

Summary
Immune‐mediated inflammatory diseases (IMIDs) are characterized by dysregulation of the normal immune response, which leads to inflammation. Together, they account for a high disease burden in the population, given that they are usually chronic conditions with associated co‐morbidities. Examples include systemic lupus erythematosus, rheumatoid arthritis, Crohn's disease and type 1 diabetes. Since the advent of genome‐wide association studies, evidence of considerable genetic overlap in the loci predisposing to a wide range of IMIDs has emerged. Understanding the genetic risk and extent of genetic overlap between IMIDs may help to determine which genes control which aspects of the different diseases; it may identify potential novel therapeutic targets for a number of these conditions, and/or it may facilitate repurposing existing therapies developed originally for different conditions. The findings show that autoantibody‐mediated autoimmune diseases cluster more closely with each other than autoantibody‐negative diseases such as psoriasis, psoriatic arthritis, Crohn's disease and ankylosing spondylitis which, instead, form a seronegative genetic cluster. The genetic clustering largely mirrors the known response to existing biological therapies, but apparent anomalies in treatment response are discussed.
Keywords: arthritis (including rheumatoid arthritis), autoinflammatory disease, genomics, inflammation, systemic lupus erythematosus
Introduction
Immune‐mediated inflammatory diseases (IMIDs) is an umbrella term that encompasses a number of common, chronic and complex disorders, characterized by a dysregulation of the normal immune response which leads to inflammation in target organs and, usually, systemic effects as well. Examples include type 1 diabetes (T1D), Crohn's disease (CD), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), systemic sclerosis (SSc), multiple sclerosis (MS) and psoriasis, but this list is not exhaustive. Together, IMIDs have an estimated incidence of 80 per 100 000 person‐years and affect 3–5% of the population 1. As these are chronic diseases, they result in significant health care, personal and societal costs.
The abnormal immune response against self‐antigens that occurs in these conditions can lead to characteristic autoantibodies that help to classify and diagnose disease. They also share evidence of systemic inflammation as a mediator of organ damage, with many showing elevated serum markers such as C‐reactive protein (CRP) and erythrocyte sedimentation rate (ESR).
Interest has grown in understanding the conditions as a whole, as treatments have emerged that target inflammatory pathways common to more than one disease, thereby providing opportunities for drug repurposing. It has also been recognized that understanding the genetic overlap between these diseases may inform which diseases should be targeted for trials of emerging immune therapies, given that genetic evidence has been found to double the chance of a new treatment being brought successfully to market 2. In this narrative review, we will consider some examples of how genetics has improved our understanding of the overlap between IMIDs, and some examples of how that might impact clinically, focusing largely upon musculoskeletal IMIDs as exemplars.
Familial genetic clustering of IMIDs
It has been recognized for some time that autoimmune diseases cluster in families. For example, a recent population‐based family study using the Taiwan National Health Research Database reported that the relative risk of SSc in first‐degree relatives of patients with SLE was 5·4 [95% confidence interval (CI) = 3·37–8·65); that of RA was 2·66 (2·28–3·11); and that of T1D was 1·68 (1·22–2·32) 3. A meta‐analysis of five IMIDs (RA, SLE, autoimmune thyroid disease, MS and T1D) conducted in 2013 found that familial clustering occurred in all five conditions, with autoimmune thyroid disease being the most frequent condition in first‐degree relatives of index cases with an IMID, followed by SLE and RA 4. Such familial clustering could arise as a result of shared genetic or environmental susceptibility factors across autoimmune diseases.
There is evidence for shared environmental susceptibility factors 5, and these include cigarette smoking, microbiota, gender and alcohol, among others. Specific examples include the fact that autoimmune diseases such as RA, SLE and SSc are more common in women, cigarette smoking increases the risk of seropositive RA 6 and SLE 7 and crystalline silica has been reported to be associated with RA 8, SLE 9 and SSc 10. An accumulation of exposure to multiple environmental risk factors could culminate in disease, but they are even more likely to trigger immune‐mediated inflammation in genetically susceptible individuals.
Twin, family and adoption studies in many IMIDs implicate a strong role for genetics in susceptibility. For example, concordance rates of RA in genetically identical monozygotic twins are four to five times higher than in dizygotic twins, who share ∼50% of the same genes 11. Indeed, numerous candidate gene studies dating back to the 1970s confirmed that the human leucocyte antigen (HLA) locus is associated with susceptibility to many IMIDs and often has the largest genetic effect 12, 13, 14. The recent advent of genome‐wide association studies (GWAS) confirmed the overlap in genetic loci contributing to disease and led to the development of the Immunochip fine‐mapping genotype array, containing 196 000 single nucleotide polymorphisms (SNPs) encompassing 186 regions of the genome that show overlap between one or more IMIDs 15.
Genetic overlap of IMIDs
The genetic overlap between IMIDs can be illustrated by the fact that of the > 100 genetic loci associated with RA, only two appear specific to RA and not associated with other IMIDs: PADI4 and CCL21 16. PADI4 encodes the protein arginine deiminase 4 (PAD4) enzyme, one of a family of enzymes involved in post‐translational citrullination of arginine amino acid residues. Antibodies to citrullinated peptides (anti‐citrullinated peptide antibodies: ACPA) are characteristic of RA and highly specific for it. A recent study suggests a mechanism by which the PADI4 gene encodes the PAD4 enzyme. PAD4 citrullinates nuclear factor‐kappa B (NF‐κB) p65 directly and enhances the interaction of p65 with importin a3, leading to nuclear localization of p65 17. The PADI4 gene, containing RA susceptibility variants, encodes a protein that interacts more efficiently with p65 to enhance NF‐κB activity, a key mediator of inflammation. Other IMIDs show specific associations; for example, variants in the insulin gene are associated specifically with T1D and no other IMIDs. However, the vast majority of associated variants are shared between IMIDs and many map to immune‐relevant genes 18.
Using results from GWAS of individual autoimmune diseases, statistical methods have been developed to quantify the extent of genetic overlap. For example, cross‐phenotype meta‐analysis detects the association of a SNP of interest to multiple phenotypes and, using this approach, 44% of SNPs associated with seven IMIDs (coeliac disease, CD, MS, psoriasis, RA, SLE and T1D) were also associated with at least one other IMID; only one variant, at the SH2B3 gene locus, was associated with all seven diseases 19. Distinct groups of proteins were present near SNPs which predispose to the same subset of diseases, thereby providing mechanistic insight into the basis of shared disease risk and pathogenesis. For example, psoriasis overlapped most closely with CD, while groups of genes associated with RA overlapped with SLE, T1D and coeliac disease.
Interestingly, for some SNPs, opposing alleles were associated with different autoimmune diseases. For example, at the IL2RA gene locus, opposite alleles associated with RA and T1D, compared with SLE. Another study used co‐localization methods in four IMIDs (RA, T1D, MS and coeliac disease) and reported that 33 of 90 regions (37%) were associated with two or more diseases and identified novel disease associations in 11 regions associated previously with one or more of the three other disorders 20. Only two SNPs (6q23, TNFAIP3 gene locus and 19p13.2, ICAM1/TYK2 locus) were associated with all four diseases, although in both cases three diseases showed co‐localization to the same SNP, with a different SNP in the same region associating with the fourth condition.
While compelling evidence exists for genetic overlap, the practical difficulties of estimating the extent of true genetic overlap should be recognized. First, different samples sizes have often been studied in different conditions, leading to variation in statistical power for drawing firm conclusions regarding associations, particularly for low‐frequency risk alleles with modest effect. This leads to difficulty in interpreting whether a particular phenotype is associated (or not) with these loci, although Bayesian statistical methods are now available that can mitigate against such issues 21. Secondly, true overlap requires the signal of association to be identical, but there are examples of differences even at the same locus; for example, different association signals are observed at the IL23R locus for psoriasis and psoriatic arthritis (PsA) 22. While the use of the Immunochip has conferred substantial benefits over previous GWAS platforms, fine‐mapping of disease‐associated regions is often still required in many cases to confirm identical association across diseases.
As described previously, many IMIDs co‐occur in the same individuals or families. For example, seronegative conditions such as ankylosing spondylitis (AS), CD and psoriasis occur more commonly together than would be expected by chance alone; such overlap of comorbidity might imply a shared genetic or molecular aetiology, but whether that is due to the same alleles predisposing to risk to more than one disease (pleiotropy) or to a subgroup of one disease carrying a higher genetic load for a second disease (heterogeneity) has been unclear. For example, patients with primary sclerosing cholangitis (PSC) develop inflammatory bowel disease (IBD) bearing similarities to UC. Possibly, the same alleles predispose to both conditions or, in a subgroup of patients with PSC, UC develops because these patients carry a higher burden of UC‐associated genetic variants. In a study designed to address this question, Ellinghaus et al. 21 used genetic risk score analysis to show that there was no evidence to support the presence of a subgroup of PSC with a higher frequency of UC risk factors but, rather, that the observed comorbidity is more likely to occur due to genetic pleiotropy.
Some of the more interesting regions of overlap are discussed below to illustrate the lessons that can be learned from understanding the shared genetic aetiology, focusing upon loci with large effect sizes in multiple IMIDs where some mechanistic understanding has been gleaned.
HLA
While most IMIDs show association with the HLA locus (and it often confers the largest effect) there are notable exceptions, including CD, where other loci have greater effect sizes 23.
Given that class II HLA genes (DR, DQ) interact with CD4+ T cells 24, these cells are considered the primary drivers of disease in IMIDs, with strong associations with such genes; examples include classic autoimmune autoantibody‐associated diseases such as RA (HLA‐DRB1) and T1D (HLA‐DQB1). By contrast, CD8 T cells are considered the primary drivers of disease where the largest associations are with HLA class 1 genes 25, examples of which are AS and PsA (HLA‐B27) and psoriasis (HLA‐Cw*0602). Support comes from studies showing enrichment of RA‐associated SNPs mapping to enhancers specific to CD4 T cells 26 and enrichment of PsA‐associated variants in CD8 T cells 22.
However, the evidence to support this is not consistent throughout diseases; for instance, despite MS being associated strongly with HLA class II genes, the predominant lymphocytes present in lesions are CD8+ T cells 27. Furthermore, multiple susceptibility variants may map to different HLA genes in the region; for example, the primary association in RA is at the HLA‐DRB1 gene at amino acids 13, 71 and 74, but variants within HLA‐B and HLA‐DP are also associated 28. Even when the same amino acid position is associated with two diseases, the amino acid residues may differ. For example, amino acid 97 within the HLA‐B gene is associated with both AS and PsA 29. While an asparagine residue at position 97 predisposes to both diseases, a serine residue (the second most common at that position) associates with PsA, but not AS.
The association with HLA molecules may imply an infectious trigger to IMIDs, but the exact mechanism and organism have yet to be defined in any condition. Furthermore, coeliac disease, an IMID with a strong HLA association, is triggered by the gluten component gliadin, meaning that an HLA association does not exclusively implicate an infectious trigger.
PTPN22
Many IMIDs have been associated with R620W of the PTPN22 gene (notable exceptions being psoriasis, coeliac disease, MS and UC). In mice, PTPN22 encodes the lymphoid tyrosine phosphatase (LYP), an important negative regulator of T cell receptor signalling 30. While a number of groups have investigated the functional consequences of the genetic variation, the exact mechanism of action remains controversial.
Some studies suggest that the R620W variation interferes with the interaction between LYP and its substrate Lck, causing reduced phosphorylation of LYP and contributing to a gain‐of‐function inhibition of T cell signalling 31. A mouse model has shown that PTPN22 –/– T cells change their cytokine profiles upon stimulation, with reduction in IL‐17 cells (disease‐inducing) and a trend towards interferon (IFN)‐γ production instead; in combination with a slight increase in regulatory T cell (Treg) numbers, the reduction in IL‐17 production decreased the disease activity of inflammatory arthritis 32. Other studies suggest that the variant is required to set the proper threshold for forkhead box protein 3 (FoxP3)+ Treg cell differentiation 33, while other work in both murine models and human subjects support a model whereby the risk variant acts throughout B cell development to promote enrichment of self‐reactive specificities into the follicular mature versus marginal zone B cell compartment 34. Recently, Schickel et al. 35 have reported that inhibition of either PTPN22 enzymatic activity or its expression by RNA interference restored defective central B cell tolerance. Clearly, further work on the functional consequences of the variant in different cell types and under different stimulatory conditions is still required to establish the most important pathways in disease aetiology.
Chromosome 6q23
The 6q23 locus harbours multiple genes, including TNFAIP3, OLIG3, IL20RA and IFNGR. Multiple variants in the region have been associated with psoriasis, RA, juvenile idiopathic arthritis (JIA), SLE, coeliac disease and MS, but genetic studies alone were unable to determine which gene in the region was being regulated by which variant in each disease. Specifically, a deletion near the TNFAIP3 gene was associated with SLE, but not RA 36. Instead, three different variants were associated with RA, but the most associated genes mapped to a large region between TNFAIP3 and three other genes 37, 38.
Intergenic variants can control genes by physical interaction with the gene due to the topology of folded DNA within the nucleus. The DNA fragment containing 6q23 and its associated SNPs does not interact exclusively with TNFAIP3, the closest gene, but also with IL20RA, located 680 kbp upstream, via chromatin looping. This indicates that the region is likely to be a super‐enhancer, controlling the regulation of multiple genes 39.
Chromosome conformation capture approaches (as per McGovern et al. 39) can be used on a genome‐wide scale, and have shown that SNPs associated with disease do not necessarily regulate the nearest gene, but sometimes skip genes 40. This has important implications for therapeutic targeting and shows the limitations of genetic studies in which SNPs are assigned arbitrarily to the nearest gene. More such studies in cells derived from patients across a variety of IMIDs need to be undertaken to annotate fully the genes and pathways underpinning IMIDs, and may lead to the identification of novel targets for drug development or opportunities for repurposing existing drugs to another indication where a common pathway is involved.
One of the difficulties of studying genetic function in cells from patients is that of potential confounding from drug‐related effects. However, there are now a number of BioResources available in many countries, in which healthy volunteers have agreed to be genotyped and recalled (according to genotype) to provide samples for cellular‐based functional assays. For example, the National Institute for Health Research BioResource 41 is a multi‐centre UK cohort of thousands of volunteers (both with and without medical conditions) who consent to be approached to participate in studies, based on genotype.
Discordant risk associations
Numerous loci have been identified where the same SNP shows strong associations between two conditions, but in opposite directions; i.e. risk versus protective 42. For example, PTPN22 and NOD2, two CD risk loci, demonstrate protective effects in UC, which may represent biological differences between the two conditions 23. Similarly, the risk allele for RA and cardiovascular disease at the IL‐6 receptor (IL6R) gene locus protects against asthma and vice versa 18. This has important implications for therapies specifically targeting the gene, as protection from one autoimmune disease could, theoretically, predispose to another. This is an emerging issue in cancer therapy, where autoimmune conditions are developing among patients receiving treatment with antibodies to cytotoxic T lymphocyte antigen‐4 (CTLA‐4, a checkpoint inhibitor) 43.
Genetic sharing between musculoskeletal IMIDs
Given that the musculoskeletal diseases share features, including a clinical predilection for joint inflammation, response to disease‐modifying drugs (including steroids) and immune‐mediated inflammation, it might be expected that these conditions would show more overlap than with other non‐musculoskeletal IMIDs. Figure 1 illustrates the overlap of genetic association found in RA with JIA, PsA and SLE and Supporting information, Table S1 lists which of the RA‐associated variants are associated with these conditions.
Figure 1.
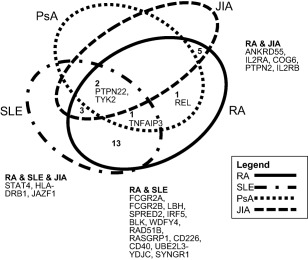
Illustration of genetic overlap between four musculoskeletal immune‐mediated inflammatory diseases (IMIDs), showing number of gene regions associated across two or more conditions, with gene names listed 16, 22, 67, 78.
The results reveal some surprising insights. For example, JIA is more genetically similar to T1D than RA 44. PsA and RA show very little genetic overlap, despite the fact that both show similar response to tumour necrosis factor (TNF) inhibition therapy 18. Similarly, a large meta‐analysis of Immunochip data from six IMIDs (AS, coeliac disease, IBD, psoriasis, T1D, RA) revealed a lack of correlation between RA and AS 42. Only five loci were found to be shared between the two conditions and the genetic enrichment score was the lowest for any pairs of diseases in the analysis. This implies that there are independent pathogenic processes leading to joint inflammation in both conditions.
A molecular taxonomy that positions an individual with respect to the major pathophysiological processes contributing to risk has been proposed recently in diabetes 45. One could hypothesize that musculoskeletal conditions could also be reclassified according to the underlying molecular pathways underpinning disease which, in turn, might better inform therapy selection; this is a goal for stratified medicine approaches.
Drug repurposing as an application of genetics
Despite advances in treatment, IMIDs remain associated with increased morbidity and mortality and pose a huge economic burden due to their chronicity. Traditional de‐novo drug discovery approaches have failed to produce large numbers of new drugs, despite large increases in funding 46. This has led pharmaceutical companies to develop new strategies to improve productivity, one such strategy being drug repurposing (see Box 1). In the remainder of this review we will discuss how, for selected biological therapies, knowledge of genetic susceptibility has or could inform the understanding of drug efficacy.
IL‐23 receptor and IL‐17 inhibition
Genetic studies have consistently shown overlap between psoriasis, PsA, AS and CD, with enrichment of common susceptibility variants over and above that expected by chance alone. For example, associations with the TRAF3IP2, IL23R, IL12B, TAGAP, ERAP1/2, TNFAIP3, TYK2, IFIH1, chromosome 22 and REL gene regions are common to psoriasis/PsA and CD 22, 23, 47.
IL23R is important in differentiation of Th17 cells, which are characterized by their production of the proinflammatory cytokine IL‐17. IL‐23 is a heterodimeric cytokine comprising two subunits: p40, which is also a subunit of IL‐12 and is targeted by ustekinumab (a human monoclonal antibody), and p19, which is expressed only in IL‐23 48. Both the p19 mRNA and p40 subunits have been found to be increased in cutaneous psoriasis and drive excessive growth and abnormal differentiation of keratinocytes 49. Ustekinumab has been found to be effective in the treatment of refractory CD (both as induction and maintenance therapy) in patients who fail anti‐TNF treatment 50 and is also effective in the treatment of psoriasis and PsA.
Given the central importance that the T helper type 17 (Th17) axis appears to have in mediating these seronegative diseases, therapies targeting the IL‐17 cytokine itself were developed. Secukinumab is a monoclonal antibody IL‐17A inhibitor and has shown to be effective in cutaneous psoriasis and autoimmune uveitis 51, AS 52 and PsA 53. As IL‐17 plays an important role in CD, it was predicted that secukinumab would also be effective in the treatment of that condition. However, a randomized double‐blind placebo‐controlled trial studying the effect of secukinumab versus placebo in 59 patients with moderate or severe CD found that secukinumab was not only ineffective, but caused worsening of disease and a higher frequency of adverse events compared to placebo 54.
The reasons proposed to explain the lack of efficacy in CD include issues with the study design and the complex biology of Th17 55. For example, IL‐17A is only one of several cytokines produced by Th17 cells, and Th17 cytokine production is not limited to T cells, with other sources of IL‐23 inducing IL‐17A 56. This suggests a complex pathway and that blockade of IL‐17A alone may not be sufficient in CD, and that other members of the IL‐17 family may be involved. However, brodalumab, which blocks additional IL‐17 family members, produced similarly disappointing results in trials in CD, despite efficacy in psoriasis 57, 58.
These findings show that genetic variants associated with a disease can highlight a gene target for therapy, but it cannot always be assumed that the downstream pathway will also produce an effective drug if targeted. Targeting downstream molecules of genetically associated disease variants works in some situations, as in the case of IL‐17 blockers and psoriasis, but not in others.
TNF‐α inhibition
Similarly, TNFα is an immunologically relevant gene encoding a proinflammatory cytokine which forms a pivotal pathway in several autoimmune diseases 59. Blockade of the pathway has proved highly successful in RA, JIA, psoriasis, PsA, CD and AS. However, anti‐TNF drugs can induce onset or worsening of MS. There are interesting genetic parallels, as the rs1800693 variant of the TNFRSF1A gene is associated with increased risk of MS but is protective of AS 60. Functional studies demonstrated that the MS risk allele directs expression of a soluble form of TNF receptor 1 (TNF‐R1), which can block TNF, mimicking the effect of anti‐TNF medications. This illustrates how manipulating a particular inflammatory pathway in one condition might have negative consequences for another.
Interestingly, large and well‐powered GWAS have not confirmed a role for genetic variants within the TNF gene with any of the conditions in which it has efficacy, although it is downstream of many susceptibility genes that lie on the same pathway. However, while RA and SLE show considerable genetic overlap 61, TNF‐inhibitor use in SLE patients remains controversial. The primary concern is the induction of lupus autoantibodies in patients treated with TNF‐inhibitors, causing drug‐induced lupus‐like syndromes, based on data derived from experimental models 62, 63, 64. TNF‐inhibitors may be beneficial in lupus patients with predominant arthritis or nephritis; however, there is a need for studies to be conducted on a larger scale to evaluate the role of TNF‐inhibitor therapy in SLE 62, and the caution exercised in this example is supported by the effect of TNF inhibition in MS above.
T cell inhibition
A further example of drug repurposing using disease‐based approaches relates to abatacept, given that variants in the CTLA‐4/CD28 gene region on 2q33 have been associated robustly with multiple autoimmune diseases such as T1D, RA and JIA 65, 66, 67. Abatacept is a recombinant fusion protein that inhibits T cell activation and comprises two domains: the extracellular domain of human CTLA‐4 and a fragment of the Fc domain of human IgG1. It competes with CD28 to bind with CD80/CD86, thereby modulating the second co‐stimulatory signal necessary for T cell activation 68.
Abatacept has been proved to be an effective treatment in both RA 69 and JIA 70. Given that CTLA‐4 is also associated with T1D, it might be predicted that abatacept would be effective and, indeed, it has been shown to have a beneficial effect at an early stage of T1D, with a slower decline in β cell function and an estimated 6–9‐month delay in reduction of C‐peptide 71. However, T1D was delayed, rather than prevented, leading to speculation that T lymphocyte activation occurs around the time of disease onset, but may lessen with time. In view of this, abatacept has been proposed as a potential candidate for research in prevention trials of T1D, or as a combination therapy.
Janus kinase inhibitors
Janus kinase (JAK) inhibitors provide a further example of disease‐based drug repurposing. The JAK family has four members – JAK1, JAK2, JAK3 and TYK2. While several autoimmune diseases are associated with a variant in the TYK2 gene, only IBD is associated additionally with JAK2 gene variants 23. Tofacitinib inhibits all the JAKs, is currently licensed for use in RA and is undergoing trials in several other IMIDs 72. While effective in these conditions, treatment is associated with adverse events, including herpes zoster reactivation, dyslipidaemia and liver function abnormalities. TYK2‐specific blockers are in development and, theoretically, may have greater efficacy, with fewer adverse events.
Repurposing based on genetic studies: IL‐6 receptor inhibition
Tocilizumab is a monoclonal antibody targeting the IL‐6 inflammatory pathway. The drug acts by blocking both membrane‐bound and circulating soluble IL‐6R and reducing its proinflammatory effects. A SNP within the IL6R gene changes an amino acid at position 358 from asparagine to alanine. The alanine variant confers protection from RA and T1D and each copy of the alanine variant increases the level of soluble IL‐6R by 35% 18, 73. The mechanism appears to be that the alanine variant increases proteolytic cleavage of the membrane‐bound receptor, and this increased shedding allows for binding and neutralization of circulating IL‐6. Given the efficacy of tocilizumab in RA, there may be potential for repurposing in T1D.
Anti‐cancer therapies
There is certainly a precedent for the repurposing of anti‐cancer therapies; for example, rituximab, a chimeric monoclonal antibody that causes B cell depletion by targeting CD20 molecules on the surface of B cells 74, was developed originally to treat B cell lymphoma, but is now an established treatment for various inflammatory conditions such as RA, JIA and SLE; i.e. conditions in which B cells are implicated in disease pathogenesis 75, 76. Furthermore, methotrexate was also used primarily as a chemotherapeutic agent until lower doses were found to be effective in RA, JIA and PsA. Interestingly, in the largest GWAS of RA to date, genes associated with RA were found to be enriched in pathways targeted by anti‐cancer therapies. For example, alvocidib and palbociclib, developed for treatment of leukaemia and lymphoma, target the CDK4 and CDK6 genes (RA‐susceptibility genes) 16. Therefore, these genetic studies may suggest new therapies for RA that have already been proven safe for use in cancers, but which now require efficacy testing in RA.
Concluding comments
In conclusion, understanding the similarities and differences in the genetic susceptibility to IMIDs is important to identify the genes and pathways that mediate different aspects of autoimmune diseases. The first challenge is to assign the associated SNPs accurately to the genes they regulate in order to define the pathways. This requires a number of functional genomics approaches including, but not limited to, expression quantitative locus (eQTL) studies, in which the genetic variants associated with disease also control gene expression; chromosome conformation studies to identify the genes under control of intergenic variants; and genome editing techniques to show the consequences of harbouring a risk variant on cellular function. All such studies need to be undertaken in different cell types and under different stimulatory conditions, as gene expression is controlled in either a tissue‐specific or condition‐specific manner; for example, shared effects were found from only approximately 25% of eQTLs examined in seven conditions (MS, CD, UC, IBD as a whole, T1D, RA and coeliac disease) 77.
An understanding of these pathways can direct research towards traditional de‐novo drug discovery and can also be used to inform drug repurposing studies, highlighting opportunities for trials of existing medications for new indications. While the development of novel therapies is a slow process, with a new drug taking an average of 17 years to reach the clinic, in the shorter term it is anticipated that better molecular stratification of conditions with overlapping clinical features could inform treatment selection and precision medicine. For example, patients with enrichment of genetic variants in genes in the B cell pathways might respond more effectively to rituximab therapy, while those with enrichment of the IL‐23/IL‐17 pathway may respond more effectively to ustekinumab. The aim for the future is to develop a predictive algorithm for patients to receive the correct treatment at the right time using a precision medicine approach, based on molecular classifiers, regardless of the specific IMID, but moving towards so‐called inflammation medicine.
Disclosure
None.
Box 1. Drug repurposing.
Also known as drug repositioning, drug reprofiling.
Drug repurposing is the identification of new therapeutic targets for drugs which are different from their original medication indication 1.
By utilization of existing knowledge, drug repurposing can offer a cheaper and faster route to market than traditional drug discovery and has also been shown to have higher success rates 2.
Methods of drug repurposing 3
Drug‐based
Predicated on the concept that similar drugs can be used in the treatment of the same condition.
-
Factors are used to identify drug similarities, e.g.:
○ Chemical/biological structure.
○ Side‐effect profile.
○ Other drug characteristics.
For example, the family of tumour necrosis factor (TNF) inhibitor drugs differ in structure, but all have similar efficacy/adverse event profiles 4.
Profile‐based
Aims to match drug target genes with disease‐associated genes obtained from genetic studies.
For example, the development of a monoclonal antibody inhibitor to proprotein convertase subtilizing/kexin 9 (PCSK9) following detection of a gain‐of‐function mutation in the gene encoding the protein in families with hypercholesterolaemia 5.
An example earlier in development is that of interferon (IFN)‐α blockers, which have been developed to treat systemic lupus erythematosus (SLE), partly on the basis of genetic evidence implicating IFN signalling pathways in the aetiology 6; a recent clinical trial has reported promising results for anifrolumab in moderate‐to‐severe lupus 7.
Disease‐based
Identify new therapeutic indications for existing drugs based on immunological, genetic and/or epidemiological overlap between diseases and phenotypical similarities 3.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Associations of 101 rheumatoid arthritis (RA) risk genes identified by Okada et al. 16 with systemic lupus erythematosus (SLE), soriatic arthritis, (PsA) and juvenile idiopathic arthritis (JIA) 8, 9, 10, 11.
Acknowledgements
This work is supported by the NIHR Manchester BRC and Arthritis Research UK (grant ref. 20385).
References
- 1. Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev 2003; 2:119–25. [DOI] [PubMed] [Google Scholar]
- 2. Nelson MR, Tipney H, Painter JL et al The support of human genetic evidence for approved drug indications. Nat Genet 2015; 47:856–60. [DOI] [PubMed] [Google Scholar]
- 3. Kuo CF, Grainge MJ, Valdes AM et al Familial aggregation of systemic lupus erythematosus and coaggregation of autoimmune diseases in affected families. JAMA Intern Med 2015; 175:1518–26. [DOI] [PubMed] [Google Scholar]
- 4. Cardenas‐Roldan J, Rojas‐Villarraga A, Anaya JM. How do autoimmune diseases cluster in families? A systematic review and meta‐analysis. BMC Med 2013; 11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anaya JM, Ramirez‐Santana C, Alzate MA, Molano‐Gonzalez N, Rojas‐Villarraga A. The autoimmune ecology. Front Immunol 2016; 7:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klareskog L, Stolt P, Lundberg K et al A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA‐DR (shared epitope)‐restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006; 54:38–46. [DOI] [PubMed] [Google Scholar]
- 7. Takvorian SU, Merola JF, Costenbader KH. Cigarette smoking, alcohol consumption and risk of systemic lupus erythematosus. Lupus 2014; 23:537–44. [DOI] [PubMed] [Google Scholar]
- 8. Stolt P, Kallberg H, Lundberg I, Sjogren B, Klareskog L, Alfredsson L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis 2005; 64:582–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barbhaiya M, Costenbader KH. Environmental exposures and the development of systemic lupus erythematosus. Curr Opin Rheumatol 2016; 28:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rubio‐Rivas M, Moreno R, Corbella X. Occupational and environmental scleroderma. Systematic review and meta‐analysis. Clin Rheumatol 2017; 36:569–82. [DOI] [PubMed] [Google Scholar]
- 11. Silman AJ, MacGregor AJ, Thomson W et al Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol 1993; 32:903–7. [DOI] [PubMed] [Google Scholar]
- 12. Brewerton D. The inherited antigen (HL‐A 27) and arthritis. Rep Rheum Dis 1974; 55. [PubMed] [Google Scholar]
- 13. Cudworth AG, Woodrow JC. Letter: HL‐A antigens and diabetes mellitus. Lancet 1974; 2:1153. [DOI] [PubMed] [Google Scholar]
- 14. Stastny P. Association of the B‐cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med 1978; 298:869–71. [DOI] [PubMed] [Google Scholar]
- 15. Cortes A, Brown MA. Promise and pitfalls of the immunochip. Arthritis Res Ther 2011; 13:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okada Y, Wu D, Trynka G et al Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014; 506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun B, Dwivedi N, Bechtel TJ et al Citrullination of NF‐kappaB p65 promotes its nuclear localization and TLR‐induced expression of IL‐1beta and TNFalpha. Sci Immunol 2017; 2:eaal3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eyre S, Orozco G, Worthington J. The genetics revolution in rheumatology: large scale genomic arrays and genetic mapping. Nat Rev Rheumatol 2017; 13:421–32. [DOI] [PubMed] [Google Scholar]
- 19. Cotsapas C, Voight BF, Rossin E et al Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet 2011; 7:e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fortune MD, Guo H, Burren O et al Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat Genet 2015; 47:839–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ellinghaus D, Jostins L, Spain SL et al Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease‐specific patterns at shared loci. Nat Genet 2016; 48:510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bowes J, Budu‐Aggrey A, Huffmeier U et al Dense genotyping of immune‐related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nat Commun 2015; 6:6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jostins L, Ripke S, Weersma RK et al Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mangalam A, Rodriguez M, David C. Role of MHC class II expressing CD4+ T cells in proteolipid protein(91–110)‐induced EAE in HLA‐DR3 transgenic mice. Eur J Immunol 2006; 36:3356–70. [DOI] [PubMed] [Google Scholar]
- 25. Chessman D, Kostenko L, Lethborg T et al Human leukocyte antigen class I‐restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity 2008; 28:822–32. [DOI] [PubMed] [Google Scholar]
- 26. Trynka G, Sandor C, Han B et al Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet 2013; 45:124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nicol B, Salou M, Vogel I et al An intermediate level of CD161 expression defines a novel activated, inflammatory, and pathogenic subset of CD8+ T cells involved in multiple sclerosis. J Autoimmun 2017; doi: 10.1016/j.aut.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 28. Raychaudhuri S, Sandor C, Stahl EA et al Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 2012; 44:291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bowes J, Ashcroft J, Dand N et al Cross phenotype association mapping of the MHC identifies genetic variants that differentiate psoriatic arthritis from psoriasis. Ann Rheum Dis 2017; 76:1774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Begovich AB, Carlton VE, Honigberg LA et al A missense single‐nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 2004; 75:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fiorillo E, Orru V, Stanford SM et al Autoimmune‐associated PTPN22 R620W variation reduces phosphorylation of lymphoid phosphatase on an inhibitory tyrosine residue. J Biol Chem 2010; 285:26506–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sood S, Brownlie RJ, Garcia C et al Loss of the protein tyrosine phosphatase PTPN22 reduces mannan‐induced autoimmune arthritis in SKG mice. J Immunol 2016; 197:429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fousteri G, Jofra T, Debernardis I et al The protein tyrosine phosphatase PTPN22 controls forkhead box protein 3 T regulatory cell induction but is dispensable for T helper type 1 cell polarization. Clin Exp Immunol 2014; 178:178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Metzler G, Dai X, Thouvenel CD et al The autoimmune risk variant PTPN22 C1858T alters B cell tolerance at discrete checkpoints and differentially shapes the naive repertoire. J Immunol 2017; 199:2249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schickel JN, Kuhny M, Baldo A et al PTPN22 inhibition resets defective human central B cell tolerance. Sci Immunol 2016; 1:aaf7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adrianto I, Wen F, Templeton A et al Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet 2011; 43:253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orozco G, Hinks A, Eyre S et al Combined effects of three independent SNPs greatly increase the risk estimate for RA at 6q23. Hum Mol Genet 2009; 18:2693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomson W, Barton A, Ke X et al Rheumatoid arthritis association at 6q23. Nat Genet 2007; 39:1431–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McGovern A, Schoenfelder S, Martin P et al Capture Hi‐C identifies a novel causal gene, IL20RA, in the pan‐autoimmune genetic susceptibility region 6q23. Genome Biol 2016; 17:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martin P, McGovern A, Orozco G et al Capture Hi‐C reveals novel candidate genes and complex long‐range interactions with related autoimmune risk loci. Nat Commun 2015; 6:10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. NIHR BioResource . Available at: https://bioresource.nihr.ac.uk/ (accessed 12 January 2018).
- 42. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune‐mediated diseases. Nat Rev Genet 2013; 14:661–73. [DOI] [PubMed] [Google Scholar]
- 43. van der Vlist M, Kuball J, Radstake TR, Meyaard L. Immune checkpoints and rheumatic diseases: what can cancer immunotherapy teach us? Nat Rev Rheumatol 2016; 12:593–604. [DOI] [PubMed] [Google Scholar]
- 44. Onengut‐Gumuscu S, Chen WM, Burren O et al Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 2015; 47:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCarthy MI. Painting a new picture of personalised medicine for diabetes. Diabetologia 2017; 60:793–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 2004; 3:673–83. [DOI] [PubMed] [Google Scholar]
- 47. Tsoi LC, Spain SL, Knight J et al Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 2012; 44:1341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bachelez H. Interleukin 23 inhibitors for psoriasis: not just another number. Lancet 2017; 390:208–10. [DOI] [PubMed] [Google Scholar]
- 49. Lee E, Trepicchio WL, Oestreicher JL et al Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 2004; 199:125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sandborn WJ, Gasink C, Gao LL et al Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med 2012; 367:1519–28. [DOI] [PubMed] [Google Scholar]
- 51. Hueber W, Patel DD, Dryja T et al Effects of AIN457, a fully human antibody to interleukin‐17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med 2010; 2:52ra72. [DOI] [PubMed] [Google Scholar]
- 52. Baeten D, Sieper J, Braun J et al Secukinumab, an interleukin‐17A inhibitor, in ankylosing spondylitis. N Engl J Med 2015; 373:2534–48. [DOI] [PubMed] [Google Scholar]
- 53. Mease PJ, McInnes IB, Kirkham B et al Secukinumab inhibition of interleukin‐17A in patients with psoriatic arthritis. N Engl J Med 2015; 373:1329–39. [DOI] [PubMed] [Google Scholar]
- 54. Hueber W, Sands BE, Lewitzky S et al Secukinumab, a human anti‐IL‐17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double‐blind placebo‐controlled trial. Gut 2012; 61:1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Raine T, Kaser A. Seventeen in Crohn's disease: less prime than we thought? Gut 2012; 61:1653–4. [DOI] [PubMed] [Google Scholar]
- 56. Buonocore S, Ahern PP, Uhlig HH et al Innate lymphoid cells drive interleukin‐23‐dependent innate intestinal pathology. Nature 2010; 464:1371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Papp KA, Leonardi C, Menter A et al Brodalumab, an anti‐interleukin‐17‐receptor antibody for psoriasis. N Engl J Med 2012; 366:1181–9. [DOI] [PubMed] [Google Scholar]
- 58. Targan SR, Feagan B, Vermeire S et al A randomized, double‐blind, placebo‐controlled phase 2 study of brodalumab in patients with moderate‐to‐severe Crohn's disease. Am J Gastroenterol 2016; 111:1599–607. [DOI] [PubMed] [Google Scholar]
- 59. Postal M, Appenzeller S. The role of tumor necrosis factor‐alpha (TNF‐alpha) in the pathogenesis of systemic lupus erythematosus. Cytokine 2011; 56:537–43. [DOI] [PubMed] [Google Scholar]
- 60. Gregory AP, Dendrou CA, Attfield KE et al TNF receptor 1 genetic risk mirrors outcome of anti‐TNF therapy in multiple sclerosis. Nature 2012; 488:508–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Orozco G, Eyre S, Hinks A et al Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 2011; 70:463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aringer M, Smolen JS. Efficacy and safety of TNF‐blocker therapy in systemic lupus erythematosus. Expert Opin Drug Saf 2008; 7:411–9. [DOI] [PubMed] [Google Scholar]
- 63. Gordon C, Ranges GE, Greenspan JS, Wofsy D. Chronic therapy with recombinant tumor necrosis factor‐alpha in autoimmune NZB/NZW F1 mice. Clin Immunol Immunopathol 1989; 52:421–34. [DOI] [PubMed] [Google Scholar]
- 64. Kontoyiannis D, Kollias G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur J Immunol 2000; 30:2038–47. [DOI] [PubMed] [Google Scholar]
- 65. Ueda H, Howson JM, Esposito L et al Association of the T‐cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003; 423:506–11. [DOI] [PubMed] [Google Scholar]
- 66. Eyre S, Bowes J, Diogo D et al High‐density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet 2012; 44:1336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hinks A, Cobb J, Marion MC et al Dense genotyping of immune‐related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat Genet 2013; 45:664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA‐4 is a second receptor for the B cell activation antigen B7. J Exp Med 1991; 174:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kremer JM, Westhovens R, Leon M et al Treatment of rheumatoid arthritis by selective inhibition of T‐cell activation with fusion protein CTLA4Ig. N Engl J Med 2003; 349:1907–15. [DOI] [PubMed] [Google Scholar]
- 70. Ruperto N, Lovell DJ, Quartier P et al Abatacept in children with juvenile idiopathic arthritis: a randomised, double‐blind, placebo‐controlled withdrawal trial. Lancet 2008; 372:383–91. [DOI] [PubMed] [Google Scholar]
- 71. Orban T, Bundy B, Becker DJ et al Co‐stimulation modulation with abatacept in patients with recent‐onset type 1 diabetes: a randomised, double‐blind, placebo‐controlled trial. Lancet 2011; 378:412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baker KF, Isaacs JD. Novel therapies for immune‐mediated inflammatory diseases: what can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn's disease and ulcerative colitis? Ann Rheum Dis 2018; 77:175–87. [DOI] [PubMed] [Google Scholar]
- 73. Ferreira RC, Freitag DF, Cutler AJ et al Functional IL6R 358Ala allele impairs classical IL‐6 receptor signaling and influences risk of diverse inflammatory diseases. PLOS Genet 2013; 9:e1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pescovitz MD. Rituximab, an anti‐cd20 monoclonal antibody: history and mechanism of action. Am J Transplant 2006; 6:859–66. [DOI] [PubMed] [Google Scholar]
- 75. Mandik‐Nayak L, Ridge N, Fields M, Park AY, Erikson J. Role of B cells in systemic lupus erythematosus and rheumatoid arthritis. Curr Opin Immunol 2008; 20:639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wiegering V, Girschick HJ, Morbach H. B‐cell pathology in juvenile idiopathic arthritis. Arthritis 2010; 2010:759868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chun S, Casparino A, Patsopoulos NA et al Limited statistical evidence for shared genetic effects of eQTLs and autoimmune‐disease‐associated loci in three major immune‐cell types. Nat Genet 2017; 49:600–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chen L, Morris DL, Vyse TJ. Genetic advances in systemic lupus erythematosus: an update. Curr Opin Rheumatol 2017; 29:423–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Associations of 101 rheumatoid arthritis (RA) risk genes identified by Okada et al. 16 with systemic lupus erythematosus (SLE), soriatic arthritis, (PsA) and juvenile idiopathic arthritis (JIA) 8, 9, 10, 11.