

Abstract
Glioblastoma (GBM) is an aggressive brain cancer with an average survival rate of 15 months. The composition of the GBM tumor microenvironment—its pH, the presence of growth and immune factors, neurotransmitters and gliotransmitters—plays an important role in GBM pathophysiology and facilitates tumor survival and growth. In particular, GBM tumor cells produce glutamate, which is toxic to healthy tissue and is associated with increased tumor invasion into adjacent brain regions. The conditions that lead to this excitotoxic release of glutamate are not completely understood. Previous studies have demonstrated that extracellular ATP is present at high levels in the tumor microenvironment, and that ATP stimulates the release of glutamate from astrocytes in culture. Here we examine the functional effects of extracellular ATP on the GL261 cell line, a model system for high-grade astrocytomas such as GBM. We show that treatment with ATP leads to an immediate, dose-dependent influx of calcium into the cell that is partially inhibited by an antagonist (o-ATP) of the ionotropic ATP receptor P2X7. In addition, GL261 cells respond to extracellular ATP with a dose-dependent release of glutamate. Consistent with other reports, we find that ATP is toxic to GL261 cells at high concentrations. Together, these results provide insight into the mechanisms responsible for glutamate production by tumor cells and inform future studies that will identify how the GBM tumor microenvironment facilitates tumor invasion into healthy areas of the brain.
Keywords: Glioblastoma, GL261, ATP, Glutamate, Tumor microenvironment, Calcium imaging, MTT
Introduction
Gliomas are aggressive tumors of the central nervous system, and more than 100,000 people in the United States currently live with a malignant glioma diagnosis [1, 2]. In brain tumors and other cancers, the microenvironment at the tumor boundary plays an important role in tumor maintenance and growth [3–6]. Necrotic cell death in particular has been implicated in a tumor’s ability to invade adjacent healthy tissue. The byproducts of necrosis promote an inflammatory response, with concomitant decrease in pH and the recruitment of immune cells which release immune factors that promote tumor growth and angiogenesis [6, 7]. In addition, extracellular ATP and glutamate—also found at the tumor boundary—are directly cytotoxic and are present in concentrations greater than 100 μM in vivo [8–10]. Together, these compounds and processes promote the destruction of healthy tissue. A hallmark of tumor cells is their ability to thrive and proliferate within this hostile environment [6, 11]. It is therefore of fundamental interest to understand how cellular signals in the tumor microenvironment are detected and what effects they have on normal and transformed cells.
As an astrocytoma, malignant glioma tumors share many of the same cellular mechanisms that in astrocytes are responsible for regulating synaptic communication and, more generally, the extracellular environment of the brain [12–14]. Among their many functions, astrocytes have been shown to respond to extracellular ATP with glutamate release [15, 16]. Under normal conditions, glutamate serves as the excitatory neurotransmitter in the majority of synapses in the central nervous system. However, abnormally increased glutamate levels are excitotoxic, causing overactivation of neuronal glutamate NMDA receptors which results in calcium influx and the initiation of mitochondria-induced apoptotic pathways [17–20]. Observations show that glutamate from tumors facilitates the destruction of nearby healthy brain tissue, a mechanism that may aid in their expansion [10, 21].
The GL261 cell line, a murine astrocyte-derived glioma tumor model system, expresses ATP receptors including the ionotropic P2X7 ATP receptor [9]. Several observations have been made regarding the effects of extracellular ATP on GL261 cells. First, GL261 cells respond to ATP with a rise in intracellular calcium [22, 23]. Second, ATP has cytotoxic effects on GL261 cells, and this cytotoxicity correlates with the degree of expression of P2X7 (as determined by Western Blot analysis) [9]. These lines of inquiry present strong evidence that ATP receptors are expressed and functional in GL261 cells. Intriguingly, a recent study used recombinant channelrhodopsin to directly modulate calcium levels in GL261 cells, and found that a rise in intracellular calcium leads directly to glutamate release [17]. The above findings suggest that extracellular ATP may stimulate glutamate release in the GL261 glioma cell line. However, a direct link remains to be established. Here we test the hypothesis that extracellular ATP leads to an increase in intracellular calcium and glutamate release from GL261 cells. An improved understanding of glioma tumor cell biology will yield insights into the functional roles of ATP and glutamate in the tumor microenvironment.
Materials and Methods
Cell Culture
GL261 cells were obtained from the NCI-Frederick Cancer Research Tumor Repository (Frederick, MD). Adherent cultures were maintained in DMEM (Corning) with 10% Fetal Bovine Serum (Atlanta Biologicals) and Penicillin/Streptomycin/Glutamine (Sigma-Aldrich), and incubated at 37.0°C with 5.0% CO2 in 75cm culture flasks. Cells were passaged by trypsinization every 3–4 days at approximately 80% confluency.
Calcium Imaging
Adherent GL261 cells were trypsinized and plated in 8-well glass chamber slides (Thermoscientific Nunc Lab-Tek) at 2 × 104 cells/well, and allowed to incubate for 24 hours before experiments. Immediately prior to plating, the glass 8-well chamber slides were coated for 60–120 minutes with poly-L-lysine (Sigma-Aldrich). Following the 24-hour incubation, cells were prepared for experimental treatments and imaging by removing growth media and washing 2X with 200 μL calcium imaging buffer to remove media and serum (CIB; 130 mM NaCl, 3.6 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 10 mM D-glucose, 10 mM HEPES, pH 7.4 adjusted with HCl and NaOH). After the washing steps, the CIB was replaced with 200 μL of 5 μM fura-2 (Invitrogen), and cells were left to incubate for 30 minutes in the dark at room temperature. After incubation, each well was washed twice with CIB and finally filled with 270 μL CIB or Ca2+-free CIB (with 2 mM EDTA as a chelating agent) for imaging. For experiments that required depletion of intracellular calcium, Fura-2-loaded GL261 cells were first incubated with 2 μM thapsigargin in calcium-free CIB for 6 minutes prior to ATP (Sigma-Aldrich) exposure. ATP-containing solutions were pH’d to physiological pH and filter-sterilized prior to use. Test solutions were delivered by pipet at 10X concentration while imaging (that is, 30 μl of solution at 10X was added to 270 μl CIB for a total of 300 μl at the final concentration). In calcium imaging experiments which used the P2X7-antogonist o-ATP, cells were exposed to 1 mM o-ATP for 30 minutes prior to addition of ATP. A Nikon Eclipse Ti-S epifluorescent inverted microscope was used to examine cells, and ratiometric pseudocolored images were captured with a dual 340/380 nm excitation filter wheel (Sutter) and associated Nikon Digital Sight DS-U3 camera. Images and videos were analyzed using the Nikon Elements software package.
Analysis
Statistical analyses, including Student’s t-tests, one-way ANOVAs, and Holm-Sidak pair-wise comparisons were performed using Sigmaplot 13. For analyzing the aggregate calcium imaging data in experiments involving calcium-free solutions and calcium depletion with thapsigargin (Figure 3), 20 cells were randomly chosen prior to analysis and marked as regions of interest (ROIs) using Nikon Elements. Ratiometric imaging data were exported to a Microsoft Excel file and cells were marked as responsive if calcium rises resulted in a 340/380 ratio 1.3 times higher than the cells’ baseline reading. Each well was counted as an independent experiment. The percentage of responding cells out of the initial 20 responding at 30 seconds was used for comparisons between treatment groups.
MTT Cell Viability Assays
MTT assays were performed using the Vybrant MTT Cell Proliferation Assay kit (Invitrogen) according to the manufacturer’s instructions. Briefly, 200 μL of cells at a density of 5×104 cells/mL were plated in wells of a 96-well plate and incubated for 24 hours. After 24 hours, cell solution was removed and replaced with 200 μL of media (control) and concentrations of ATP at 200 μM, 1 mM, or 5 mM and allowed to incubate for 48 hours. ATP-containing solutions were pH’d to physiological pH and filter-sterilized prior to use. Following incubation, media was removed and replaced with MTT reagent. Cells were then left to incubate for another 4 hours after which all of the solution was removed and replaced with 100 μl DMSO to dissolve the purple formazan crystals created by reduction of the MTT reagent during the incubation period. After 10 minutes, plates were shaken for 1 minute in a BioRad Benchmark Plus microplate reader and analyzed at a wavelength of 570 nm. Co-treatment toxicity studies were done using the above protocol for treatments with 100 μM o-ATP + media (as vehicle control), 100 μM o-ATP + 200 μM ATP, and 100 μM o-ATP + 1 mM ATP.
Glutamate Detection Assay
Glutamate assays were performed using the Sigma-Aldrich Glutamate Assay Kit (MAK004) according to the manufacturer’s instructions. Briefly, 2 mL of cells at 1 × 105 cells/mL per well were plated in a 6-well plate and incubated for 24 hours. After 24 hours, the media was removed and replaced with 2 mL treatment media with vehicle control or ATP at concentrations of 200 μM or 1 mM and allowed to incubate for 48 hours. ATP-containing solutions were pH’d to physiological pH and filter-sterilized prior to use. Following incubation, 50 μL samples of extracellular solution were placed in a 96-well plate and the kit’s enzymatic reaction mix was added to induce a glutamate-dependent color change. Solutions were left to incubate for 30 minutes after which plates were analyzed using a BioRad Benchmark Plus microplate reader at a wavelength of 450 nm.
Results
GL261 cells respond to ATP with an increase in intracellular calcium
Previous reports indicate that GL261 cells respond to extracellular ATP and capsaicin (CAP) with an immediate and robust intracellular increase in calcium [23, 22]. We sought to determine whether this functional response can be observed in GL261 cells cultured adherently. To determine if GL261 cells respond to ATP and CAP with an intracellular calcium influx, GL261 cells were loaded with the ratiometric calcium probe Fura-2 and monitored for fluorescence changes. While we observed a robust calcium response to 1 mM ATP, very few or no cells were observed to respond to 100 μM capsaicin, a finding that is consistent with previous work in our laboratory and that may result from phenotypic differences between cells cultured adherently or as neurospheres (Fig. 1) [22]. Photomicrographs show brightfield images and pseudocolored fluorescent images of a representative experiment. All images are at 200X magnification, and in the pseudocolored fluorescent images warm colors indicate higher concentrations of calcium as given by an increase in the Fura-2 ratio (Fig. 1a). Individual cells were monitored for changes to calcium concentration, as indicated by changes in the Fura-2 ratio, and traces representing cellular responses to CAP and ATP are depicted in Fig. 1b. Next, we sought to characterize the functional responses of GL261 cells. We constructed a dose-response curve by applying two treatments of ATP (that is, infusions of ATP into the extracellular solution) in succession: a test dose and a 1 mM ATP dose, with responses measured at 30 seconds post-treatment. We chose 1 mM ATP to normalize our treatments because calcium responses were observed to be saturated at 1 mM ATP. Cellular responses (intracellular calcium rise) were observed at doses as low as 1 μM ATP, which yielded a response of 4.2% of the maximal response (Fig. 2b). For other concentrations, 10 μM ATP was found to produce a response of 32.1% relative to the maximum, and 100 μM ATP was found to produce a response 85.5% of maximal (n = 4 for each) (Fig. 2c). The half-maximal effective concentration (EC50) was found to be 20.9 μM when these values were fitted with a dose-response curve generated by a Hill equation (Hill coefficient = 1.06).
Fig. 1.
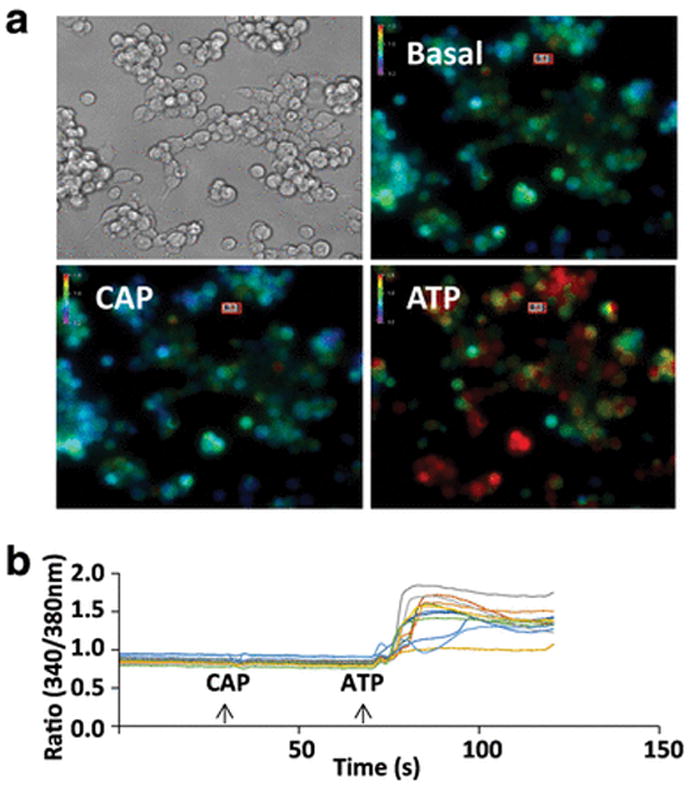
Extracellular ATP, but not capsaicin, leads to an intracellular calcium rise in GL261 cells cultured adherently. a Brightfield and pseudocolored photomicrographs of GL261 cells (×200 magnification). Pseudocolored images depict calcium levels under basal conditions and approximately 30 seconds after exposure to 100 μM capsaicin (CAP) or 1 mM ATP. In each image, bars display the pseudocolor scale, from relatively low (cool colors) to relatively high (warm colors) levels of intracellular calcium. b Traces depict the fura-2 fluorescence ratio over the course of 2 minutes for 12 cells exposed to 100 μM capsaicin (CAP) and 1 mM ATP (arrows)
Fig. 2.
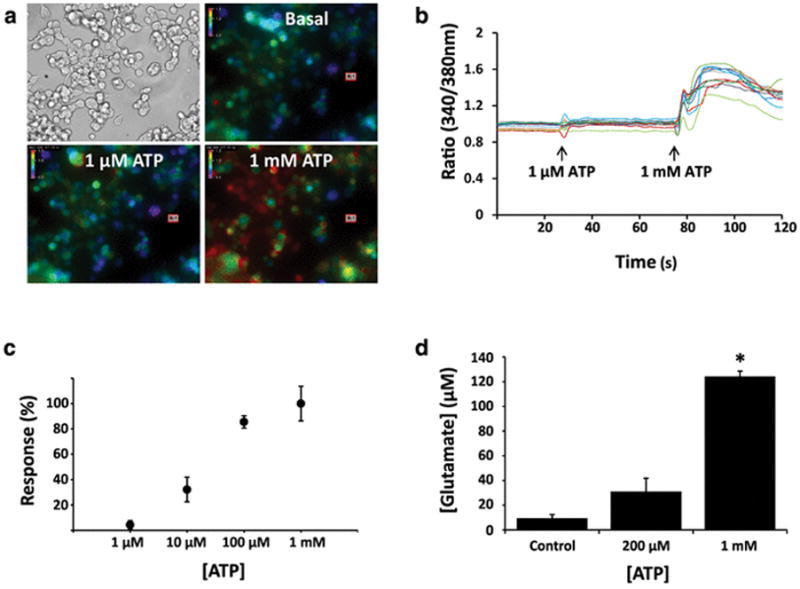
GL261 cells exposed to extracellular ATP exhibit dose-dependent intracellular calcium responses and glutamate release. a Brightfield and pseudocolored photomicrographs of GL261 cells (×200 magnification). Pseudocolored images depict intracellular calcium levels under basal conditions and approximately 30 seconds after exposure to 1 μM or 1 mM ATP. In each image, bars display the pseudocolor scale, from relatively low (cool colors) to relatively high (warm colors) levels of intracellular calcium. b Traces depict the fura-2 fluorescence ratio over the course of 2 minutes for 12 cells exposed to 1 μM ATP and 1 mM ATP (arrows). c ATP dose-response curve (n = 4 wells for each treatment). Response % values are relative to the maximum response seen relative to a saturating dose of 1 mM. Error bars represent the standard error of the mean. d) Glutamate was measured in the culture medium following a 48-hour exposure of GL261 cells to vehicle control, 200 μM ATP, and 1 mM ATP (n = 6 for each treatment). Star (*) indicates statistical significance; the 1 mM ATP treatment group shows significant increases in extracellular glutamate concentrations as compared to other treatment groups (p < 0.01)
GL261 cells respond to ATP by releasing glutamate
Next, we investigated functional responses of GL261 cells (an astrocyte-derived tumor) to extracellular ATP. Two observations guided this line of inquiry. First, previous studies have demonstrated ATP-induced glutamate release by astrocytes [15]. Second, a recent report indicates that GL261 cells exocytose glutamate in response to intracellular calcium increases [17]. Because we determined that intracellular calcium levels rise following ATP exposure in a dose-dependent manner, we hypothesized that if we exposed the cells to varying concentrations of ATP, we would observe a dose-dependent release of glutamate into the culture media surrounding GL261 cells. We found that glutamate levels did in fact increase in proportion to ATP exposure, and that after 48 hours glutamate levels rose from a basal concentration of 9.3 μM in both of two treatment groups, to 31.1 μM (200 μM ATP exposure; n = 6) or 124.3 μM (1 mM ATP; n = 6) (Fig. 2d). The 1 mM ATP treatment was significant as compared to all other treatment groups as determined by a one-way ANOVA and post-hoc testing using the Holm-Sidak method (p < 0.01).
ATP-induced calcium responses are diminished in the absence of external and internal calcium
To determine the source of calcium during ATP-mediated calcium increases in GL261 cells, we used live cell calcium imaging under conditions where extracellular calcium and/or intracellular calcium was depleted. For each experiment in our analysis, we calculated the proportion of 20 cells that responded to ATP (Fig. 3). In each trial, cells were selected without prior knowledge of their responsiveness. Fig. 3a and Fig. 3b depict representative responses under the different treatment conditions for an experiment. Under conditions where physiological calcium was present, we generally observed a robust aggregate response in our cell population and found that 59.1% of all cells responded (n = 8) (Fig. 3c). Next, we measured intracellular calcium responses to ATP when no calcium was present in the extracellular imaging solution and the solution was buffered with 2 mM EDTA to bring the concentration to a calculated 40 nM. This concentration is less than the free calcium in most cell types, including astrocytes [24, 25]. Thus in our preparation we do not predict that an electrochemical driving force would permit calcium influx into GL261 cells when the external solution is chelated with EDTA. In the near-absence of extracellular calcium, ATP-mediated calcium increases were attenuated, and in each experiment we observed a diminished aggregate cellular response and a reduction in the proportion of cells responding to 27.6% of controls (n = 16). Lastly, we examined ATP responses when intracellular stores had been first depleted by thapsigargin. We observed that treatment of GL261 cells with 2 μM thapsigargin led to an immediate rise in cytosolic calcium that slowly diminished over a period of 2–5 minutes and returned to near-baseline (data not shown). This depletion likely represents calcium being actively removed from the cytosol as thapsigargin inhibits the action of the SERCA Ca2+ ATP-ase in the endoplasmic reticulum [26]. Following pre-treatment with 2 μM thapsigargin, we stimulated cells with ATP when the extracellular solution was buffered as before with 2 mM EDTA. In this treatment, we found that on average, cells displayed a greatly diminished response to ATP and only 16.0% of cells responded with any appreciable calcium rise (n = 26). A one-way ANOVA indicated a difference among groups (p < 0.01), and post-hoc pairwise testing using the Holm-Sidak method showed that both treatment groups were significantly different from control.
Fig. 3.
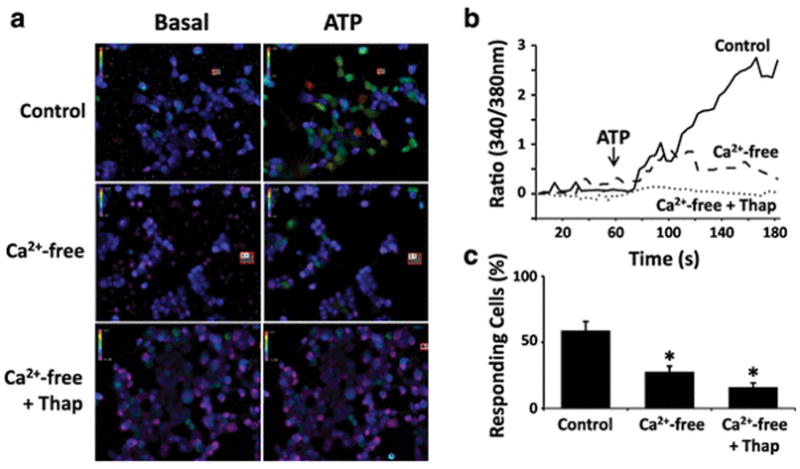
Calcium responses to ATP are attenuated in calcium-free solutions and after depletion of intracellular calcium stores. a Pseudocolored images depict intracellular calcium at basal levels and approximately 30 seconds following application of 200 μM ATP. In the experimental treatments, the extracellular solution contains physiological calcium (control), calcium-free solution, or calcium-free solution where intracellular calcium stores were first depleted by pre-treatment with 2 μM thapsigargin. b Graph depicting the average calcium responses of GL261 cells exposed to 200 μM ATP in control solution (solid line), calcium-free solution (dashed line), or calcium-free solution where intracellular calcium stores were first depleted by pre-treatment with 2 μM thapsigargin (dotted line). During analysis, 20 cells were selected for each experiment with no prior knowledge of the cells’ responsiveness to ATP. c Percentage of GL261 cells responding to ATP in control solution (n = 8), calcium-free solution (n = 16), and calcium-free solution where cells were first pre-treated with 2 μM thapsigargin (n = 26). Bars indicate standard error of the mean. Differences among groups were statistically significant (p < 0.01, one-way ANOVA). Pairwise comparisons between treatment groups were tested using the Holm-Sidak method; star (*) indicates groups that are significantly different from control
The P2X7 antagonist o-ATP attenuates ATP-induced calcium responses
It has been suggested that GL261 cells respond to ATP via the purinergic receptor P2X7, as GL261 cells express P2X7 and RNA interference knockdown of P2X7 attenuates ATP toxicity in GL261 cells [9]. P2X7 is an ionotropic ATP receptor isoform, and subsequent to ATP binding P2X7 permits the influx of calcium and other cations into the cell[27]. Building on these observations, we next investigated whether P2X7 is responsible for the extracellular calcium influx into GL261 cells following ATP exposure. To investigate, we used a pharmacological approach and performed live cell calcium imaging as before to examine ATP responses, but first pre-treated cells using periodate-oxidized ATP (o-ATP), a P2X7-specific antagonist that has been used in a number of studies to inhibit P2X7 function in many cell types, including microglia, hippocampal neurons, and GL261 cells [9, 28, 29]. We found that the proportion of cells responding to ATP decreased from 54.8% (n = 5) to 33.3% (n = 6) when cells were first pre-treated for 30 minutes with the P2X7 antagonist (Fig. 4). This result was statistically significant as determined by a Student’s t-test (p < 0.02).
Fig. 4.
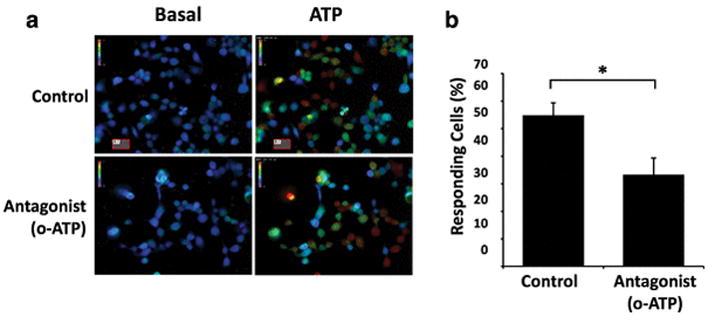
Calcium responses to ATP are attenuated in the presence of the P2X7 antagonist o-ATP. a Pseudocolored images depict intracellular calcium at basal levels and approximately 30 seconds following application of 200 μM ATP. In experimental treatments, cells were pre-incubated in 1 mM o-ATP for 30 minutes prior to imaging. b Percentage of GL261 cells responding to ATP in control solution (n = 5) and when pre-incubated with 1 mM o-ATP (n = 6). Bars indicate standard error of the mean. Star (*) indicates a statistically significant difference between treatments (p < 0.02, Student’s t-test)
ATP and o-ATP decrease GL261 cell viability after 48 hours
ATP is known to be toxic to many cell types, including GL261 cells, though many glioma tumors are resistant to ATP-mediated cell death [9, 11]. Because we found that the P2X7 receptor antagonist o-ATP decreases acute calcium responses in GL261 cells following ATP exposure, we asked if o-ATP could also attenuate ATP-mediated toxicity. We first examined whether ATP was toxic to cells in a dose-dependent manner using MTT cell viability assays, which rely on the reduction of formazan to provide a colorimetric readout of cell metabolic activity [30–33]. Following treatment of GL261 cells with ATP for 48 hours, we found that 200 μM ATP increased cell viability to 105.0% of control (n = 7), 1 mM ATP decreased cell viability to 83.2% of control (n = 7), and 5 mM ATP decreased cell viability to 33.8% of control (n = 5). Of these, only the highest dose of ATP (5 mM) resulted in a statistically significant change in cell viability (one-way ANOVA, p < 0.05) (Fig. 5a). We next assessed whether o-ATP itself is toxic to GL261 cells, as our goal was to investigate its ability to rescue GL261 cells from ATP-mediated toxicity. We found that at concentrations of 10 μM and 100 μM, o-ATP did not have a significant effect on cell viability when compared to control, with a decrease in cell viability of 81.2% and 70.1% relative to control. At o-ATP concentrations of 1 mM and 5 mM however, there was a significant decrease in cell viability compared to control, with a reduction of cell viability to 43.5% and 0.7% (data not shown, n = 5 for each treatment) (one-way ANOVA, p < 0.05). Thus we determined that we could treat cells for 48 hours with 100 μM o-ATP without a substantial reduction in cell-viability. Finally, we were able to test the hypothesis that o-ATP could rescue GL261 cells from ATP-mediated toxicity. Using MTT cell viability assays, we co-treated cells with 100 μM o-ATP in conjunction with either 200 μM, 1 mM, or 5 mM ATP (Fig. 5b). Co-treatments where the ATP concentration was 1 mM or less (n = 5 for all treatments) were not significantly different than vehicle control (one-way ANOVA, p>0.05). However the co-treatment with the highest dose of ATP (100 μM o-ATP + 5 mM ATP) showed a significant decrease in cellular viability compared to control and thus, contrary to our hypothesis, suggests that o-ATP does not rescue GL261 cells from ATP-mediated cytotoxicity at this dose and time-point.
Fig. 5.
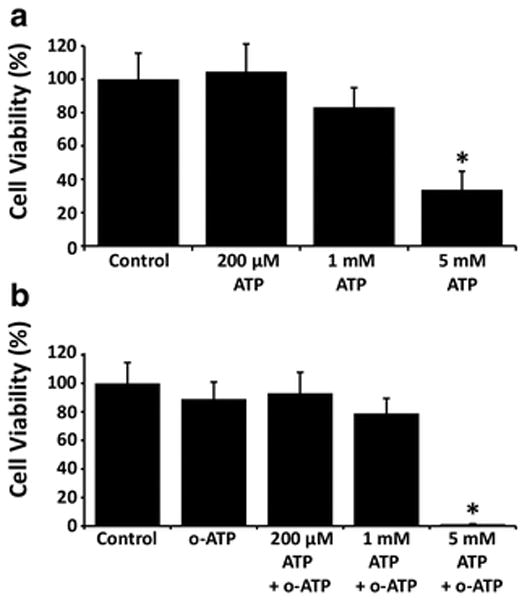
Extracellular ATP decreases GL261 cell viability in a dose-dependent manner. a GL261 cells were incubated for 48 hours in vehicle control or the indicated concentrations of ATP and cell viability was assessed using the MTT assay (n = 5 experiments per treatment). Differences among groups were statistically significant, indicated by star (*) (p < 0.05, one-way ANOVA). Pairwise comparisons between treatment groups were tested using the Holm-Sidak method. There was a significant decrease in cell viability when cells were exposed to 5 mM ATP when compared to control and lower concentrations of ATP. b GL261 cells were co-treated with ATP and the P2X7-antagonist o-ATP for 48 hours and cell viability was assessed with MTT assays. GL261 cells were incubated with 100 μM o-ATP along with either vehicle control or 200 μM, 1 mM, or 5 mM ATP. The 100 μM o-ATP + 5 mM ATP dose showed a decrease in viability as compared with all other treatment groups (one-way ANOVA, p < 0.05)
Discussion
In astrocytes, extracellular ATP leads to a rise in intracellular calcium and subsequent glutamate release [15]. In GL261 cells, an increase in intracellular calcium leads to glutamate exocytosis [17]. Here we sought to determine whether ATP can induce GL261 cells to release glutamate, which has implications for understanding both tumor pathobiology and the tumor microenvironment’s effect on adjacent healthy tissue. In the current work, we have shown that ATP, but not capsaicin, results in an almost immediate rise in intracellular calcium in adherently-cultured GL261 cells (Figure 1). These results are consistent with previous findings in our laboratory, and the fact that our cells are cultured adherently (rather than as neurospheres) explains the discrepancy with other published reports that demonstrate a calcium response to capsaicin in neurosphere-cultured cells [23, 22]. After establishing that GL261 cells respond to ATP with an increase in calcium, we established its dose-dependence (EC50 = 20.9 μM) (Figure 2). In further experiments, we chose to use a dose of 200 μM for calcium imaging. We selected this dose because it 1) induces a near maximal calcium response, 2) approximates the physiological conditions of tumor microenvironments and 3) because it is well below a cytotoxic dose (see Figure 5) [8, 9, 23].
Glutamate exocytosis from glial tumors is associated with excitotoxicity and may facilitate the invasion of tumor cells into healthy brain tissue [10, 21, 34]. Increased extracellular glutamate may result from a number of mechanisms, including glutamate exocytosis and the action of the cysteine-glutamate transporter; other astrocyte-associated functions (such as glutamate reuptake) may play a role as well [17, 16, 35]. In the present work, we found that extracellular ATP induces dose-dependent glutamate release by GL261 cells, with extracellular concentrations following 48-hour treatments of 200 μM or 1 mM ATP leading to glutamate increases of approximately 3- or 12-fold, respectively (Figure 2). Because ATP was found not to be cytotoxic to GL261 cells at 1 mM (Figure 5), we can conclude the increased glutamate release is not simply a function of glutamate being released from unhealthy cells into the extracellular space. Though previous studies have shown that ATP- and calcium-induced glutamate release occurs via exocytotic mechanisms, we cannot formally exclude other possibilities [15, 17]. Our study provides evidence that GL261 cells release glutamate when exposed to ATP at a 48 hour time-scale, and the magnitude of the response is consistent with previous work and at levels shown to be cytotoxic to healthy neurons [15, 17].
Detection of extracellular ATP is mediated by two major classes of purinergic receptors, the ionotropic (P2X) receptors and metabotropic (P2Y) receptors [36]. Thus the calcium response observed may result from a direct influx of external calcium via P2X receptors, mobilization of calcium from the endoplasmic reticulum via downstream signaling from P2X and/or P2Y receptors, or more complex signaling pathways where combinations of the purinergic receptors or even other calcium-permeable channels are involved. We found that in the absence of external calcium, the ATP response was significantly diminished both in terms of its magnitude and the number of cells that responded (Figure 3). Pre-treating with thapsigargin to deplete intracellular stores abolished the ATP response entirely. From this, we conclude that internal stores cannot be responsible for the entirety of the ATP-induced calcium response, and that some or all of the calcium supplied must be influx via calcium-permeable channel(s). Experiments that deplete intracellular calcium stores while leaving extracellular calcium at physiological levels would provide additional insight into the relative contribution of external calcium; if responses remained near those of controls after depletion of intracellular stores we would conclude that the source of calcium is predominately external.
P2X7 has been suggested as a candidate molecule for mediating ATP sensitivity in GL261 cells. Studies show that inhibiting protein synthesis via RNA silencing decreases ATP-induced toxicity [9]. Here, we used a pharmacological approach—with the irreversible P2X7 antagonist o-ATP—to test whether P2X7 activity is necessary for ATP-induced calcium influx. A decrease in the number of responding cells was observed in the presence of this inhibitor, indicating that P2X7 is indeed necessary for ATP-induced calcium responses (Figure 4).
A hallmark of gliomas is their ability to survive in an environment that is normally toxic to healthy tissue [6, 11]. Degradation of extracellular nucleotides is slowed in many glioma cell lines as compared to astrocytes, suggesting one mechanism for generating and maintaining the increased ATP observed in the tumor microenvironment [8, 37]. Still, at very high concentrations ATP remains toxic to GL261 cells, with Tamajusuku et al. reporting toxicity at doses above 1 mM after 2 hours [9]. We find, consistent with these studies, that at longer time points (48 hours) ATP is toxic to GL261 cells at a concentration of 5 mM (Figure 5). This longer time point more closely approximates the physiological challenge presented to tumors in the tumor microenvironment. These findings also validate the use of ATP at concentrations of 1 mM and below (for example in our calcium imaging and glutamate release studies) as they are below toxic levels (Figure 2).
Lastly, we characterized GL261 cell viability in the presence of the P2X7 antagonist o-ATP. In o-ATP dose-response toxicity studies, we found no significant changes in cellular viability at 48 hours at concentrations of 100 μM or less (data not shown). Thus 100 μM o-ATP was used for MTT assays. When GL261 cells were treated with 100 μM o-ATP and co-treated with either 200 μM or 1 mM ATP there was no significant effect on cellular viability (Figure 5). Thus we find no rescue effect for o-ATP in this assay, and in fact, o-ATP was itself toxic to cells at high doses, an effect that was additive with the toxic effect of ATP. This finding was in contrast to previous reports that demonstrate o-ATP rescue from ATP toxicity in shorter treatments [9]. Because of this finding (that o-ATP itself is toxic to GL261 cells at 48-hours) our experimental paradigm cannot directly address whether P2X7 mediates ATP-induced toxicity or glutamate release at longer time-points. In the future it may be useful to investigate the pharmacology of ATP detection using more specific P2X7 antagonists (such as A438079 and/or A740003) to assess the role of P2X7 in mediating these responses, especially if these antagonists are less cytotoxic.
A complete understanding of the signaling pathways responsible for ATP-induced calcium responses, glutamate release, and toxicity will require understanding the relative contributions of various purinergic signaling pathways. Extracellular ATP is degraded by ectonucleotidases into derivatives such as ADP, AMP and adenosine, each of which can activate its own downstream signaling pathways via a myriad of purinergic receptors [9, 36, 38–40]. Thus completely elucidating these pathways will require pharmacological studies that inhibit or activate specific purinergic receptors and/or gene silencing of the various receptor subtypes. In addition, it is important to recognize that studies using cell culture to understand human cancers require validation across multiple experimental systems because of the great heterogeneity found in GBM tumors and tumor cells. Gene expression studies indicate that GBM tumors classified based on morphology may vary considerably with regard to their gene expression profile and phenotypes [41]. Even within the same patient, tumor cells may exhibit considerable variation, including large gene expression differences among tumor cells in important drug targets such as the epidermal growth factor (EGF) receptor and tumor stem cell markers such as CD133 [42, 43]. Cell culture-based model systems also exhibit differences in gene expression and phenotype. For example, specific differences have been noted for TRPV1 expression among various glioma cells lines, with U373 cells showing increased TRPV1 expression as compared to normal human astrocyte (NHA) cells, whereas the U87, FC1, and FLS model systems show decreased TRPV1 expression compared to NHA [44]. Lastly, cell culture conditions can influence the phenotypes of cells even from the same model system. Previous results from our laboratory have shown that GL261 cells cultured as neurospheres display increased functional responses to calcium channel agonists such as capsaicin as compared to cells cultured adherently [22]. Therefore, because of the known heterogeneity in primary gliomas and in glioma cell lines, it will be important to determine whether our findings can be validated in other glioma models. This includes, importantly, using GL261 cells cultured as neurospheres (which represent the tumor stem cell population) in order to validate our results.
In summary, this study shows that extracellular ATP is associated with a short term rise in intracellular calcium and subsequent glutamate release from GL261 cells. Our work extends previous research by examining the effects of ATP over several days. We also provide evidence that the calcium rise associated with glutamate release is due in part to influx of external calcium, possibly via the ionotropic P2X7 channel. Together, these studies will add to our understanding of glioma pathobiology and provide insight into the maintenance of the tumor microenvironment.
Acknowledgments
David Dunn provided assistance with establishing glutamate detection assays in our laboratory. Funding for this work was provided by the M.J. Murdock Charitable Trust (Research Start-up Grant for New Science Faculty), the Mountain States Tumor Institute (Small Projects Grant and Summer Fellows Program), and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grant #P20GM103408. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding organizations. NIH, M.J. Murdock Charitable Trust, and MSTI had no role in study design, data collection, data analysis and interpretation, decision to publish, or preparation of the manuscript.
Footnotes
Authors’ contributions: ADS, MCI and NH performed calcium imaging experiments. ADS performed glutamate detection assays and cell viability studies. ADS, MCI, NH and RLD contributed to study design, data analysis, and drafts of figures. ADS and RLD contributed to drafting the manuscript. RLD edited and finalized figures, figure legends, and manuscript text. ADS, MCI, NH and RLD read and approved the final manuscript.
Conflict of interest: The authors declare that they have no competing interests.
Research involving human and animal rights: This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. J Am Med Assoc. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, Bigner DD. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs. 2009;18:1061–1083. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- 3.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–12. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koontongkaew S. The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. J Cancer. 2013;4:66–83. doi: 10.7150/jca.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60:502–514. doi: 10.1002/glia.21264. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg Ra. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS One. 2008;3:1–9. doi: 10.1371/journal.pone.0002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamajusuku ASK, Villodre ES, Paulus R, Coutinho-Silva R, Battasstini AMO, Wink MR, Lenz G. Characterization of ATP-induced cell death in the GL261 mouse glioma. J Cell Biochem. 2010;109:983–91. doi: 10.1002/jcb.22478. [DOI] [PubMed] [Google Scholar]
- 10.de Groot J, Sontheimer H. Glutamate and the biology of gliomas. Glia. 2011;59:1181–9. doi: 10.1002/glia.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrone FB, Horn AP, Stella J, Spiller F, Sarkis JJF, Salbego CG, Lenz G, Battastini AMO. Increased resistance of glioma cell lines to extracellular ATP cytotoxicity. J Neurooncol. 2005;71:135–140. doi: 10.1007/s11060-004-1383-1. [DOI] [PubMed] [Google Scholar]
- 12.Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Ben Achour S, Pascual O. Glia: The many ways to modulate synaptic plasticity. Neurochem Int. 2010;57:440–445. doi: 10.1016/j.neuint.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227–238. doi: 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- 15.Jeremic A, Jeftinija K, Stevanovic J, Glavaski A, Jeftinija S. ATP stimulates calcium-dependent glutamate release from cultured astrocytes. J Neurochem. 2001;77:664–675. doi: 10.1046/j.1471-4159.2001.00272.x. [DOI] [PubMed] [Google Scholar]
- 16.Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–54. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ono K, Suzuki H, Higa M, Tabata K, Sawada M. Glutamate release from astrocyte cell-line GL261 via alterations in the intracellular ion environment. J Neural Transm. 2014;121:245–257. doi: 10.1007/s00702-013-1096-8. [DOI] [PubMed] [Google Scholar]
- 18.Leon R, Wu H, Jin Y, Wei J, Buddhala C, Prentice H, Wu JY. Protective function of taurine in glutamate-induced apoptosis in cultured neurons. J Neurosci Res. 2009;87:1185–1194. doi: 10.1002/jnr.21926. [DOI] [PubMed] [Google Scholar]
- 19.Uberti D, Grilli M, Memo M. Contribution of NF-κB and p53 in the glutamate-induced apoptosis. Int J Dev Neurosci. 2000;18:447–454. doi: 10.1016/s0736-5748(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 20.Manev H, Favaron M, Guidotti a, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36:106–112. [PubMed] [Google Scholar]
- 21.Takano T. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 22.Strong AD, Daniels RL. Live-cell calcium imaging of adherent and non-adherent GL261 cells reveals phenotype-dependent differences in drug responses. BMC Cancer. 2017;17:516. doi: 10.1186/s12885-017-3507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stock K, Kumar J, Synowitz M, Petrosino S, Imperatore R, Smith ESJ, Wend P, Purfürst B, Nuber Ua, Gurok U, Matyash V, Wälzlein JH, Chirasani SR, Dittmar G, Cravatt BF, Momma S, Lewin GR, Ligresti A, De Petrocellis L, Cristino L, Di Marzo V, Kettenmann H, Glass R. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat Med. 2012;18:1232–1238. doi: 10.1038/nm.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clapham DE. Calcium Signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 26.Michelangeli F, East JM. A diversity of SERCA Ca2+ pump inhibitors. Biochem Soc Trans. 2011;39:789–797. doi: 10.1042/BST0390789. [DOI] [PubMed] [Google Scholar]
- 27.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–67. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 28.Cavaliere F, Amadio S, Sancesario G, Bernardi G, Volonte C. Synaptic P2X 7 and Oxygen / Glucose Deprivation in Organotypic Hippocampal Cultures. 2004 doi: 10.1097/01.WCB.0000113528.73852.E3. [DOI] [PubMed] [Google Scholar]
- 29.Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J Biol Chem. 2003;278:13309–13317. doi: 10.1074/jbc.M209478200. [DOI] [PubMed] [Google Scholar]
- 30.Lobner D. Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis? J Neurosci Methods. 2000;96:147–52. doi: 10.1016/s0165-0270(99)00193-4. [DOI] [PubMed] [Google Scholar]
- 31.Shen Y, Hu WW, Fan YY, Dai HB, Fu QL, Wei EQ, Luo JH, Chen Z. Carnosine protects against NMDA-induced neurotoxicity in differentiated rat PC12 cells through carnosine-histidine-histamine pathway and H(1)/H(3) receptors. Biochem Pharmacol. 2007;73:709–17. doi: 10.1016/j.bcp.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Weyermann J, Lochmann D, Zimmer A. A practical note on the use of cytotoxicity assays. Int J Pharm. 2005;288:369–76. doi: 10.1016/j.ijpharm.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 33.Lin LH, Chan HL, Chou HC. DNA-dependent protein kinase regulated glioblastoma survival in doxorubicin-induced cytotoxicity. Genomic Med Biomarkers, Heal Sci. 2012;4:54–56. [Google Scholar]
- 34.Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008;105:287–95. doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bridges RJ, Natale NR, Patel SA. System xc− cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. 2009;32:19–29. doi: 10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Wink MR, Lenz G, Braganhol E, Tamajusuku ASK, Schwartsmann G, Sarkis JJF, Battastini AMO. Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett. 2003;198:211–8. doi: 10.1016/s0304-3835(03)00308-2. [DOI] [PubMed] [Google Scholar]
- 38.Wink MR, Lenz G, Braganhol E, Tamajusuku aSK, Schwartsmann G, Sarkis JJF, Battastini aMO. Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett. 2003;198:211–218. doi: 10.1016/s0304-3835(03)00308-2. [DOI] [PubMed] [Google Scholar]
- 39.Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36:293–303. doi: 10.1038/onc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burnstock G. Purinergic signalling: from discovery to current developments. Exp Physiol. 2014;99:16–34. doi: 10.1113/expphysiol.2013.071951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vitucci M, Hayes DN, Miller CR. Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. Br J Cancer. 2011;104:545–53. doi: 10.1038/sj.bjc.6606031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006 doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella a, Giangaspero F, Santoni G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J Neurochem. 2007;102:977–90. doi: 10.1111/j.1471-4159.2007.04582.x. [DOI] [PubMed] [Google Scholar]