

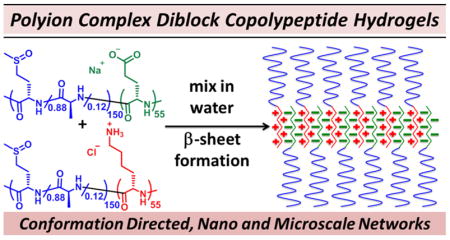
Abstract
Synthetic diblock copolypeptides were designed to incorporate oppositely charged ionic segments that form β-sheet-structured hydrogel assemblies via polyion complexation when mixed in aqueous media. The observed chain conformation directed assembly was found to be required for efficient hydrogel formation and provided distinct and useful properties to these hydrogels, including self-healing after deformation, microporous architecture, and stability against dilution in aqueous media. While many promising self-assembled materials have been prepared using disordered or liquid coacervate polyion complex (PIC) assemblies, the use of ordered chain conformations in PIC assemblies to direct formation of new supramolecular morphologies is unprecedented. The promising attributes and unique features of the β-sheet-structured PIC hydrogels described here highlight the potential of harnessing conformational order derived from PIC assembly to create new supramolecular materials.
Graphical Abstract
INTRODUCTION
The concept of conformation-directed assembly has been widely utilized in the design and preparation of ordered supramolecular structures.1–3 In particular, assemblies containing peptides and polypeptides often take advantage of hydrophobic and H-bonding interactions between ordered chain conformations in these molecules, e.g., α-helices and β-strands, to produce materials with advantageous properties, such as defined morphology or enhanced stability.4 Polypep-tide-containing block copolymer assemblies, such as micelles, vesicles, and hydrogels, have also been prepared using polyion complexes (PIC),5,6 where oppositely charged chain segments aggregate and phase separate upon mixing in aqueous media. In these systems, as well as those based on other synthetic polymers, formation of unstructured, liquid PIC coacervate domains is common and often desired. Fluidity in PIC coacervates can assist rapid complex formation and equilibration, while formation of solid β-sheet structures can lead to irregular assemblies with less desirable properties.5–8 In a contrarian approach, we sought to generate conformational order in polypeptides using PIC assembly to direct supra-molecular assemblies into unprecedented morphologies with desirable properties. Using knowledge that certain homochiral polypeptide PICs form β-sheet aggregates,9–11 we designed diblock copolypeptides incorporating such oppositely charged ionic segments that were able to form supramolecular hydrogels via PIC assembly in aqueous media. Since these hydrogels assemble via ordered chain conformations, different from other PIC hydrogels that require triblock copolymers and utilize disordered confromations,12–15 they possess distinct properties such as rapid recovery after stress and microporous architecture. Furthermore, these PIC diblock copolypeptide hydrogels (DCHPIC) possess certain advantages over hydro-phobically assembled diblock copolypeptide hydrogels (DCH)16 in that they are resistant to dilution in aqueous media and are readily prepared at high concentrations for increased hydrogel stiffness.
Aqueous PIC assembly of block copolymers containing both nonionic and oppositely charged ionic segments has been used to prepare a diverse array of micelles, vesicles, and hydro-gels.5,6,17 Most polypeptide-containing PIC assemblies utilize polyethylene glycol (PEG) chains as hydrophilic nonionic segments, as well as ionic polypeptide segments that form disordered or liquid coacervate immiscible phases.5,6,12–15,18,19 The resulting lack of internal order in the complexes tends to favor formation of spherical assemblies as found in diblock copolymer micelles and triblock copolymer hydro-gels.5,6,12–15,18,19 Kataoka’s laboratory has further shown that use of polypeptide segments that are long relative to PEG chain lengths results in formation of PICsome vesicular membrane assemblies.20,21 In the few examples where internal order has been incorporated into polypeptide PIC assemblies, via use of ionic α-helical segments8 or by β-sheet formation during assembly,7,22 only minimal perturbation of spherical micelle formation or slowed formation of micelles with increased polydispersity was observed. While there are examples of peptides and polypeptides where β-sheet structures are used to direct formation of self-assembled materials, these all rely on other components, such as hydrophobic and nonionic residues to drive β-sheet formation.23,24 To the best of our knowledge, there are no reports demonstrating formation of new supramolecular morphologies that rely on assembly of polyelectrolyte segments into β-sheet-rich domains.
The complexation of oppositely charged polypeptide electrolytes in aqueous media has been under study for many decades.5,6 Blout and Idelson were the first to report that a mixture of homochiral poly(L-lysine-HCl), K, and poly(L-glutamate-Na), E, in water gave rise to phase separation of PICs rich in β-sheet content.9 Later studies confirmed this result and showed that while the mixture of L-lysine and L-glutamate homopolymers formed β-sheets, other combinations with L-ornithine or L-aspartate homopolymers gave only disordered PICs.10 This observation led to the hypothesis that interactions between hydrophobic side chains, which are longer in lysine and glutamate, helped stabilize formation of the observed PIC β-sheet structure. More recently, the importance of chirality was shown by studies where replacement of one or both of the homochiral K and E components with racemic equivalents, i.e., rac-K or rac-E, led to formation of liquid coacervates instead of solid β-sheets.7,22 Although most designs for polypeptide PIC assemblies favor formation of complexes with disordered internal structure,5,6 we sought to take advantage of the ability of homochiral K- and E-based PICs to form ordered β-sheet assemblies.
The design of DCHPIC was developed from our understanding of hydrophobically assembled amphiphilic DCH. In these materials, the combination of long, hydrophilic segments with disordered conformations, and shorter hydrophobic segments with either α-helical or β-sheet ordered conformations, e.g., poly(L-lysine-HCl)180-block-poly(L-leucine)20, K180L20, or poly(L-lysine-HCl)160-block-poly(L-valine)40, K160V40, respectively, was found to direct their assembly in water into anisotropic structures that branch and entangle to give 3D hydrogel networks.16 The key molecular elements required for hydrogel formation are the conformation-directed assembly of ordered hydrophobic segments into anisotropic structures instead of spherical micelles and long, disordered hydrophilic chains that sterically limit packing of hydrophobic segments into tapelike assemblies as opposed to flat 2D membranes. To create DCHPIC, we sought to test if the ordered hydrophobic component in amphiphilic DCH, e.g., β-sheet-forming V segments, could be replaced with β-sheet PICs formed from mixing of K and E chains (Figure 1). Concurrently, it was also necessary to replace the disordered, ionic hydrophilic component of amphiphilic DCH, e.g., charged K segments, with a disordered, nonionic hydrophilic component. While there are a few candidate water-soluble, disordered, nonionic polypeptides,25 we chose to use segments based on water-soluble poly(L-methionine sulfoxide), MO, as this polymer is readily prepared, avoids the need to use racemic amino acid monomers, and is a naturally occurring residue that shows minimal toxicity.26–28
Figure 1.
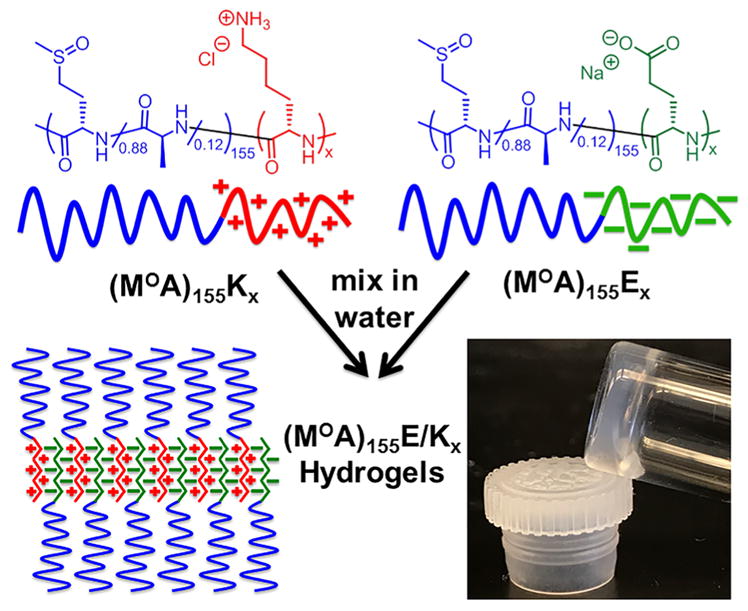
Schematic showing the assembly process for preparation of polyion complex (MOA)155E/Kx diblock copolypeptide hydrogels.
RESULTS AND DISCUSSION
Candidate DCHPIC compositions were designed to include long, disordered hydrophilic segments as well as oppositely charged ionic segments able to form β-sheet complexes upon mixing (Figure 1). For shorter chain lengths (ca. 60 residues), nonionic, hydrophilic MO segments have been conveniently prepared from poly(L-methionine), M, precursors by post-polymerization oxidation.28 However, at longer chain lengths, i.e., >100 residues, M polymers aggregate during polymerization, which limits the ability to control the chain length and prepare well-defined block copolymers.29 This issue was circumvented by mixing a small amount of L-alanine N-carboxyanhydride (NCA) monomer (ca. 12 mol %) with L-methionine NCA to prepare statistical copolymer segments that did not aggregate during polymerization due to disrupted side-chain packing.29 The incorporation of a small amount of minimally hydrophobic alanine was found to allow efficient copolypeptide synthesis without adversely affecting the water solubility or disordered conformation of the resulting poly(L-methionine sulfoxide-stat-L-alanine), MOA, segments compared to the MO homopolymer (see the Supporting Information, Figure S13).26,28
Using this strategy, we prepared diblock copolypeptides containing poly(L-methionine-stat-L-alanine), MA, segments ca. 155 residues long, followed by side-chain-protected K or E segments of different lengths (Scheme 1).16,28 Subsequent oxidation of M residues, followed by side-chain deprotection of K and E residues and purification, gave the target copolypeptides poly(L-methionine sulfoxide-stat-L-alanine)155-block-poly(L-lysine-HCl)x, (MOA)155Kx, and poly(L-methionine sulfoxide-stat-L-alanine)155-block-poly(L-glutamate-Na)x, (MOA)155Ex, where x = 30, 60, 90, and 120 (Scheme 1). All copolymers were isolated in high yield with compositions that closely matched the expected values (see the Supporting Information, Table S1). The MOA segment length was chosen on the basis of values known, from previous studies on amphiphilic DCH,16 to be sufficiently long to promote hydrogel formation. The K and E lengths were varied to study the role of the structured PIC domain size on the hydrogel formation and properties.
Scheme 1.

Synthesis of Oppositely Charged, Dual Hydrophilic Diblock Copolypeptides (MOA)155Kx and (MOA)155Exa
aReaction conditions: (a) TBHP, CSA, H2O, 20 °C, 1 d; (b) K2CO3, MeOH/H2O, 50 °C, 8 h; (c) MSA, TFA, anisole, 20 °C, 1.5 h.
For initial evaluation, matching length (MOA)155Kx and (MOA)155Ex samples were separately dissolved in aqueous 1× PBS buffer (5.0 wt % concentration of each copolypeptide) to give clear solutions. These solutions were then combined in equal volumes at essentially stoichiometric E to K ratios (ca. 1.02–1.04 to 1) and agitated briefly in a vortex mixer, whereupon all samples ((MOA)155E/Kx, x = 30, 60, 90, and 120; 5.0 wt % total copolypeptide after mixing) were observed to form hydrogels within 15 s to 1 min. These observations were confirmed by oscillatory rheology measurements where storage moduli (G′) were found to dominate over loss moduli (G ), indicating elastic behavior for all samples (Figure 2A; see the Supporting Information, Table S2).16 Mismatched mixtures, prepared either using nonstoichiometric E to K ratios or by maintaining the E to K stoichiometry but combining samples of different lengths (e.g., three (MOA)155K30 molecules to one (MOA)155E90 molecule), were found to give substantially weaker hydrogels compared to corresponding stoichiometric and length-matched samples. Stoichiometric hydrogels prepared using longer E/Kx segments (90 and 120) were opaque, likely due to microscopic aggregate precipitation. Hydrogels prepared using shorter E/Kx segments (30 and 60) were translucent, with only slight turbidity. In general, hydrogel stiffness (G′) was found to increase with the E/Kx segment length, yet aggregate precipitation with longer segments diminished this trend, as can be seen in G′ for the E/K90 sample (Figure 2A). The minimum total copolypeptide concentration required for hydrogel formation was found to be ca. 4.0 wt % for the (MOA)155E/K30 sample, and decreased with increasing E/Kx segment length.
Figure 2.

PIC diblock copolypeptide hydrogel properties. (A) Storage modulus, G′ (Pa, blue), and loss modulus, G″ (Pa, red), of hydrogels formed from stoichiometric (MOA)155E/Kx with different ionic segment lengths (x = 30, 60, 90, and 120). Samples (5.0 wt % total combined cationic and anionic copolypeptide) were prepared in 1× PBS buffer at 20 °C. (B) G′ (Pa, blue) and G″ (Pa, red) of (MOA)155E/K60 hydrogels at different concentrations in PBS buffer at 20 °C. (C) G′ (Pa, blue) and G″ (Pa, red) of 5.0 wt % (MOA)155E/K60 hydrogels prepared in different buffers at 20 °C. DMEM = Dulbecco’s modified Eagle’s medium, and FBS = fetal bovine serum. For (A), (B), and (D), all G′ and G″ values were measured at an angular frequency of 5 rad/s and a strain amplitude of 0.05. (D) Recovery of 5.0 wt % in PBS (MOA)155E/K60 hydrogel properties (G′, filled circles; G , open circles) over time after large-amplitude oscillatory breakdown (strain amplitude of 10 at 5 rad/s for 200 s), followed by linear recovery measurement (strain amplitude of 0.05 at 5 rad/s).
Due to its desirable combination of hydrogel stiffness and minimal turbidity, (MOA)155E/K60 was chosen as an optimized DCHPIC composition for further study. Preparation of hydrogels using different concentrations of (MOA)155E/K60 in 1× PBS was found to be a convenient means to adjust the hydrogel stiffness (Figure 2B). All samples formed elastic hydrogels of similar clarity (G′ ≫ G″ over a range of frequency; see the Supporting Information, Figure S14), and their stiffness was found to increase with higher copolypeptide concentrations. The lack of visible aggregates in these samples suggests that polymer chains were able to assemble into the desired structures even with fast mixing at high concentrations. Since the precursor solutions of two-component DCHPIC are low-viscosity liquids, it is noteworthy that these hydrogels can be readily prepared at significantly higher concentrations relative to one-component, amphiphilic DCH, where dissolution of copolypeptide at higher concentration (i.e., >5.0 wt %) is hindered by spontaneous gel formation.16 (MOA)155E/K60 hydrogels could also be prepared in a variety of aqueous media (Figure 2C). Solution ionic strengths in the range of ca. 100–300 mM were found to be superior for PIC hydrogel formation, while deionized water and higher salt concentrations (e.g., 500 mM NaCl) resulted in weaker hydrogels. For 5.0 wt % (MOA)155E/K30 in 500 mM NaCl, a hydrogel did not form, and for 5.0 wt % (MOA)155E/K90, hydrogels were able to form up to 1 M NaCl, but did not form in 2 M NaCl. These results suggest that a lack of charge screening in pure water prevents annealing of PIC chains into ordered complexes (vide infra), while conversely excess charge screening in higher salt concentrations leads to weakened complexation and is PIC length dependent.5,6 Finally, elevated temperature (80 °C for 1.5 h) was found to have no visible effect on a 5.0 wt % (MOA)155E/K60 hydrogel in 1× PBS, showing that DCHPIC possess good thermal stability.
The self-healing properties of DCHPIC after mechanical breakdown were studied by subjecting a 5.0 wt % (MOA)155E/K60 sample in 1× PBS to high-amplitude oscillatory strain, and then monitoring the recovery of elasticity over time by measuring G′ at a much smaller strain amplitude (Figure 2D). During the initial 200 s of high strain amplitude, G′ dropped by 2 orders of magnitude to below the level of G , indicating the sample had become a viscous liquid. Upon switching to a low strain amplitude, the sample began recovering its elastic properties, with most of the original gel stiffness regained within the brief period (ca. 10 s) needed to switch between strain amplitudes. Full recovery of DCHPIC elasticity continued to occur over a time scale of minutes. The rapid self-healing ability of DCHPIC, which allows delivery of DCHPIC via injection through small-bore needles, is remarkably similar to that observed in amphiphilic DCHs, such as K180L20 or K160V40, despite their substantial differences in chemical composition and mode of assembly.16 The observed similarity in physical properties may arise from the modular design of these block copolymers that likely leads to formation of similar supramolecular assembled structures.
To better understand the assembly of DCHPIC, the influence of polyelectrolyte chirality on hydrogel formation was studied. We designed DCHPIC such that the K and E segments were envisioned to form structured PICs, rich in β-sheet content, upon complexation.7,9,10 To test this hypothesis, a new copolypeptide component, (MOA)155(rac-E)60, was prepared, where the rac-E segment was composed of racemic residues that should inhibit β-sheet formation in PICs.5–7,22 When equivalent amounts of (MOA)155(rac-E)60 and (MOA)155K60 were mixed (total 5.0 wt % in 1× PBS), the resulting sample did not form a hydrogel and gave only a low-viscosity liquid (see the Supporting Information, Figure S15). This result confirmed the importance of chirality in the formation of the (MOA)155E/K60 hydrogel structure. To directly verify the formation of β-sheet assembly in (MOA)155E/Kx DCHPIC’s, the hydrogels were also analyzed using FTIR, since different polypeptide conformations possess characteristic stretching frequencies for their amide I and amide II bands.30 In FTIR analysis of lyophilized (MOA)155E/Kx hydrogels (x = 30, 60, 90, and 120), all samples possessed strong 1653 cm−1 amide I bands due to the disordered chain conformations of the (MOA)155 segments (see the Supporting Information, Figures S13 and S16). The samples also possessed 1630 cm−1 amide I bands, characteristic of β-sheet chain conformations, which increased in intensity as the E/Kx segment length increased, suggesting that this band resulted from PIC formation (see the Supporting Information, Figure S16). The β-sheet amide I band at 1630 cm−1 was only present in the homochiral (MOA)155E/Kx PICs, and was absent in the individual components as well as the (MOA)155(rac-E)/K60 PIC formed with a racemic component (see the Supporting Information, Figure S17). Together, these data confirmed that the K and E segments in (MOA)155E/Kx PIC are assembling as β-sheets, and that this organized assembly is required for hydrogel formation.
The supramolecular structure of (MOA)155E/K60 hydrogels was analyzed at both microscale and nanoscale resolution. To visualize the microscopic structure, chains of (MOA)155E60 and (MOA)155K60 were separately conjugated with different fluorescent probes (tetramethylrhodamine and fluorescein, respectively) and then mixed to form DCHPIC. Laser scanning confocal microscopy (LSCM) was then used to visualize the labeled chains and the hydrogel network (Figure 3A,B). Both K-labeled (TRITC, red) and E-labeled (FITC, green) channels showed DCHPIC are composed of microporous networks containing interconnected polypeptide-rich domains that coexist with domains primarily composed of water, seen as dark regions in the images.31 An overlay of red and green channels revealed that K and E segments are colocalized, indicating good mixing of the components within the DCHPIC domains (Figure 3C). Cryo electron microscopy (cryoEM) imaging of a thin layer of vitrified (MOA)155E/K60 hydrogel showed structures resembling “plumber’s nightmare” morphologies, which consist of membrane-like regions interconnected with tapelike struts, and contain many defects that form a nanoporous network (Figure 3D). The combination of microscale and nanoscale structure in DCHPIC is reminiscent of the structure observed in amphiphilic DCH.16 The design of DCHPI appears to have been successful in mimicking amphiphilic DCH by replicating many of their structural features and properties even though DCHPIC utilize PICs instead of hydrophobic segments for self-assembly.
Figure 3.

Laser scanning confocal and cryoelectron microscopy (cryoEM) images of (MOA)155E/K60 hydrogels. (A–C) LSCM images (z-thickness 0.78 μm) of TRITC-labeled (MOA)155K60 (red) and FITC-labeled (MOA)155E60 (green) hydrogel mixtures showing a microporous structure (3.0 wt % in PBS): (A) FITC channel, (B) TRITC channel, (C) merge of (A) and (B). (D) CryoEM image of (MOA)155E/K60 hydrogel showing a nanoporous structure (2.0 wt % in PBS). Scale bars: (A–C) 25 μm, (D) 200 nm.
Compared to amphiphilic DCH, DCHPIC were found to possess a significant difference in hydrogel stability against dilution in aqueous media. Amphiphilic DCH are formed by direct dissolution of solid copolypeptide in aqueous media, where samples, e.g., K180L20, swell to fill the volume and spontaneously form hydrogels. If additional media are then added, the copolypeptides will continue to disperse to give diluted samples that fill the larger volume.16 While this property allows facile preparation of DCH formulations for a number of applications,32 it does limit the ability to suspend hydrogels in excess media, as could be desired for in vitro cell culture.33 To study the stability of DCHPIC against dilution in aqueous media, a 5.0 wt % (MOA)155E/K60 hydrogel in PBS was prepared, and then an equal volume of DMEM cell culture media was added on top of the hydrogel (Figure 4A). For comparison, a similar experiment was performed using a 2.0 wt % K180L20 hydrogel in PBS (Figure 4B), where its concentration was chosen to match the stiffness of the (MOA)155E/K60 hydrogel. Initially, the DMEM solutions formed clear layers above both hydrogels. After 3 days, the K180L20 sample had fully mixed with the DMEM layer and the diluted sample was a viscous liquid. With (MOA)155E/K60, although the DMEM solutes were able to diffuse into the sample over 3 days, the hydrogel was able to retain its shape and stiffness (Figure 4C,D). Compared to the hydrophobic interactions of amphiphilic DCH, the combination of H-bonding and electrostatic interactions present in the assemblies of DCHPIC was found to impart superior resistance against dissolution in aqueous media.
Figure 4.

Stability of diblock copolypeptide hydrogels against dilution. Polyion complex (MOA)155E/K55 (A, C; 5.0 wt %) and hydrophobic assembled K180L20 (B, D; 2.0 wt %) hydrogels of equivalent stiffness (G′ ≈ 120 Pa) in 1× PBS were each diluted with an equal volume of DMEM cell culture media. (A, B) Layer of cell media formed over both gels at the beginning of the experiment (time 0). (C, D) After 3 days, the (MOA)155E/K60 hydrogel remained intact, while K180L20 had dispersed into the full volume of media and was a liquid.
The ability of DCHPIC to resist dissolution or swelling once formed provides a means to cast hydrogel shapes from precursor solutions and then use these stable hydrogels for various applications in aqueous media. To showcase their potential utility, we encapsulated primary neural stem progenitor cells (NSPCs)33 in an (MOA)155E/K60 hydrogel (Figure 5). The NSPCs were encapsulated by mixing a suspension of cells in media with an equal volume of (MOA)155E60 solution in media, which was then combined with an equal volume of (MOA)155K60 solution in media to rapidly form the cell-containing hydrogel. This sample, as well as cell-only and cells in K180L20 hydrogel controls in media, was incubated for 1 day, and then the cell viability was quantified using a live/dead assay (Figure 5).34 The cationic K180L20 hydrogel was found to be highly cytotoxic, as expected from the literature,33 and served as a good negative control. The (MOA)155E/K60 hydrogel provided good cell viability, similar to the cells in the media-only control, which suggests that DCHPIC may be promising for use as cell carriers. Although DCHPIC contain long, charged polypeptide segments, these are effectively sequestered by PIC formation and steric shielding from the uncharged MOA hydrophilic segments, resulting in hydrogels that are effectively nonionic.35 Although cells were exposed to the noncomplexed, charged components of DCHPIC during the mixing process, this brief exposure, regardless of the mixing order, was found to have minimal adverse effects on the cell viability. More detailed studies on cell suspension in DCHPIC will be the subject of future investigations.
Figure 5.

Viability of neural stem progenitor cells (NSPCs) encapsulated in hydrogels. (A) NSPC viability after 1 day of incubation in different conditions: cells in media-only control, in media plus 2.0 wt % K180L20 hydrogel, or in media plus 5.0 wt % (MOA)155E/K60 hydrogel. (B–D) Fluorescence microscopy images of NSPC cells after 1 day of incubation under different conditions and then stained using the live/dead viability/ cytotoxicity assay where green is due to calcein (live cells) and red is due to EthD-1 (dead cells). (B) Cells in media-only control. (C) Cells in media plus 2.0 wt % K180L20 hydrogel. (D) Cells in media plus 5.0 wt % (MOA)155E/K60 hydrogel. Scale bars 100 μm. The asterisk indicates p < 0.0001 (unpaired Student’s t test for K180L20 with either cell control or (MOA)155E/K60).
CONCLUSIONS
A new class of cell-compatible copolypeptide hydrogels, DCHPIC, that possess chain-conformation-directed PIC supra-molecular architectures was reported. The use of polypeptide components that form β-sheets upon polyion complexation, in combination with nonionic disordered segments, led to anisotropic supramolecular assembly into anisotropic structures that form extended hydrogel networks capable of rapid self-healing after deformation. Unlike amphiphilic DCH, the use of PIC assembly in DCHPIC was found to impart these materials with stability against dilution in aqueous media, a valuable feature for downstream applications. While there are many examples of useful materials based on liquid coacervate or disordered PIC assemblies, the promising attributes and unique features of DCHPIC highlight the potential for use of conformational order in PIC assembly to create new supra-molecular materials.
Supplementary Material
Acknowledgments
We thank Eric Raftery for assistance with the ATR-IR measurements and Professor Pirouz Kavehpour and Kelly Connelly for access to equipment and assistance with the rheology measurements. This work was supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and National Science Foundation (Grant DMR-1548924 to Z.H.Z.). We acknowledge the use of instruments at the Electron Imaging Center for NanoMachines supported by the National Institutes of Health (Grant 1S10RR23057) and California NanoSystems Institute at the University of California, Los Angeles.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08190.
Experimental procedures, spectral data, rheology data, circular dichroism spectra, and methods for all hydrogel characterization and cell culture (PDF)
References
- 1.Boekhoven J, Stupp SI. Adv Mater. 2014;26:1642–1659. doi: 10.1002/adma.201304606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webber MJ, Appel EJ, Meijer EW, Langer R. Nat Mater. 2016;15:13–26. doi: 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]
- 3.Woolfson DN. Fibrous Proteins: Structures and Mechanisms. In: Parry DAD, Squire JM, editors. Subcellular Biochemistry. Vol. 82. Springer; Cham, Switzerland: 2017. pp. 35–61. [Google Scholar]
- 4.Deming TJ, editor. Top Curr Chem. 2012;310:1–171. doi: 10.1007/128_2011_173. [DOI] [PubMed] [Google Scholar]
- 5.Blocher WC, Perry SL. WIREs Nanomed Nanobiotechnol. 2017;9:e1442. doi: 10.1002/wnan.1442. [DOI] [PubMed] [Google Scholar]
- 6.Marciel AB, Chung EJ, Brettmann BK, Leon L. Adv Colloid Interface Sci. 2017;239:187–198. doi: 10.1016/j.cis.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry SL, Leon L, Hoffmann KQ, Kade ML, Priftis D, Black KA, Wong D, Klein RA, Pierce CF, III, Margossian KO, Whitmer JK, Qin J, de Pablo JJ, Tirrell M. Nat Commun. 2015;6:6052. doi: 10.1038/ncomms7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Priftis D, Leon L, Song Z, Perry SL, Margossian KO, Tropnikova A, Cheng J, Tirrell M. Angew Chem, Int Ed. 2015;54:11128–11132. doi: 10.1002/anie.201504861. [DOI] [PubMed] [Google Scholar]
- 9.Blout ER, Idelson M. J Am Chem Soc. 1958;80:4909–4913. [Google Scholar]
- 10.Hammes GG, Schullery SE. Biochemistry. 1968;7:3882–2887. doi: 10.1021/bi00851a014. [DOI] [PubMed] [Google Scholar]
- 11.Pacalin NM, Leon L, Tirrell M. Eur Phys J: Spec Top. 2016;225:1805–1815. [Google Scholar]
- 12.Lemmers M, Sprakel J, Voets IJ, van der Gucht J, Cohen Stuart MA. Angew Chem, Int Ed. 2010;49:708–711. doi: 10.1002/anie.200905515. [DOI] [PubMed] [Google Scholar]
- 13.Hunt JN, Feldman KE, Lynd NA, Deek J, Campos LM, Spruell JM, Hernandez BM, Kramer EJ, Hawker CJ. Adv Mater. 2011;23:2327–2331. doi: 10.1002/adma.201004230. [DOI] [PubMed] [Google Scholar]
- 14.Cui H, Zhuang X, He C, Wei Y, Chen X. Acta Biomater. 2015;11:183–190. doi: 10.1016/j.actbio.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Chassenieux C, Tsitsilianis C. Soft Matter. 2016;12:1344–1359. doi: 10.1039/c5sm02710a. [DOI] [PubMed] [Google Scholar]
- 16.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 17.Yoon H, Dell EJ, Freyer JL, Campos LM, Jang WD. Polymer. 2014;55:453–464. [Google Scholar]
- 18.Harada A, Kataoka K. Macromolecules. 1995;28:5294–5299. [Google Scholar]
- 19.Harada A, Kataoka K. Science. 1999;283:65–67. doi: 10.1126/science.283.5398.65. [DOI] [PubMed] [Google Scholar]
- 20.Koide A, Kishimura A, Osada K, Jang WD, Yamasaki Y, Kataoka K. J Am Chem Soc. 2006;128:5988–5989. doi: 10.1021/ja057993r. [DOI] [PubMed] [Google Scholar]
- 21.Anraku Y, Kishimura A, Oba M, Yamasaki Y, Kataoka K. J Am Chem Soc. 2010;132:1631–1636. doi: 10.1021/ja908350e. [DOI] [PubMed] [Google Scholar]
- 22.Mutaf OF, Kishimura A, Mochida Y, Kim A, Kataoka K. Macromol Rapid Commun. 2015;36:1958–1964. doi: 10.1002/marc.201500368. [DOI] [PubMed] [Google Scholar]
- 23.Wu LC, Yang J, Kopeček J. Biomaterials. 2011;32:5341–5353. doi: 10.1016/j.biomaterials.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rombouts WH, de Kort DW, Pham TTH, van Mierlo CPM, Werten MWT, de Wolf FA, van der Gucht J. Biomacromolecules. 2015;16:2506–2513. doi: 10.1021/acs.biomac.5b00766. [DOI] [PubMed] [Google Scholar]
- 25.Deming TJ. Chem Rev. 2016;116:786–808. doi: 10.1021/acs.chemrev.5b00292. [DOI] [PubMed] [Google Scholar]
- 26.Aiba SI, Minoura M, Fujiwara Y. Makromol Chem. 1982;183:1333–1342. [Google Scholar]
- 27.Pitha J, Szente L, Greenberg J. J Pharm Sci. 1983;72:665–668. doi: 10.1002/jps.2600720618. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez AR, Kramer JR, Deming TJ. Biomacromolecules. 2013;14:3610–3614. doi: 10.1021/bm400971p. [DOI] [PubMed] [Google Scholar]
- 29.Kramer JR, Deming TJ. Biomacromolecules. 2010;11:3668–3672. doi: 10.1021/bm101123k. [DOI] [PubMed] [Google Scholar]
- 30.Miyazawa T. In: Poly-α-Amino Acids. Protein Models for Conformational Studies. Fasman GD, editor. Dekker; New York: 1967. pp. 69–104. [Google Scholar]
- 31.Srivastava S, Andreev M, Levi AE, Goldfeld DJ, Mao J, Heller WT, Prabhu VM, de Pablo JJ, Tirrell M. Nat Commun. 2017;8:14131. doi: 10.1038/ncomms14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang S, Burda JE, Anderson MA, Zhao Z, Ao Y, Cheng Y, Sun Y, Deming TJ, Sofroniew MV. ACS Biomater Sci Eng. 2015;1:705–717. doi: 10.1021/acsbiomaterials.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng T, Chen M, Chang W, Huang M, Wang T. Biomaterials. 2013;34:2005–2016. doi: 10.1016/j.biomaterials.2012.11.043. [DOI] [PubMed] [Google Scholar]
- 35.Black KA, Priftis D, Perry SL, Yip J, Byun WY, Tirrell M. ACS Macro Lett. 2014;3:1088–1091. doi: 10.1021/mz500529v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.