

Abstract
In this study, we investigated the biological function of pyruvate kinase M2 (PKM2) and its regulation by deregulated microRNAs (miRNAs) in thyroid cancer (TC). The mRNA and protein expression of PKM2 was examined by quantitative reverse transcription PCR and western blot. The biological role of PKM2 was demonstrated through small interfering RNA-mediated knockdown experiments. The regulation of PKM2 by miR-148a and miR-326 was confirmed by western blot, dual luciferase activity assays, and rescue experiments. PKM2 was overexpressed in TC tissues and cell lines. The knockdown of PKM2 in TC cells suppressed cell proliferation, reduced colony formation, and inhibited cell invasion and migration significantly. Luciferase reporter assays revealed that PKM2 is a direct target of two tumor-suppressive miRNAs, miR-148a and miR-326. Re-expressed PKM2 rescued the anticancer effects of miR-148a. Taken together, these data strongly suggest that, apart from gene amplification and mutation, the activation of PKM2 in TC is partly due to the down-regulation of the tumor-suppressive miRNAs miR-148a and miR-326. Thus, PKM2 is overexpressed and plays an oncogenic role in thyroid carcinogenesis.
Keywords: PKM2, thyroid cancer, miR-148a, miR-326
Introduction
Thyroid cancer (TC), located at the base of the neck, is one of the most common endocrine malignancies [1] and one of the fastest rising malignant tumors, owing to widespread environmental and dietary changes in recent years [2,3]. Although most TCs can be managed successfully with a combination of radioactive iodide and levothyroxine suppression therapy after complete surgical intervention, a proportion of TCs remain irresponsive to treatment and result in comorbidities and mortality [4]. Diverse TCs have been identified, and the variability of TCs has been confirmed in molecular, cellular, and clinical studies, which have suggested an imbalance in related regulatory genes as a risk factor for tumorigenesis [5]. Understanding the molecular mechanisms involved in the formation and development of TC is important for preventing and treating TC as well as improving patient survival.
Pyruvate kinase (PK), a rate-limiting enzyme in glycolysis, exists in four isoforms (M1, M2, L, and R). Among these isoforms, PKM2 is predominantly expressed in the fetus and is replaced by PKM1 during development [6]. This leads to increased glucose uptake and lactate production and decreased O2 consumption by cancer cells. A large body of evidence indicates that cancers predominantly express PKM2, which acts as a critical regulator of cancer cell metabolism [7]. Usually, high levels of PKM2 are associated with a poor prognosis for patients with cancer [8]. Additionally, PKM2 acts as a transcriptional coactivator to promote gene transcription in cancer cells and is believed to play a crucial role in tumor progression. Most physiological studies have focused on the regulation of cell migration by PKM2 and found that PKM2 is a key determinant of cell proliferation and migratory behavior [9]. Xu et al. [10] reported that the PKM2/NF-κB/miR-148a/152 pathways regulate angiogenesis and cancer progression. However, the mechanism by which PKM2 up-regulation and activation are involved in TC has not been elucidated. Thus, we investigated the mechanism of PKM2 up-regulation and how PKM2 activation promotes TC.
Materials and methods
Clinical samples
Twenty pairs of TC tissues and adjacent non-tumor tissues were collected from The First Affiliated Hospital of Bengbu Medical College and The Second Hospital of Anhui Medical University. All patients did not receive any radiotherapy, chemotherapy or radiofrequency ablation before surgical resection. Written informed consents were obtained from all enrolled patients.
Cell lines
The human TC cell lines (TPC-1, K1 and BCPAP), and the normal human thyroid cell line HT-ori3 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All the cell lines were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS, Hyclone, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, US) in a humidified 5% (v/v) CO2 atmosphere at 37°C.
RNA isolation and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
According to the manufacturer’s protocol, total RNA was isolated from homogenised tissues and cell lines using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). RNA quantity and quality were evaluated by Nano Drop ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA synthesis and qRT-PCR as previously introduced [11]. U6 and GAPDH were used to normalize the level of miR-148a, miR-326 and PKM2 mRNA expression, respectively. The primers used in the present study were as follows: miR-148a, 5’-TCA GTG CAC TAC AGA ACT TTG T-3’ (forward) and 5’-TCT GTC AAC GAT ACG CTA CGT-3’ (reverse); miR-326, 5’-GTC TCC CAG AUA AUG CG-3’ (forward) and 5’-CGT TCA GGG TCC-3’ (reverse); U6, 5’-TCT TCG TCA TCA CAT ATA CTA AAA T-3’ (forward) and 5’-CTC TTC ACG AAT TTT CGT GTC AT-3’ (reverse); PKM2, 5’-GTC GAA TCC CCA TAG TGA AG-3’ (forward) and 5’-GTG AAT CAA TGT CCA GTC GG-3’ (reverse); and GAPDH, 5’-CGG AGT CAA CGG ATT TGG TCG TAT-3’ (forward) and 5’-ATC CTT CTC CAT GGT GGT GAA GAC-3’ (reverse).
Western blot analysis
To determine the expression of PKM2, equal amounts of protein (about 30 mg) per lane were separated by 8% SDS-polyacrylamide gel and transferred to PVDF membrane. The membranes were blocked in 5% skim milk for 1 h and then incubated with a specific antibody (PKM2, ab38237, 1:1000; Abcam, Cambridge, MA, USA) for 2 h. GAPDH (Santa Cruz, USA) was selected for an internal control. The band density of specific proteins was quantified after normalization with the density of GAPDH.
miRNA/siRNA transfection
The miRNA precursors, miR-148a, miR-326 were from Life Technologies, and siPKM2, PKM2 were from Qiagen (Valencia, CA, USA). All transfection were performed in a 30 nM concentration for 36 hours followed with functional study and RNA/protein analysis.
MTT cell proliferation was assessed by Cell Titer 96 NonRadioactive Cell Proliferation Assays (Promega, Madison, WI) and colony formation assays were used to detect the proliferation activity according to manufacturer’s instruction. The cell migration assays were performed by Transwell chamber (Corning Glass Works, Corning, New York, USA) and the cell invasion assay using BD Biocoat Matrigel Invasion Chambers (BD Biosciences, Franklin Lakes, NJ) has been described previously [12]. The functional study of siPKM2, PKM2 (Qiagen, Valencia, CA) was also performed as above except that a concentration of 25 nM siRNA was used.
Luciferase reporter assay
The cells were seeded into 48-well plates followed by transfection with the Renilla luciferase pRL-TK plasmid (100 ng/ml, Promega, USA) plus the recombinant Firefly luciferase pGL3 reporters contained 3’UTR region of human PKM2 in combination with miR-148a or miR-326 by using Lipofectamine 2000. For the luciferase reporter assay, cells were cultured in 96-well plates, transfected with the plasmids and miRNA mimics using Lipofectamine 2000. 48 h after transfection, relative luciferase activities were measured using the Luciferase Assay System (Promega).
In vivo tumorigenicity model
TPC-1 cells (5×106 cells suspended in 0.1 ml PBS) were transfected with PKM2 or scramble control then were injected subcutaneously into the left flank of 4-week old Balb/c nude mice respectively. Diameters of tumors were measured and documented each 5 days with a total of 25 days. The animal care and experimental protocols were approved by Bengbu Medical College. And parts of tumours were evaluated by immunohistochemistry with indicated antibodies (Ki67).
Rescue experiments
For the rescue experiments, TCP-1 and K1 cells were first transfected with miR-148a, miR-326 precursor or negative control respectively. After 24 h incubation, PKM2 expression plasmid together with empty vector (pcDNA3.1, Life Technologies) was transfected to the cells using Transfection Reagent (Roche, Nutley, NJ). After another 24 h, cells were harvested for functional study (MTT proliferation, monolayer colony formation, and cell invasion assays).
Statistical analysis
All data were analyzed by GraphPad Prism version 6 statistical software. Measurement data were expressed by mean ± standard deviation (SD). The Student’s t-test test was used for comparisons between two groups. One-way analysis of variance was applied for comparisons between multiple groups. Statistical significance was assumed for P < 0.05.
Results
PKM2 is up-regulated in TC
To investigate whether PKM2 is dysregulated in TC, we firstly detected the expression level of PKM2 in 20 pairs of human TC tissues and the matched normal tissues by RT-qPCR assays. As shown in Figure 1A, PKM2 expression levels were significantly up-regulated in human TC tissues compared with matched normal tissues. Moreover, the mRNA expression of PKM2 in the TC cell lines also was determined. As shown in the Figure 1B, we showed significant up-regulation of PKM2 in the TC cell lines compared to the normal human thyroid cell HT-ori3.
Figure 1.
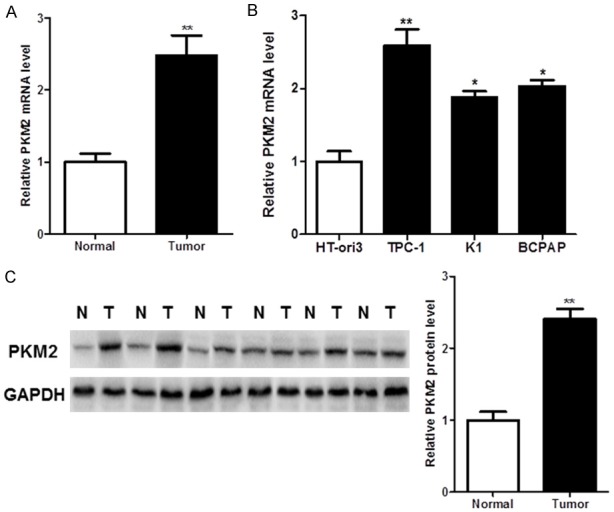
PKM2 is overexpressed in thyroid cancer (TC). A, B. PKM2 mRNA was up-regulated in TC tissues and cell lines. C. PKM2 protein was overexpressed in TC tissues.
In further to determine the role of PKM2 in the TC, we also detected the protein level of PKM2 in the human TC tissues and the matched normal tissues by western blot. The TC tissues showed an up-regulated PKM2 expression compared with the matched normal tissues (Figure 1C).
The oncogenic role of PKM2 in vitro and vivo
Given the abundance of PKM2 in TC, siRNA-mediated knockdown was performed in TPC-1 and K1 cells for functional studies. A significantly decreased cell proliferation was detected in PKM2-depleted TC cells (P < 0.01, Figure 2A). Also, PKM2 knockdown significantly reduced colony formation in these cell lines (P < 0.01, Figure 2B). In addition, siRNA-mediated knockdown of PKM2 decreased TC cell migration (P < 0.01, Figure 2C) and invasion (P < 0.01, Figure 2D). Moreover, the oncogenic role of PKM2 was validated in vivo. PKM2 overexpression significantly enhanced the growth of tumor xenografts in nude mice (P < 0.01, Figure 2E). In addition, we also detected the expression of Ki67 in the tumor tissues. Compared with the NC group, PKM2 overexpression group showed a significant increase of Ki67.
Figure 2.
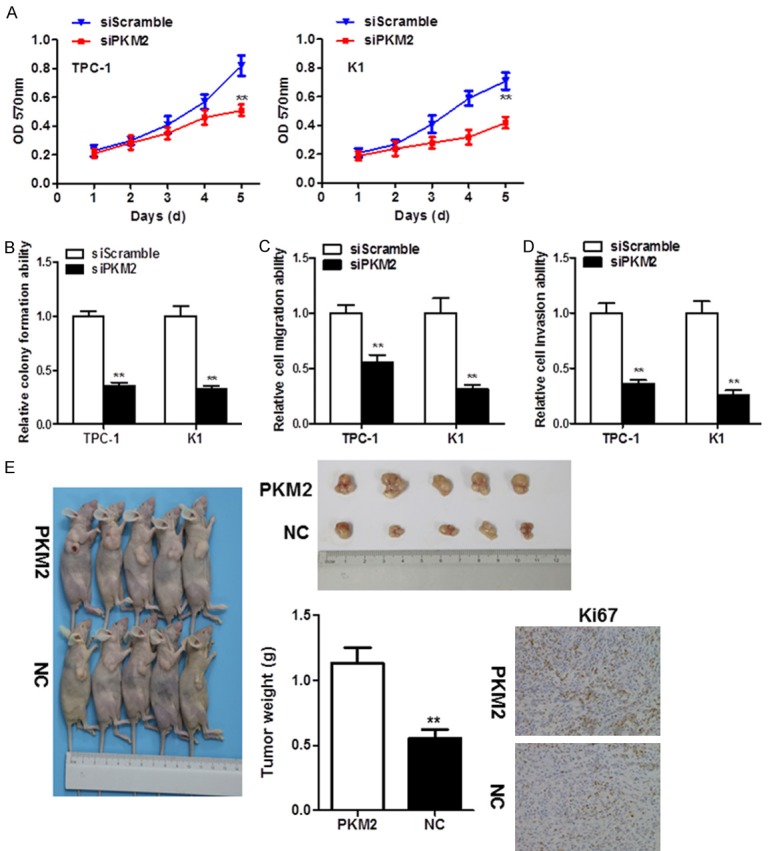
PKM2 silencing in TC cells exhibited a tumor-suppressive function. A. MTT assays revealed that PKM2 knockdown by short interfering RNA significantly suppressed TC cell proliferation. B. Monolayer colony formation assays suggested that PKM2 depletion reduced anchorage-dependent colony formation. C. PKM2 knockdown decreased the cell migration ability of TC cells. D. Cell invasion was significantly inhibited in PKM2-depleted TC cells. E. PKM2 overexpression promoted xenograft formation in nude mice and Ki67 levels in tumors, as assessed by immunohistochemistry.
PKM2 is negatively regulated by miR-148a and miR-326 in TC
In the first, we searched the TargetScan database (http://www.targetscan.org/) to predict the potential mRNA targets, miR-148a and miR-326 were noticed as they have multiple putative targets on PKM2 3’UTR (Figure 3A). In TC cell lines, miR-148a or miR-326 reduce PKM2 expression in both mRNA and protein levels (Figure 3B). Moreover, luciferase assays were performed to validate the direct binding affinity between PKM2 3’UTR and these two miRNAs. Reporter assays revealed that induction of miR-148a or miR-326 triggered a marked decrease of luciferase activity from pGL3-WT-PKM2-3’UTR but produced no change in the luciferase activity from pGL3-MUT-PKM2-3’UTR (Figure 3C). These data indicated that miR-148a or miR-326 directly modulated PKM2 expression by binding to the respective 3’UTR.
Figure 3.
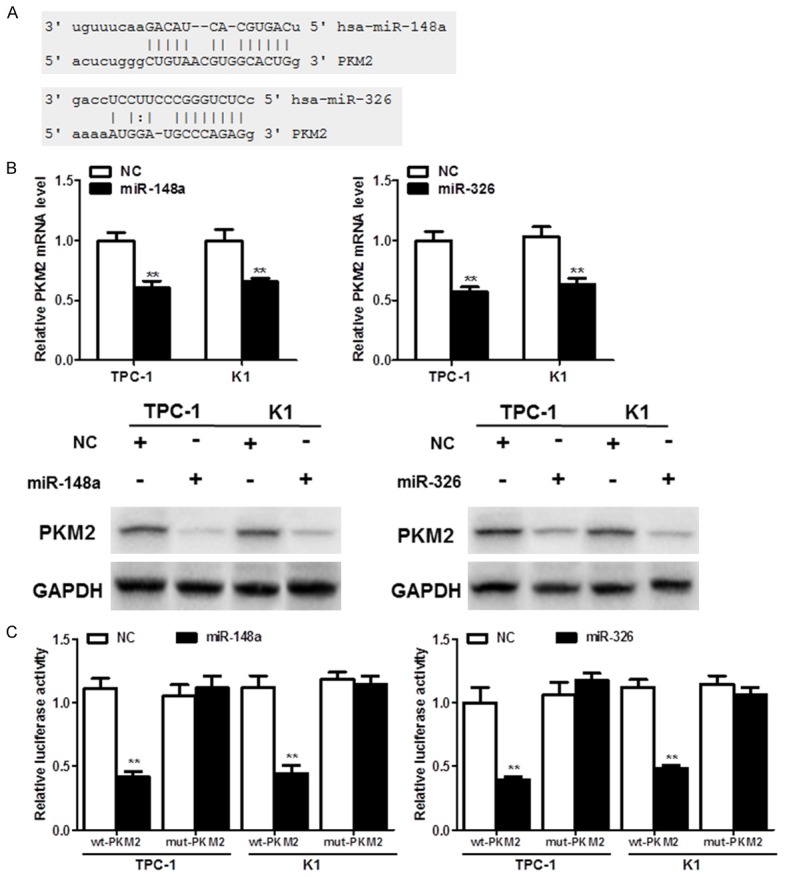
PKM2 is directly regulated by miR-148a and miR-326 in TC. A. Predicted putative miR-148a and miR-326-binding sites in the 3’-untranslated region (UTR) of PKM2. B. The mRNA and protein expression of PKM2 after miR-148a or miR-326 overexpression in TPC-1 and K1 cells. C. miR-148a or miR-326 suppressed the relative luciferase activity in constructs that contained the 3’-UTR of PKM2.
miR-148a and miR-326 are down-regulated and display anti-tumor effect in TC
Expression of miR-148a and miR-326 were down-regulated in multiple TC cell lines compared with normal human thyroid cell HT-ori3 (Figure 4A). Given that miR-148a and miR-326 are often down-regulated in TC, ectopic expression of miR-148a and miR-326 was applied to identify their functional roles. Overexpressed miR-148a and miR-326 suppressed TC cell growth accordingly (P < 0.01, Figure 4B). Moreover, a significant reduction of colony numbers in miRNAs’ transfectants was detected compared with scramble miRNA groups (P < 0.01, Figure 4C). In addition, ectopic expression of miR-148a and miR-326 significantly diminished cell migration (P < 0.01, Figure 4D) and cell invasion (P < 0.05, P < 0.01, Figure 4E), suggesting their tumor-suppressive role in TC.
Figure 4.

miR-148a and miR-326 are decreased and play anti-tumor roles in TC. A. miR-148a and miR-326 expression was down-regulated in TC cell lines. B. MTT proliferation results with ectopic miR-148a and miR-326 expression in TC cells. C. Ectopic expression of miR-148a and miR-326 inhibited monolayer colony formation by TCP-1 and K1 cells. D, E. Migration and invasion were significantly impaired in miR-148a- and miR-326-treated cells compared with their scrambled miRNA counterparts.
PKM2 is the bona fide functional target of miR-148a in thyroid carcinogenesis
Rescue experiments were also conducted to justify whether PKM2 is the functional target of miR-148a. And PKM2 overexpression was confirmed by western blot analysis (Figure 5A). The re-expression of PKM2 partly diminished the tumor-suppressive effect of miR-148a (MTT proliferation assays, P < 0.01, Figure 5B; monolayer colony formation, P < 0.01, Figure 5C; cell invasion, P < 0.01, Figure 5D).
Figure 5.
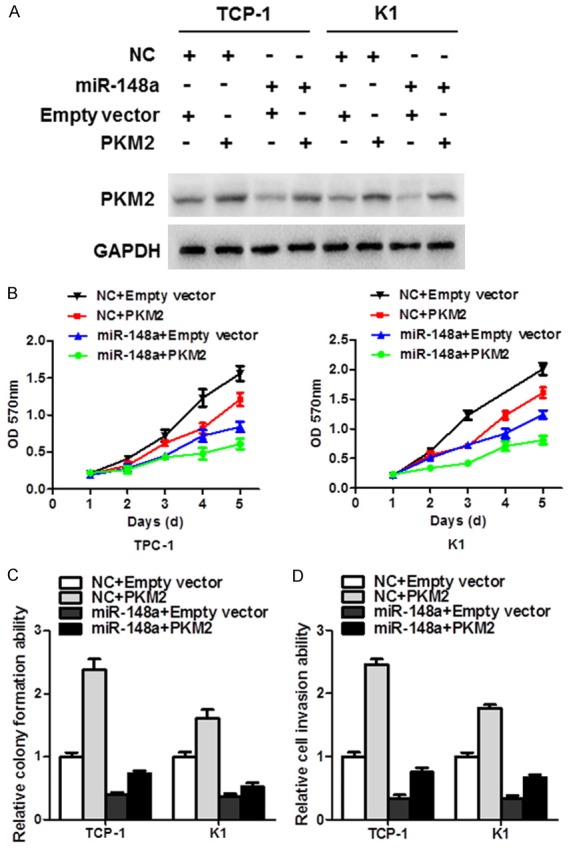
PKM2 re-expression partly restored the anti-cancer effect of miR-148a. A. The levels of PKM2 protein among miR-148a-treated TC cell lines after re-expression. B. MTT proliferation assays revealed that overexpression of PKM2 partially diminished the tumor-suppressive effect of miR-148a. C. Colony formation was partly restored in miR-148a transfectants after the re-expression of PKM2. D. PKM2 re-expression also revived cell invasion, which was impaired by miR-148a.
Discussion
In this study, PKM2 was up-regulated in TC tissues compared to adjacent non-tumor tissues. In addition, miR-148a and miR-326 were consistently down-regulated across a panel of TC cells compared with a normal thyroid cell line, suggesting their potential roles in TC tumorigenesis. TC cells were also treated with miR-148a or miR-326 to determine whether epigenetic modification is responsible for the down-regulation of miR-148a/326 in TC. Ectopic expression of miR-148a and miR-326 resulted in tumor-suppressive functions by inhibiting TC cell proliferation, invasion, and migration. These functional studies suggest that PKM2 plays a crucial role in the development and progression of TC.
As some cases of TC exhibit PKM2 mRNA up-regulation without gene amplification, the miRNA-mediated regulation on PKM2 was investigated. We identified miR-148a and miR-326 as two promising miRNAs that may target PKM2 in TC. The activation of PKM2 is partly due to epigenetic silencing of these two miRNAs, which have been identified as tumor suppressors. Expression of these miRNAs inhibited cell proliferation, promoted the apoptosis of cancer cells, and suppressed tumorigenicity. miR-148a is silenced in cancers and exerts a tumor-suppressive function in esophageal cancer [13], colorectal tumorigenesis [14], liver cancer [15], and pancreatic cancer [16]. In TC cells, miR-148a functions as a tumor suppressor by repressing the expression of CDK8 [17] and INO80 [18], suggesting that PKM2 is a key target of miR-148a in many cancers. miR-326 is abnormally expressed in various human cancers and acts as a tumor suppressor in melanoma [19], osteosarcoma [20], and cervical cancer [21]. Moreover, miR-326 is underexpressed in lung tumor tissues and lung cancer cell lines [22]. Low miR-326 expression is correlated with disease progression and metastasis. Li et al. [23] found that down-regulation of miR-326 was associated with advanced stage and low overall survival of patients with gastric cancer. Together, these findings suggest that aberrant down-regulation of miR-148a and miR-326 represents a prognostic marker of human cancer. Our study enriches the target pool for miR-148a and miR-326 in TC. Aberrant regulation of PKM2 by this dysregulated miRNA network is novel. PKM2 is reported to have a relatively high mutation rate in cancers. A risk variant of PKM2 in ovarian cancer was revealed by a genome-wide association study. PKM2 missense variants were uncovered in papillary thyroid carcinoma and are proposed to impair its Cdc42 inhibitory ability. We conclude that aberrant up-regulation of PKM2, mediated by the epigenetic silencing of two miRNAs, may also contribute to TC development in the absence of genetic mutations.
In conclusion, PKM2 functions as an oncogene in thyroid tumorigenesis, especially by promoting metastasis. Multiple mechanisms contribute to PKM2 up-regulation and activation, including gene amplification, gain-of-function mutations, and miRNA regulation. Our study provides a novel therapeutic target and describes the clinical translational potential for TC.
Acknowledgements
This study is supported by Science and Technology Development Foundation of Bengbu Medical College (BYKF1795).
Disclosure of conflict of interest
None.
References
- 1.Kan Q, Su Y, Yang H. MicroRNA-335 is downregulated in papillary thyroid cancer and suppresses cancer cell growth, migration and invasion by directly targeting ZEB2. Oncol Lett. 2017;14:7622–7628. doi: 10.3892/ol.2017.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramirez-Vick M, Nieves-Rodriguez M, Lugaro-Gomez A, Perez-Irizarry J. Increasing incidence of thyroid cancer in Puerto Rico, 1985-2004. P R Health Sci J. 2011;30:109–115. [PubMed] [Google Scholar]
- 3.Malaguarnera R, Vella V, Nicolosi ML, Belfiore A. Insulin resistance: any role in the changing epidemiology of thyroid cancer? Front Endocrinol (Lausanne) 2017;8:314. doi: 10.3389/fendo.2017.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russo D, Damante G, Puxeddu E, Durante C, Filetti S. Epigenetics of thyroid cancer and novel therapeutic targets. J Mol Endocrinol. 2011;46:73–81. doi: 10.1530/JME-10-0150. [DOI] [PubMed] [Google Scholar]
- 5.Giunti S, Antonelli A, Amorosi A, Santarpia L. Cellular signaling pathway alterations and potential targeted therapies for medullary thyroid carcinoma. Int J Endocrinol. 2013;2013:803171. doi: 10.1155/2013/803171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 7.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Shen L, Chen X, Qian Y, Zhou Q, Wang Y, Li K, Liu M, Zhang S, Huang X. High expression of PKM2 as a poor prognosis indicator is associated with radiation resistance in cervical cancer. Histol Histopathol. 2015;30:1313–1320. doi: 10.14670/HH-11-627. [DOI] [PubMed] [Google Scholar]
- 9.Mendez-Lucas A, Li X, Hu J, Che L, Song X, Jia J, Wang J, Xie C, Driscoll PC, Tschaharganeh DF. Glucose catabolism in liver tumors induced by c-MYC can be sustained by various PKM1/PKM2 ratios and pyruvate kinase activities. Cancer Res. 2017;77:4355. doi: 10.1158/0008-5472.CAN-17-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Q, Liu LZ, Yin Y, He J, Li Q, Qian X, You Y, Lu Z, Peiper SC, Shu Y. Regulatory circuit of PKM2/NF-. B/miR-148a/152-modulated tumor angiogenesis and cancer progression. Oncogene. 2015;34:5482–5493. doi: 10.1038/onc.2015.6. [DOI] [PubMed] [Google Scholar]
- 11.Yu G, Jia B, Cheng Y, Zhou L, Qian B, Liu Z, Wang Y. MicroRNA-429 sensitizes pancreatic cancer cells to gemcitabine through regulation of PDCD4. Am J Transl Res. 2017;9:5048–5055. [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N, Jiang F, Han XY, Li M, Chen WJ, Liu QC, Liao CX, Lv YF. MiRNA-155 promotes the invasion of colorectal cancer SW-480 cells through regulating the Wnt/beta-catenin. Eur Rev Med Pharmacol Sci. 2018;22:101–109. doi: 10.26355/eurrev_201801_14106. [DOI] [PubMed] [Google Scholar]
- 13.Chen Q, Luo G, Zhang X. MiR-148a modulates HLA-G expression and influences tumor apoptosis in esophageal squamous cell carcinoma. Exp Ther Med. 2017;14:4448–4452. doi: 10.3892/etm.2017.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hibino Y, Sakamoto N, Naito Y, Goto K, Oo HZ, Sentani K, Hinoi T, Ohdan H, Oue N, Yasui W. Significance of miR-148a in colorectal neoplasia: downregulation of miR-148a contributes to the carcinogenesis and cell invasion of colorectal cancer. Pathobiology. 2015;82:233–241. doi: 10.1159/000438826. [DOI] [PubMed] [Google Scholar]
- 15.Cheng L, Zhu Y, Han H. MicroRNA-148a deficiency promotes hepatic lipid metabolism and hepatocarcinogenesis in mice. Cell Death Dis. 2017;8:e2916. doi: 10.1038/cddis.2017.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng L, Liu Z, Xiao J, Tu Y, Wan Z, Xiong H, Li Y, Xiao W. MicroRNA-148a suppresses epithelial-mesenchymal transition and invasion of pancreatic cancer cells by targeting Wnt10b and inhibiting the Wnt/beta-catenin signaling pathway. Oncol Rep. 2017;38:301–308. doi: 10.3892/or.2017.5705. [DOI] [PubMed] [Google Scholar]
- 17.Han C, Zheng W, Ge M, Wang K, Xiang Y, Wang P. Downregulation of cyclin-dependent kinase 8 by microRNA-148a suppresses proliferation and invasiveness of papillary thyroid carcinomas. Am J Cancer Res. 2017;7:2081–2090. [PMC free article] [PubMed] [Google Scholar]
- 18.Sheng W, Chen Y, Gong Y, Dong T, Zhang B, Gao W. miR-148a inhibits self-renewal of thyroid cancer stem cells via repressing INO80 expression. Oncol Rep. 2016;36:3387–3396. doi: 10.3892/or.2016.5203. [DOI] [PubMed] [Google Scholar]
- 19.Kang K, Zhang J, Zhang X, Chen Z. MicroRNA-326 inhibits melanoma progression by targeting KRAS and suppressing the AKT and ERK signalling pathways. Oncol Rep. 2018;39:401–410. doi: 10.3892/or.2017.6074. [DOI] [PubMed] [Google Scholar]
- 20.Cao L, Wang J, Wang PQ. MiR-326 is a diagnostic biomarker and regulates cell survival and apoptosis by targeting Bcl-2 in osteosarcoma. Biomed Pharmacother. 2016;84:828–835. doi: 10.1016/j.biopha.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Zhang P, Kong F, Deng X, Yu Y, Hou C, Liang T, Zhu L. MicroRNA-326 suppresses the proliferation, migration and invasion of cervical cancer cells by targeting ELK1. Oncol Lett. 2017;13:2949–2956. doi: 10.3892/ol.2017.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang R, Chen X, Xu T, Xia R, Han L, Chen W, De W, Shu Y. MiR-326 regulates cell proliferation and migration in lung cancer by targeting phox2a and is regulated by HOTAIR. Am J Cancer Res. 2016;6:173–186. [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Gao Y, Xu Y, Ma H, Yang M. Down-regulation of miR-326 is associated with poor prognosis and promotes growth and metastasis by targeting FSCN1 in gastric cancer. Growth Factors. 2015;33:267–274. doi: 10.3109/08977194.2015.1076406. [DOI] [PubMed] [Google Scholar]