

SUMMARY
Although breast cancer stem cells (BCSCs) display plasticity transitioning between quiescent mesenchymal- (M) and proliferative epithelial-like (E) states, how this plasticity is regulated by metabolic/oxidative stress remains poorly understood. Here, we show that M- and E-BCSCs rely on distinct metabolic pathways and display markedly different sensitivities to inhibitors of glycolysis and redox metabolism. Metabolic/oxidative stress generated by 2DG/H2O2 or hypoxia promotes the transition of ROSlo M-BCSCs to a ROShi E-state. This transition is reversed by N-acetyl cysteine and mediated by activation of the AMPK-HIF1α axis. Moreover, E-BCSCs exhibit robust NRF2-mediated antioxidant responses, rendering them vulnerable to ROS-induced differentiation and cytotoxicity following suppression of NRF2 or downstream thioredoxin (TXN) and glutathione (GSH) antioxidant pathways. Co-inhibition of glycolysis and TXN/GSH pathways suppresses tumor growth, tumor-initiating potential and metastasis by eliminating both M- and E-BCSCs. Exploiting metabolic vulnerabilities of distinct BCSC states provides a novel therapeutic approach targeting this critical tumor cell population.
In Brief
Luo et al. report that metabolic stressors modulate breast cancer stem cell (BCSC) state dynamics through ROS-mediated activation of the AMPK-HIF1α axis. They further describe the metabolic pathways and vulnerabilities of epithelial- and mesenchymal-like BCSCs and build a conceptual framework to effectively target both BCSC states in PDX and systemic metastasis models of TNBC.
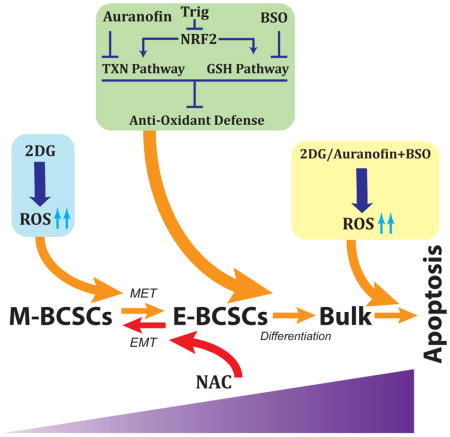
INTRODUCTION
Breast cancer (BC) is a complex disease, in which six different subtypes have been defined based on distinct gene expression signatures and histological characteristics (Cancer Genome Atlas, 2012; Prat and Perou, 2011). While therapeutics targeting estrogen receptor (ER) and epidermal growth factor receptor family member HER2/ErbB2 have provided substantial clinical benefits for ER+ and HER2+ BC, treatment of patients with triple-negative BC (TNBC) has been challenging due to disease heterogeneity and the absence of effective molecularly targeted therapeutics. One reason for the lack of efficacy of current therapies for TNBC may be their inability to effectively target “cancer stem cells” or “tumor initiating cells”. These cells, residing at the apex of tumor heterogeneity, are inherently resistant to chemotherapy and ionizing radiation, leading to treatment resistance and metastases (Balic et al., 2006; Creighton et al., 2009; Dean et al., 2005; Diehn et al., 2009).
Recent studies demonstrate that breast cancer stem cells (BCSCs) exhibit plasticity enabling them to transition between two phenotypic states: a proliferative epithelial-like (E) state, characterized by high expression of aldehyde dehydrogenase (ALDH), and a quiescent, invasive mesenchymal-like (M) state, characterized by CD24−CD44+ expression (Liu et al., 2014). The transition of BCSCs from the E to M state closely resembles the epithelial-to-mesenchymal transition (EMT), which is associated with the acquisition of stem cell properties (Mani et al., 2008). The equilibrium of these BCSC states is regulated by the tumor microenvironment via multifaceted mechanisms including cytokine/chemokine signaling and genetic/epigenetic regulation of key transcription factors, growth factor receptors and microRNA/LncRNAs (Brooks et al., 2015; Luo et al., 2015a; Zhu et al., 2014). For example, HER2 overexpression drives the self-renewal of ALDH+ E-BCSCs that are sensitive to the HER2 antibody trastuzumab (Ithimakin et al., 2013). In contrast, resistance to the HER2 blockade is associated with an increase in CD24−CD44+ M-BCSCs resulting from the activation of an IL6 driven inflammatory loop (Korkaya et al., 2012). In trastuzumab-resistant HER2+ BC, a combinatory approach targeting the IL6 receptor (by tocilizumab) and HER2 (by trastuzumab) synergistically abrogates tumor growth and metastases by eliminating both M- and E-BCSCs (Korkaya et al., 2012), illustrating a novel treatment approach targeting both BCSC states. However, combinatory approaches targeting distinct CSC states in TNBC have not been developed.
The plasticity of BCSCs allowing them to transition between proliferative E and invasive M states facilitates their ability to initiate and grow primary tumors, invade the basement membrane, traverse tissue vasculature, and ultimately colonize distant organs to form clinically significant metastases (Luo et al., 2015a; Luo et al., 2015b). This model of BCSC plasticity complements the current model of cancer metastasis where EMT drives tumor cell invasion and dissemination and the converse mesenchymal-to-epithelial transition (MET) drives proliferation and metastatic colonization (Brabletz, 2012; Nieto et al., 2016). The dynamic equilibrium of CSCs in E- and M-like states suggests that therapeutic approaches targeting either state alone may not be sufficient to eliminate CSCs, since the targeted cell population could be rapidly regenerated by CSCs in alternating states.
Historically, Otto Warburg reported that cancer cells preferentially utilized aerobic glycolysis to generate copious amounts of lactate, regardless of the presence of oxygen (Warburg et al., 1927). This increased glycolysis is beneficial not only for cellular bioenergetics, but also for the generation of metabolic intermediates important for tumor macromolecular biosynthesis. Despite intensive studies documenting that cancers are addictive to glycolysis, glycolytic inhibitors such as 2-Deoxyglucose (2DG) and Lonidamine have little or no effect on solid tumor growth in clinical settings (Papaldo et al., 2003; Prasanna et al., 2009). Although the mechanisms accounting for the lack of sensitivity of cancer cells to glycolytic inhibition remain to be characterized, a recent study suggested that mTORC1-dependent metabolic rewiring underlies the escape of cancer cells to glycolytic addiction (Pusapati et al., 2016). Currently, little is known about the metabolic heterogeneity of distinct CSC states and how these states respond to metabolic stressors such as glycolytic inhibition, nutrient deprivation, hypoxia, and oxidative stress. These metabolic differences in M- and E-like CSCs may contribute to the resistance of cancers to glycolytic inhibition and play critical roles in tumor cell survival and tumor progression under metabolic stress. Furthermore, elucidation of the metabolic differences of distinct CSC states may help to define and exploit their metabolic vulnerabilities and provide a conceptual framework to effectively target this critical tumor cell population.
Here, we examined how metabolic/oxidative stress modulates the state dynamics of BCSCs and identified markedly different responses of M- and E-BCSCs to oxidant stress closely linked to their distinct metabolic pathways and redox potential. We developed a combinatory strategy to exploit metabolic vulnerabilities of distinct BCSC states by co-inhibition of glycolysis and NRF2-TXN/GSH mediated antioxidant pathways. Co-inhibition of these metabolic pathways effectively targeted BCSCs resulting in reduced tumor growth, tumor-initiating capacity and metastasis.
RESULTS
The Glycolytic Inhibitor 2DG Selectively Decreases M-, while Increasing E-BCSCs
To determine if glycolytic inhibition by 2DG affects BCSC state equilibria, we treated basal BC cell lines SUM149 (Fig. 1A) and HCC1806 (Fig. 1B) with 2DG, which significantly decreased the content of CD24−CD44+ M-BCSCs while increasing ALDH+ E-BCSCs in a dose-dependent manner. These different responses of M- and E-BCSCs to 2DG were also observed in luminal BC cell lines MCF7 (Fig. 1C) and T47D (Fig. 1D). 2DG treatment also decreased the absolute number of M-BCSCs in SUM149 (Fig. S1A) and T47D (Fig. S1B) in a dose-dependent manner, while the total number of E-BCSCs was significantly increased in SUM149 treated with 20mM 2DG (Fig. S1A). In T47D cells, 2DG was toxic at 20mM, but 10mM 2DG significantly decreased the total number of M-, but not E-BCSCs (Fig. S1B). This specific reduction of M-BCSCs by 2DG was not due to apoptotic cell death as assessed by Annexin V labeling, although 2DG at 10 or 20 mM significantly induced apoptosis in the differentiated tumor cells which constitute the CD24+CD44− population (Fig. 1E).
Figure 1. BCSC State Equilibrium Is Tightly Controlled by Changes of Redox States.

(A–E) E- and M-BCSCs in SUM149 (A), HCC1806 (B), MCF7 (C) and T47D (D) BC cells treated with 2DG (10 and 20 mM, 40h) and effects of 2DG to induce apoptosis in M-BCSCs or bulk tumor cells (E). *, **: P<0.05 or 0.01 (vs. no 2DG).
(F) SUM149 treated with or without 2DG (20mM, 2h) were stained with CellROX orange (5 nM, 30min) and MFI of CellROX orange was analyzed in each cell subset. **P<0.01 vs. untreated.
(G) CD24−CD44+ M-BCSCs and CD24+CD44− bulk cells were plated and treated with 2DG (20mM), H2O2 (200 μM), NAC (1mM), or 2DG+NAC for 40h and analyzed for ALDH+ cell content. *, **, ***: P< 0.05, 0.01 and 0.001 respectively (vs. untreated).
(H) ALDH+ and ALDH− cells from SUM149 were plated and treated with NAC for 24h and then analyzed for E- and M-BCSCs. *, **: P< 0.05 or 0.01 (vs. no NAC).
(I) Schematic model of redox-regulated BCSC state equilibria.
Glycolytic Inhibition, via Oxidant Stress, Promotes the Transition of BCSCs from the M to E State; an Effect Reversible by NAC
The reduction of M- associated with an increase of E-BCSCs by 2DG led us to investigate if these results could be accounted for by a BCSC state transition driven by oxidant stress, since 2DG selectively increases oxidative stress in cancer vs. normal cells (Aykin-Burns et al., 2009; Li et al., 2015; Shutt et al., 2010). In untreated SUM149 BC cells, both CD24−CD44+ M- and ALDH+ E-BCSCs display significantly lower reactive oxygen species (ROS) levels than ALDH−CD24+CD44− bulk tumor cells (Fig. 1F). However, E-BCSCs exhibited significantly higher ROS levels than M-BCSCs. ROS levels were significantly increased in all cell populations following treatment with 2DG (Fig. 1F).
We next examined the source of ROS in E-BCSCs. MitoSOX Red oxidation, an indicator of mitochondrial superoxide generation in SUM149 (Fig. S1C) and HCC1806 (Fig. S1D) was significantly higher in ALDH+ vs. ALDH− cells that contain M-BCSCs and bulk tumor cells. Thus, increased oxidative phosphorylation (OXPHOS), a major source of ROS, may contribute to the higher ROS levels in E-BCSCs, consistent with a recent study which demonstrated that ALDH+ E-BCSCs displayed increased mitochondrial mass and activity (Lamb et al., 2015). Treatment with 2DG (20mM, 16h) significantly increased mitochondrial superoxide generation in both ALDH− and ALDH+ cells (Fig. S1E), suggesting that glycolytic inhibition promotes OXPHOS, leading to increased mitochondrial ROS.
To determine if oxidant stress generated by 2DG or exogenous H2O2 promotes transition of ROSlo M-BCSCs to a ROShi E-state, we seeded CD24−CD44+ M-BCSCs and CD24+CD44− bulk cells (ALDH+ cells not excluded) and treated them with 2DG, H2O2, NAC or NAC+2DG for 40h. Examination of ALDH+ cell content in untreated M-BCSCs and bulk cells indicated that the proportion of ALDH+ cells was significantly higher in bulk cells (4.4±0.1%) vs. M-BCSCs (0.53±0.03%, Fig. 1G)), reflecting that E- and M-BCSCs are two distinct populations (Ginestier et al., 2007). Although 2DG or H2O2 slightly increased ALDH+ content in pre-seeded bulk cells, the fold increase of ALDH+ cells in pre-seeded M-BCSCs after 2DG (456% increase) or H2O2 (564% increase) treatment was significantly higher (Fig. 1G). This suggests that a state transition from M- to E-BCSCs largely accounts for the significant increase of ALDH+ cells in pre-seeded M-BCSCs following 2DG or H2O2 treatment. A similar state transition of M- to E-BCSCs induced by 2DG or H2O2 was also observed in HCC1806 (Fig. S1F). Treatment with the thiol antioxidant NAC significantly reduced the proportion of ALDH+ cells in pre-seeded M-BCSCs and bulk cells, and combined treatment with NAC and 2DG partially inhibited the increase of ALDH+ cells induced by 2DG (Fig. 1G), confirming the role of oxidant stress in mediating CSC state transition.
The reduction of E-BCSCs by NAC prompted us to examine if NAC promotes the transition of ROShi E-BCSCs to a ROSlo M state. ALDH+ E-BCSCs and ALDH− bulk cells from SUM149 were sorted, plated and treated with or without NAC for 24h. NAC significantly decreased E-, but increased M-BCSCs in pre-seeded ALDH+ and ALDH− cells in a dose-dependent fashion (Fig. 1H), suggesting an E to M transition of BCSCs induced by NAC.
We next examined how changing mitochondrial ROS modulates BCSC state dynamics. Treatment of SUM149 with MitoPQ, a compound selectively increasing superoxide production in mitochondria (Robb et al., 2015), significantly enhanced E- but reduced M-BCSCs in a dose-dependent manner (Fig. S1G). In contrast, MitoTEMPO, a specific scavenger of mitochondrial superoxide (Dikalova et al., 2010), dose-dependently decreased E-BCSCs and partially inhibited the increase of E-BCSCs induced by 2DG (Fig. S1H). Unlike NAC (Fig. 1H), MitoTEMPO slightly decreased M-BCSCs (Fig. S1H), suggesting multiple mechanisms of antioxidant regulation of BCSC state dynamics. The differential effects of NAC and MitoTEMPO on M-BCSC may explain their divergent roles in promoting (Piskounova et al., 2015) or suppressing (Porporato et al., 2014) metastasis respectively. Together, our studies demonstrated that increased oxidant stress generated by 2DG/H2O2 promotes ROSlo M-BCSCs to undergo a state transition resembling MET, generating E-BCSCs. In contrast, NAC induces ROShi E-BCSCs to undergo an EMT-like transition, generating M-BCSCs (Fig. 1I).
E- and M-BCSCs Are Driven by Distinct Metabolic Pathways and Display Enhanced Metabolic Plasticity in Response to Glycolytic Inhibition
To investigate the metabolic pathways active in E- and M-BCSCs, we performed RNA sequencing using E- and M-BCSCs and bulk tumor cells isolated from two PDX models of TNBC, Vari068 and MC1. iPathway analysis of the RNA-Seq data identified pathways significantly enriched in E- and M-BCSCs vs. bulk tumor cells in Vari068 (Fig. 2, A&B) and MC1 (Fig. S2, A&B). Remarkably, three independent pathways including cell cycle, OXPHOS and glutathione metabolism were commonly enriched in E-BCSCs of Vari068 (Fig. 2A) and MC1 (Fig. S2A). Further dissection of OXPHOS regulatory genes indicated that genes from the electron transport chain (ETC) complex I to V were systemically elevated in E-BCSCs of Vari068 (Fig. 2C) and MC1 (Fig. S2C). This suggests that proliferative E-BCSCs have increased mitochondrial OXPHOS coupling with enhanced glutathione metabolism and antioxidant defenses. In contrast to E-BCSCs, the pathways enriched in M-BCSCs of Vari068 (Fig. 2B) and MC1 (Fig. S2B) appeared to be more heterogeneous as only the cytokine-cytokine receptor interaction pathway was shared by both PDXs. However, the glycolysis/gluconeogenesis pathway was significantly enriched in M-BCSCs of Vari068 (Fig. 2B), consistent with previous studies indicating increased glycolysis in M-BCSCs (Ciavardelli et al., 2014; Dong et al., 2013).
Figure 2. E- and M-BCSCs Exhibit Distinct Metabolic Pathways and Enhanced Metabolic Plasticity.

(A–C) Pathways enriched in E- (A) and M- (B) BCSCs and ETC complex genes elevated in E-BCSCs (C) of Vari068.
(D, E) E- (ALDH+) and M- (ALDH−CD24−CD44+) BCSCs and bulk cells (ALDH−CD24+CD44−) from HCC1806 were plated, treated with or without 2DG and measured by FLIM immediately or after 1h culture (D) and NADH Lifetimes calculated from 400–500K cells in each condition (E). Bar: 40 μm, all p<3×10−18.
We also constructed heat maps of annotated metabolic pathway genes including glycolysis and OXPHOS in Vari068 and MC1. As shown in Fig. S2D, systemic elevation of glycolytic pathway including glucose transporters (Gluts) and a spectrum of enzymes of glycolysis and pentose phosphate pathway (PPP) was found in E- and M-BCSCs of Vari068. In MC1, elevation of the glycolytic pathway, including PPP, was more pronounced in E-BCSCs, albeit a number of glycolytic genes (i.e., Glut3, 6, 9, HK2, PGM2, PFKP, PFKFB4, PGAM5 and ENO3) were also elevated in M-BCSCs (Fig. S2G). Despite the robust elevated expression of genes involving the glycolytic pathway, different patterns of glycolysis and PPP gene expression in M- and E-BCSCs were also evident in Vari068. Specifically, a wide variety of glycolytic enzymes ranging from HK1 to LDHD were highly elevated in M- but less robustly in E-BCSCs. In contrast, various PPP enzyme genes, including the rate-limiting G6PD, were highly elevated in E- but less robustly in M-BCSCs (Fig. S2D). This suggests that E-BCSCs may have elevated PPP shunting. Consistent with the iPathway analyses, mitochondrial genes of the TCA cycle (Fig. S2, E&H) and ETC complexes I, III, IV, and V (Fig. S2, F&I) were all systemically upregulated in E- and less robustly in M-BCSCs of Vari068 (Fig. S2, D-F) and MC1 (Fig. S2, G-I). This robust expression of OXPHOS regulatory genes in E-BCSCs supports the idea that mitochondrial respiration is important to maintain this proliferative CSC state.
To characterize the metabolic states of E- and M-BCSCs in glycolysis enabled and inhibited conditions, we utilized fluorescence lifetime imaging microscopy (FLIM) of the endogenous metabolic cofactor NADH, which exhibits long or short fluorescent lifetimes in its mitochondrial protein bound (OXPHOS) or unbound (glycolysis) state respectively (Kolenc and Quinn, 2018; Stringari et al., 2012). This allows measurement of the relative balance of short vs. long lifetimes of NADH to examine the transitions between glycolysis and oxidative metabolism at single cell resolution. As shown in Fig. 2D, untreated E- and M-BCSCs as well as bulk tumor cells from HCC1806 displayed much shorter NADH lifetimes as compared to 2DG-treated cells; indicative of a higher glycolytic state in glycolysis-enabled conditions. Calculation of NADH lifetimes revealed that E- and M-BCSCs in 2DG-untreated condition displayed significantly higher and lower NADH lifetimes vs. bulk tumor cells respectively (Fig. 2E), suggesting that E-BCSCs are more oxidative while M-BCSCs are more glycolytic in glucose-rich medium. Interestingly, when these cells were treated with 2DG and measured by FLIM (2DG 30min_t0), we observed significantly increased NADH lifetimes for all three cell subsets, but the E-BCSCs exhibited the longest NADH lifetimes significantly higher than that of bulk and M-BCSCs (Fig. 2E). When 2DG-treated cells were cultured for 1 hour in fresh medium and then measured by FLIM (2DG 30min_t60), NADH lifetimes of M-BCSCs were further increased (Fig. 2E), suggesting that M-BCSCs are able to gradually boost OXPHOS in response to glycolytic inhibition. These results, together with increased expression of glycolysis and OXPHOS regulatory genes in E- and M-BCSCs, strongly support the notion that E- and less robustly M-BCSCs possess enhanced metabolic plasticity in response to metabolic stress (i.e., 2DG).
ROS-Induced BCSC Transition from the M to E State Is Facilitated by Activation of the AMPK-HIF1α Axis
To investigate the molecular mechanisms underlying redox-regulated BCSC state transition, we examined activation of the AMPK-HIF1α axis, which is regulated by metabolic/oxidative stress (Jeon et al., 2012; Jung et al., 2008). Modest levels of HIF1α were found to be constitutively expressed in three TNBC cell lines including SUM149 (Fig. 3A), HCC1937 (Fig. S3A) and HCC1806 (Fig. S3B). Treatment with 2DG or H2O2 for 1.5h resulted in enhanced HIF1α protein expression, accompanied by increased Ser79 phosphorylation of Acetyl-CoA carboxylase (ACC), a marker of AMP-activated protein kinase (AMPK) activation (Fig. 3A and Fig. S3, A&B). In contrast, treatment with NAC blunted basal HIF1α expression, accompanied by reduced Ser79 phosphorylation of ACC (Fig. 3B). This suggests that increased ROS, through activation of AMPK, stabilized HIF1α while NAC decreased HIF1α stability by reducing AMPK activity. We also compared HIF1α stabilization induced by 2DG/H2O2 and by hypoxia-mimetic cobalt chloride (CoCl2) in HCC1806. CoCl2 induced HIF1α stabilization without activation of AMPK (Fig. S3B), suggesting different mechanisms of HIF1α stabilization by ROS and CoCl2.
Figure 3. The ROS-AMPK-HIF1α Axis Regulates BCSC Phenotypic Plasticity.

(A, B) SUM149 BC cells treated with 2DG/H2O2 for 1.5h (A) or NAC for 20h (B) was analyzed by p-ACC (S79) and HIF1α antibodies.
(C, D) HIF1α protein expression in SUM149 treated with different doses of compound C alone or with 2DG (C) or H2O2 (D) for 2h.
(E) Contents of E- and M-BCSCs in SUM149 after 20h treatment with H2O2 with or without compound C. *, **: P< 0.05 or 0.01 (vs. untreated or corresponding cells without Compound C).
(F) Characterization of two HIF1α knockdown (3808 and 3811) lines without apparent HIF2α compensatory responses after H2O2 stimulation.
(G, H) E- (G) and M- (H) BCSCs in SCR and HIF1α knockdown cell lines with or without H2O2 treatment. **P< 0.01 (vs. untreated).
(I, J) SUM149 (I) and MCF7 (J) BC cells cultured under normoxic (N) or hypoxic (H) conditions for 48h were analyzed for E- and M-BCSCs. **, ***: P<0.01 or 0.001 (vs. N).
(K–N) NOTCH1 (K), HES1 (L), ALDH1A1 (M) and ALDH1A3 (N) expression in HIF1α knockdown or SCR control cells induced by H2O2.*, **: P< 0.05 or 0.01 (vs. untreated).
(O) Moderate levels of H2O2 (0.1–0.2 mM) stimulate NRF2 reporter activity. *, **: P< 0.05 or 0.01 (vs. no H2O2).
(P) Three independent mechanisms for ROS induced propagations of E-BCSCs.
We next investigated to what extent AMPK activation is required for ROS-induced HIF1α stabilization. Treatment of SUM149 with Compound C, an AMPK inhibitor, effectively blocked basal HIF1α expression and HIF1α induction by 2DG (Fig. 3C) or H2O2 (Fig. 3D) dose-dependently. This Compound C-mediated inhibition of AMPK-HIF1α significantly compromised H2O2-induced transition of BCSCs from the M to E state (Fig. 3E). As Compound C may affect HIF1α stability independent of AMPK (Emerling et al., 2007), we employed AICAR, an AMP analog activating AMPK independent of ROS, to further address if activation of AMPK-HIF1α modulates BCSC state equilibria. Treatment with AICAR dose-dependently activated the AMPK-HIF1α axis in SUM149 (Fig. S3C) and HCC1806 (Fig. S3D), leading to significantly increased E- but decreased M-BCSCs (Fig. S3E). This suggests that AICAR is able to induce the transition of BCSCs from the M to E state independent of ROS.
To determine if AMPK exerts its effect on CSC state transition through HIF1α, we next utilized SUM149 to express five different shRNA against human HIF1α or a scrambled sequence (SCR). In parental SUM149 cells, constitutive expression of HIF1α and HIF2α was observed (Fig 3F, line 1). Following H2O2 stimulation, HIF1α in parental and SCR lines was enhanced while HIF2α was decreased (Fig 3F, line 2&3), indicating markedly different regulation of HIF1α and HIF2α in response to oxidant stress. In all five HIF1α knockdown lines, a significant reduction of HIF1α protein levels was observed following H2O2 stimulation (Fig 3F), but HIF2α was significantly elevated in three HIF1α knockdown lines (3809, 3810 and 10819). The establishment of two HIF1α knockdown lines without apparent HIF2α compensatory responses (3808 &3811) provides a unique tool to assess the role of HIF1α in ROS-induced BCSC state transition. Further studies of the HIF1α knockdown lines 3808 & 3811 indicated that HIF1α knockdown significantly decreased E-BCSCs (Fig. 3G), suggesting a role for HIF1α in maintaining E-BCSCs. We also observed compromised transition of BCSCs from the M to E state following H2O2 stimulation in the HIF1α knockdown but not SCR lines (Fig. 3H), suggesting that HIF1α knockdown results in BCSCs persistently residing in the M state. Of note, although HIF1α knockdown blocked H2O2-induced M to E transition, the content of E-BCSCs was still significantly increased by H2O2 (Fig. 3G), suggesting that oxidant stress propagates E-BCSCs independent of HIF1α.
We next examined if HIF1α knockdown blocks AICAR-mediated BCSC state transition and observed similarly compromised M to E transition of BCSCs in HIF1α knockdown but not SCR cells (Fig. S3F). Unlike H2O2, AICAR treatment did not significantly increase E-BCSCs in HIF1α knockdown lines 3808 and 3811, suggesting divergent mechanisms of AICAR and ROS to modulate BCSC state dynamics. Both AICAR and ROS induced HIF1α-dependent transition of BCSCs from the M to E state (Fig. 3, G&H and Fig. S3F). However, ROS also elicited a HIF1α-independent process propagating E-BCSCs (Fig. 3G).
Hypoxia Promotes M to E Transition of BCSCs in a HIF1α-dependent Manner
Intratumoral hypoxia is commonly found in advanced cancers, which promotes tumor metastasis and therapeutic resistance through stabilization of HIF1α (Wilson and Hay, 2011). We previously showed that tumor hypoxia generated by anti-angiogenic agents enriched E-BCSCs in tumor hypoxic zones in a HIF1α-dependent manner (Conley et al., 2012). As hypoxia stabilizes HIF1α by ROS-dependent and -independent mechanisms (Chandel et al., 2000; Maxwell et al., 1999), we determined whether hypoxia promotes the enrichment of E-BCSCs by inducing M to E state transition. Culturing SUM149 (Fig. 3I) or MCF7 (Fig. 3J) in a hypoxic chamber (1% O2) for 48h led to a significant increase in E- with a concomitant decrease in M-BCSCs vs. cells cultured under normoxic conditions. This suggests that hypoxia induces BCSC phenotypic transition from M to E states.
We next determined if knockdown of HIF1α compromises hypoxia-induced BCSC state transition. Culturing HIF1α SCR cells under hypoxic vs. normoxic conditions led to a significant increase in E- (Fig. S3G, upper panel) but decrease in M- (Fig. S3G, lower panel) BCSCs. This reduction of M-BCSCs was significantly reduced in HIF1α knockdown lines 3808 and 3811 following hypoxia treatment, suggesting a compromised M to E state transition. Similar to H2O2 treatment, hypoxia significantly increased E-BCSCs in HIF1α knockdown lines (Fig. S3G, upper panel), confirming that hypoxic/oxidant stress propagates E-BCSCs independent of its effect on HIF1α.
ROS-Stimulated NOTCH and NRF2 Pathways Contribute to the Propagation of E-BCSCs
To elucidate the alternative pathways contributing to ROS-induced propagation of E-BCSCs, we next conducted quantitative real-time PCR (qRT-PCR) analyses using SUM149 cells treated with or without 2DG (20mM, 8h) and found that the expression of NOTCH pathway genes NOTCH1 and its downstream target HES1 was significantly upregulated by 2DG (Fig. S3H). Further analysis of SUM149 SCR or HIF1α knockdown lines confirmed that treatment with H2O2 (200 μM, 8h) significantly induced NOTCH1 (Fig. 3K) and HES1 (Fig. 3L) expression in a HIF1α-dependent manner, consistent with our previous study where activation of NOTCH pathway was associated with hypoxia-induced ALDH+ BCSCs (Conley et al., 2012). Treatment of SUM149 BC cells with an inhibitor of NOTCH signaling, gamma secretase inhibitor (GSI), at doses of 1–10 μM for 24h significantly inhibited E-BCSCs (Fig. S3I, left panel). Moreover, GSI at the concentrations of 3–10, but not 1μM, also reduced M-BCSCs (Fig. S3I, right panel).
We next determined to what extent the expression of ALDH1A1 and ALDH1A3, two major isoforms expressed in ALDH+ E-BCSCs, are affected by oxidant stress. qRT-PCR analyses revealed that ALDH1A3, the major ALDH isoform expressed in SUM149, was significantly upregulated by 2DG (Fig. S3J). Further studies with SUM149 SCR or HIF1α knockdown cells confirmed that H2O2 treatment moderately induced ALDH1A1 (Fig. 3M) and more robustly ALDH1A3 (Fig. 3N) expression, an effect that was not significantly affected by the knockdown of HIF1α. This HIF1α-independent activation of ALDH1A1/3 gene expression by oxidant stress may account for the fact that H2O2/hypoxia propagates E-BCSCs independent of HIF1α. The elevated expression of ALDH genes by oxidative stress also indicates that these genes play functional roles for the protection/maintenance of E-BCSCs.
The family of ALDH genes including ALDH1A1/3 is among the many target genes of NRF2 mediated antioxidant responses (Gorrini et al., 2013; Hayes and Dinkova-Kostova, 2014). To determine if H2O2 induces NRF2 activity, we established a NRF2 reporter cell line expressing the antioxidant response element (ARE) driven firefly luciferase (ARE-LUC). H2O2 at 100–200 μM significantly induced NRF2 reporter activity while H2O2 at 400–800 μM reduced or abolished NRF2 activity, presumably due to the cytotoxic effect of high H2O2 concentrations (Fig. 3O). Together, our studies identify three different mechanisms of regulation of BCSC state dynamics: 1) conversion of M- to E-BCSCs mediated by AMPK-HIF1α, 2) activation of the HIF1α-NOTCH self-renewal pathway, 3) activation of NRF2-mediated ALDH1A1/3 expression independent of HIF1α (Fig. 3P).
E-BCSCs Display Robust NRF2 Antioxidant Responses, which Support their Maintenance, Sphere-forming Activity and Tumor Initiating Potential
The resistance of E-BCSCs to oxidative stress prompted us to explore the mechanisms underlying their ability to survive stressful metabolic conditions. Gene profiling of antioxidant genes in Vari068 and MC1 PDX models revealed that NRF2 (NFE2L2) and a wide variety of NRF2 antioxidant responsive genes including the family of drug transporters and detoxification enzymes, NADPH production, TXN and GSH antioxidant pathways were all robustly elevated in E-BCSCs vs. M-BCSCs or bulk tumor cells (Fig. 4A). This suggested that elevated NRF2 antioxidant defenses may be critical for E-BCSCs to cope with higher ROS levels and to survive under metabolic/oxidative stress.
Figure 4. E-BCSCs Are Endowed with Robust NRF2 Antioxidant Responses, which Support their Maintenance and Sphere-forming Capacity.

(A) Heat map of NRF2 antioxidant responsive genes in E- and M-BCSCs vs. bulk cells in Vari068 and MC1 PDXs.
(B) Relative expression of NRF2 in E- and M-BCSCs vs. bulk cells in SUM149. *: P< 0.05 vs. bulk.
(C, D) A NRF2 mCherry reporter in SUM149 indicating 46% cells with NRF2 activity (C) and mCherry MFI in ALDH− vs. ALDH+ cells (D).
(E, F) ALDH+ E- (E) and CD24−CD44+ M- (F) BCSCs treated with various doses of Trig for 40h, *, **: P< 0.05 or 0.01 (vs. untreated).
(G–K) NRF2 protein levels in SUM149 BC cells expressing SCR or different shNRF2 sequences (G), and contents of E- (H) and M- (I) BCSCs as well as primary (J) and secondary (K) sphere formation in SCR vs. NRF2 knockdown lines. *, **: P< 0.05 or 0.01 (vs. SCR).
To determine if the increased NRF2 expression in E-BCSCs observed in two PDXs of TNBC is recapitulated in the prototype basal BC cell line SUM149, we performed qRT-PCR analysis of isolated E (ALDH+) and M (ALDH−CD24−CD44+) BCSCs and bulk tumor cells (ALDH−CD24+CD44−). NRF2 mRNA expression was significantly higher in E-BCSCs vs. M-BCSCs and bulk tumor cells (Fig. 4B), while the latter two populations displayed no significant difference in NRF2 expression.
To determine if E-BCSCs exhibit constitutively higher NRF2 activity, we established NRF2 fluorescent reporter cells expressing mCherry driven by an NRF2-binding ARE. Flow cytometry of the cells expressing 1xARE_mCherry vs. empty vector revealed that 46% of SUM149 BC cells exhibited NRF2 reporter activity (Fig. 4C). Further analysis of mCherry mean fluorescent intensity (MFI) indicated that ALDH+ cells displayed significantly higher NRF2 reporter activity vs. ALDH− cells (Fig. 4D).
Recent studies indicated that NRF2 is necessary to maintain tumor cell proliferation by preventing oxidative inactivation of protein translation machinery (Chio et al., 2016). To investigate the role of NRF2 in maintaining BCSC activity, we treated SUM149 with a NRF2 inhibitor, Trigonelline (Arlt et al., 2013; Syu et al., 2016) for 40h. Treatment with Trig led to significantly decreased E- (Fig. 4E), but not M- (Fig. 4F) BCSCs, in a dose dependent manner. This abrogation of E- but not M-BCSCs by Trig was also observed in T47D luminal BC cells (Fig. S4A). We also utilized SUM149 BC cells to express scrambled (SCR) and different shRNA sequences against human NFE2L2/NRF2. Expression of two distinct NRF2 hairpins, 7555 or 7557, led to significantly decreased basal NRF2 expression by over 80% (Fig. 4G), and significantly decreased E- (Fig. 4H) but not M- (Fig. 4I) BCSCs, confirming the specific effect of targeting NRF2 to abrogate proliferative E-BCSCs.
We next assessed tumorsphere formation in low-attachment conditions (a property of CSCs) to examine to what extent NRF2 knockdown impairs the proliferative and self-renewal activities of BCSCs. NRF2 knockdown by 7555 or 7557 significantly decreased SUM149 primary (Fig. 4J) and secondary (Fig. 4K) sphere formation, and the size of spheres (Fig. S4B), indicating a role for NRF2 in supporting the proliferation of BCSCs/progenitor cells. The impaired sphere-forming capacity following NRF2 knockdown was rescued by NAC (Fig. S4C), confirming the role of NRF2 in supporting BCSC activity through antioxidant defenses. As NRF2-mediated antioxidant responsive genes were more robustly expressed in E- vs. M-BCSCs, we also examined how NRF2 knockdown affects sphere formation of E- and M-BCSCs. Surprisingly, sphere formation of CD24−CD44+ (M) or ALDH+ (E) BCSCs were both significantly decreased by NRF2 knockdown (Fig. S4D). The ability of NRF2 knockdown to inhibit sphere formation of M-BCSCs suggests that these quiescent CSCs may have to transition to their proliferative E-state in order to form spheres in suspension culture, rendering them sensitive to knockdown of NRF2 mediated antioxidant responses. This requirement for NRF2 in maintaining the self-renewal and proliferative activity of BCSCs was further confirmed by tumor-initiating studies in immune-deficient mice, as knockdown of NRF2 by 7555 significantly impaired tumor-initiating potential of SUM149 BC cells (Fig. S4E).
Inhibition of TXN and GSH Antioxidant Pathways downstream of NRF2 Abrogates E- but not M-BCSCs
Previous studies indicated that the GSH and TXN antioxidant pathways downstream of NRF2 synergize to drive tumor initiation and progression (Harris et al., 2015). Treatment of SUM149 BC cells with Auranofin (AUR), a specific inhibitor of TXN reductase (TR), at 0.3 but not 0.1 μM, led to significantly reduced proportion and absolute number of E- but not M-BCSCs (Fig. 5, A&B and Fig. S5, A&B). In contrast, treatment with 10μM of L-buthionine-sulfoximine (BSO), an inhibitor of GSH biosynthesis, significantly increased the content and absolute number of E- but not M-BCSCs. This stimulatory effect of BSO on E-BCSCs reflects a compensatory increased TXN pathway when GSH biosynthesis is inhibited (Fath et al., 2011; Harris et al., 2015). Although 0.1μM AUR or 10 μM BSO did not inhibit E-BCSCs, combination of AUR (0.1μM) and BSO (10μM) significantly decreased the content (Fig. 5, A&B) and absolute number (Fig. S5, A&B) of E- but not M-BCSCs. This suggests a complementary role of AUR and BSO in disrupting antioxidant defenses, rendering E-BCSCs vulnerable to the oxidant stress elicited by the combined treatment. Indeed, the inhibitory effect of AUR or AUR+BSO on E-BCSCs was reversed by NAC (Fig. 5A and Fig. S5A), confirming a role for oxidant stress in mediating the effects of AUR or AUR+BSO on E-BCSCs.
Figure 5. Inhibition of TXN and GSH Antioxidant Pathways downstream of NRF2 Abrogates E- but not M-BCSCs.

(A–C) Contents of E- (A) and M- (B) BCSCs in SUM149 treated with AUR, BSO or AUR+BSO with or without NAC for 24h and the effects of Trig to sensitize E-BCSCs to AUR (C). *, **: P<0.05 or 0.01 vs. untreated. NS: not significant.
(D–G) Sphere formation of SUM149 (D, E) and H2Kd− Vari068 tumor cells (F, G) subjected to various treatments. Scale Bar: 100 μm. **, ***: P< 0.01 or 0.001 vs. untreated.
We also determined if introducing exogenous antioxidant enzymes rescues the decreased E-BCSCs by AUR in SUM149 pretreated with or without PEGylated catalase (Cat, which removes H2O2) or superoxide dismutase (SOD). Only PEGylated SOD inhibited the cytotoxic effect of AUR on E-BCSCs (Fig. S5C). This suggests that mitochondrial superoxide production is harmful for E-BCSCs when the TXN antioxidant pathway is inhibited.
We next tested if NRF2 inhibition by Trig sensitizes E-BCSCs to AUR treatment. Treatment of SUM149 with AUR at 200 nM or Trig at 3 μM each significantly reduced E-BCSCs, while AUR at 50 nM or Trig at 0.3 μM each had no effect (Fig. 5C). When low doses of AUR (50 nM) and Trig (0.3 μM) were employed, E-BCSCs was reduced to the same extent as in cells treated with 200 nM AUR, indicating a sensitizing effect of Trig on AUR in targeting E-BCSCs. Of note, treatment with higher doses of Trig (3 μM) and AUR (200 nM) did not result in further reduction of E-BCSCs, supporting the roles of Trig and AUR in abrogating E-BCSCs through this pathway.
To substantiate the roles of TXN/GSH pathways in BCSCs, we examined how treatments with AUR and/or BSO affect sphere formation. As shown in Fig. 5, D&E, AUR at 0.3μM significantly decreased sphere formation of SUM149 BC cells, while 0.1μM of AUR reduced the size (from 164.4±41.38 to 62.84±12.75 μm in dimeter) but not the number of spheres formed, suggesting a role of low dose of AUR in inhibiting stem/progenitor cell proliferation. This inhibitory effect of AUR on BCSC/progenitor cell activity was rescued by NAC (Fig. 5D, g & h). In contrast to AUR, BSO at 10 or 30μM each had no inhibitory effect on sphere formation (Fig. 5D, e & f). Of note, although BSO alone exerted no inhibitory effect, co-treatment with 10μM BSO and 0.1μM AUR exhibited a significant additive effect in suppressing sphere formation (Fig. 5D, i), an effect that was fully inhibited by NAC (Fig. 5D, k). Combined treatment with 30μM BSO and 0.3μM AUR dramatically inhibited sphere formation (Fig. 5D, j), which was not fully rescued by NAC (Fig. 5D, l), suggesting an increase in toxicity while combining higher doses of AUR and BSO. These studies provide functional evidence that inhibition of the TXN pathway suppresses self-renewal/proliferative activity of BCSCs, and co-inhibition of TXN and GSH antioxidant pathways generates additive effect to target BCSC’s
We also tested the effect of 2DG, AUR, and BSO on primary (Fig. 5F) and secondary (Fig. 5G) sphere formation of Vari068 primary tumor cells. AUR at 0.3μM, but not 0.1μM, significantly inhibited primary and secondary sphere formation, while 2DG (5 or 10 mM) or BSO (10 or 30μM) had no significant effect. Combined treatment with 0.1μM AUR and 10μM BSO additively inhibited Vari068 sphere formation. We also observed an additive effect of 2DG (10mM) combined with AUR (0.1μM) and BSO (10μM) in inhibiting primary and secondary sphere formation.
Suppression of TXN Antioxidant Pathways Renders E-BCSCs Vulnerable to ROS-induced Differentiation and Cytotoxicity
To explore how inhibition of the TXN pathway abrogates sphere formation and reduces E-BCSCs, we determined whether AUR induces apoptosis directly in E-BCSCs. Treatment of SUM149 with AUR at 0.5 μM significantly decreased ALDH+ E-BCSCs, whereas BSO at 1mM had no significant effect (Fig. 6A). This reduction of E-BCSCs by AUR was not due to the induction of apoptotic cell death (Fig. 6B, and Fig. S5E), although a significant increase of Annexin V+ apoptotic cells in ALDH− differentiated tumor cells was observed upon treatment with AUR, but not BSO (Fig. 6B and Fig. S5E).
Figure 6. Inhibition of TXN Pathway Induces Differentiation and Apoptosis of E-BCSCs and Co-inhibition of Glycolysis and TXN/GSH Pathways Additively Suppresses Tumor Growth and Tumor-Initiating Potential by Abrogating both M- and E-BCSCs.

(A, B) SUM149 treated with AUR (0.5 μM) or BSO (1mM) for 24h were labeled with Annexin V and examined for the content of E-BCSCs (A) and Annexin V+ cell ratio in ALDH+ and ALDH− cell populations (B).
(C, D) SUM149 treated with Vehicle, 0.5μM of AUR, or 30μM of BSO for 24h were stained for CD24 and CK8/18 (C) and the ratio of CD24+ or CK8/18+ cells over total DAPI+ cells were plotted (D). Scale Bar: 20 μm. **: P<0.01 vs. Mock.
(E) SUM149 treated with Vehicle, 0.5 μM AUR, or 20 mM 2DG were stained with antibodies against human ALDH1A1 and Ki67. Scale Bar: 50 μm.
(F–H) Vari068 tumor growth following treatment with Vehicle, 2DG, AUR+BSO, and 2DG+AUR+BSO
(F), and contents of E- (G) and M- (H) BCSCs (G) in each cohort of tumors after 7-week treatment. *, **, ***: P<0.05, 0.01 or 0.001 vs. Vehicle.
(I) Tumors of Vehicle and AUR+BSO group after 7-week treatment were examined for TR activity and GSH/GSSG ratio. *: P<0.05 vs. Vehicle.
(J) Tumor-initiating potential of Vari068 tumor cells in 4 cohorts of tumors after 7-week treatment were examined in secondary NOD/SCID mice.
We next determined whether AUR induces differentiation of E-BCSCs, rendering them sensitive to ROS-induced apoptosis. Treatment of SUM149 with AUR, but not vehicle or BSO, significantly increased the portion of cells expressing the luminal differentiation markers CD24 and CK8/18 (Fig. 6, C&D), suggesting that AUR reduced E-BCSCs by promoting their differentiation. Further examination of the cell proliferation marker Ki67 in E-BCSCs (Fig. 6E) revealed that, in vehicle-treated SUM149 BC cells, Ki67 was found in almost 100% of cells expressing ALDH1A1, and a significant portion of ALDH1A1+ cells were found in mitosis, reflecting the highly proliferative nature of E-BCSCs. After AUR treatment, the expression of Ki67 was lost in more than 90% cells expressing ALDH1A1 and none of these cells were found in mitosis, suggesting that AUR treatment blocked the proliferation of E-BCSCs, a phenomenon that may accompany terminal differentiation. We also determined if 2DG affects the proliferation of E-BCSCs and observed that 2DG at 20mM increased ALDH1A1 and Ki67 double positive cells. As 2DG or H2O2 promotes the transition of ROSlo M-BCSCs to a ROShi E-state vulnerable to the inhibition of TXN/GSH antioxidant pathways, our studies provide a conceptual framework for targeting M- and E-BCSCs by co-inhibition of glycolysis and TXN/GSH pathways.
Co-inhibition of Glycolysis and TXN/GSH Antioxidant Pathways Additively Suppresses Tumor Growth, Metastasis and Tumor Initiating Potential by Targeting both M- and E-BCSCs
We next determined if combining the glycolytic inhibitor 2DG with inhibitors of TXN/GSH pathways effectively targets M- and E-BCSCs in two PDXs of TNBC. NOD/SCID mice bearing Vari068 tumors were randomized into four cohorts and treated with Vehicle, 2DG (400mg/kg), AUR (1.5mg/kg)+BSO(30mg/kg), and 2DG+AUR+BSO respectively. After 7 weeks of treatment, 2DG did not suppress tumor growth while AUR+BSO significantly reduced tumor growth (Fig. 6F), associated with reduced TR activity and ratio of GSH/GSSG (Fig. 6I) in the tumors, indicating an effect of inhibition of antioxidant defenses mediated by AUR+BSO on tumor growth. Although 2DG alone had no effect, treatment with 2DG together with AUR+BSO additively suppressed tumor growth (Fig. 6F), cell proliferation (Fig. S6A), and the content of both E- (Fig. 6G) and M- (Fig. 6H) BCSCs. The additive effect of 2DG and AUR+BSO in suppressing BCSCs was validated by tumor-initiating studies in secondary NOD/SCID mice (Fig. 6J). Only the combination of 2DG+AUR+BSO significantly decreased the frequency of tumor-initiating cells in Vari068.
As tumor growth inhibition was apparent only at the late phase of treatment (week 4–7, Fig. 6F), we tested the antitumor effects of single or combinatory treatment in another TNBC PDX model, MC1, utilizing an increased dose of AUR (3mg/kg). This study demonstrated that AUR or BSO alone or combined with 2DG had no significant effect on the tumor growth (Fig. S6B). However, combinatory treatment using 2DG+AUR+BSO significantly suppressed MC1 tumor growth at earlier time points (day 17 and 23, Fig. S6C), associated with significant reduction of both M- and E-BCSCs (Fig. S6, D&E) and reduced frequency of tumor-initiating cells (Fig. S6F).
As Vari068 and MC1 PDX mice are not metastatic, we assessed if the combinatory approach by co-inhibition of glycolysis and TXN/GSH antioxidant pathways suppresses metastasis of luciferase-labelled SUM159 BC cells introduced into NOD/SCID mice by cardiac injection. The formation of systemic metastasis was monitored by bioluminescent imaging after 4 and 7 weeks of treatment. As shown in Fig. 7A, compared to vehicle-treated mice, which exhibited metastasis formation in all mice at week 4 and 7 (3 mice died from week 4–7 due to severe tumor complications), treatment with AUR (1.5mg/kg)+BSO(30mg/kg) or 2DG(400mg/kg)+AUR+BSO for 7 weeks suppressed metastasis formation over 70% and 85% respectively. In contrast, a separate study of 2DG (400mg/kg) or vehicle treated mice for 7 weeks revealed that 2DG alone did not significantly inhibit metastasis formation (Fig. S7, A&B). Together, these studies build a conceptual framework to collectively target M- and E-BCSCs in TNBC by co-inhibition of glycolysis and NRF2-mediated antioxidant responses. This combinatory approach, by disrupting redox homeostasis of two distinct BCSC states, promotes differentiation and subsequent apoptosis of BCSCs (Fig. 7B).
Figure 7. Co-inhibition of Glycolysis and TXN/GSH Antioxidant Pathways Suppresses Metastasis and Expression of NRF2 or TXN Antioxidant Pathway Is Correlated with Poor Survival of BC Patients.

(A) NOD/SCID mice with 100K SUM159-Luc cells injected into the left ventricle were treated for 7 weeks and examined for metastasis formation by bioluminescent imaging.
(B) A schematic model illustrating co-inhibition of glycolysis and NRF2 mediated antioxidant responses in disrupting BCSC state equilibrium.
(C, D) NRF2 (C) and 7 out of 10 TXN antioxidant pathway genes (D) expression correlate to poor patient survival of BC.
(E) The PRDX5 and TXN genes correlate to poor patient survival of basal BC.
Elevated Expression of NRF2 and TXN Antioxidant Pathway Genes Significantly Correlates with Poor Survival of BC Patients
To determine if NRF2 and its downstream TXN/GSH antioxidant pathways play a role in affecting the clinical course of BC, we performed Kaplan-Meier analysis using a 3951 BC patient database (Gyorffy et al., 2010). Remarkably, NRF2 (Fig. 7C) and 7 out of 10 TXN antioxidant pathway genes that were elevated in E-BCSCs of Vari068 and MC1 PDXs, including PRDX1, 4, 5, 6, TXN, TXNRD1 and 3 (Fig. 7D), were all inversely correlated with patient survival. In contrast, of the 19 GSH pathway genes that were mostly elevated in E-BCSCs (Fig. 4A), only GCLM (Fig. S7C) and GSTO1 (Fig. S7D) were inversely correlated with patient survival.
We next determined if expression of NRF2 and its downstream TXN antioxidant pathway genes are correlated with disease progression across the specific molecular subtypes of BC. High expression of NRF2 correlates to the poor patient survival of luminal BC including luminal A and B, but not other molecular subtypes (Fig. S7E), potentially due to the relatively small number of basal and HER2+ BC patients in the database. Of the seven TXN pathway genes that correlate to poor patient survival, PRDX1, 4 and 6 (Fig. S7, F-H) and TXNRD1 (Fig. S7I) were significantly correlated with poor survival of luminal A or B subtypes, while PRDX5 (Fig. 7E, upper panel) is specifically correlated with poor patient survival of basal, but not luminal A/B or HER2+ subtypes. High TXN expression also correlates to poor patient survival of the basal and luminal A subtype (Fig. 7E, lower panel). These data suggest further prognostic value of the TXN pathway gene expression, especially PRDX5 and TXN, in the clinical course of basal BC.
DISCUSSION
In this study, we report distinct redox states and divergent sensitivities of E- and M-BCSCs to metabolic/oxidant stress. These differential responses of E- and M-BCSCs to metabolic/oxidative stress are linked to their distinct metabolic pathways, which are identified by extensive gene profiling and pathway analysis of the RNA-Seq data derived from E- and M-BCSCs and bulk tumor cells isolated from two independent PDXs of TNBC. We demonstrate that, although both BCSC states exhibit elevated glycolysis regulatory genes, the proliferative E-BCSCs are endowed with robust mitochondrial OXPHOS capacity and metabolic plasticity coupled with highly elevated NRF2 antioxidant responses, which allow them to survive metabolic/oxidative stress.
In addition to identifying metabolic differences between E- and M-BCSCs, we show that metabolic/oxidant stress promotes the transition of BCSCs from the ROSlo M to a ROShi E state, and that E-BCSCs are endowed with elevated NRF2 antioxidant defenses. These findings also identify NRF2 and its downstream TXN and GSH antioxidant pathways as potential metabolic targets in these cells. We demonstrated that inhibition of the TXN pathway had a prominent inhibitory effect on E-BCSCs, and co-inhibition of the TXN and GSH antioxidant pathways additively abrogated E-BCSCs and tumorsphere formation, suggesting synthetic lethality for E-BCSCs when both pathways are collectively targeted. These studies are consistent with recent studies showing that blocking GSH and TXN pathways synergistically inhibits tumor growth (Fath et al., 2011; Harris et al., 2015). As metabolic/oxidative stress such as glycolytic inhibition by 2DG promotes the transition of quiescent M-BCSCs to their proliferative E-state susceptible to the blockade of NRF2 or its downstream TXN/GSH pathways, we tested a combinatory treatment approach utilizing 2DG together with inhibitors of TXN (AUR) and GSH (BSO) pathways in two PDXs and a systemic metastasis model of TNBC. We demonstrated that this combinatory approach significantly suppressed tumor growth, tumor-initiating potential and metastasis by abrogating both M- and E-BCSCs. Although statistically significant, we acknowledge that the antitumor effects of the combination therapy in suppressing tumor growth and BCSCs in two PDXs of TNBC are modest. One reason for this may be due to the low dosage of AUR utilized in order to avoid toxicity observed in NOD/SCID mice when combining higher doses of AUR with BSO (Fig. S6B). This toxicity warrants further studies using better inhibitors of NRF2-mediated antioxidant pathways to target ALDH+ E-BCSCs. Nevertheless, our preclinical studies suggested a novel pro-oxidant based therapeutic approach utilizing 2DG or other ROS-inducing agents in combination with inhibitors of NRF2-mediated antioxidant responses (i.e., AUR/BSO) to treat metastatic cancer (i.e., TNBC), by targeting CSCs of different phenotypic states.
Besides defining the metabolic vulnerabilities of BCSCs, Kaplan-Meier analysis of a BC gene expression database suggested that elevated NRF2-TXN antioxidant pathway correlates to poor BC patient survival, and that high levels of PRDX5 and TXN gene expression also correlates with poor patient survival of TNBC. The association of genes involved in the regulation of cancer stem cells which constitute only a small proportion of tumor cells in whole tumor samples to patient outcome suggests a role of “stemness” gene signature in predicting tumor “dedifferentiation” state and poor clinical outcome. Strong support for this association was recently reported by Malta et al. who utilized TCGA data involving 12,000 primary tumors representing 33 different cancer types to demonstrate that a “stemness index” calculated on total tumor mRNA expression reflected the proportion of stem-like cells and predicted development of metastases and patient outcome (Malta et al., 2018).
In this report, we documented that proliferative E-BCSCs exhibit significantly higher ROS levels than that of quiescent M-BCSCs. The different redox levels observed in E- and M-BCSCs suggest that adequate levels of ROS in E- and M-BCSCs are required for maintaining each BCSC state. This idea is further supported by our findings that 2DG or H2O2 treatment propagated E-BCSCs whereas treatment of presorted E-BCSCs with the thiol antioxidant NAC significantly decreased E- while increasing M-BCSCs. We identified divergent mechanisms by which ROS stimulates E-BCSCs via HIF1α dependent and independent pathways (Fig. 3P). These studies are in agreement with a recent report documenting that MYC and MCL1, two genes frequently amplified in TNBC, cooperatively increase ROS generation by promoting mitochondrial OXPHOS, leading to increased HIF1α stabilization and enrichment of ALDH+ BCSCs (Lee et al., 2017).
One of the key findings in this study is that metabolic/oxidative stress, through modulation of the AMPK-HIF1α axis, regulates BCSC state equilibrium and phenotypic plasticity. This tight association of cell redox potential with BCSC state dynamics sheds light on the controversial roles of antioxidants in cancer, as studies have documented context-dependent roles of antioxidants in promoting (DeNicola et al., 2011; Glasauer et al., 2014; Harris et al., 2015; Sayin et al., 2014) or suppressing (Gao et al., 2007; Glasauer and Chandel, 2014) cancer development. On one hand, antioxidants help to scavenge ROS from the body, preventing their cancer-causing effects in damaging DNA, as well as proteins and lipids important for tissue homeostasis. On the other hand, once cells are transformed, exposure to antioxidants may promote the transition of cancer stem cells to a quiescent M-like state prone to escape traditional therapy and form distal micro-metastases that can then re-enter a proliferative state. Indeed, treatment of melanoma cells with the antioxidant NAC has been shown to promote metastasis (Piskounova et al., 2015). Together, our studies reveal novel cellular and molecular mechanisms demonstrating how perturbation of redox signaling modulates the equilibrium of BCSC states. These studies define metabolic vulnerabilities of M- and E-BCSCs, and also provide a feasible therapeutic approach to collectively target these distinct CSC states. As CSC state equilibria may be similarly regulated across a spectrum of tumors with diverse oncogenic drivers, this approach may have broad therapeutic applicability.
Limitations of the study
One limitation of this study is that it utilizes PDX models established in immune-deficient mice to investigate BCSC metabolic pathways and evaluate the effects of pro-oxidant based combination therapy to target BCSCs. These preclinical models are appropriate to assess antitumor efficacies of the therapeutic strategies directly on patient-derived tumor cells. However, as the immune system plays important roles in regulating CSCs and treatment responses, future studies are needed to assess how modulation of glucose and redox metabolism affects distinct CSC states in immune-competent animal models. This limitation must be taken into account when interpreting the results and translating the therapeutic approaches to patients.
STAR METHODS
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ALDH1A1 | LSBio | Cat# LS-C156250 |
| Anti-beta-actin-Peroxidase | Sigma-Aldrich | Cat# A3854; RRID:AB_262011 |
| Anti-CK8/18 | American Research Products | Cat# 24-M6-691 |
| Anti HIF1α | Cell Signaling Technology | Cat# 14179S; RRID:AB_2622225 |
| Anti HIF2α | Abcam | Cat# ab179825 |
| Anti-Ki67 | Cell Signaling Technology | Cat# 9129S |
| Anti-Mouse IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2005; RRID:AB_631736 |
| Anti NRF2 | GeneTex | Cat# GTX103322; RRID:AB_1950993 |
| Anti P-ACC | Cell Signaling Technology | Cat# 3661S; RRID:AB_330337 |
| Anti-Rabbit IgG, HRP Linked Antibody | Cell Signaling Technology | Cat# 7074S; RRID:AB_2099233 |
| APC Annexin V | BD Biosciences | Cat# 550474 |
| APC Anti-human CD44 | BD Biosciences | Cat# 559942; RRID:AB_398683 |
| APC anti-mouse H-2K[d] | BioLegend | Cat# 116620; AB_10645328 |
| FITC Anti-human CD24 | BD Biosciences | Cat# 560992; RRID:AB_10562033 |
| FITC Anti-human CD44 | BD Biosciences | Cat# 555478; RRID:AB_395870 |
| PE anti-mouse H-2K[d] | BD Biosciences | Cat# 553566; RRID:AB_394924 |
| PE/Cy7 Anti-human CD24 | BD Biosciences | Cat# 561646; |
| PE/Cy7 Anti-human CD24 | BioLegend | Cat# 311120; AB_2259843 |
| Bacterial and Virus Strains | ||
| Cignal Lentiviral Reporter (ARE-LUC) | QIAGEN | CLS-2020L |
| Biological Samples | ||
| MC1 Patient-derived xenografts | University of Michigan | N/A |
| Vari068 Patient-derived xenografts | Van Andel Research Institute | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) | Sigma-Aldrich | Cat# A9978; CAS: 2627-69-2 |
| Auranofin | Enzo Life Sciences | Cat# BML-EI206-0100; CAS: 34031-32-8 |
| Catalase-Polyethylene Glycol | Sigma-Aldrich | Cat# C4963 |
| CellROX® Orange Reagent | Thermo Fisher Scientific | Cat# C10443 |
| 2-Deoxy-D-glucose (2DG) | Sigma-Aldrich | Cat# D8375; CAS: 154-17-6 |
| Dorsomorphin dihydrochloride (Compound C) | TOCRIS | Cat# 3093; CAS: 1219168-18-9 |
| γ-Secretase Inhibition I | Millipore Sigma | Cat# 565750 |
| Hydrogen peroxide solution, 30% (w/w) | Sigma-Aldrich | Cat# H1009; CAS: 7722-84-1 |
| L-Buthionine-sulfoximine (BSO) | Sigma-Aldrich | Cat# B2515; CAS: 83730-53-4 |
| Matrigel® Basement Membrane Matrix | Corning Life Sciences | Cat# 354234 |
| MitoPQ | Cayman Chemical | Cat# 18808 CAS: 1821370-28-8 |
| MitoSOX™ Red | Thermo Fisher Scientific | Cat# M36008 |
| MitoTEMPO (hydrate) | Cayman Chemical | Cat# 16621 CAS: 1569257-94-8 |
| N-Acetyl-L-cysteine (NAC) | Sigma-Aldrich | Cat# A7250; CAS: 616-91-1 |
| Superoxide Dismutase-Polyethylene Glycol (SOD) | Sigma-Aldrich | Cat# S9549 |
| Trigonelline hydrochloride | Sigma-Aldrich | Cat# T5509 |
| Critical Commercial Assays | ||
| Aldefluor Assay | STEMCELL Technologies | Cat# 01700 |
| Luciferase Reporter Gene Assay | Sigma-Aldrich | Cat# 11669893001 |
| Deposited Data | ||
| Data files for RNA sequencing | This paper | GSE115302 |
| Experimental Models: Cell Lines | ||
| Human: HCC1937 | ATCC | CRL-2336™ |
| Human: HCC1806 | ATCC | CRL-2335™ |
| Human: MCF7 | ATCC | HTB-22™ |
| Human: SUM149 and SUM159 | Stephen P. Ethier, Ph.D. | Medical University of South Carolina |
| Human: T47D | ATCC | HTB-133™ |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD/SCID | University of Michigan | N/A |
| Oligonucleotides | ||
| ALDH1A1 | ThermoFisher Scientific | Hs00946916_m1 |
| ALDH1A3 | ThermoFisher Scientific | Hs00167476_m1 |
| GAPDH | ThermoFisher Scientific | Hs02786624_g1 |
| HES1 | ThermoFisher Scientific | Hs00172878_m1 |
| NOTCH1 | ThermoFisher Scientific | Hs01062014_m1 |
| NRF2 | IDT | Forward primer: TGCCAACTACTCCCAGGTTG Reverse primer: AAGTGACTGAAACGTAGCCGA |
| Recombinant DNA | ||
| HIF1α shRNA clone TRCN0000003808 | Sigma-Aldrich | SHCLNG-NM_001530 |
| HIF1α shRNA clone TRCN0000003809 | Sigma-Aldrich | SHCLNG-NM_001530 |
| HIF1α shRNA clone TRCN0000003810 | Sigma-Aldrich | SHCLNG-NM_001530 |
| HIF1α shRNA clone TRCN0000003811 | Sigma-Aldrich | SHCLNG-NM_001530 |
| HIF1α shRNA clone TRCN00000010919 | Sigma-Aldrich | SHCLNG-NM_001530 |
| NRF2 shRNA clone TRCN0000007555 | Sigma-Aldrich | SHCLND-NM_006164 |
| NRF2 shRNA clone TRCN0000007557 | Sigma-Aldrich | SHCLND-NM_006164 |
| PLKO_Scrambled | University of Michigan | N/A |
| Software and Algorithms | ||
| ELDA | WEHI Bioinformatics Resources | http://bioinf.wehi.edu.au/software/elda/index.html |
| FlowJo | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo/ |
| GraphPad Prism | GraphPad Software Inc | http://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Illumina Casava1.8.2 | Illumina | https://www.illumina.com/ |
| STAR 2.3.1 | Open Source | http://code.google.com/p/rna-star/ |
| R | Open Source | www.r-project.org |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to the Lead Contact, Max S. Wicha (mwicha@med.umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
SUM149 and SUM159-Luc cells were cultured in Ham’s F-12 (Invitrogen) supplemented with 5% FBS (ThermoFisher Scientific, Pittsburgh, PA), 5 μg/mL insulin, and 1 μg/mL hydrocortisone (both from Sigma, St. Louis, MO), 10 μg/ml gentimycin, and 1% antibiotic-antimycotic (both from Invitrogen). MCF-7 was maintained in EMEM medium (ThermoFisher Scientific) supplemented with 5% FBS, 1% antibiotic-antimycotic (both from ThermoFisher Scientific), and 5 μg/ml insulin (Sigma, St Louis, MO). HCC1806, HCC1937 and T47D were maintained in RPMI1640 medium (ThermoFisher Scientific) supplemented with 10% FBS and 1% antibiotic-antimycotic. All cell lines were developed from female breast cancer patients and are cultured in an incubator at 37°C with 5% CO2.
Mouse Models
Vari068 xenograft model was recently established by injecting dissociated tumor cells derived from a female Caucasian patient with TNBC into the No. 4 mammary fat pads of NOD/SCID mice. A section of tumor from a lumpectomy of the Caucasian patient with TNBC was submitted to Van Andel Research Institute. MC1 xenograft model was established from an ER−PR−ERBB2− tumor derived from a female patient at the University of Michigan Hospital, and currently is at the 20th passage in NOD/SCID mice. Patient-derived xenograft models of TNBC including Vari068 and MC1 were established with informed patient consent and approved by the institutional review board at the University of Michigan under the IRB protocol IRBMED: 2001–0344.
Animal Studies and Drug Treatment
Female NOD/SCID mice were bred at the Unit for Laboratory Animal Medicine (ULAM) of University of Michigan and housed in AAALAC-accredited specific pathogen-free rodent facilities in standard cages at constant temperature (24 °C) and humidity (29.1%), with 12 hour light-dark cycles and standard chow. Tumor growth was determined by injecting primary breast cancer cells (Vari068: 5×105 cells per site; MC1: 2000 cells per site) with 30% Matrigel (BD Biosciences) into the #4 mammary fat pads of 6-week-old female NOD-SCID mice with 8–10 mice per cohort. Drug treatments were started when the tumors reached to 3–5mm in diameter. To detect the antitumor effect on systemic metastasis, luciferase-labeled SUM159 BC cells (100K in 100 μl of PBS) were injected into the left ventricle of 6–8-week-old female NOD/SCID mice. Drug treatment started at the following day of cardiac injection. Metastasis formation was detected by bioluminescent imaging after 4 and 7 weeks of treatment. 2DG (400mg/kg), Auranofin (1.5–5 mg/kg) and BSO (30 mg/kg) were given by I.P., every two days. Tumor size was measured once a week with a caliper, and calculated as tumor volume = Length x Width2/2. Animals were euthanized at the end of treatments and tumor cells were dissociated into single cells and analyzed by Aldeflour or CD24CD44 flow cytometry. Tumor cells of human origin in tumor xenografts were obtained by gating out mouse H2Kd+ cells. For secondary transplantation, H2Kd− primary tumor cells sorted by flow cytometry were prepared in 30% Matrigel (BD Biosciences) with 2 or 3 different dilutions, and injected bilaterally into the #4 mammary fat pads of 6-week-old female NOD-SCID mice with 3–4 mice per dilution. Tumor appearance was monitored for 3 months and frequency of tumor initiating cells following transplantation was calculated using the ELDA software (Walter + Eliza Hall Bioinformatics, Institute of Medical research). All mouse experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of University of Michigan.
METHOD DETAILS
RNA Sequencing, Gene Profiling and iPathway Analysis
Total RNA was extracted from E- (H2Kd−ALDH+), M- (H2Kd−CD44+/CD24−) BCSC-enriched and bulk (H2Kd−ALDH−CD44−/CD24+) cell populations freshly sorted from dissociated tumor cells of Vari068 and MC1 PDXs. RNA samples were sent to Tgen (Arizona, USA) for processing and sequencing. Expression data for annotated metabolic and antioxidant pathway genes were extracted. For each PDX, data of M- and E-BCSCs were normalized to Bulk. The statistical program R was then used to create a heatmap with the heatmap.2 command with no row or column clustering and with row data scaling, which is shown as the Row Z-Score. For iPathway analysis (https://ipathwayguide.advaitabio.com), 3759 differentially expressed genes were identified out of a total of 9983 genes with measured expression. These were obtained using a threshold of 0.05 for statistical significance (p-value) and a log fold change of expression with absolute value of at least 0.6. These data were analyzed in the context of pathways obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Release 81.0+/01–20, Jan 17), gene ontologies from the Gene Ontology Consortium database (2016–Sep26), miRNAs from the miRBase (Release 21) and TARGETSCAN (Targetscan version: 7.1) databases, and diseases from the KEGG database (Release 81.0+/01–20, Jan 17).
Cell Labeling and Flow Cytometry
To determine the effect of drug treatment on M- and E-BCSCs in basal BC cell lines, SUM149 or HCC1806 following various treatment were digested by 0.25% Trypsin-EDTA, re-suspended in HF buffer (HBSS plus 2% FBS) at 2×105 cells/100 μl and incubated with antibodies against human CD24 (PE-Cy7-conjugated, 1:100 from BD for SUM149 or 1:75 from Biolegend for HCC1806) and CD44 (APC-conjugated, 1:200, from BD) in cold room for 30 minutes. Content of ALDH+ E-BCSCs was determined by Aldefluor assay (StemCell Technologies) per manufacturer’s instructions. After labeling, or assay buffer 6-cells were washed twice, re-suspended in HF Aldefluor containing 1μg/mL of 4′, diamidino-2-phenylindole (DAPI) (sigma) to discriminate live from dead cells. To label M-BCSCs from luminal BC cells (MCF7 and T47D), antibodies against human CD24 (FITC-conjugated, 1:200, BD) and CD44 (APC-conjugated, 1:200, BD) were used. To obtain M- and E-BCSCs and bulk tumor cells from tumor xenografts, tumors of Vari068 and MC1 PDX models grown in NOD/SCID mice were digested into single cells by collagenase for an hour and briefly shaken every 15 minutes. Cells debris was removed by filtration through a 40μm cell strainer (BD Biosciences) to obtain single cell suspensions. Red blood cells were removed with 0.8% ammonium chloride solution. The dissociated single tumor cells were re-suspended in HF buffer and incubated first with antibodies (all from BD Biosciences) against mouse H2Kd (PE conjugated, 1:100, BD) and human CD24 (PE-Cy7-conjugated, 1:100, BD) and CD44 (APC-conjugated, 1:100, BD) in cold room for 30 minutes. Tumor cells labeled with H2Kd, CD24 and CD44 antibodies were further subjected to Aldefluor assay and tumor cells of human origin in tumor xenografts were obtained by gating out mouse H2Kd+ cells. Flow cytometry analysis or sorting was performed on a MoFlow XDP or Astrios Cell Sorter (Beckman Coulter) at the University of Michigan Flow Cytometry Core facility.
Analysis of ROS and Mitochondrial Superoxide
To measure cellular ROS and mitochondrial superoxide levels, cells were stained with CellROX™ Orange or MitoSOX Red (all from Thermo Fisher Scientific), which generates florescent signals when oxidized by reactive oxygen species (ROS) or mitochondrial superoxide in the cells. Cells were incubated with pre-warmed CellROX or MitoSOX Red (diluted in PBS to a final concentration of 2.5 μM) staining solution for 30 min at 37 °C. All subsequent steps were performed in the dark. Cells were washed in PBS, harvested, and further labeled with Aldefluor assay or markers of M-BCSCs (CD24 and CD44 conjugated with corresponding fluorophores). Cells were then analyzed by flow cytometry. Data analysis was performed using the FlowJo software.
Metabolic States of BCSCs and Differentiated Tumor Cells by FLIM
To quantify the metabolism of distinct BCSC states vs. bulk tumor cells, we choose HCC1806 as a model system as the sorted E- (ALDH+), M- (ALDH−CD24−CD44+) as well as bulk tumor cells (ALDH−CD24+CD44−) fully adhere and spread in 2h when plated in complete medium, which is necessary for measuring their metabolic states and keeping their original stem or differentiated status. We conducted fluorescence lifetime imaging (FLIM) of the endogenous fluorophore NADH, a metabolic cofactor with long or short lifetimes reflecting its mitochondrial protein bound (OXPHOS) or unbound (glycolysis) state respectively. We performed all studies with a two-photon Olympus FVMPE RS microscope outfitted with a 25X/1.05 numerical aperture objective and Insight DS+ (Spectra-Physics) laser combined with an ISS FastFLIM system for phase-resolved lifetime measurements. We first calibrated the FLIM system using a fluorescein dye slide with a single component lifetime of 4.1 ns, 740nm excitation wavelength, 552nm dichroic mirror, and an open filter. For imaging samples, we used a 740nm excitation wavelength, 552nm dichroic mirror, and 435/50 band pass filter for emitted light from NADH. We scanned each plane 16 times with 2x zoom at 512×512 pixels with a pixel dwell time of 4μs at 15% laser to acquire sufficient counts to reach the threshold needed for reliable FLIM data based on recommendations of the manufacturer.
To analyze FLIM data, we exported the modulation lifetime and DC counts from the ISS VistaVision software into MATLAB. Using custom built software, we omitted data points that did not reach our count limit of 10 or did not have a modulation lifetime between a 0.5 and 5.5 ns. We then applied a Gaussian filter on the images and pseudo colored them to display their NADH lifetime. Data are presented as box and whisker plots plotted in GraphPad Prism with error bars representing the 5th and 95th percentiles, respectively. Significance testing was performed using one-way analysis of variance (ANOVA) and multiple comparison functions in MATLAB.
Apoptosis, Cell proliferation and Differentiation
Apoptotic cells in cultured SUM149 breast cancer cells following 2DG, AUR or BSO treatment were determined by labeling with APC-AnnexinV (BD, 1:50) and subjected to flow cytometry to detect AnnexinV-staining cells in E- (ALDH+), M- (CD24−CD44+) BCSCs and bulk cells (CD24+CD44− or ALDH−). To detect proliferative status of tumor cells in tissue sections or E-BCSCs following AUR, BSO or 2DG treatment, dewaxed and antigen retrieved tumor sections or 4% paraformaldehyde fixed SUM149 breast cancer cells cultured on chamber slides were labeled by Ki67 (1:100, Cell Signaling Technology) together without or with ALDH1 (LSBio, 1:100) antibodies. SUM149 BC cell differentiation following AUR or BSO treatment was determined by fluorescent staining of luminal epithelial cell markers cytokeratin8/18 (American Research Products, 1:300) and CD24 (BD Biosciences, 1:100). All tissue/cell samples were mounted with VECTASHIELD mounting medium with DAPI (Vector Laboratories). Slides were examined under a BX41 microscope (Olympus) using UplanF1 20x-40x/0.5 objective lenses. Images were captured with a DP70 camera with DP Controller version 1.2.1.108 (Olympus).
RNA Extraction and qRT-PCR
Total RNA was extracted using an RNeasy Mini kit (Qiagen), and 10ng to1 μg of RNA was used for cDNA generation with the High-Capacity cDNA Reverse Transcription Kit (Life Technologies). cDNA was analyzed using TaqMan Real-Time PCR Master Mix (ThermoFisher Scientific) on ABI PRISM 7900HT Real-Time PCR system (Applied Biosystems) according to the manufacturer’s instructions. Commercial primer pairs of NOTCH1, HES1, ALDH1A1, ALDH1A3 and GAPDH as well as NRF2 RT-PCR primers are listed in key resource table. The relative gene expression level of mRNAs was generated by normalization to internal control GAPDH.
Western Immunoblotting
Total cell protein was extracted with RIPA buffer (10mM Tris.Cl, 100mM NaCl, 2mM EDTA, 20mM NaF, 5 mM Na3VO4, 0.5% sodium deoxycholate, 0.1% SDS, 1% Triton X-100) supplemented with protease inhibitor cocktail (Sigma). Cell lysates were subjected to 10% SDS-PAGE, transferred to nitrocellulose membrane and probed with different primary antibodies as listed in the resource table. Secondary antibodies against rabbit and mouse antibodies were purchased from Cell Signaling Technology and Santa Cruz Biotechnology respectively.
Tumorsphere Assay
For tumorsphere culture of SUM149 BC cell lines transduced with shNRF2 7555, 7557 or SCR, 20 cells were allocated into each well of 96-well ultralow attachment plate (Corning) in 120 μl of completed human MammoCult medium (StemCell Technologies) supplemented with 4 μg/mL heparin, 1μg/mL hydrocortisone, 1% antibiotic-antimycotic and 20 μg/mL gentamycin (all from Invitrogen) and cultured for 7–10 days. Tumorspheres with diameter over 40 μM were counted under an optical microscope with 10x optical lens (EVOS all-in-one digital inverted microscope). For secondary tumorsphere formation, spheres in all wells of each 96-well ultralow attachment plate were pooled and dissociated into single cells by 0.25% trypsin-EDTA. Cells (20 cells/well) derived from primary spheres were cultured at the same conditions for 7–10 days as described above, counted and plotted for mean sphere formation per 100 cells. Tumorspheres of parental SUM149 BC cells under various drug treatment regimens were cultivated at the condition of10 cells/well. For sphere formation of Vari068 xenograft tumor cells, H2Kd− tumor cells of human origin were sorted and placed at a density of 20,000 cells/mL (first passage, P1) or 5,000 cells/mL (second passage, P2) in 6 well ultralow attachment plates (Corning). Cells were cultured in completed human MammoCult medium for 7–10 days. To count tumorspheres, spheres formed in each well of 6-well plate were collected and transferred to one well of regular 96-well flat-bottomed plate. Tumorspheres settled in these conditions were counted under an optical microscope at low magnification.
shRNAs and Lentiviral Infection
Lentiviral mission shRNA clones against HIF1α (TRCN0000003808, TRCN0000003809, TRCN0000003810, TRCN0000003811 and TRCN0000010819), NRF2 (TRCN0000007555 and TRCN0000007557) were purchased from Sigma. Recombinant lentiviruses expressing HIF1α or NRF2 shRNA as well as scrambled sequence were packaged at the University of Michigan Vector Core. Parental SUM149 BC cells were infected with recombinant lentiviruses at the presence of polybrene (8μg/ml, Millipore) overnight and the medium containing viruses was changed and replaced with fresh medium after 20h of infection. Puromycin (Invitrogen) selection was performed using 2.5μg/ml for 1 week to select lentiviral vector transduced cells.
Tumor Glutathione Assay
To detect GSH/GSSG ratios in tumor tissues following Vehicle or AUR+BSO treatment, tumor samples were stored frozen at −80 for less than 2 weeks before processing. Approximately 2 mg sample of 10 xenografts were homogenized in 150 μL DETAPAC buffer [1.34 mM Diethylenetriaminepenta-Acetic acid (Sigma D6518) in potassium phosphate buffer]. After centrifugation 50 μL of supernatant was combined with 50 μL of 5% sulfosalicylic acid (Sigma S2130). Total GSH content was determined by mixing 700 μl working buffer [0.298 mM NADPH (Sigma N6505) in sodium phosphate buffer], 100 μl Ellman’s Reagent [6 mM 5,5 - dithio-bis-2-nitrobenzoic acid (Sigma D8130) in sodium phosphate buffer], 100 μl dH2O, 5–25 μl of sample and 50 μl glutathione reductase (Sigma G4759 made to ~0.023 U/μL in water) in a plastic cuvette and the absorbance read at 412 nm, every 15 seconds for 2.5 min in a 6 sample automatically controlled DU670 Beckman spectrophotometer. The baseline rate obtained using the equivalent amount of sample buffer was subtracted from each sample rate. Glutathione disulfide (GSSG) was determined by adding 3 μL of a 1:1 mixture of 2-vinylpyridine (Sigma-Aldrich 132292) and ethanol to 50 μL of sample and incubating for 2 hours before adding the 5% sulfosalicylic acid and assaying as described. The rates of the reactions were compared with similarly prepared GSH and GSSG standard curves. GSH determinations were normalized to the protein content of 2 μL of homogenate in DETAPAC buffer by Lowry method.
Thioredoxin Reductase Assay
Approximately 2 mg sample of 10 xenografts were homogenized in thioredoxin reductase assay kit buffer with the addition of mini-protease inhibitor (Roche 04693159001) and centrifuged for 10,000g for 15 minutes. 30–50 μL of the supernatants were then processed using the thioredoxin reductase assay kit (Sigma CS0170). A standard curve was made using pure thioredoxin reductase and all sample’s subtracted rates (with and without inhibitor) were within the range of the standard curve. Each sample was normalized to protein using the Lowry protein assay.
QUANTIFICATION AND STATISTICAL ANALYSIS
Kaplan-Meier Analysis
Kaplan-Meier analysis of the TXN and GSH antioxidant pathway genes in 3951 breast cancer patient database (including all molecular subtypes) are performed using Kaplan-Meier Plotter (http://kmplot.com/analysis/index.php?p=service&default=true). Selected TXN antioxidant pathway genes with correlation to poor patient survival were further analyzed using restricted analysis to different molecular subtypes.
Statistical Analysis
Results are presented as the Mean ± SD (standard deviation) for representative experiments with 2–3 independent biological repeats. The statistical parameters used for the specific sets of data are described in the figure legends. In most cases, the unpaired, two-tailed Student’s t test was used to compare two groups. A P value of < 0.05 was considered as statistically significant. No statistical method was used to determine whether the data met assumptions of the statistical approach. Statistical analysis was performed using the Prism 6 (GraphPad) software.
DATA AND SOFTWARE AVAILABILITY
The processed and raw data files of RNA-Seq reported in this paper have been deposited at https://www.ncbi.nlm.nih.gov/geo with access number: GSE115302.
Supplementary Material
Highlights.
E/M-BCSCs have divergent sensitivities to glycolysis/redox metabolism inhibition
Hypoxic/oxidant stress promotes M to E state transition by activating AMPK-HIF1α
E-BCSCs are more oxidative (OXPHOS) and reliant on NRF2 antioxidant responses
Co-inhibition of glycolysis and TXN/GSH pathways targets both M- and E-BCSCs
Acknowledgments
We thank UMCCC Flow Cytometry Core for assistance in flow analysis and sorting, Jill Granger for editing the manuscript. This research was supported by R01 CA101860 and R35 CA197585 from the NIH and from the BCRF to M.S.W.; R01 CA182804 and P30 CA086862 to D.R.S.; and R01 CA196018 and U01 CA210152 to G.D.L.
Footnotes
AUTHOR CONTRIBUTIONS
M.L. designed and executed each experiment and wrote the manuscript. J.M.B performed and analyzed FLIM experiments. M.S.W. G.D.L, J.M.B. and D.R.S. reviewed experimental data and edited the manuscript. L.S. helped for western blotting, flow cytometry and animal studies, M.B. for heatmap and graphics design, Y.Z. for qRT-PCR, M.A.F for TR/GSH analysis, A.D., E.G, Y.W., A.L. and D.T. for cell culture and flow cytometry, R.H. for fluorescent imaging, E.J., S.M. and S.D.M for RNA-Seq and S.C. for helpful ideas.
DECLARATION OF INTERESTS
M.S.W. holds equity in OncoMed Pharmaceuticals.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arlt A, Sebens S, Krebs S, Geismann C, Grossmann M, Kruse ML, Schreiber S, Schafer H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene. 2013;32:4825–4835. doi: 10.1038/onc.2012.493. [DOI] [PubMed] [Google Scholar]
- Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. The Biochemical journal. 2009;418:29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, Datar RH, Cote RJ. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:5615–5621. doi: 10.1158/1078-0432.CCR-06-0169. [DOI] [PubMed] [Google Scholar]
- Brabletz T. To differentiate or not--routes towards metastasis. Nature reviews Cancer. 2012;12:425–436. doi: 10.1038/nrc3265. [DOI] [PubMed] [Google Scholar]
- Brooks MD, Burness ML, Wicha MS. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell stem cell. 2015;17:260–271. doi: 10.1016/j.stem.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. The Journal of biological chemistry. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- Chio II, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Ohlund D, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell. 2016;166:963–976. doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, De Cola A, Scavo E, Carollo R, D’Agostino D, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell death & disease. 2014;5:e1336. doi: 10.1038/cddis.2014.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature reviews Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circulation research. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer cell. 2013;23:316–331. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Viollet B, Tormos KV, Chandel NS. Compound C inhibits hypoxic activation of HIF-1 independent of AMPK. FEBS letters. 2007;581:5727–5731. doi: 10.1016/j.febslet.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath MA, Ahmad IM, Smith CJ, Spence J, Spitz DR. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:6206–6217. doi: 10.1158/1078-0432.CCR-11-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer cell. 2007;12:230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochemical pharmacology. 2014;92:90–101. doi: 10.1016/j.bcp.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandel NS. Targeting SOD1 reduces experimental non-small-cell lung cancer. J Clin Invest. 2014;124:117–128. doi: 10.1172/JCI71714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature reviews Drug discovery. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends in biochemical sciences. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault S, Davis A, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer research. 2013;73:1635–1646. doi: 10.1158/0008-5472.CAN-12-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SN, Yang WK, Kim J, Kim HS, Kim EJ, Yun H, Park H, Kim SS, Choe W, Kang I, et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis. 2008;29:713–721. doi: 10.1093/carcin/bgn032. [DOI] [PubMed] [Google Scholar]
- Kolenc OI, Quinn KP. Evaluating Cell Metabolism Through Autofluorescence Imaging of NAD(P)H and FAD. Antioxidants & redox signaling. 2018 doi: 10.1089/ars.2017.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D’Angelo R, Paulson AK, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Molecular cell. 2012;47:570–584. doi: 10.1016/j.molcel.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R, Bonuccelli G, Ozsvari B, Peiris-Pages M, Fiorillo M, Smith DL, Bevilacqua G, Mazzanti CM, McDonnell LA, Naccarato AG, et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget. 2015;6:30453–30471. doi: 10.18632/oncotarget.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sanchez V, Sanders ME, et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell metabolism. 2017;26:633–647.e637. doi: 10.1016/j.cmet.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Fath MA, Scarbrough PM, Watson WH, Spitz DR. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox biology. 2015;4:127–135. doi: 10.1016/j.redox.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Reports. 2014;2:78–91. doi: 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015a;21:1301–1310. doi: 10.2174/1381612821666141211120604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Clouthier SG, Deol Y, Liu S, Nagrath S, Azizi E, Wicha MS. Breast cancer stem cells: current advances and clinical implications. Methods Mol Biol. 2015b;1293:1–49. doi: 10.1007/978-1-4939-2519-3_1. [DOI] [PubMed] [Google Scholar]
- Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, Kaminska B, Huelsken J, Omberg L, Gevaert O, et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell. 2018;173:338–354.e315. doi: 10.1016/j.cell.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Papaldo P, Lopez M, Cortesi E, Cammilluzzi E, Antimi M, Terzoli E, Lepidini G, Vici P, Barone C, Ferretti G, et al. Addition of either lonidamine or granulocyte colony-stimulating factor does not improve survival in early breast cancer patients treated with high-dose epirubicin and cyclophosphamide. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003;21:3462–3468. doi: 10.1200/JCO.2003.03.034. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, et al. A mitochondrial switch promotes tumor metastasis. Cell reports. 2014;8:754–766. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- Prasanna VK, Venkataramana NK, Dwarakanath BS, Santhosh V. Differential responses of tumors and normal brain to the combined treatment of 2-DG and radiation in glioablastoma. Journal of cancer research and therapeutics. 2009;5(Suppl 1):S44–47. doi: 10.4103/0973-1482.55141. [DOI] [PubMed] [Google Scholar]
- Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Molecular oncology. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, Baudy AR, Hatzivassiliou G, Evangelista M, Settleman J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer cell. 2016;29:548–562. doi: 10.1016/j.ccell.2016.02.018. [DOI] [PubMed] [Google Scholar]
- Robb EL, Gawel JM, Aksentijevic D, Cocheme HM, Stewart TS, Shchepinova MM, Qiang H, Prime TA, Bright TP, James AM, et al. Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free radical biology & medicine. 2015;89:883–894. doi: 10.1016/j.freeradbiomed.2015.08.021. [DOI] [PubMed] [Google Scholar]
- Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra215. doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- Shutt DC, O’Dorisio MS, Aykin-Burns N, Spitz DR. 2-deoxy-D-glucose induces oxidative stress and cell killing in human neuroblastoma cells. Cancer biology & therapy. 2010;9:853–861. doi: 10.4161/cbt.9.11.11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringari C, Nourse JL, Flanagan LA, Gratton E. Phasor fluorescence lifetime microscopy of free and protein-bound NADH reveals neural stem cell differentiation potential. PloS one. 2012;7:e48014. doi: 10.1371/journal.pone.0048014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syu JP, Chi JT, Kung HN. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget. 2016;7:14659–14672. doi: 10.18632/oncotarget.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nature reviews Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Luo M, Brooks M, Clouthier SG, Wicha MS. Biological and clinical significance of cancer stem cell plasticity. Clin Transl Med. 2014;3:32. doi: 10.1186/s40169-014-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.