

Summary
Natural killer (NK) cells play a major role in host immunity against leukaemia and lymphoma. However, clinical trials applying NK cells have not been as efficient as hoped for. Patients treated with rapidly accelerated fibrosarcoma (RAF) inhibitors exhibit increased tumour infiltration by immune cells, suggesting that a combination of RAF inhibitors with immunotherapy might be beneficial. As mitogen‐activated protein kinases (MAPKs) such as raf‐1 proto‐oncogene, serine/threonine kinase (CRAF) regulate NK cell functions, we performed an in‐vitro investigation on the potential of clinically relevant short‐acting tyrosine kinase inhibitors (TKIs) as potential adjuvants for NK cell therapy: NK cells from healthy human blood donors were thus treated with sorafenib, sunitinib or the pan‐RAF inhibitor ZM336372 during ex‐vivo expansion. Functional outcomes assessed after washout of the drugs included cytokine production, degranulation, cytotoxicity, apoptosis induction and signal transduction with/without target cell contact. Paradoxically, sorafenib enhanced NK cell effector functions in a time‐ and dose‐dependent manner by raising the steady‐state activation level. Of note, this did not lead to NK cell exhaustion, but enhanced activity against target cells such as K562 or Daudis mediated via the RAS/RAF/extracellular‐regulated kinase (ERK) pathway, but not via protein kinase B (AKT). Our data will pave the path to develop a rationale for the considered use of RAF inhibitors such as sorafenib for pre‐activation in NK cell‐based adoptive immune therapy.
Keywords: immunotherapy, NK cells, tyrosine kinase inhibitor, ZM336372
Introduction
Because initial clinical trials infusing autologous natural killer (NK) cells did not show the desired clinical effects against leukaemia/lymphoma 1, 2, 3, there is an urgent need for pharmacotherapeutic approaches to enhance autologous NK cell activity ex vivo that might lead to better clinical activity against leukaemia and lymphoma. Currently, different NK cell manufacturing and activating strategies are in evaluation, e.g. ex‐vivo expansion with irradiated autologous feeder cells, administration of interleukin (IL)‐2, IL‐15 and anti‐CD3 during expansion, magnetic affinity cell sorting (MACS)‐based CD3 depletion with or without CD56 enrichment or the use of genetically engineered NK cells 3. Here we demonstrate a different approach: short‐acting tyrosine kinase inhibitors (TKIs) might be suitable candidates, as they modulate important signal transduction pathways in both tumour and immune cells. Different studies, however, reported somewhat contradictory findings. On one hand, in‐vitro pretreatment with clinically relevant concentrations of the multi‐targeting tyrosine kinase inhibitor sorafenib, developed originally as the rapidly accelerated fibrosarcoma (RAF) inhibitor, leads to impaired NK cell effector functions 4, while treatment with the more receptor tyrosine kinase‐specific drug sunitinib during functional assays does not compromise NK cell functions 5, 6. On the other hand, we have shown activating effects on NK cells to be induced by long‐term pretreatment with other TKIs, such as the bcr‐abl/SRC inhibitor dasatinib followed by removal of the drug prior to functional assays 7. While these TKIs all target various intra‐ and extracellular kinases, including vascular endothelial growth factor receptor (VEGFR)‐2, platelet‐derived growth factor receptor (PDGFR)‐beta, fms‐like tyrosine kinase‐3 (Flt‐3) and c‐Kit 8, 9, 10, one major difference between sorafenib and sunitinib is that sorafenib also targets intracellular signalling pathways, such as the RAS/RAF/extracellular‐regulated kinase (ERK) pathway 5, 11. For NK cell effector functions, the protein kinase B (AKT)/mammalian target of rapamycin (mTor) signalling represents another important signalling pathway that is also modulated by sorafenib 12. Of note, a paradoxical activation of raf‐1 proto‐oncogene, serine/threonine kinase (CRAF) by sorafenib has been reported in cancer cell lines 13. In addition, large granular lymphocyte (LGL) expansion with enhanced leukaemic control and increased response in dasatinib‐treated acute lymphocytic leukaemia patients has been reported by us and others 14, 15. To investigate these somehow contradictory findings further and eventually provide a rationale and hierarchy for the use or limitation of RAF inhibitors as enhancers for NK cell‐based immunotherapy, we investigated the effects of sorafenib on polyclonally expanded human NK cells and compared these effects to those induced by sunitinib or the specific RAF inhibitor ZM336372. Our data regarding paradoxical RAF activation in human NK cells in vitro should set the path to develop a rationale for the considered use of RAF inhibitors for pre‐activation in NK cell‐based adoptive immune therapy.
Materials and methods
Reagents
Sorafenib (sorafenib tosylate, Nexavar®, Bayer 43–9006) and sunitinib (sunitinib malate, Sutent®) were obtained from LC Laboratories (Woburn, MA, USA). ZM336372 was purchased from Calbiochem (CAS 208260‐29‐1; Calbiochem, Merck, Billerica, MA, USA). Stock solutions were prepared with dimethyl sulphoxide (DMSO) and stored at −20°C until use. Working solutions were prepared immediately before use. DMSO 0·075% was included as solvent control in all assays.
Cell lines
The target cell line K562 [major histocompatibility complex (MHC)‐negative] was a kind gift from Professor Dr U. Kämmerer (University Hospital Würzburg, Germany) and Daudi JP (MHC‐defective) was a kind gift from Professor Dr P. Fisch (University of Freiburg, Germany). For cytotoxicity assays, cell lines were transfected with the ffluc:zeo mammalian expression vector at 300 V, as described previously 16. RPMI‐8866 (Sigma‐Aldrich, Taufkirchen, Germany) was used as the feeder cell line for NK cell expansion. K562, Daudi JP and RPMI‐8866 were cultivated in R10 medium, consisting of RPMI‐1640 medium supplemented with 10% heat‐inactivated fetal calf serum (FCS), 2 mM L‐glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin (all by PAN Biotech, Aidenbach, Germany). K562 ffLuc+ and Daudi ffLuc+ cells were cultivated in R10 selection media containing 0·6 mg/ml ZeocinTM (InvivoGen, San Diego, CA, USA).
Peripheral blood mononuclear cells (PBMCs)
PBMCs were isolated from leucocyte reduction chambers of healthy human blood donors (Department of Transfusion Medicine University Hospital of Würzburg) by Ficoll density centrifugation after written informed consent. Approval was obtained from the Ethics Committee of the University of Würzburg for these studies (reference 198/08).
Polyclonal NK cell expansion and culture
NK cells were expanded ex vivo either by using a feeder cell system applying RPMI‐8866 17, 18, 19 or a commercial NK cell isolation and activation/expansion kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Feeder cell‐based expansion was performed as described previously 7. Briefly, peripheral blood lymphocytes (PBL) were purified by plastic adherence, washed and reconstituted in R10 medium. RPMI‐8866 feeder cells were irradiated (30 Gy), washed and co‐cultured with PBLs at a ratio of 1 : 4 for 6–8 days with intermediate substitution of R10 media.
Ex‐vivo expansion of NK cells using the kit was performed according to the manufacturer's instructions. Briefly, NK cells were isolated and resting NK cells were expanded by negative selection of PBMCs and stimulation via CD2 and CD335 (NKp46) for 12–14 days.
In both cases, NK cell purity was determined using a fluorescence activated cell sorter (FACS)Calibur (BD Biosciences, Heidelberg, Germany) by gating on CD16+CD56+CD3– cells. On average, the purity of NK cells was 75 ± 7% [n = 11, mean ± standard error of the mean (s.e.m.)] for feeder cell‐ and 97 ± 2% (n = 3, mean ± s.e.m.) for kit‐expanded NK cells.
Long‐term pretreatment of polyclonal NK cells
For long‐term pretreatment, NK cells were cultured in the presence of DMSO (0·075% solvent control), sorafenib (1 µg/ml, 3 µg/ml, 5 µg/ml), sunitinib (50 ng/ml, 200 ng/ml) or ZM336372 (0·05 µg/ml, 0·5 µg/ml, 1·5 µg/ml) during expansion. Prior to functional assays and analyses, cells were washed twice with phosphate‐buffered saline (PBS) to remove the drugs.
Antibodies
Antibodies against CD107a [fluorescein isothiocyanate (FITC), lysosome‐associated membrane protein 1 (LAMP‐1) antibody (H4A3)], interferon (IFN)‐γ [phycoerythrin (PE), B27], tumour necrosis factor (TNF)‐α (PE, Mab11), CD56 [allophycocyanin (APC), NCAM 16], CD3 [peridinin chlorophyll (PerCP), SK7] (all BD Biosciences), CD56 (APC, AF12‐7H3), CD16 (APC, VEP13), TNF‐α (FITC, cA2) (all Miltenyi Biotec), CD16 (PerCP, 3G8) and human leucocyte antigen (HLA)‐E (PE, 3D12) (all BioLegend, San Diego, CA, USA) were used for flow cytometric analysis.
Antibodies against phospho‐CRAF (Ser338, 56A6), phospho‐p44/42 mitogen‐activated protein kinase (MAPK) (Thr202/Tyr204), phospho‐AKT (Ser473, 193H12) p44/42 MAPK (ERK1/2), AKT (all rabbit, polyclonal; Cell Signaling, Danvers, MA, USA), CRAF (C‐12, rabbit; Santa Cruz Biotechnology, Dallas, TX, USA), β‐actin (AC‐74, mouse; Sigma‐Aldrich) and secondary goat anti‐rabbit horseradish peroxidase (HRP) and goat anti‐mouse HRP antibodies (GE Healthcare, Freiburg, Germany) were used for Western blot analysis.
Apoptosis/necrosis assay
The toxic effect of long‐term TKI pretreatment on NK cells was determined by staining with annexin V (AnnV; BD Biosciences) and 7‐aminoactinomycin D (7‐AAD; Sigma‐Aldrich) and measured on a FACS Calibur (BD Biosciences). CD56+CD16+CD3– cells were considered to be apoptotic or late apoptotic/necrotic when they were AnnV+/7‐AAD– or AnnV+/7‐AAD+, respectively.
Degranulation and cytokine assay
Ex‐vivo expanded NK cells (3 × 105) were stimulated with K562 cells (4 × 104) in the presence of brefeldin A (Sigma‐Aldrich), GolgiStopTM (BD Biosciences) and anti‐CD107a‐FITC antibody. After 6 h of incubation, cells were stained with anti‐CD16‐APC, anti‐CD56‐APC and anti‐CD3‐PerCP antibodies. Afterwards, cells were fixed with 4% PFA and permeabilized with 0·5% saponin solution (Roth, Karlsruhe, Germany), followed by incubation with anti‐TNF‐α‐PE and anti‐IFN‐γ‐PE antibodies.
NK cell cytotoxicity assay
For the modified biophotonic luciferase assay 16, K562 ffLuc+ and Daudi ffLuc+ cells were applied as target cells. NK cells were pretreated with sorafenib or DMSO during expansion and washed prior to assay. NK cells and target cells were incubated at effector : target cell ratio of 2.5 : 1 for 4 h. Each condition was measured in triplicate using a microplate reader (Genios Basic; Tecan, Crailsheim, Germany). Effector cell cytolytic activity resulted in target cell lysis, which was monitored by a decrease in detected luminescence flux (RLU). RLU in the condition without effector cells was set as 100% and all other results referred to it.
Protein extraction
NK cell purity was determined with a FACSCalibur (BD Biosciences) as above; 1 × 107 polyclonally expanded NK cells were pretreated with sorafenib (1 µg/ml, 3 µg/ml), ZM336372 (0·5 µg/ml, 1·5 µg/ml) or DMSO for 6 and 24 h or during the time of expansion, washed twice with ice‐cold PBS and lysed directly in radioimmunoprecipitation assay (RIPA) lysis buffer. Protein concentrations were determined by Bio‐Rad protein assay (Bio‐Rad, München, Germany), according to the manufacturer's instructions.
Western blot
Whole cell lysates were boiled with sodium dodecyl sulphide (SDS) sample buffer and reducing agent (all by Invitrogen, Carlsbad, CA, USA) for 10 min at 70°C and run on 10% Bis–Tris protein gels. Proteins were transferred to nitrocellulose membranes and exposed for 2 h to the respective blocking buffer, as recommended by the manufacturer. After overnight incubation with primary phospho‐specific antibodies, membranes were washed four times in Tris‐buffered saline ‐Tween 20 (TBST) followed by 1–2‐h incubation with HRP‐conjugated secondary antibodies. Equal loading was confirmed by re‐probing the membranes with antibodies directed against total CRAF, ERK1/2, AKT and beta actin. Western blot was developed by exposing membranes to ECL substrate and X‐ray films using a CP1000 (Agfa, Düsseldorf, Germany). Densitometric quantification of protein phosphorylation was evaluated using ImageJ (NIH/DHHS, Bethesda MD, USA).
Statistical analysis
Calculation of mean, s.e.m. and establishment of statistical significance was performed with GraphPad Prism version 6.0 (GraphPad Software, Inc., San Diego, CA, USA) using the paired Student's t‐test. P‐values < 0·05 were considered statistically significant (*P < 0·05, **P < 0·01 and ***P < 0·001).
Results
Effects of sorafenib on NK cell expansion and apoptosis/necrosis induction
NK cells were expanded selectively over 8–10 days in a co‐culture system with RPMI‐8866 cells 17, 18, 19 in the presence of sorafenib. On day 8, DMSO‐treated NK cells showed a 58·3 ± 3·0‐fold increase compared to day 1, while cells treated with sorafenib (3 µg/ml) showed only an 8·9 ± 1·1‐fold increase on day 8. NK cell expansion was inhibited completely by sorafenib concentrations of 5 and 10 µg/ml (n = 7, Supporting information, Fig. S1a). Suppression of NK cell proliferation by the lower concentrations was not the result of reduced NK cell viability, as apoptosis/necrosis rates were not increased at 1 µg/ml and still below 20% at 3 µg/ml. In contrast, sorafenib concentrations above 5 µg/ml led to a significant increase in apoptosis rates (n = 5, Supporting information, Fig. S1b).
Long‐term pretreatment of NK cells with sorafenib paradoxically increases their steady state activation level, resulting in enhanced cytokine production and degranulation marker expression
Inhibitory effects of 5 and 10 µg/ml sorafenib on NK cell function have been described previously 5. Indeed, we could confirm a significant inhibition of cytokine production (IFN‐γ/TNF‐α) in NK cells when the TKI was present during the functional assay (data not shown). However, the impact of long‐term treatment with sorafenib has not yet been determined, but is relevant for NK‐cell based immunotherapeutic approaches. Therefore, we expanded human NK cells using irradiated RPMI‐8866 feeder cells in the presence of subtoxic concentrations of sorafenib (see Supporting information, Fig. S1). Afterwards, sorafenib was washed out and production of cytokines (IFN‐γ/TNF‐α) as well as expression of CD107a as surrogate marker for degranulation was determined as described 20. This long‐term pretreatment during NK cell expansion led to a markedly enhanced cytokine production by NK cells, with levels increasing by approximately fivefold when expanded with 3 µg/ml and by approximately fourfold when expanded with 1 µg/ml sorafenib (vehicle: 0·97 ± 0·24; 1 µg/ml sorafenib: 4·12 ± 1·16; 3 µg/ml sorafenib: 5·03 ± 1·24, mean ± s.e.m., n = 3) (Fig. 1a). In addition, long‐term pretreatment during expansion led to a dose‐dependent increase in expression of the degranulation marker CD107a (vehicle: 0·91 ± 0·28, 1 µg/ml sorafenib: 1·20 ± 0·28; 3 µg/ml sorafenib: 2·06 ± 0·27, mean ± s.e.m., n = 3) (Fig. 1b). As reported previously, a treatment with the TKI sunitinib did not alter the NK cell reactivity in vitro 5. Both TKIs are multi‐kinase inhibitors, but only sorafenib also targets proto‐oncogene protein B‐raf (BRAF) and CRAF. Therefore, we performed experiments using NK cells that had been expanded with non‐toxic concentrations of the specific RAF inhibitor ZM336372 21.
Figure 1.
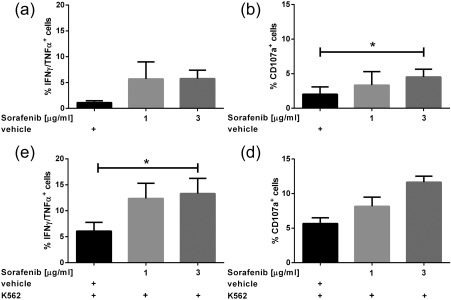
Effects of sorafenib pretreatment on natural killer (NK) cell effector functions. Polyclonally expanded NK cells were treated with sorafenib (1 µg/ml, 3 µg/ml) or dimethylsulphoxide (DMSO) (0·075%, vehicle control) during the time of expansion. Drugs were washed out before the functional assays were performed. Expanded NK cells were left unstimulated (a,b) or stimulated with the target cell line K562 (effector : target ratio 7·5 : 1) (c,d). After 6 h of incubation, secretion of cytokine [tumour necrosis factor (TNF)‐α/interferon (IFN)‐γ] (a,c) expression of CD107a (b,d) was determined by multi‐colour flow analysis. Data represent mean ± standard error of the mean (s.e.m.) from three independent experiments. Paired Student's t‐test was used for statistical evaluation. *P < 0·05.
To determine optimal concentrations of ZM336372, NK cells were expanded in the presence of this compound. Treatment with 0·5 µg/ml ZM336372 did not reduce NK cell proliferation, while expansion rates were reduced at 1·5 µg/ml. Both concentrations did not lead to induction of apoptosis/necrosis, while application of higher concentrations (above 3·75 µg/ml) resulted in significantly higher apoptosis rates (n = 3, Supporting information, Fig. S2a,b).
Cytokine production in NK cells was increased by 3·6‐fold when expanded with 1·5 µg/ml ZM336372 and CD107a expression was more than doubled, although the differences were not statistically significant (n = 3, Supporting information, Fig. S3a,b).
Long‐term pretreatment of NK cells with sorafenib or ZM336372 enhances their cytokine expression and degranulation marker expression after contact with target cells
After investigating the steady state activation of NK cells pretreated with sorafenib or ZM336372, we aimed to determine whether NK cell effector function in response to target cells is affected by pretreatment with sorafenib or ZM336372. Cytokine production (n = 3, Fig. 1c), as well as degranulation marker expression (n = 3, Fig. 1d), was enhanced significantly by sorafenib with an optimum at 3 µg/ml. Figure 2 shows a representative example of three independent experiments. Pretreatment with ZM336372 led to a similar enhancement of NK cell effector functions; namely, a significantly enhanced cytokine production and further increased expression of degranulation markers when stimulated with K562 cells compared to a DMSO‐treated control (n = 3, Supporting information, Fig. S3c,d). Similar results were obtained when applying Daudi JP as target cells (n = 3, data not shown).
Figure 2.
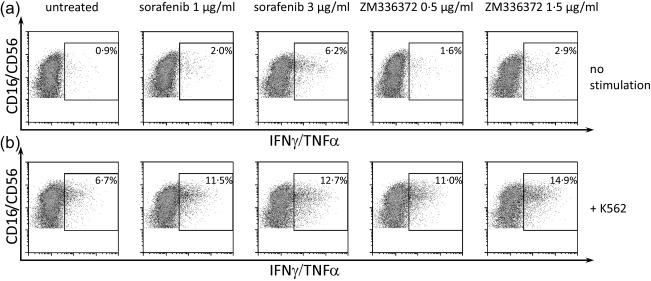
Effects of sorafenib and ZM336372 treatment on cytokine secretion. Polyclonally expanded natural killer (NK) cells were treated with sorafenib or ZM336372 during expansion (concentrations as indicated). NK cells were left untreated (a) or were pre‐incubated with the target cell line K562 (b) for 6 h before functional analysis. After incubation drugs were washed out, gates were set on lymphocytes and CD3– cells. A representative example of three independent experiments is shown.
Long‐term pretreatment of NK cells with sorafenib enhances their lytic activity against target cells
As the expression of CD107a is a surrogate marker for degranulation 20, 22, we wanted to exclude that NK cells pretreated long‐term with the TKIs become exhausted by ineffective spontaneous degranulation. Therefore, we measured the cytotoxic activity of expanded NK cells with a modified biophotonic luciferase assay 16 using ffLuc‐transfected K562 (K562 ffLuc+) and Daudi (Daudi ffLuc+) cells.
NK cells showed a profound cytotoxic effect, as the percentage of viable K562 ffLuc+ cells was reduced to 51% in the presence of NK cells expanded without any TKI. A pretreatment with sorafenib enhanced this cytotoxic effect further ; K562 viability was decreased significantly to 40% (1 µg/ml sorafenib) and 26% (3 µg/ml sorafenib) (n = 5, Fig. 3a). In addition, sorafenib pretreatment primed lytic activity of NK cells towards a cell line more resistant to NK cell effector function 23, whereas untreated NK control cells lysed only approximately 15% of Daudi ffLuc+ cells, and numbers of viable Daudi were reduced to 67 and 42% when co‐cultured with NK cells that had been pretreated with 1 and 3 µg/ml sorafenib during expansion, respectively (n = 7, Fig. 3b).
Figure 3.
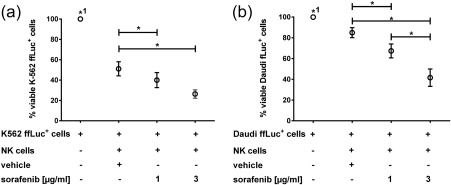
Effect of long‐term sorafenib pretreatment on natural killer (NK) cell cytotoxicity. NK cells were expanded without treatment or in the presence of sorafenib (1 µg/ml, 3 µg/ml) or dimethylsulphoxide (DMSO) (vehicle control). After expansion, cells were washed twice to remove the drugs and co‐cultured with either K562 ffLuc+ cells (n = 5) (a) or Daudi ffLuc+ cells (n = 7) (b) at an E : T ratio of 2·5 : 1 for 4 h, ffLuc+ cells alone served as controls. Each condition was measured in triplicate. Data were normalized, setting luciferase activity of ffLuc+ target cells as 100%. Data shown represent means of triplicates with standard error of the mean (s.e.m.) of n = 5 (a) or n = 7 (b) independent experiments as indicated. Paired Student's t‐test using raw data was used for statistical evaluation. *P < 0·05; *1 P < 0·05 in comparison to all other groups.
Sorafenib activates phosphorylation of CRAF and ERK1/2 in NK cells
To elucidate the underlying mechanisms for the enhanced NK cell effector functions, we performed analysis of signal transduction using lysates from NK cells that had been pretreated with sorafenib or ZM336372. We investigated the phosphorylation level of CRAF and downstream pathway components in pretreated NK cells. Immunoblotting confirmed a dose‐dependent enhancement of the phosphorylation status of CRAF and ERK1/2 in NK cells treated with sorafenib and ZM336372, whereas phosphorylation of AKT was unaffected by incubation with these drugs (Fig. 4). An enhancing effect on the phosphorylation of ERK1/2 was observed to a lesser extent also after 6 and 24 hours of pretreatment with sorafenbib or ZM336372, while a treatment with sunitinib did not alter the phosphorylation status of CRAF or ERK1/2 in any treatment scheme, (n = 3, data not shown), confirming that the sorafenib‐mediated effects are driven by RAF inhibition. To ensure that the observed effects were based on modulation of NK cell activity and not due to effects related to contaminating T cells (mean purity of NK cells: 75%), we repeated the Western blot experiments with lysates from highly purified NK cells (CD16/56+CD3– of 97 ± 2%, n = 3, mean ± s.e.m.), which had been obtained using an isolation and activation kit. In line with the results from the feeder cell expansions, ERK1/2 phosphorylation was increased similarly by treatment with sorafenib and ZM336372 (data not shown).
Figure 4.

Effects of sorafenib and ZM336372 on raf‐1 proto‐oncogene, serine/threonine kinase (CRAF), extracellular‐regulated kinase (ERK)1/2 and protein kinase B (AKT) phosphorylation of natural killer (NK) cells. (a) Simplified model for induction of effector functions in NK cells. Effector functions can be induced by via several activating receptors on the NK cell surface, including NKG2D and the natural cytotoxicity receptors NKp30, NKp44 and NKp46 that are coupled to proteins carrying immunoreceptor tyrosine‐based activation motifs (ITAM) [FcRγ, CD3ζ and DNAX activation protein of 12kDa (DAP12)]. Signalling pathways leading to NK cell effector functions include the activation of ERK or AKT. Mitogen‐activated protein kinase kinase (MEK1/2) and ERK1/2 can be triggered either via the GTPase RAS and its effector molecule RAF downstream of ITAM‐coupled receptors or via phosphoinositide 3‐kinase (PI3K)‐Rac family small GTPase 1 (Rac1)‐p21‐activated kinase‐1 (PAK) downstream of NKG2D coupled to the transmembrane adaptor DAP10. In addition, PI3K also mediates signal transduction via activation of AKT and mTOR. All pathways lead to activation of transcription factors and thereby to synthesis and regulation of proteins mediating NK cell effector functions. Scheme based on 27, 40. (b) Polyclonally expanded NK cells were treated with the indicated concentrations of sorafenib and ZM336372 during the time of expansion. Both treatments led to a dose‐dependent increase of phosphorylated CRAF and ERK1/2. No obvious differences of phosphorylation were detected for AKT. Representative Western blot for four independent experiments; NK cell purity median 77%, range 75–84%.
Discussion
Our data regarding paradoxical RAF activation in polyclonally expanded human NK cells ex vivo should set the path for the consideration of CRAF inhibitors for pre‐activation of NK cells in NK cell‐based adoptive immune therapy. Our results indicate that NK cell effector functions can be stimulated by RAF inhibitor treatment. The signalling cascade leading to NK cell activation is somewhat complex; in addition to MHC‐I‐dependent signalling, NK cells possess a broad spectrum of activating and inhibitory receptors. The complicated balance of activating and inhibitory signals determines NK cell functions 24, 25. One of the major intracellular signalling pathways represents the MAPK/ERK pathway 25, 26, 27. We could demonstrate clearly that the stimulatory effects of sorafenib or ZM336372 are related to alteration of the MAPK/ERK pathway, while the alternative AKT pathway was not affected. Our results indicate that treatment might shift the balance towards activation by constitutively activating the MAPK/ERK pathway. This hypothesis can be strengthened further by work performed by other groups 13, 28: Poulikakos et al. demonstrated that six different RAF inhibitors – including sorafenib and ZM336372 – lead to a dephosphorylation of MEK/ERK1/2 in the Calu‐6 cell line, a human cancer cell line with a BRAFV600E mutation, but lead to an increase of ERK1/2 phosphorylation in cells with wild‐type RAS and wild‐type RAF. Hatzivassiliou et al. showed that cancer cell lines not carrying the BRAFV600E mutation had an increased tumour growth reliant upon CRAF signalling when treated with RAF inhibitors. In BRAFV600E cells, BRAF monomers are constitutively active while RAS is suppressed. Due to the suppression of RAS, no RAF dimers are formed. In these BRAF mutated cells the inhibitors bind the ATP binding site of the RAF monomer and thereby inhibit signalling. In contrast, BRAF/BRAF homodimers or BRAF/CRAF heterodimers are formed in a RAS‐dependent manner in BRAF wild‐type cells. The binding of sorafenib or ZM336372 to the ATP binding site of one monomer leads to a transactivation of the second catalytic centre. While one catalytic centre is blocked by the inhibitor, the catalytic centre of the second RAF monomer becomes constitutively active leading to tonic signalling. As these inhibitors are ATP competitive, increasing concentrations of the drugs lead to an increase of transactivated dimers in the first place. When the concentration reaches a level where both ATP binding sites are occupied by the inhibitor, the signalling is blocked 13, 28. In addition, the results of a Phase II study of sorafenib in children with low‐grade astrocytoma showed an unexpectedly high rate of early and rapid progression, which led to early termination of the study. The authors stated paradoxical ERK activation as the probable underlying pathomechanism. However, the effects were not related to BRAF mutational status 29. In cholangiocytes, sorafenib‐induced cAMP/protein kinase A (PKA)‐dependent Raf/MEK/ERK signalling activation has been described 30. Hereby, sorafenib seems to activate AMPK indirectly by inhibiting mitochondrial metabolism and increasing cellular AMP : ADP and/or ADP : ATP ratios 31.
Considering these observations, we can formulate two diverging clinical propositions: on one hand, RAF inhibitors seem to be potent enhancement tools for the pre‐activation of NK cells prior to autologous NK cell transfusion. Careful dosing during the time of expansion is necessary, as high concentrations inhibit proliferation and lead to immunosuppression with decreased anti‐tumour efficacy. However, whether the ex‐vivo pre‐activation results in better clinical response rates has yet to be determined. Especially in Flt‐3‐positive refractory AML, treatment with sorafenib seems appealing, as both sorafenib as well as NK cell‐based immunotherapy/lymphokine‐activated killer cells are already tested clinically and seem to be efficient and well‐tolerated treatment strategies 32, 33. A Phase I study in patients with Flt‐3‐positive AML, which applied sorafenib (200–400 mg twice daily) after allogenic stem cell transplantation, showed promising results with an overall 1‐year progression‐free survival of 85% 34. A clinical trial comparing chemotherapy alone versus chemotherapy + donor lymphocyte infusion (DLI) versus sorafenib alone versus sorafenib + DLI in AML patients with relapse after allogenic transplant and Flt‐3 mutation has been concluded in six of 16 patients; the results are pending [SIRA trial, http://ClinicalTrials.gov Identifier: NCT02867891]. In these patients, the use of low‐dose sorafenib + pre‐activated DLIs might enhance NK cell effector functions and also target effectively the FLT‐3 mutated AML blasts. Moreover, an enhanced number of tumour‐infiltrating NK cells have been observed in melanoma after administration of BRAF inhibitor PLX4720 35.
On the other hand, our results indicate that treatment with 400 mg sorafenib twice daily might lead to immunosuppression and reduced anti‐tumour response by NK cells in vivo. This hypothesis can be strengthened by in‐vivo experiments in mouse models 36, 37, 38, 39, while the clinical relevance of these findings remains unclear so far.
Our data thus indicate that timing and dosing have to be considered carefully if RAF inhibitor pretreatment improves the efficacy of adoptive immune therapies. While certain dosage levels applied for long‐term treatment can lead to activating effects, higher doses show an immune‐inhibitory effect while low‐dose sorafenib treatment may have no effect at all. In the setting of autologous NK cell therapy, where expansion and pre‐activation will be performed in vitro, these conditions can be adjusted carefully. In this context, our in‐vitro test system may have provided a starting‐point to find the most suitable dose and exposure time. For patients treated systemically with sorafenib, the narrow therapeutic window between activation of the immune response and proper anti‐tumour effect of the drug will be very challenging to define.
To gain further understanding of the underlying mechanisms, phospho‐proteomic approaches revealing the crucial signalling events during NK cell pre‐activation might prove helpful. Especially with regard to NK cell chimeric antigen receptor (CAR)‐based immunotherapy, knowledge of the signalling landscape with the ability to stimulate NK cell activation selectively is essential. Furthermore, our data indicate that thoughtful dosing and surveillance of plasma levels during therapy is necessary to maximize anti‐tumour effects while simultaneously maintaining immunological integrity.
Disclosure
None of the authors has any potential conflict of interest relating to this paper.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Effects of sorafenib treatment on natural killer (NK) cell apoptosis/necrosis and expansion: NK cells were expanded for 8 days by co‐culturing peripheral blood lymphocyte (PBLs) with irradiated RPMI‐8866 cells at a ratio of 4 : 1 in the presence of sorafenib. On days 1, 5 and 8, NK cell number was calculated based on the counted total number of viable cells and percentage of CD56+CD16+CD3– cells, which was determined using BD FACSCalibur (a). At day 8 of expansion, apoptotic and late apoptotic/necrotic cells were determined based on the amount of annexinV+/7‐amino actinomycin (7AAD–) and annexinV+/7AAD+CD56+CD16+CD3– cells, respectively. For vehicle control, cells were treated with dimethyl sulphoxide (DMSO) (b). Shown are the mean ± standard error of the mean (s.e.m.) of seven (a) or five (b) independent experiments.
Fig. S2. Effects of ZM336372 treatment of natural killer (NK) cell apoptosis/necrosis and expansion: NK cells were expanded for 8 days by co‐culturing peripheral blood lymphocyte (PBLs) with irradiated RPMI‐8866 cells at a ratio of 4 : 1 in the presence of ZM336372. On days 1, 5 and 8, NK cell number was calculated based on the counted total number of viable cells and percentage of CD56+CD16+CD3– cells which was determined using BD FACSCalibur (a). At day 8 of expansion, apoptotic and late apoptotic/necrotic cells were determined based on the amount of annexinV+/7‐amino actinomycin (7AAD–) and annexinV+/7AAD+CD56+CD16+CD3– cells, respectively. For vehicle control, cells were treated with dimethyl sulphoxide (DMSO) (b). Shown are the mean ± standard error of the mean (s.e.m.) of three independent experiments.
Fig. S3. Effects of ZM pretreatment on natural killer (NK) cell effector functions. Polyclonally expanded NK cells were treated with ZM336372 (0·5 µg/ml, 1·5 µg/ml) or dimethyl sulphoxide (DMSO) (0·075%, vehicle control) during the time of expansion. Drugs were washed out before the functional assays were performed. Expanded NK cells were left unstimulated (a,b) or stimulated with the target cell line K562 (effector to target ratio 7·5 : 1) (c and d). After 6 h of incubation, secretion of cytokines [tumour necrosis factor (TNF)‐α/interferon (IFN)‐γ] (a,c) expression of CD107a (b,d) was determined by multi‐colour flow analysis. Data represent mean ± standard error of the mean (s.e.m.) from three independent experiments. Paired Student's t‐test was used for statistical evaluation. *P < 0·05.
Supplementary Legend
Author contributions
J. L., T. N. and J. D. performed the research, R. S. B. and H. E. designed the research study, T. N., J. L. and J. D. analysed the data and T. N., J. L. and R. S. B. wrote the paper. All authors read and approved the final manuscript.
Acknowledgements
We thank Carolin Koechel and Irina Eichelbroenner for technical assistance with functional assays. This work was supported in part by IZKF, Würzburg, Germany (grant Z‐2/27 to R.S. B.), Deutsche Krebshilfe e. V. (grant no. 109205 to R. S. B.). We thank Professor Jörg Wischhusen for critical reading of the manuscript.
References
- 1. Zamai L, Ponti C, Mirandola P et al NK cells and cancer. J Immunol 2007; 178:4011–6. [DOI] [PubMed] [Google Scholar]
- 2. Bachanova V, Miller JS. NK cells in therapy of cancer. Crit Rev Oncog 2014; 19:133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pittari G, Filippini P, Gentilcore G, Grivel JC, Rutella S. Revving up natural killer cells and cytokine‐induced killer cells against hematological malignancies. Front Immunol 2015; 6:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin del Campo SE, Levine KM, Mundy‐Bosse BL et al The Raf kinase inhibitor sorafenib inhibits JAK–STAT signal transduction in human immune cells. J Immunol 2015; 195:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krusch M, Salih J, Schlicke M et al The kinase inhibitors sunitinib and sorafenib differentially affect NK cell antitumor reactivity in vitro . J Immunol 2009; 183:8286–94. [DOI] [PubMed] [Google Scholar]
- 6. Salih J, Hilpert J, Placke T et al The BCR/ABL‐inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int J Cancer 2010; 127:2119–28. [DOI] [PubMed] [Google Scholar]
- 7. Hassold N, Seystahl K, Kempf K et al Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int J Cancer 2012; 131:E916–27. [DOI] [PubMed] [Google Scholar]
- 8. Kumar R, Crouthamel MC, Rominger DH et al Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer 2009; 101:1717–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roskoski R Jr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun 2007; 356:323–8. [DOI] [PubMed] [Google Scholar]
- 10. Karaman MW, Herrgard S, Treiber DK et al A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008; 26:127–32. [DOI] [PubMed] [Google Scholar]
- 11. Packer LM, Rana S, Hayward R et al Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug‐resistant chronic myeloid leukemia. Cancer Cell 2011; 20:715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhai B, Hu F, Jiang X et al Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther 2014; 13:1589–98. [DOI] [PubMed] [Google Scholar]
- 13. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature 2010; 464:427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mustjoki S, Ekblom M, Arstila TP et al Clonal expansion of T/NK‐cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia 2009; 23:1398–405. [DOI] [PubMed] [Google Scholar]
- 15. Kreutzman A, Juvonen V, Kairisto V et al Mono/oligoclonal T and NK cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood 2010; 116:772–82. [DOI] [PubMed] [Google Scholar]
- 16. Brown CE, Wright CL, Naranjo A et al Biophotonic cytotoxicity assay for high‐throughput screening of cytolytic killing. J Immunol Methods 2005; 297:39–52. [DOI] [PubMed] [Google Scholar]
- 17. Valiante NM, Rengaraju M, Trinchieri G. Role of the production of natural killer cell stimulatory factor (NKSF/IL‐12) in the ability of B cell lines to stimulate T and NK cell proliferation. Cell Immunol 1992; 145:187–98. [DOI] [PubMed] [Google Scholar]
- 18. Baltz KM, Krusch M, Bringmann A et al Cancer immunoediting by GITR (glucocorticoid‐induced TNF‐related protein) ligand in humans: NK cell/tumor cell interactions. FASEB J 2007; 21:2442–54. [DOI] [PubMed] [Google Scholar]
- 19. Mainiero F, Soriani A, Strippoli R et al RAC1/P38 MAPK signaling pathway controls beta1 integrin‐induced interleukin‐8 production in human natural killer cells. Immunity 2000; 12:7–16. [DOI] [PubMed] [Google Scholar]
- 20. Betts MR, Brenchley JM, Price DA et al Sensitive and viable identification of antigen‐specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 2003; 281:65–78. [DOI] [PubMed] [Google Scholar]
- 21. Hall‐Jackson CA, Eyers PA, Cohen P et al Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol 1999; 6:559–68. [DOI] [PubMed] [Google Scholar]
- 22. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med 2005; 202:1001–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hermann GG, Zeuthen J, Claesson MH. LAK‐cell‐mediated cytotoxicity against tumor cell targets used to monitor the stimulatory effect of interleukin‐2: cytotoxicity, target recognition and phenotype of effector cells lysing the Daudi, T24 and K562 tumor cell lines. Nat Immun 1992; 11:7–16. [PubMed] [Google Scholar]
- 24. Miller JS. The biology of natural killer cells in cancer, infection, and pregnancy. Exp Hematol 2001; 29:1157–68. [DOI] [PubMed] [Google Scholar]
- 25. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Farag SS, Caligiuri MA. Human natural killer cell development and biology. Blood Rev 2006; 20:123–37. [DOI] [PubMed] [Google Scholar]
- 27. Vivier E, Ugolini S, Nunes JA. ADAPted secretion of cytokines in NK cells. Nat Immunol 2013; 14:1108–10. [DOI] [PubMed] [Google Scholar]
- 28. Hatzivassiliou G, Song K, Yen I et al RAF inhibitors prime wild‐type RAF to activate the MAPK pathway and enhance growth. Nature 2010; 464:431–5. [DOI] [PubMed] [Google Scholar]
- 29. Karajannis MA, Legault G, Fisher MJ et al Phase II study of sorafenib in children with recurrent or progressive low‐grade astrocytomas. Neuro Oncol 2014; 16:1408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spirli C, Morell CM, Locatelli L et al Cyclic AMP/PKA‐dependent paradoxical activation of Raf/MEK/ERK signaling in polycystin‐2 defective mice treated with sorafenib. Hepatology 2012; 56:2363–74. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Ross FA, Hawley SA, Auciello FR et al Mechanisms of paradoxical activation of AMPK by the kinase inhibitors SU6656 and sorafenib. Cell Chem Biol 2017; 24:813–24.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tarlock K, Chang B, Cooper T et al Sorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemia. Pediatr Blood Cancer 2015; 62:1048–54. [DOI] [PubMed] [Google Scholar]
- 33. Antar A, Kharfan‐Dabaja MA, Mahfouz R, Bazarbachi A. Sorafenib maintenance appears safe and improves clinical outcomes in FLT3‐ITD acute myeloid leukemia after allogeneic hematopoietic cell transplantation. Clin Lymphoma Myeloma Leuk 2015; 15:298–302. [DOI] [PubMed] [Google Scholar]
- 34. Chen YB, Li S, Lane AA et al Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms‐like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Biol Blood Marrow Transplant 2014; 20:2042–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Knight DA, Ngiow SF, Li M et al Host immunity contributes to the anti‐melanoma activity of BRAF inhibitors. J Clin Invest 2013; 123:1371–81. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Chen Y, Duda DG. Targeting immunosuppression after standard sorafenib treatment to facilitate immune checkpoint blockade in hepatocellular carcinoma – an auto‐commentary on clinical potential and future development. Oncoimmunology 2015; 4:e1029703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Porta C, Paglino C, Imarisio I, Ganini C, Pedrazzoli P. Immunological effects of multikinase inhibitors for kidney cancer: a clue for integration with cellular therapies? J Cancer 2011; 2:333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hipp MM, Hilf N, Walter S et al Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 2008; 111:5610–20. [DOI] [PubMed] [Google Scholar]
- 39. Zhang QB, Sun HC, Zhang KZ et al Suppression of natural killer cells by sorafenib contributes to prometastatic effects in hepatocellular carcinoma. PLOS ONE 2013; 8:e55945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ali AK, Nandagopal N, Lee SH. IL‐15–PI3K–AKT–mTOR: a critical pathway in the life journey of natural killer cells. Front Immunol 2015; 6:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Effects of sorafenib treatment on natural killer (NK) cell apoptosis/necrosis and expansion: NK cells were expanded for 8 days by co‐culturing peripheral blood lymphocyte (PBLs) with irradiated RPMI‐8866 cells at a ratio of 4 : 1 in the presence of sorafenib. On days 1, 5 and 8, NK cell number was calculated based on the counted total number of viable cells and percentage of CD56+CD16+CD3– cells, which was determined using BD FACSCalibur (a). At day 8 of expansion, apoptotic and late apoptotic/necrotic cells were determined based on the amount of annexinV+/7‐amino actinomycin (7AAD–) and annexinV+/7AAD+CD56+CD16+CD3– cells, respectively. For vehicle control, cells were treated with dimethyl sulphoxide (DMSO) (b). Shown are the mean ± standard error of the mean (s.e.m.) of seven (a) or five (b) independent experiments.
Fig. S2. Effects of ZM336372 treatment of natural killer (NK) cell apoptosis/necrosis and expansion: NK cells were expanded for 8 days by co‐culturing peripheral blood lymphocyte (PBLs) with irradiated RPMI‐8866 cells at a ratio of 4 : 1 in the presence of ZM336372. On days 1, 5 and 8, NK cell number was calculated based on the counted total number of viable cells and percentage of CD56+CD16+CD3– cells which was determined using BD FACSCalibur (a). At day 8 of expansion, apoptotic and late apoptotic/necrotic cells were determined based on the amount of annexinV+/7‐amino actinomycin (7AAD–) and annexinV+/7AAD+CD56+CD16+CD3– cells, respectively. For vehicle control, cells were treated with dimethyl sulphoxide (DMSO) (b). Shown are the mean ± standard error of the mean (s.e.m.) of three independent experiments.
Fig. S3. Effects of ZM pretreatment on natural killer (NK) cell effector functions. Polyclonally expanded NK cells were treated with ZM336372 (0·5 µg/ml, 1·5 µg/ml) or dimethyl sulphoxide (DMSO) (0·075%, vehicle control) during the time of expansion. Drugs were washed out before the functional assays were performed. Expanded NK cells were left unstimulated (a,b) or stimulated with the target cell line K562 (effector to target ratio 7·5 : 1) (c and d). After 6 h of incubation, secretion of cytokines [tumour necrosis factor (TNF)‐α/interferon (IFN)‐γ] (a,c) expression of CD107a (b,d) was determined by multi‐colour flow analysis. Data represent mean ± standard error of the mean (s.e.m.) from three independent experiments. Paired Student's t‐test was used for statistical evaluation. *P < 0·05.
Supplementary Legend