

Abstract
Cell-based therapies hold great promise for a myriad of clinical applications. However, as these therapies move from phase I to phase II and III trials, there is a need to improve scale-up of adherent cells for the production of larger good manufacturing practice (GMP) cell banks. As we advanced our neural stem cell (NSC)-mediated gene therapy trials for glioma to include dose escalation and multiple treatment cycles, GMP production using cell factories (CellStacks) generated insufficient neural stem cell (NSC) yields. To increase yield, we developed an expansion method using the hollow fiber quantum cell expansion (QCE) system. Seeding of 5.2 × 107 NSCs in a single unit yielded up to 3 × 109 cells within 10 days. These QCE NSCs showed genetic and functional stability equivalent to those expanded by conventional flask-based methods. We then expanded the NSCs in 7 units simultaneously to generate a pooled GMP-grade NSC clinical lot of more than 1.5 × 1010 cells in only 9 days versus 8 × 109 over 6 weeks in CellStacks. We also adenovirally transduced our NSCs within the QCE. We found the QCE system enabled rapid cell expansion and increased yield while maintaining cell properties and reducing process time, labor, and costs with improved efficiency and reproducibility.
Keywords: neural stem cells, HB1.F3.CD, manufacturing, clinical grade, quantum cell expansion system, bioreactor, adherent cells, GMP
Introduction
Cell-based therapies, including stem cell therapies, are uniquely poised for a wide range of clinical applications in the near future. However, a major bottleneck in moving adherent cell-based therapies toward phase II and III clinical trials and commercialization is cost-effective scale-up for manufacture and production of larger good manufacturing practice (GMP)-grade cell banks. Factors that must be considered when scaling up cell production include the seeding surface area and environment, cell type, growth media, cell growth rate, time limitations, harvest and recovery, and reproducibility. Current scale-up options for adherent cells include multiplate-based technologies, microcarrier systems, and packed-bed bioreactors.1, 2, 3, 4 The most common method used for adherent cell expansion is multilayer cell factories. However, this approach has several drawbacks, including the amount of time required for expansion, need for multiple passages, use of an open system, and a limited final cell yield due to availability of incubator space and specialized personnel.
An alternative scale-up method is the use of hollow fiber bioreactor systems, which overcome many of these drawbacks by providing a greater cell culture surface in a smaller footprint, as well as a closed, automated system for controlled temperature and gas exposure and an adjustable flow rate of media to maintain optimal cell culture conditions.5, 6, 7, 8, 9 One of the leading closed automated bioreactors available on the market is the quantum cell expansion (QCE) system from Terumo BCT.10 The QCE system is composed of a 3-dimensional, functionally closed bioreactor containing 11,500 hollow fibers (2.1 m2 surface area), with a flexible protocol management allowing for process optimization for individual cell types. The surface area is equivalent to that of 120 T-175 flasks. This system allows for continuous control over gases, temperature, cell medium feeding, and waste removal, and it has shown reproducibility for generating the final cell product. Several preclinical studies using the QCE system have reported successful expansion and genetic stability of adherent adipose stromal cells7 and mesenchymal stem cells (MSCs).6, 8, 9, 11 Rojewski and others isolated and expanded bone marrow-derived MSCs using a QCE system for clinical-scale production of GMP-compliant MSCs.11, 12 Recently, Sheu et al.13 reported large-scale lentiviral vector production using a QCE system, which offers the potential for manufacturing large quantities of gene therapy vector products.
Another adherent cell type that can benefit from production using this system is neural stem cells (NSCs). Due to their inherent pathotropism to sites of damage in the CNS, NSCs are currently being evaluated in clinical trials for the repair of damage associated with stroke, multiple sclerosis, Parkinson’s disease, and other neurodegenerative diseases.14, 15, 16, 17, 18, 19, 20, 21, 22 We are using an adherent, genetically modified allogeneic NSC line that has demonstrated tumor-tropic properties as a delivery vehicle to target enzyme/prodrug gene therapies selectively to glioma foci throughout the brain. To evaluate this approach, we are conducting two first-in-human NSC-mediated gene therapy clinical trials for recurrent glioma patients (ClinicalTrials.gov: NCT02015819 and NCT02192359).23 As these trials progressed from a single-dose regimen to phase I dose escalation and multiple treatment cycles, we found that producing NSC clinical lots through cell factories was no longer sufficient to meet the needs of the trials. Limitations included inability to produce sufficient cell numbers to complete a single study, long expansion times (3–4 weeks) that resulted from limited incubator space, and limited GMP facility availability. In addition, the repeated manual passaging of cells exposed them to metabolic stress and increased the risk of contamination and human error. This mode of production also incurred high production costs due to extended use of GMP production suites and specialized personnel, as well as release testing of multiple clinical lots to complete a given trial. Furthermore, each clinical lot required U.S. FDA approval. Because the QCE system has the potential to overcome these expansion limitations and to allow in unit ex vivo adenoviral transduction, we sought to develop a bioreactor-based manufacturing approach to meet the growing clinical production demands of our adherent NSCs.
We now report methods for using the QCE system to optimize laboratory and GMP expansion of an allogeneic, genetically modified NSC line that stably expresses the prodrug-activating enzyme cytosine deaminase (CD-NSCs, HB1.F3.CD21),7 as well as successful adenoviral transduction of these NSCs within the QCE system to express a modified human carboxylesterase (CE-NSCs, hCE1m6).22 We reproducibly demonstrated expansion of our clinical-grade CD-NSCs from an initial seeding of a single QCE unit with 5.2 × 107 cells to a harvest of 1.4–3 × 109 CD-NSCs in 7–10 days. This CD-NSC product was equivalent to CD-NSCs produced through conventional flask-based expansion with regard to viability, genetic stability, growth kinetics, tumor tropism, and transgene expression. We then expanded the CD-NSCs in 7 QCE units simultaneously to generate a pooled GMP-grade NSC clinical lot of more than 1.5 × 1010 cells in only 9 days from seeding to harvest, versus production of a clinical lot of only 8 × 109 cells in 3–4 weeks in 30 10-layer CellStacks. This QCE-produced GMP CD-NSC clinical lot was approved by the FDA for use in our phase I trial of CD-NSC and 5-flucyotosine for localized production of 5-fluorouracil in recurrent brain tumor patients (ClinicalTrials.gov: NCT02015819). This trial was completed with the QCE-produced CD-NSC clinical lot, and, to our knowledge, was the first patient use of a QCE-manufactured cell product.
Results
Expansion of Adherent NSCs in the QCE System
CD-NSCs grown in conventional flasks were tested for tumor tropism, stability, and viability. To ensure that QCE production did not alter the CD-NSC growth and tumor-tropic properties, we compared CD-NSCs expanded in the QCE system with CD-NSCs expanded by conventional flask-based cultures. For all experiments, we used CD-NSC clinical equivalent research cell banks (stable passages 22–28). The standard protocol for growing HB1.F3.CD NSCs in flasks uses a plating density of 2 × 104 cells/cm2.24 However, following recommendations from Terumo scientists, we seeded CD-NSCs into the QCE system at a plating density of 2 × 103cells/cm2. Freshly thawed CD-NSCs were seeded into cell culture flasks per standard protocol and grown for 48 hr (initial seeding of 2 × 104 cells/cm2).25 Pre-plating of cells in culture for 48 hr was used to ensure the best possible outcome for cell viability and attachment following standard procedures. After 48 hr, CD-NSCs were harvested and seeded into the QCE system (5.2 × 107 NSCs/unit using a plating density of 2 × 103cells/cm2). After initial CD-NSC seeding, lactic acid levels were monitored in the conditioned media daily (days 3–7). As the number of cells in the bioreactor increased and the lactate levels increased, we adjusted the medium feed rate (perfusion rate) to the cells daily to maintain optimal growth conditions (i.e., lactic acid levels between 8 and 10 mmol/L) (Figure 1A). After 7 days of growth in the QCE system, cells were then detached using Accutase and collected to assess yield, viability, and doubling time (run A). CD-NSC yield with QCE was 1.4 × 109 cells with >95% viability and an average doubling time of 33.9 ± 5.4 hr (mean ± SD, n = 4). In comparison, similarly seeded and expanded CD-NSCs harvested on day 9 (run B) yielded 3.0 × 109 CD-NSCs, doubling the cell number from run A, with 98% viability (Table 1).
Figure 1.

Lactic Acid Monitoring of Run A and Characterization of CD-NSCs that Were Propagated in the QCE System
(A) Lactic acid concentrations in culture media collected from CD-NSCs grown using the QCE system (run A). Lactic acid levels were maintained at 8–12 mmol/L by increasing the feed rate in the QCE system to limit metabolic stress during the propagation of CD-NSCs. (B) Comparison of QCE-grown or flask-grown CD-NSCs expressing the cell identity marker human nestin (red bars) and CD transgene (blue bars), as assessed by flow cytometry. Samples were run in triplicate (percent positive cells, mean ± SD: 99.94% ± 0.08% [nestin]; 93.75% ± 1.3% [CD]). These identity tests are part of the cell bank release criteria for CD-NSCs.
Table 1.
Properties of QCE-Produced CD-NSCs
| Run | Cell Source | Cell Passage at Seeding | Harvest (Day) | Cell Passage at Harvest | Cell Yield | Viability (%) |
|---|---|---|---|---|---|---|
| A | cultured cellsa | p26 | 7 | p27 | 1.4 × 109 | 95 |
| B | cultured cellsa | p22 | 9 | p23 | 3 × 109 | 98 |
| C | from thawb | p23 | 7 | p24 | 1.9 × 109 | 95 |
| D | from thawb | p23 | 11 | p24 | 3.2 × 109 | 98 |
Each run had initial seeding of 5.2 × 107 NSCs/unit resulting in a plating density of 2 × 103 cells/cm2.
NSCs were cultured in flasks prior to loading into QCE system.
NSCs were thawed and directly loaded to QCE system.
We next tested the expansion of CD-NSCs seeded directly into the QCE system from thaw, pursuant to our clinical protocol optimized for expansion efficiency. CD-NSCs were thawed, resuspended in media, counted, and loaded into the QCE system at the same seeding density (2 × 103 cells/cm2) as for cells grown for 48 hr post-thaw. CD-NSCs were then propagated for 7 days (run C) or 11 days (run D) in the QCE system to determine the maximum length of time and yield of CD-NSCs (based on time to confluence, as determined by lactic acid levels). Lactic acid levels were monitored daily, as above, and the medium flow rate was adjusted accordingly. Runs C and D yielded 1.9 × 109 and 3.2 × 109 cells, respectively, and both runs had cell viability >95% (Table 1). No significant differences in terms of viability and chromosomal stability (normal karyotype) were found between the seeding of CD-NSCs in the QCE system when propagated in vitro for 48 hr versus directly from thaw.
For clinical release tests, we confirmed cell identity by immunostaining by flow cytometry using anti-human nestin and E. coli (bacterial) cytosine deaminase (bCD) antibodies. Immunostaining was performed on cells from all QCE runs (A–D) and compared to that of cells grown in flasks. Flow cytometry revealed that cells harvested from the QCE system had similar expression of human nestin and bCD as cells plated in flasks (Figure 1B). We performed short tandem repeat (STR) loci profiling of cell products from runs B–F. STR results showed identical locus identification and a 100% match for a reference CD-NSC cell bank propagated in flasks. Human leukocyte antigen (HLA) typing of HB1.F3.CD cells also confirmed an identical match to the reference cell bank that was propagated by conventional cell culture. This suggests that growing cells in the QCE system is a valid and more efficient approach for clinical cell expansion.
Growth of cells in the QCE system is continuous from cell seeding to cell harvest, whereas cells grown in flasks must be counted at set time points to determine cell doubling. To calculate the population doubling time (PDT) for cells grown in the QCE system, we used a formula developed by Lambrechts et al.5 This formula uses initial and final cell numbers that include lag phase and possible stationary growth phase. Growth kinetics of cells grown in the QCE system were calculated as follows: expansion factor (EF) = number of cells harvested/number of cells seeded; number of population doublings per passage (PD) = log2 (EF); PDT = culture time/PD. Cells grown in the QCE system had an average doubling time ranging from 30 to 40 hr, similar to conventional culture (or cell factories), for which doubling time of CD-NSCs was 30–35 hr.26 Therefore, the collective promising results obtained from optimizing CD-NSC propagation using the QCE system led us to apply this method to GMP manufacture of a CD-NSC clinical lot.
GMP Manufacture of a CD-NSC Clinical Lot Using the QCE System
A GMP-compliant clinical lot of CD-NSCs cells was produced in the QCE system in accordance with our established standard operating procedures (SOPs). For this production, seven Quantum bioreactors (manufacturing plan summarized in Figure 2A) were prepared per an established protocol (see the Materials and Methods), loading 5.2 × 107 cells per each QCE unit. Of these 5.2 × 107 cells, 4 × 107 cells adhered to the inside of the hollow fibers of the bioreactor, and the remaining 1.2 × 107 cells adhered to the tubing set outside of the bioreactor due to the dispersion of cells in the QCE bioreactor during loading (per Terumo QCE setup). Lactic acid readings were recorded daily for 7 days starting on day 1 post-seeding, and lactic acid levels were maintained between 8 and 12 mmol/L by adjusting medium flow rate daily until cell harvest (Figure 2B). Each of the 7 QCE systems was harvested individually, and cells were stained with trypan blue to determine cell count and viability. Cells were then pooled.
Figure 2.

QCE-Based Production of a Clinical Bank of CD-NSCs under GMP Conditions
(A) Schematic of production flow for CD-NSCs manufactured under cGMP (ISO-7) conditions. (B) Lactic acid levels for CD-NSCs for 36–171 hr after cell loading in 7 QCE units that were used to generate a pooled clinical cell bank. Multiple t test of repeated treatments showed no statistical differences among lactic acid levels for the 7 reactors.
This yielded 1.5 × 1010 CD-NSCs with a viability of >98% (Table 2). A total of 260 vials of CD-NSCs (6 × 107 cells/2.5 mL Cryostor10) was frozen down in liquid nitrogen. This QCE bank underwent working cell bank (WCB) release tests, as was done for our previous CellStacks-generated WCB.25 WCB tests included CD-NSC viability, cell recovery post-thaw, identity tests for human nestin and bCD, and karyotype analysis. CD-NSCs produced using CellStacks or QCE systems were comparable in terms of key characteristics, and all the required criteria were passed (Table 3). However, there was a substantial difference in production time—8 days for QCE systems versus 21 days for conventional CellStacks (Table 3). Following review and approval by the U.S. FDA, this QCE-produced clinical lot was used for 7 patients, completing our phase I trial for patients with recurrent glioma.
Table 2.
Cell Yield and Viability of CD-NSCs from QCE Units Producing GMP Clinical Lot
| QCE Unit Number | Cell Count | Viability (%) |
|---|---|---|
| 1 | 2.5 × 109 | 97 |
| 2 | 2.1 × 109 | 98 |
| 3 | 2.4 × 109 | 97 |
| 4 | 2.1 × 109 | 96 |
| 5 | 1.9 × 109 | 97 |
| 6 | 1.9 × 109 | 94 |
| 7 | 2.5 × 109 | 97 |
| Pooled 1–7 | 1.5 × 1010 | 98 |
Table 3.
Analytical Summary of Cell Banks Made by Conventional Methods as Compared to QCE Bank
| GMP WCBs | Lot 1 | Lot 2 | Lot 3 |
|---|---|---|---|
| Manufacturing | cell factory | cell factory | QCE |
| Sterility USP | no growth | no growth | no growth |
| Bacteriostasis and fungistasis-qualification of sterility testing | non-inhibitory | non-inhibitory | non-inhibitory |
| Mycoplasma USP | negative | negative | negative |
| Mycoplasmastasis | non-inhibitory | non-inhibitory | non-inhibitory |
| Endotoxin | 0.082 EU/mL | <0.050 EU/mL | 0.207 EU/mL |
| In vitro viral assay | negative | negative | negative |
| Cell line identity | human origin | human origin | human origin |
| Viability at thaw (%) | 92 | 91 | 94 |
| Viable cell concentration on thaw | 2.4 × 107 cells/mL | 2.9 × 107 cells/mL | 2.2 × 107 cells/mL |
| Transgene expression (%) (CD) | 96.30 | 95.52 | 99.61 |
| RCR assay using Mus Dunni cells | negative | negative | negative |
| RCR assay using SC1 cells | negative | negative | negative |
| Karyotype | 46 XX [20] | 46 XX [20] | 46 XX [20] |
| Production time (days) | 21 | 21 | 8 |
| Total product yield | 4.9 × 109 | 8.6 × 109 | 1.5 × 1010 |
Adenoviral Transduction of Adherent NSCs in the QCE System
CD-NSCs were further modified by adenoviral transduction to secrete a modified human carboxylesterase (CE), hCE1m6 (CE-NSCs).26, 27 CE converts irinotecan (CPT-11) to its 1,000× more potent topoisomerase-1 inhibitor SN-38.26 Based on our previous SOPs for adenoviral transduction in conventional culture, we established a protocol for adenoviral transduction of the CD-NSCs within QCE systems. Freshly thawed CD-NSCs (passage 24) were loaded directly into a QCE bioreactor and propagated. On day 7, cells were transduced with adenovirus encoding hCE1m6 (Adv.hCE1m6; MOI of 13.2 or 20) to generate CE-NSCs (Table 4, runs E and F).26 Throughout QCE propagation, the medium flow rate was adjusted based on lactic acid levels, as described above. On day 8, cells were harvested and frozen in cryovials at a density of 2.6 × 107 cell/mL in Cryostor10. Upon thaw, CE-NSCs were analyzed for recovery, viability, and bCD expression, as was done previously for runs A–D (Figure 3). For run E (CD-NSCs transduced with an Adv.hCE1m6 MOI of 13.2), we harvested 1.5 × 109 cells with a viability of 89%. For run F (CD-NSCs transduced with an Adv.hCE1m6 MOI of 20), we harvested 1.8 × 109 cells with a viability of 96%, demonstrating similar outcomes for these parameters (Table 4). In addition, CE transgene activity was assessed on days 2–4 post-plating using an established o-Nitropenyl acetate (o-NPA) functional assay.28 We have previously determined that CE activity reaches a maximum at day 4 after thaw in culture.26 CE activity on day 4 was 750 nmol/min/mL and 882 nmol/min/mL for runs E and F, respectively (Figure 4). These results show that transduction is possible in the QCE system and gives comparable results to flask-based transductions.
Table 4.
Properties of QCE-Adenovirally Transduced CE-NSCs
| Run | Cell Passage at Seeding | Adenoviral Transduction 24 hr prior to Harvest | Harvest (Day) | Cell Passage at Harvest | Cell Yield | Viability (%) | Doubling Time (hr) |
|---|---|---|---|---|---|---|---|
| E | p24 | hCE1m6 MOI 13.2 | 8 | p25 | 1.5 × 109 | 89 | 36.7 |
| F | p24 | hCE1m6 MOI 20 | 8 | p25 | 1.8 × 109 | 96 | 35.1 |
Figure 3.
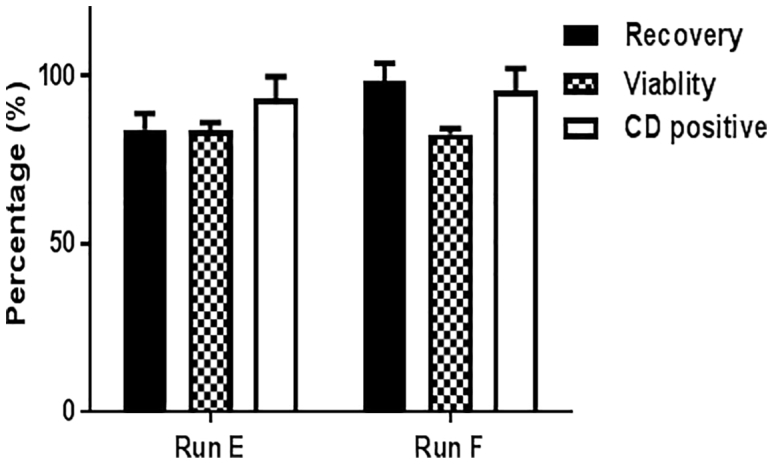
Adenoviral Transduction of NSCs Using the QCE System
CD-NSCs were transduced with Adv.hCE1m6 at MOIs of 13.2 and 20 (runs E and F, respectively) on day 7 and harvested the next day. Percent cell recovery (91.12% ± 10.2%), viability (83.1% ± 0.74%), and cell identity test, CD expression (94.3% ± 1.6%) are shown (values mean ± SD). Cell products from runs E and F passed the cell bank release criteria of recovery (>80%), viability (>70%), and CD expression (>70%).
Figure 4.
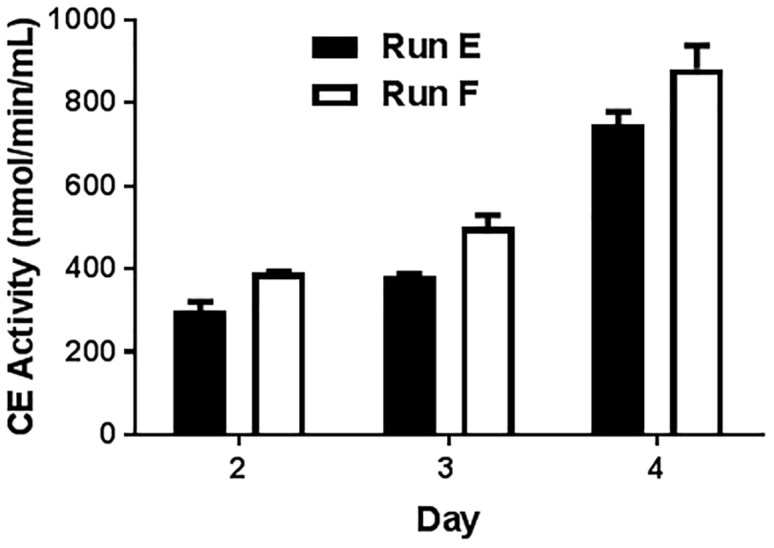
Functional CE Activity of CE-NSCs
CD-NSCs were transduced with adenovirus-hCE1m6 at MOIs of 13.2 and 20 (runs E and F, respectively) on day 7 and harvested the next day. Functional CE activity of CE-NSCs expressing the transgene protein hCE1m6 from days 2 to 4 after thaw in culture is shown (CE activity, mean ± SD: 749 ± 28 [run E] and 882.1 ± 56.7 [run F] on day 4). The CE activity levels of both cell products corresponded to MOIs used during transduction. Both runs yielded cell products that passed the cell bank release criteria for CE expression of 300 nmol/min/mL on day 4.
To test stability, three frozen vials from runs A–F were randomly selected for release testing, including percent cell recovery and viability upon thaw, percent positive staining for human nestin and bCD, karyotype, and CE functional activity for runs E and F (Table 5). These tests were done to compare QCE results to those of CD-NSCs grown in flasks under GMP conditions. To date, all cells banks propagated using QCE systems have successfully passed the release criteria (Table 5).
Table 5.
Release Test Results for QCE Runs A–F
| Release Test | Passing Criteria | A | B | C | D | E | F |
|---|---|---|---|---|---|---|---|
| Recovery post thaw (%) | >80 | 88 | 90 | 91 | 84 | 103 | 98.3 |
| Viability post thaw (%) | >70 | 93 | 91 | 92 | 81 | 85.6 | 82.6 |
| Nestin positive (%) | >90 | 100 | 93 | 95 | 99.9 | 99.9 | 99.7 |
| bCD positive (%) | >70 | 95 | 81 | 92 | 98 | 98 | 95.5 |
| Karyotype | normal | normal | normal | normal | normal | normal | normal |
GMP Manufacture of a Clinical Lot of CE-NSCs Using the QCE System
For the GMP manufacturing of a CE-NSC clinical lot, CD-NSCs from our GMP WCB were thawed at passage 26, and 5.2 × 107 cells were loaded into each of 8 QCE units. Lactic acid levels were maintained between 8 and 12 mmol/L, as described above, by adjusting medium flow rate daily until harvest. Two QCE units were excluded from the clinical production on day 9 due to low lactic acid levels, which suggested the cell proliferation was not as robust as for the other 6 units. On day 9, cells from one QCE unit were harvested for cell count to calculate the amount of virus that is needed to transduce the cells. Cells in the remaining 5 QCE units were then transduced with Adv.hCE1m6 at an MOI of 15. On day 10, cells were harvested from the QCE units using the same protocol as described for runs E and F above. Viability and cell count of harvested CE-NSCs were determined for each QCE unit, and then cells were pooled. A total yield of 8 × 109 cells was frozen at 7 × 107 cells/2.5 mL in Cryostor10 (Table 6), with functional CE activity of pooled cells at 550 nmol/min/mL. This CE-NSC clinical bank was established for a proposed clinical study for the treatment of patients with metastatic neuroblastoma. The clinical cell banks are used in clinical trials under the oversight of the institutional review board.
Table 6.
Cell Yield and Viability of CD-NSCs Transduced with Adv.hCE1m6 in QCE Units Producing GMP Clinical Lot
| QCE Unit Number | Cell Count | Viability (%) |
|---|---|---|
| 1 | 1.7 × 109 | 94 |
| 2 | 1.3 × 109 | 93 |
| 3 | 1.5 × 109 | 92 |
| 4 | 1.9 × 109 | 95 |
| 5 | 1.8 × 109 | 94 |
| Pooled 1–5 | 8 × 109 | 95 |
Cell Bank Release Tests
Three frozen vials from runs A–F were randomly selected for release testing. The cell bank release tests included: cell recovery, cell viability upon thaw, percent positive staining for human nestin and bCD, karyotype, and CE transgene activity (for runs E and F) (Figure 4). All QCE-produced cell banks from runs A–F passed the research cell bank release criteria (Table 5). Furthermore, all GMP clinical cell banks underwent comprehensive testing to fulfill standards in accordance with 21CFR.210, 21.CFR.211, and FDA Guidance.29
Discussion
GMP cell expansion using QCE units can overcome many of the limitations of current scale-up methods for adherent cells, thus meeting the growing demand for larger clinical lots as cell-based clinical trials move toward phase II and III studies and commercialization.7 This approach is relatively simple; does not involve cell (re)-plating; and gases, temperature, and culture media can be controlled and optimized for any adherent cell type.6, 7, 8, 9 Cells harvested from multiple QCE bioreactor units can be combined to produce one clinical lot of 1.5 × 1010 cells in a smaller footprint than conventional cell factory-based cultures. This improved efficiency in manufacturing reduces the need for multiple cell banks to be produced and release tested for a given trial, thereby reducing costs. The processing time is reduced due to automated procedures within the system, limiting the undesirable labor-intensive aspect of conventional cell factory-based cultures. The entire cell expansion system, consisting of cartridges and tubing, is single use and disposable, ensuring no contamination from one run to the next.
For our study, CD-NSCs were propagated as an adherent culture in fibronectin-coated hollow fiber membranes, with fully automated medium flow and gas exchange providing a constant supply of nutrients and optimal physiologic conditions for growth of the CD-NSCs. There is a 30% loss of volume due to the tubing network, which is connected to the bioreactor; therefore, 5.2 × 107 cells were loaded for an actual seeding of 4 × 107 cells (2 × 103 cells/cm2). NSC proliferation was measured via lactate levels, and medium flow rate was adjusted as lactate levels increased. For flask-based proliferation, cells must be expanded by passaging when they exceed the growth area of the flasks. This requires manipulation of the cells thru enzymatic detachment, which increases the chances of contamination and genomic instability. However, using the QCE system, CD-NSCs were easily expanded and maintained for 7–11 days. Moreover, the CD-NSCs maintained their genetic, functional, and morphologic properties as compared to cells grown in conventional culture flasks. Our results were reproducible across 6 independent runs, including two in which we adenovirally transduced CD-NSCs, and they were consistent with other studies using the QCE system for propagation of other adherent lines.5, 6, 11, 12
Overall, we have developed a novel method for QCE of NSCs, which can be refined for other adherent cell types that can then be used for a multitude of clinical applications by optimizing variables, including cell plating density, medium flow, and propagation time. Our GMP production of a clinical lot of CD-NSCs, by using multiple QCE reactors simultaneously, generated a cell bank 2–3 times larger than that produced using conventional flask-based methods. Following FDA approval for an amendment to the Chemistry Manufacturing and Control section of our investigational new drug (IND) application, we have successfully used this bank in our CD-NSC phase I clinical trial for patients with recurrent glioma. Although two other USA trials have also received FDA approval for cell banks generated using QCE systems,8, 9, 30 we were the first to transplant the final cell product into patients. Finally, having now developed SOPs to adenovirally transduce our HB1.F3.CD NSCs to express a modified human CE (hCE1m6), the GMP clinical lot of hCE1m6-adenovirally transduced NSCs is currently being release tested and reviewed by the FDA for use in an upcoming NSC gene therapy trial for the treatment of metastatic neuroblastoma.14 In conclusion, the QCE system offers a novel, advanced alternative to conventional methods for the production of scaled-up clinical cell banks for patient use.
Materials and Methods
Propagation of CD-NSCs in the QCE System: Quantum Protocol Breakdown
All steps in the preparation and running of the QCE System (Terumo BCT, Lakewood, CO) were performed using the pre-programmed tasks, which are part of an automated operation system in which all tasks can be modified to meet the needs of the project. These pre-programmed tasks are described with technical details below.
Load Cell Expansion
The QCE system single-use disposable set (bioreactor with connections) was loaded onto the machine per the manufacturer’s instructions, with multiple cross-checks to ensure proper installation (one technician needed to perform tasks and one to verify).
Prime Cell Expansion
The bioreactor was then primed with 2.5 L PBS (1× PBS) (Mediatech, Manassas, VA).
Coat Bioreactor and Washout
The hollow fibers of the bioreactor were coated overnight with 100 mL fibronectin (0.05 mg/mL) (R&D Systems, Minneapolis, MN), after which all fluids in the internal capillary (IC) and external capillary (EC) lines were replaced with 2.5 L NSC culture media (composition provided below).
Condition Media
In preparation for cell seeding, the media in the QCE system was conditioned by circulating media through the gas transfer module.
Load Cells with Uniform Suspension
For all runs, 5.2 × 107 cells in 100 mL NSC media were loaded into a cell inlet bag using a 60-mL Luer-Lok syringe (BD Biosciences, San Jose, CA) in a biosafety cabinet. The number of cells loaded was 30% greater than the 4 × 107 cells we considered seeded (plating density 2 × 103 cells/cm2) to account for cell adhesion to the tubing surrounding the bioreactor in the IC loop. The cell bag was then welded to the cell inlet line using the TSCD – Q tubing welder (Terumo BCT).
Custom 1, Attach Cells
Cells that had been loaded into the lumen of the hollow fibers in the IC loop were allowed to attach for 24 hr (no medium circulation in the IC loop). During this time, circulation of the EC loop supplied the needed glucose feed (4.5 g/L), removed waste products, managed gas exchange (21% O2, 5% CO2, and 74% nitrogen), and maintained the bioreactor temperature at 37°C.
Custom 1, Feed Cells
After the 24-hr attachment period, cells were fed by pumping media over the cells through the IC. The rate of perfusion of fresh media was adjusted to keep the lactate measurements between 8 and 10 mmol/L. Lactate levels were measured once or twice daily. Adjustments were made manually by increasing the medium flow rate when lactic acid levels rose above 10 nmol/L.
Release and Harvest of Adherent Cells
Cells were harvested from the QCE system 7–11 days after being loaded into the system. The day of harvest was selected based on observation of a plateau in the total lactate production. Harvesting was accomplished by using the IC/EC washout program, which uses 1.5 L PBS to wash out and replace the media. After the washout, 180 mL Accutase (eBioscience, San Diego, CA) was added to the IC line to displace the PBS in the bioreactor. Accutase was circulated for 10 min to allow for enzymatic release of the cells. The cells inside the IC loop were then flushed with an equal volume of complete NSC media and collected into a cell harvest bag (final volume, 375–400 mL). Cells were collected by centrifugation at 450 × g; cell counts and viability were determined using a Guavacyte (Millipore, Billerica, MA) or by trypan blue (product) staining (Gibco, Grand Island, NY).31
NSC Culture Media
HB1.F3.CD21 NSCs were cultured using NSC media prepared from DMEM high glucose (Gibco, Grand Island, NY), with 10% fetal bovine serum (Gemini Biosciences, West Sacramento, CA) for the manufacturing of research cell banks or 10% fetal bovine serum (Hyclone, GE Healthcare, Logan, UT) for manufacturing of clinical cell banks, and 1% L-glutamine (Invitrogen, Grand Island, NY). NSC medium was filtered through a 1-L sterile filter unit (Millipore) prior to use. For the QCE system, 4 L media was prepared and pumped into a Terumo medium bag, which contained an inline 0.2-μm filter (Terumo BCT), using a peristaltic pump (Cole Palmer, Vernon Hills, IL) at 16 L/hr. Medium bags were prepared and replaced as needed.
Preparation of CD-NSCs for the QCE System
For runs A and B, CD-NSCs were plated at 2 × 104 cell/cm2 into T182 flasks (Genesee, San Diego, CA) and incubated (37°C, 6% CO2) for 2 days, after which they were harvested and counted. For all other runs, frozen CD-NSCs were thawed in NSC media and adjusted to a concentration of 5.2 × 107 cells/100 mL. Cells were then loaded into a cell inlet bag using a 60-mL Luer-Lok syringe. The cell bag was then welded to the cell inlet line and loaded onto the QCE system.
CD-NSC Clinical Bank Production
To produce the current GMP (cGMP) clinical bank of CD-NSCs, eight frozen vials of clinical-grade CD-NSCs (frozen at passage 26) were thawed at 37°C using an established SOP (SOP-1955A). Cell number was determined using trypan blue staining, and cells were diluted to 5.2 × 107 cells/100 mL NSC medium, transferred into eight cell inlet bags, and expanded in the QCE system according to SOP-1955A. Harvested cells were pooled and frozen at 6 × 107cells/2.5 mL Cryostor10 (Biolife Solutions, Bothell, WA). Cells were aliquoted (2.5 mL per vial) into 4-mL externally threaded polypropylene cryogenic vials with a conical bottom (Corning 430662) and frozen in a controlled rate freezer (Planer PLC, Middlesex, UK) at cooling rates of −0.1°C/min to 10°C/min until cells reached −80°C, at which point they were transferred to the vapor phase of a liquid nitrogen tank. In total, 260 vials were frozen and stored.
Monitoring of Cell Metabolic Activity
To measure the metabolic activity of proliferating NSCs in the QCE system, lactate concentrations were measured twice daily using a Nova lactate meter and test strips (Nova Biomedical, Waltham, MA). As the number of cells in the bioreactor increased, as judged by the lactate levels, the feed rate (perfusion rate) to the cells was increased to maintain optimal growth conditions (flow rates were adjusted to keep lactate concentrations between 8 and 10 nmol/L).
Post-production Processing
The products of the QCE system runs were frozen in Cryostor10 (Biolife Solutions, Bothell, WA) and stored at −120°C in lN2. Representative vials for each run were release tested as described below to ensure cells met all relevant release criteria for a clinical cell bank.
Release Tests
One frozen vial was thawed, as per our current clinical SOP, to determine cell viability and recovery. Briefly, CD-NSCs were thawed at 37°C for 3 min, washed, and resuspended in 2% HSA/PFCNS (human serum albumin [Baxter, West Lake Village, CA] in CNS Perfusion Fluid [M Dialysis, Johanneshov, Sweden]). Cell viability and numbers were quantified using a Guava EasyCyte (EMD Millipore, Billerica, MA), and recovery was calculated based on the number of cells that were pre-determined to be in each vial. Cell proliferation was determined by plating 1 × 104 cell/well into each well of a 24-well plate and counting replicate wells daily over 7 days, as previously described.26
Flow Cytometry Identity Assays
Cell suspensions were stained for human nestin (Nestin antibody, R&D Systems, Minneapolis, MN) and bCD (bCD antibody, BD Pharmingen, San Jose, CA) and analyzed by flow cytometry to measure percentages of Nestin-positive and bCD-positive cells.24, 25
Karyotype Analysis
Karyotype analysis was performed by the clinical research cytogenetic core facility at City of Hope.
CE-NSC Clinical Bank Production
To enable adenoviral transduction of NSCs in the QCE system, a custom task was designed. This task included the following steps: adding virus to the system in transduction media, 6-hr incubation of cells with virus, followed by the feed-cell protocol overnight. Prior to cell release and harvest, an additional wash step using 2.5 L PBS with Ca (Calcium Chloride, 0.1 g/L), Mg (Magnesium Chloride Hexahydrate, 0.1 g/L), and 10 U/mL heparin was added to the protocol, followed by a wash with 2.5 L PBS without Ca and Mg but containing 10 U/mL heparin. The first wash with Ca and Mg removed all of the media containing virus while maintaining cell adhesion; washes with PBS without Ca and Mg aided in release of cells during enzymatic treatment. Cells were seeded into the QCE system, as described above, and then transduced with replication-deficient adenovirus containing a modified human CE gene (Adv.hCE1m6) on day 7, at an MOI of 13.2 for run E and an MOI of 20 for run F in NSC media containing 10 μg/mL protamine sulfate (APP Pharmaceuticals, Schaumburg, IL).26 Cells were harvested as described above and frozen in Cryostor10 (Biolife Solutions, Bothell, WA) at 2.4 × 107 cells/mL. Frozen cells were release tested as described above, and CE activity (nmol/min/mL) was measured as previously described.26 Briefly, a thawed vial of cells was plated at 2 × 104 cell/cm2 into a T175 cell culture flask in 12 mL NSC media. Aliquots of 200 μL media were taken daily from day 2 to day 4 and the conversion of o-NPA to o-Nitrophenol (o-NP) was measured on a SpectraMax M3 spectrophotometer (Molecular Devices, Sunnyvale, CA).
Statistical Analyses
Statistical analyses and graphs of functions were produced using Prism 6. Analyses of percent cell recovery, viability, and cell identity were measured in triplicate for each run. Column statistics, such as percent positive cells (mean ± SD), were performed for each measurement. Multiple t test of repeated treatments was used to measure statistical difference among samples tested for lactic acid levels.
Author Contributions
M.Z.M., R.T., and Z.L. carried out experiments and manufacturing, analyzed data, and developed and executed methods for testing the QCE system for the preparation of clinical cell banks. C.H. and D.O. performed experiments and manufacturing. D.H., J.B., and A.J.A. provided expertise in cell manufacturing, experimental design, and development of QCE of CD-NSCs. M.G. and K.S.A. led the project, conceptual design, data analysis, and interpretation and prepared the final manuscript.
Conflicts of Interest
K.S.A. and A.J.A. are directors, officers, and shareholders of Therabiologics, Inc.
Acknowledgments
The authors acknowledge the technical expertise of the City of Hope GMP facility and the editorial assistance of Dr. Keely L. Walker (City of Hope). This research was supported by grants from the NIH-NINDS cooperative program in translational research of the NIH U01NS082328, NIH-NCI R01CA198076, FDA R01FD004816, NIH-NCI P30CA033572, CIRM DR1-01421, The Rosalinde and Arthur Gilbert Foundation, and City of Hope. Additionally, research reported in this publication included work performed in the following City of Hope core facilities, which are supported by the NCI under award number P30CA033572: Cytogenetics, Center for Biomedicine and Genetics, and the Office of IND Development and Regulatory Affairs. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Contributor Information
Margarita Gutova, Email: mgutova@coh.org.
Karen S. Aboody, Email: kaboody@coh.org.
References
- 1.Sabatino M., Ren J., David-Ocampo V., England L., McGann M., Tran M., Kuznetsov S.A., Khuu H., Balakumaran A., Klein H.G. The establishment of a bank of stored clinical bone marrow stromal cell products. J. Transl. Med. 2012;10:23. doi: 10.1186/1479-5876-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mirfeizi L., Stratton J.A., Kumar R., Shah P., Agabalyan N., Stykel M.G., Midha R., Biernaskie J., Kallos M.S. Serum-free bioprocessing of adult human and rodent skin-derived Schwann cells: implications for cell therapy in nervous system injury. J. Tissue Eng. Regen. Med. 2017;11:3385–3397. doi: 10.1002/term.2252. [DOI] [PubMed] [Google Scholar]
- 3.Rafiq Q.A., Hanga M.P., Heathman T.R.J., Coopman K., Nienow A.W., Williams D.J., Hewitt C.J. Process development of human multipotent stromal cell microcarrier culture using an automated high-throughput microbioreactor. Biotechnol. Bioeng. 2017;114:2253–2266. doi: 10.1002/bit.26359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tozetti P.A., Caruso S.R., Mizukami A., Fernandes T.R., da Silva F.B., Traina F., Covas D.T., Orellana M.D., Swiech K. Expansion strategies for human mesenchymal stromal cells culture under xeno-free conditions. Biotechnol. Prog. 2017;33:1358–1367. doi: 10.1002/btpr.2494. [DOI] [PubMed] [Google Scholar]
- 5.Lambrechts T., Papantoniou I., Rice B., Schrooten J., Luyten F.P., Aerts J.M. Large-scale progenitor cell expansion for multiple donors in a monitored hollow fibre bioreactor. Cytotherapy. 2016;18:1219–1233. doi: 10.1016/j.jcyt.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Jones M., Varella-Garcia M., Skokan M., Bryce S., Schowinsky J., Peters R., Vang B., Brecheisen M., Startz T., Frank N., Nankervis B. Genetic stability of bone marrow-derived human mesenchymal stromal cells in the Quantum System. Cytotherapy. 2013;15:1323–1339. doi: 10.1016/j.jcyt.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haack-Sørensen M., Follin B., Juhl M., Brorsen S.K., Søndergaard R.H., Kastrup J., Ekblond A. Culture expansion of adipose derived stromal cells. A closed automated Quantum Cell Expansion System compared with manual flask-based culture. J. Transl. Med. 2016;14:319. doi: 10.1186/s12967-016-1080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin-Manso G., Hanley P.J. Using the quantum cell expansion system for the automated expansion of clinical-grade bone marrow-derived human mesenchymal stromal cells. Methods Mol. Biol. 2015;1283:53–63. doi: 10.1007/7651_2014_164. [DOI] [PubMed] [Google Scholar]
- 9.Hanley P.J., Mei Z., Durett A.G., Cabreira-Hansen Mda.G., Klis M., Li W., Zhao Y., Yang B., Parsha K., Mir O. Efficient manufacturing of therapeutic mesenchymal stromal cells with the use of the Quantum Cell Expansion System. Cytotherapy. 2014;16:1048–1058. doi: 10.1016/j.jcyt.2014.01.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts I., Baila S., Rice R.B., Janssens M.E., Nguyen K., Moens N., Ruban L., Hernandez D., Coffey P., Mason C. Scale-up of human embryonic stem cell culture using a hollow fibre bioreactor. Biotechnol. Lett. 2012;34:2307–2315. doi: 10.1007/s10529-012-1033-1. [DOI] [PubMed] [Google Scholar]
- 11.Rojewski M.T., Fekete N., Baila S., Nguyen K., Fürst D., Antwiler D., Dausend J., Kreja L., Ignatius A., Sensebé L., Schrezenmeier H. GMP-compliant isolation and expansion of bone marrow-derived MSCs in the closed, automated device quantum cell expansion system. Cell Transplant. 2013;22:1981–2000. doi: 10.3727/096368912X657990. [DOI] [PubMed] [Google Scholar]
- 12.Barckhausen C., Rice B., Baila S., Sensebé L., Schrezenmeier H., Nold P., Hackstein H., Rojewski M.T. GMP-Compliant Expansion of Clinical-Grade Human Mesenchymal Stromal/Stem Cells Using a Closed Hollow Fiber Bioreactor. Methods Mol. Biol. 2016;1416:389–412. doi: 10.1007/978-1-4939-3584-0_23. [DOI] [PubMed] [Google Scholar]
- 13.Sheu J., Beltzer J., Fury B., Wilczek K., Tobin S., Falconer D., Nolta J., Bauer G. Large-scale production of lentiviral vector in a closed system hollow fiber bioreactor. Mol. Ther. Methods Clin. Dev. 2015;2:15020. doi: 10.1038/mtm.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutova M., Goldstein L., Metz M., Hovsepyan A., Tsurkan L.G., Tirughana R., Tsaturyan L., Annala A.J., Synold T.W., Wan Z. Optimization of a Neural Stem-Cell-Mediated Carboxylesterase/Irinotecan Gene Therapy for Metastatic Neuroblastoma. Mol. Ther. Oncolytics. 2016;4:67–76. doi: 10.1016/j.omto.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z., Oganesyan D., Mooney R., Rong X., Christensen M.J., Shahmanyan D., Perrigue P.M., Benetatos J., Tsaturyan L., Aramburo S. L-MYC Expression Maintains Self-Renewal and Prolongs Multipotency of Primary Human Neural Stem Cells. Stem Cell Reports. 2016;7:483–495. doi: 10.1016/j.stemcr.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S.U. Human neural stem cells genetically modified for brain repair in neurological disorders. Neuropathology. 2004;24:159–171. doi: 10.1111/j.1440-1789.2004.00552.x. [DOI] [PubMed] [Google Scholar]
- 17.Vishwakarma S.K., Bardia A., Tiwari S.K., Paspala S.A., Khan A.A. Current concept in neural regeneration research: NSCs isolation, characterization and transplantation in various neurodegenerative diseases and stroke: A review. J. Adv. Res. 2014;5:277–294. doi: 10.1016/j.jare.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kishimoto N., Shimizu K., Sawamoto K. Neuronal regeneration in a zebrafish model of adult brain injury. Dis. Model. Mech. 2012;5:200–209. doi: 10.1242/dmm.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binan L., Ajji A., De Crescenzo G., Jolicoeur M. Approaches for neural tissue regeneration. Stem Cell Rev. 2014;10:44–59. doi: 10.1007/s12015-013-9474-z. [DOI] [PubMed] [Google Scholar]
- 20.Hermann A., Gerlach M., Schwarz J., Storch A. Neurorestoration in Parkinson’s disease by cell replacement and endogenous regeneration. Expert Opin. Biol. Ther. 2004;4:131–143. doi: 10.1517/14712598.4.2.131. [DOI] [PubMed] [Google Scholar]
- 21.Xiao L., Tsutsui T. Human dental mesenchymal stem cells and neural regeneration. Hum. Cell. 2013;26:91–96. doi: 10.1007/s13577-013-0069-4. [DOI] [PubMed] [Google Scholar]
- 22.Aboody K., Capela A., Niazi N., Stern J.H., Temple S. Translating stem cell studies to the clinic for CNS repair: current state of the art and the need for a Rosetta stone. Neuron. 2011;70:597–613. doi: 10.1016/j.neuron.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 23.Portnow J., Synold T.W., Badie B., Tirughana R., Lacey S.F., D’Apuzzo M., Metz M.Z., Najbauer J., Bedell V., Vo T. Neural stem cell-based anticancer gene therapy: a first-in-human study in recurrent high-grade glioma patients. Clin. Cancer Res. 2017;23:2951–2960. doi: 10.1158/1078-0432.CCR-16-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aboody K.S., Najbauer J., Schmidt N.O., Yang W., Wu J.K., Zhuge Y., Przylecki W., Carroll R., Black P.M., Perides G. Targeting of melanoma brain metastases using engineered neural stem/progenitor cells. Neuro-oncol. 2006;8:119–126. doi: 10.1215/15228517-2005-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aboody K.S., Najbauer J., Metz M.Z., D’Apuzzo M., Gutova M., Annala A.J., Synold T.W., Couture L.A., Blanchard S., Moats R.A. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci. Transl. Med. 2013;5:184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metz M.Z., Gutova M., Lacey S.F., Abramyants Y., Vo T., Gilchrist M., Tirughana R., Ghoda L.Y., Barish M.E., Brown C.E. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: implications for clinical use. Stem Cells Transl. Med. 2013;2:983–992. doi: 10.5966/sctm.2012-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wierdl M., Tsurkan L., Hyatt J.L., Edwards C.C., Hatfield M.J., Morton C.L., Houghton P.J., Danks M.K., Redinbo M.R., Potter P.M. An improved human carboxylesterase for enzyme/prodrug therapy with CPT-11. Cancer Gene Ther. 2008;15:183–192. doi: 10.1038/sj.cgt.7701112. [DOI] [PubMed] [Google Scholar]
- 28.Wierdl M., Morton C.L., Weeks J.K., Danks M.K., Harris L.C., Potter P.M. Sensitization of human tumor cells to CPT-11 via adenoviral-mediated delivery of a rabbit liver carboxylesterase. Cancer Res. 2001;61:5078–5082. [PubMed] [Google Scholar]
- 29.Center for Biologics Evaluation and Research (2008). Guidance for FDA Reviewers and Sponsors: Content and Review of Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). Guidance from the U.S. Department of Health and Human Services, Food and Drug Administration, April 2008. https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm078694.pdf.
- 30.Hanley P.J. Therapeutic mesenchymal stromal cells: where we are headed. Methods Mol. Biol. 2015;1283:1–11. doi: 10.1007/7651_2014_175. [DOI] [PubMed] [Google Scholar]
- 31.Abduvaliev A.A., Gil’dieva M.S. [Differential trypan blue staining of tumor cells for the determination of apoptosis] Klin. Lab. Diagn. 2006;(2):36–38. [PubMed] [Google Scholar]