LC, Anderson CD. Autophagy is involved in endoplasmic reticulum stress-induced cell death of rat hepatocytes. J Surg Res. 2013;183:929–935. doi: 10.1016/j.jss.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, Ma J, Kemper B, Kemper JK. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108–111. doi: 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Manley S, Ni HM, Kong B, Apte U, Guo G, Ding WX. Suppression of autophagic flux by bile acids in hepatocytes. Toxicological sciences: an official journal of the Society of Toxicology. 2014;137:478–490. doi: 10.1093/toxsci/kft246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galman C, Angelin B, Rudling M. Bile acid synthesis in humans has a rapid diurnal variation that is asynchronous with cholesterol synthesis. Gastroenterology. 2005;129:1445–1453. doi: 10.1053/j.gastro.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 75.Lundasen T, Galman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530–536. doi: 10.1111/j.1365-2796.2006.01731.x. [DOI] [PubMed] [Google Scholar]
- 76.Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, Fukamizu A. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem. 2004;279:23158–23165. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 77.Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, Xu HE, Shulman GI, Kliewer SA, Mangelsdorf DJ. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331:1621–1624. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, Suino-Powell K, Xu HE, Gerard RD, Finck BN, Burgess SC, Mangelsdorf DJ, Kliewer SA. FGF15/19 Regulates Hepatic Glucose Metabolism by Inhibiting the CREB-PGC-1alpha Pathway. Cell Metab. 2011;13:729–738. doi: 10.1016/j.cmet.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim S, Han SY, Yu KS, Han D, Ahn HJ, Jo JE, Kim JH, Shin J, Park HW. Impaired autophagy promotes bile acid-induced hepatic injury and accumulation of ubiquitinated proteins. Biochem Biophys Res Commun. 2018;495:1541–1547. doi: 10.1016/j.bbrc.2017.11.202. [DOI] [PubMed] [Google Scholar]
- 80.Tang Y, Fickert P, Trauner M, Marcus N, Blomenkamp K, Teckman J. Autophagy induced by exogenous bile acids is therapeutic in a model of alpha-1-AT deficiency liver disease. Am J Physiol Gastrointest Liver Physiol. 2016;311:G156–165. doi: 10.1152/ajpgi.00143.2015. [DOI] [PubMed] [Google Scholar]
- 81.Chen L, Yao X, Young A, McNulty J, Anderson D, Liu Y, Nystrom C, Croom D, Ross S, Collins J, Rajpal D, Hamlet K, Smith C, Gedulin B. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab. 2012;302:E68–76. doi: 10.1152/ajpendo.00323.2011. [DOI] [PubMed] [Google Scholar]
- 82.Rao A, Kosters A, Mells JE, Zhang W, Setchell KD, Amanso AM, Wynn GM, Xu T, Keller BT, Yin H, Banton S, Jones DP, Wu H, Dawson PA, Karpen SJ. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Science translational medicine. 2016;8:357ra122. doi: 10.1126/scitranslmed.aaf4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med. 1994;121:416–422. doi: 10.7326/0003-4819-121-6-199409150-00004. [DOI] [PubMed] [Google Scholar]
- 84.Wu Y, Aquino CJ, Cowan DJ, Anderson DL, Ambroso JL, Bishop MJ, Boros EE, Chen L, Cunningham A, Dobbins RL, Feldman PL, Harston LT, Kaldor IW, Klein R, Liang X, McIntyre MS, Merrill CL, Patterson KM, Prescott JS, Ray JS, Roller SG, Yao X, Young A, Yuen J, Collins JL. Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J Med Chem. 2013;56:5094–5114. doi: 10.1021/jm400459m. [DOI] [PubMed] [Google Scholar]
- 85.Li T, Francl JM, Boehme S, Chiang JY. Regulation of cholesterol and bile acid homeostasis by the cholesterol 7alpha-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology. 2013;58:1111–1121. doi: 10.1002/hep.26427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu J, Dang Y, Ren YR, Liu JO. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc Natl Acad Sci U S A. 2010;107:4764–4769. doi: 10.1073/pnas.0910872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khera AV, Rader DJ. Future therapeutic directions in reverse cholesterol transport. Curr Atheroscler Rep. 2010;12:73–81. doi: 10.1007/s11883-009-0080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klucken J, Buchler C, Orso E, Kaminski WE, Porsch-Ozcurumez M, Liebisch G, Kapinsky M, Diederich W, Drobnik W, Dean M, Allikmets R, Schmitz G. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci U S A. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J, Jr, Hayden MR. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 91.Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 92.Kennedy MA, Venkateswaran A, Tarr PT, Xenarios I, Kudoh J, Shimizu N, Edwards PA. Characterization of the human ABCG1 gene: liver X receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J Biol Chem. 2001;276:39438–39447. doi: 10.1074/jbc.M105863200. [DOI] [PubMed] [Google Scholar]
- 93.Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol. 2012;32:575–581. doi: 10.1161/ATVBAHA.111.240705. [DOI] [PubMed] [Google Scholar]
- 94.Huang LH, Melton EM, Li H, Sohn P, Rogers MA, Mulligan-Kehoe MJ, Fiering SN, Hickey WF, Chang CC, Chang TY. Myeloid Acyl-CoA:Cholesterol Acyltransferase 1 Deficiency Reduces Lesion Macrophage Content and Suppresses Atherosclerosis Progression. J Biol Chem. 2016;291:6232–6244. doi: 10.1074/jbc.M116.713818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest. 2001;107:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shibuya Y, Chang CC, Huang LH, Bryleva EY, Chang TY. Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Abeta1-42 clearance. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:14484–14501. doi: 10.1523/JNEUROSCI.2567-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol. 2014;34:1942–1952. doi: 10.1161/ATVBAHA.114.303342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carr CS, Sharp PA. A helix-loop-helix protein related to the immunoglobulin E box-binding proteins. Mol Cell Biol. 1990;10:4384–4388. doi: 10.1128/mcb.10.8.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 101.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS, Javaheri A, Crowley JR, Ballabio A, Schilling JD, Epelman S, Weihl CC, Diwan A, Fan D, Zayed MA, Razani B. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nature communications. 2017;8:15750. doi: 10.1038/ncomms15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu H, Fan Y, Qiao C, Liang W, Hu W, Zhu T, Zhang J, Chen YE. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Science signaling. 2017:10. doi: 10.1126/scisignal.aah4214. [DOI] [PubMed] [Google Scholar]
- 104.Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, Dent P, Spiegel S, Shi R, Xu W, Liu X, Bohdan P, Zhang L, Zhou H, Hylemon PB. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012;55:267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wan ZY, Tian JS, Tan HW, Chow AL, Sim AY, Ban KH, Long YC. Mechanistic target of rapamycin complex 1 is an essential mediator of metabolic and mitogenic effects of fibroblast growth factor 19 in hepatoma cells. Hepatology. 2016;64:1289–1301. doi: 10.1002/hep.28639. [DOI] [PubMed] [Google Scholar]