

Abstract
Mitoquinone (MitoQ) is a powerful mitochondrial-targeted antioxidant whose neuroprotective effects have been shown in a variety of animal models of neurological diseases. However, its roles in traumatic brain injury (TBI) remain unexplored. The primary objective of this study was to investigate the neuroprotection afforded by MitoQ in a mouse model of TBI, and the involvement of the Nrf2-ARE signaling pathway in the putative neuroprotective mechanism. Mice were randomly divided into four groups: sham group, TBI group, TBI + vehicle group, and TBI + MitoQ group. MitoQ (4 mg/kg, administered intraperitoneally) or an equal volume of vehicle was given at 30 min after TBI. After 24 h, brain samples were harvested for analysis. The results demonstrated that treatment with MitoQ significantly improved neurological deficits, alleviated brain edema and inhibited cortical neuronal apoptosis. Furthermore, MitoQ administration increased the activity of antioxidant enzymes, including superoxide dismutase (SOD) and glutathione peroxidase (GPx), whereas it decreased the malondialdehyde (MDA) content. In addition, MitoQ treatment reduced Bax protein translocation to mitochondria and cytochrome c release into the cytosol. Moreover, MitoQ greatly accelerated the Nrf2 nuclear translocation and subsequently upregulated the expression of Nrf2 downstream proteins, including heme oxygenase-1 (HO-1) and quinone oxidoreductase 1 (Nqo1). In conclusion, the results in the study demonstrate that MitoQ exerts neuroprotective effects in the mouse model of TBI, possibly by activating the Nrf2-ARE pathway.
Keywords: Mitoquinone (MitoQ), traumatic brain injury, nuclear factor erythroid 2-related factor 2, oxidative stress, apoptosis, neuroprotection
Introduction
Traumatic brain injury (TBI) remains a serious socio-economic problem with high morbidity and mortality. To date, there is no effective therapeutic strategy for TBI due to its heterogeneity and complexity. Evidence strongly indicates that oxidative stress plays a key role in the mechanisms of secondary brain damage, such as excitotoxicity, mitochondrial disorder, autophagy, apoptosis and inflammatory response, following TBI [1]. Oxidative stress generates excessive reactive oxygen species (ROS), and causes redox imbalance, resulting in lipid peroxidation, nucleic acids oxidation and DNA damage, that lead to neural impairment and death [2,3]. Mitochondria are the major cellular source of ROS and are particularly susceptible to oxidative injury. Mitochondrial impairment leads to ROS overproduction, which damage mitochondrial proteins, lipids and DNA, that in turn triggers apoptosis and lead to metabolic disorders [4,5]. The available research data show that decreased mitochondrial oxidative stress may suppress or delay the progression of TBI [6]. Accordingly, delivering mitochondrial-targeted antioxidants to mitochondria could protect mitochondria against oxidative stress and inhibit neuronal death in a mouse model of TBI.
The mitochondrial-targeted antioxidant mitoquinone (MitoQ) is a derivative of ubiquinone that is conjugated to the lipophilic triphenylphosphonium cation [7-9]. Because of the strong mitochondrial membrane potential, this cation is accumulated inside mitochondria. The conjugated antioxidant moiety makes it hundreds of times more potent than conventional untargeted antioxidant as regards preventing mitochondrial oxidative insult [10]. Increasing evidence shows that MitoQ plays a protective role in various diseases, including neurodegenerative diseases, cardiac hypertrophy, and liver fibrosis [11-14]. Among these beneficial roles, its antioxidative activity is remarkable in the neuroprotective roles of MitoQ. However, the mechanisms underlying its effects on TBI remain unknown.
Nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor, plays an indispensable part in antioxidative stress. Normally bound to the Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm, Nrf2 is separated from Keap1 and translocates from the cytoplasm to the nucleus in response to oxidative stress [15]. Nrf2 is able to reverse the harmful effects of oxidative stress by activating the antioxidant response and inducing the transcription of a wide variety of genes, thus restoring intracellular homeostasis [16]. By binding to antioxidant response element (ARE), Nrf2 enhances the expression of many phase II detoxifying and antioxidant enzyme genes, such as heme oxygenase-1 (HO-1), quinine oxidoreductase 1 (Nqo1), superoxide dismutase (SOD), and glutathione peroxidase (GPx) [15,17,18]. More and more studies have reported that the Nrf2-ARE signaling pathway is activated in nervous system diseases including TBI, and provides a defense against the oxidative insult induced by TBI.
Currently, no data are available about the neuroprotective effect of MitoQ in TBI. The primary objective of our study was to investigate the neuroprotective efficacy of MitoQ in a mouse model of TBI, and the involvement of the Nrf2-ARE signaling pathway in the putative neuroprotective machanism in the weight-drop model of TBI.
Materials and methods
Animal ethical approval
Our animal experiments were conducted with the approval of our regional animal ethical committee at Nanjing University (Jiangsu, China). Experimental protocols conformed to the Guide for the Care and Use of Laboratory Animals set by the National Institutes of Health (NIH). Adult male ICR mice weighing 28 to 32 g were housed under standard conditions: constant temperature (22±1°C) and relative humidity (30%) and a 12-h light/dark cycle. Mice were given ad libitum access to food and water.
TBI model in mice
The mouse model of TBI used in our study is that according to Marmarou’s weight-drop model as previously described by Flierl et al., and in a study we conducted previously [19,20]. In short, mice were anesthetized by isoflurane inhalation (induced at 4%, maintained at 1.5%), and subsequently placed on the platform directly under the weight-drop unit. A 1.5 cm long longitudinal midline scalp incision was made, then the fascia was separated to expose the skull. After locating the left frontal region (3 mm posterior the coronal suture, 1.5 mm lateral to the midline), selected as the impact region, we dropped a 200-g weight onto the left frontal bone from a 2.5-cm height. The scalp incision was sutured by standard procedure. Then, the mice were returned to the cages. The sham group underwent the same procedure without the brain injury.
Experimental design
A total of 204 mice were randomly divided into the following groups: Sham, TBI, TBI + vehicle, and TBI + MitoQ (3 subgroups: 2 mg/kg, 4 mg/kg, 8 mg/kg). MitoQ was purchased from Focus Biomolecules LLC (Plymouth Meeting, PA, USA) and freshly prepared in saline containing 1% dimethyl sulfoxide (DMSO) just before injection. Mice in the TBI + MitoQ group were injected with 4 mg/kg MitoQ intraperitoneally 30 min after TBI. Mice in the TBI + vehicle group were given equal volumes of vehicle (1% DMSO) at the corresponding time points. The dosage used in the present study was chosen based on reported safety and efficacy data [7].
Neurobehavioral evaluation
The mice neurobehavior was assessed at 1 day, 3 days and 7 days after TBI using a 10-point neurological severity score (NSS) [21]. The NSS was used to evaluate the ability of the mice to perform ten different tasks including motor ability, balance and alertness. The tasks comprised an exit circle, monoparesis/hemiparesis, straight walk, startle reflex, seeking behavior, beam balancing, round stick balancing, and beam walk at 1, 2 and 3 cm. A score of one point indicates each of the tasks could not be performed. All neurobehavioral evaluations were performed by two investigators who were blinded to the experimental groups.
Measurement of the brain water content
Brain water content was evaluated in light of a previous study [20]. In brief, the mouse brain was quickly harvested and placed on a cooled brain matrix 24 h after TBI. The cerebellum and brainstem were removed, and the ipsilateral cortical tissue was harvested and immediately weighed to obtain the wet weight (WW). Then this tissue was dried for 72 h at 80°C and weighted to obtain the dry weight (DW) of the specimen. The percentage of brain water content was calculated using the following formula: (WW-DW)/WW × 100%.
Tissue processing
Animals were deeply anesthetized via inhalation of isoflurane at 24 h after TBI and perfused transcardially with 0.9% normal saline solution. The ipsilateral cortex pericontusion was harvested. Then, the brain samples were rapidly frozen in liquid nitrogen, and stored at -80°C. For immunofluorescence assay and terminal deoxynucleotidyl transferase-mediated biotinylated deoxyuridine triphosphate nickend labeling (TUNEL) staining, animals were also euthanized with isoflurane inhalation 24 h after TBI, then perfused transcardially with 0.9% normal saline solution followed by ice-cold 4% buffered paraformaldehyde. Next, the brains were resected and immersed in 4% buffered paraformaldehyde for the night. Routinely paraffin-embedded blocks were archived, coronal sections that were cut at 5 μm thickness consecutively.
Determination of malondialdehyde (MDA), superoxide dismutase (SOD), glutathione peroxidase (GPx) activity
The ipsilateral cortex tissue was homogenized in 2 ml of 10 mM phosphate-buffer (PH 7.4). Following centrifugation at 12,000 g for 20 min, the levels of MDA, SOD and GPx in the supernatant were quantitated spectrophotometrically according to the manufacturer’s instructions (Nanjing Jiancheng Biochemistry Co., Nanjing, China). The protein concentrations were measured using the Bradford method.
TUNEL staining
TUNEL staining was used to evaluate neuronal apoptosis. The procedures were performed according to protocol of the in situ cell death detection Kit POD (ISCDD, Boehringer Mannheim, Germany). After washing 3 times with PBS, the slides were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for 15 min. Following rinsing in PBST, the slides were coverslipped with anti-fade mounting medium for further study. Quantitative analysis of TUNEL-positive cells in the ipsilateral cortex were conducted in six random non-overlapping fields (× 400) under epifluorescence microscopy. To evaluate the extent of brain injury, the apoptotic index was determined by using the mean number of TUNEL-positive neurons in each slice. Cell counts were made by two pathologists blinded to the experiment.
Western blot analysis
Protein extraction was conducted by using a Total Protein Extraction Kit, Nuclear-Cytosol Extraction Kit and Tissue Mitochondria Isolation Kit (Beyotime Biotech, Nantong, China) following the protocol provided by the manufacturer. Protein concentrations were measured by the Bradford method. Protein samples (50 ug/lane) were separated on a 12% sodium dodecyl sulfate-polyacrylamide gel, electrophoretically transferred to polyvinylidene-difluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk for 2 hours at room temperature and then incubated with the appropriate primary antibody at 4°C overnight respectively using the following dilution ratio: Histone 3 (1:1000, #9715s; Cell Signaling Technology, Danvers, MA, USA), cytochrome c (1:1000, ab53056; Abcam, Cambridge, UK), Bax (1:200, sc-4239; Santa Cruz Biotechnology, Santa Cruz, CA, USA), COX IV (1:1000, #11967s; Cell Signaling Technolog), β-actin (1:5000, AA32121; Bioworld Technology, Saint Louis Park, MN, USA), Nrf2 (1:1000, ab31163; Abcam), HO-1 (1:1000, ab137749; Abcam), Nqo1 (1:1000, ab341173, Abcam). Subsequently, the membranes were incubated with the appropriate horseradish peroxidase-linked secondary antibodies for 2 h. The protein bands were visualized using enhanced chemiluminescence (ECL) Western blot detection reagents (Millipore, Billerica, MA, USA). The band relative density was analyzed using the Image J software.
Real-time quantitative polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the ipsilateral cortex with the RNAiso Plus kit (Takara Bio., Dalian, China). The concentration and purity of the total RNA was determined with a spectrophotometer (OD 260/280 1.8-2.0) and by 1% agarose gel electrophoresis. To avoid RNA degradation, some of the RNA was immediately reverse-transcribed to cDNA using the PrimeScript RT reagent kit (Takara Bio); the rest of the RNA was stored at -80°C. The primers were designed according to the sequences in PubMed GenBank and synthesized by Invitrogen Life Technologies (Shanghai, China). The sequences were as follows: HO-1: F, 5’-ATCGTGCTCGCATGAACACT-3’; R, 5’-CCAACACTGC-ATTTACATGGC-3’; Nqo1: F, 5’-CATTCTGAAAGGCTGGTTTGA-3’; R, 5’-CTAGCTTTGATCTGGTT-GTCAG-3’; β-actin: F, 5’-AGTGTGACG-TTGACATCCGTA-3’; R, 5’-GCCAGAGCAGTAATCTCCTTCT-3’.
qRT-PCR analysis was performed on the Mx3000P System (Stratagene, San Diego, CA, USA), using real-time SYBR Green PCR technology. All samples were analyzed in triplicate. β-actin was used as an internal reference “housekeeping” gene.
Immunofluorescence assay for Nrf2
We performed double staining of Nrf2 and NeuN, to determine expression of Nrf2 in neurons. The paraffin-embedded tissue slides (5 μm thick) were subjected to deparaffinization, dehydration and antigen retrieval. Then, the slides were rinsed in blocking buffer (PBS, BSA, 0.3% Triton X-100) for 15 min followed by overnight incubation at 4°C with rabbit anti-Nrf2 (1:100, ab31163, Abcam) and mouse anti-NeuN (1:100, MAB377X, Millipore). After sequential rinse steps with PBST, the slides were incubated with the appropriate secondary antibodies (Alexa Fluor® 594, 1:200; Molecular Probe, Eugene, OR, USA) for 1 h at room temperature. Following the washes with PBST, the slides were counterstained with DAPI for 2 min. After rinsing with PBST, slides were coverslipped with mounting medium. Slides were observed by fluorescence microscopy (Olympus, IX71, Olympus Co., Tokyo, Japan). The relative fluorescence intensity was analyzed by Image J software.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, CA, USA) software. Values are expressed as the mean ± SEM. Statistical comparisons between groups were analyzed with one-way ANOVA and Tukey’s test. P-value less than 0.05 was considered statistically significant.
Results
MitoQ improves the neurobehavioral function and alleviates cerebral edema after TBI
To investigate the protective effect of MitoQ on neurological function, we set six groups as follows: Sham, TBI, TBI + vehicle, TBI + MitoQ (three different dose groups: 2 mg/kg, 4 mg/kg, 8 mg/kg). The neurological severity scores (NSS) at 1 day, 3 days and 7 days after TBI were used. All experimental animals were prepared 24 hours before TBI. Within 3 days after injury, there was no statistical relationship between time points in the sham group (data not shown), because the NSS in the sham group were nearly zero in this experiment. No obvious difference was observed between the TBI group and the vehicle-treated group. The group of mice that received 2 mg/kg MitoQ exhibited a slightly improved motor performance after TBI, however, there was no statistically evident change compared to the vehicle-treated group of mice on days 1 and 3 (P > 0.05, Figure 1A). The NSS of the mice treated with 4 mg/ kg MitoQ were significantly improved compared to that of the vehicle-treated mice after TBI (P < 0.05, Figure 1A). A high dose of MitoQ (8 mg/kg) did not exerted better results than a 4 mg/kg dose (P > 0.05; Figure 1A). However, there was no significant difference appeared between the vehicle-treated group and TBI + MitoQ group at 7 d (P > 0.05).
Figure 1.
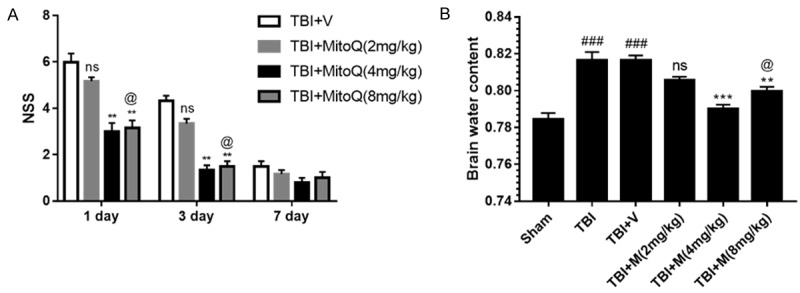
MitoQ administration ameliorated secondary brain insult in TBI models. A. Neurobehavioral function was evaluated by neurological severity scores (NSS) at day 1 and 3 after TBI. B. Brain water content was significantly increased after TBI. Data represent as mean ± SEM (n=6). nsP > 0.05, ** P < 0.01 vs. vehicle-treated group; ### P < 0.001 vs. sham group; @ P > 0.05 vs. TBI + MitoQ (4 mg/kg).
Brain water content was examined to evaluate the neuroprotective effect of MitoQ. The brain water content was remarkably increased in the TBI group compared with the sham group. There was no significant difference between the vehicle-treated group and TBI + 2 mg/kg MitoQ group (P > 0.05, Figure 1B). Mice treated with 4 and 8 mg/kg MitoQ exerted decreased brain edema compared to the vehicle-treated group (P < 0.001 and P < 0.01, respectively; Figure 1B). Consistent with the NSS, the 4 mg/kg dose had a markedly greater effect in attenuating the TBI-induced brain edema than the vehicle-treated group (P < 0.05; Figure 1B). Collectively, these results revealed that MitoQ has a neuroprotective effect against TBI. Importantly, these findings suggested that MitoQ at a dose of 4 mg/kg might be an effective treatment and thus was used in the subsequent studies.
MitoQ attenuates oxidative stress caused by TBI
To investigate the neuroprotective effect of MitoQ on TBI-induced oxidative stress, the production of MDA, and activity of GPx and SOD were analyzed in brain tissue. The activities of GPx and SOD were greatly decreased after TBI compared with the sham group (both P < 0.001). In comparison, the activities of GPx and SOD in the MitoQ-treated group were considerably upregulated (both P < 0.01; Figure 2A and 2B). MDA levels in the TBI and vehicle-treated groups were increased relative to the sham group (P < 0.001) (Figure 2C), and there was no marked difference between the TBI and vehicle-treated groups (P > 0.05). However, MDA content in MitoQ-treated mice was significantly lower than in the vehicle-treated group (P < 0.001) (Figure 2C).
Figure 2.
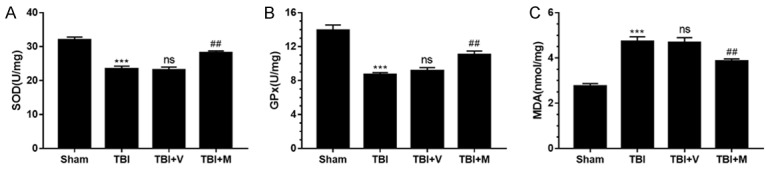
MitoQ attenuated oxidative stress after TBI. A, B. The activities of GPx and SOD were greatly reduced after TBI, while treatment with MitoQ increased the activities of GPx and SOD. C. The levels of MDA in the cortex were significantly elevated 24 h after TBI. MitoQ administration markedly decreased MDA levels after TBI. Data are presented as mean ± SEM. (n=6). *** P < 0.001 vs. sham group; ns P > 0.05 vs. TBI group; ## P < 0.01 vs. vehicle-treated group.
MitoQ administration ameliorated neuronal apoptosis in the brain following TBI
To ascertain whether MitoQ administration could ameliorate apoptosis in the injured cortex 24 h after TBI, we used NeuN/TUNEL double immunofluorescence staining to evaluate neuronal apoptosis. As shown in (Figure 3), only a few TUNEL-positive cells were observed in the sham group, whereas the number of TUNEL-postive cells was significantly increased in the TBI and vehicle-treated groups after TBI. No difference in the number of apoptotic cells was found between the TBI group and the vehicle-treated group. The number of TUNEL-positive neurons was decreased after MitoQ administration, which is consistent with the NSS and brain water content results. Taken together, these data suggest that MitoQ potentially reduces neuronal apoptosis following TBI.
Figure 3.
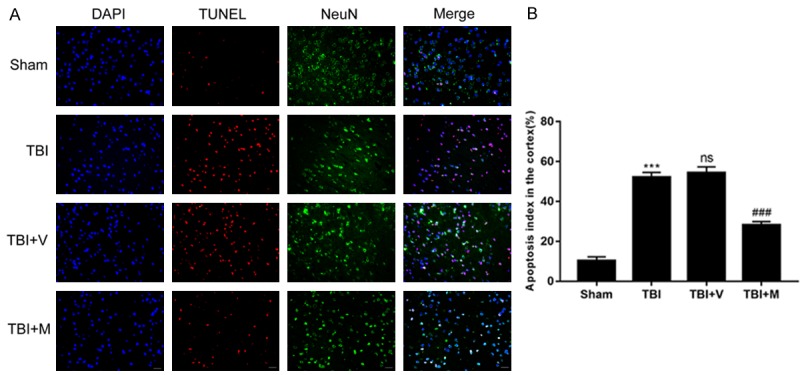
The effect of MitoQ on TBI-induced neuronal apoptosis was evaluated by TUNEL following TBI. A. Representing immunofluorescence images demonstrated that MitoQ administration obviously reduced cortex neuron apoptosis at 24 h after TBI. B. The apoptosis index was denoted by ratio of TUNENL-positive cells against neural cells. The number of TUNEL-positive neurons was reduced significantly by MitoQ administration following TBI. Quantification analysis indicated that MitoQ administration markedly decreased TUNEL-positive neurons compared with the vehicle-treated group. Data are presented as mean ± SEM (n=6). Bar =50 μm. *** P < 0.001 vs. Sham group; ns P > 0.05 vs. TBI group; ### P < 0.001 vs. vehicle-treated group.
To further elucidate the effects of MitoQ on TBI-induced apoptosis, we quantitated the expression of mitochondrial apoptosis-related proteins such as Bax and cytochrome c, after TBI. The results showed that the levels of mitochondrial and cytosolic Bax protein were increased and decreased, respectively, after TBI, compared with the sham group, whereas mitochondrial and cytosolic cytochrome c levels were decreased and increased, respectively, after TBI, relative to the sham group (Figure 4). These effects were reversed by MitoQ treatment, which inhibited mitochondrial translocation of Bax and subsequent cytosolic release of cytochrome c. These results confirmed that MitoQ can reduce neuronal apoptosis in the cortical contusion after TBI.
Figure 4.
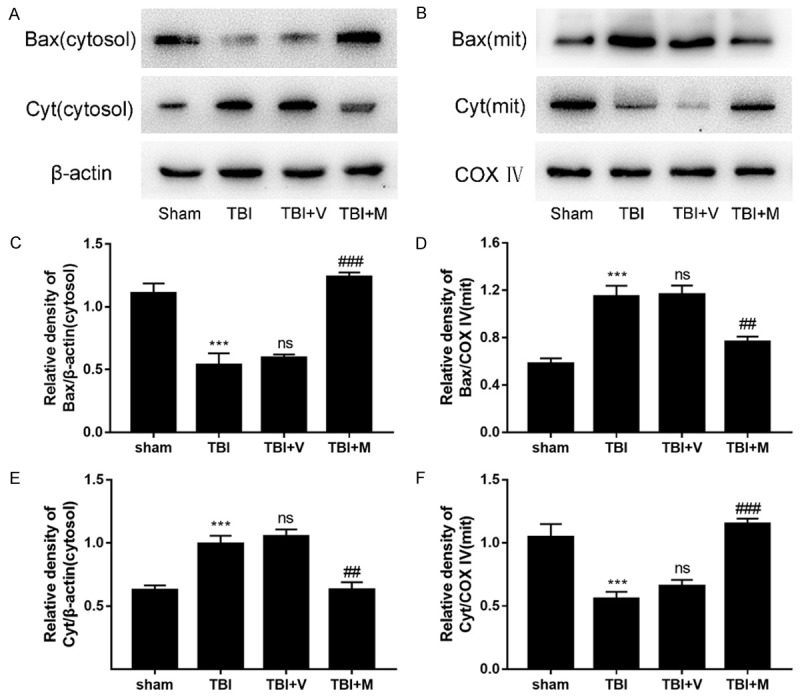
Effect of MitoQ on pro-apoptotic protein expression 24 h after TBI. (A, B) Western blotting was used to evaluate the expression of Bax and cytochrome c in the left cortex after TBI. Representative bands indicate the relative intensity of (D, F) mitochondrial and (C, E) cytosolic Bax and cytochrome c. The intensity of the target bands was standardized with respect to β-actin protein or COX IV protein. Data are represented as mean ± SEM (n=6). *** P < 0.001 vs. sham group; ns P > 0.05 vs. TBI group; ## P < 0.01 and ### P < 0.001 vs. vehicle-treated group.
MitoQ accelerated translocation of Nrf2 from cytoplasm to nucleus after TBI
The evidence obtained in this study indicated that MitoQ has substantially increased the activities of antioxidant enzymes following TBI. However, the underlying molecular mechanisms remain unclear. Nrf2 plays an orchestrating role in the regulation of oxidative stress. Accordingly, it was reasonable to hypothesize that the effect of MitoQ might be mediated through activation of Nrf2, which elevates the activity of the antioxidant enzymes. The well-established, classic activation pattern of Nrf2 is the translocation from the cytoplasm to the nucleus. We investigated the expression of cytoplasmic Nrf2, nuclear Nrf2 and total Nrf2. These results revealed that both TBI and MitoQ administration promoted Nrf2 nuclear translocation compared with the sham group (Figure 5A). Additionally, the MitoQ treatment significantly increased the levels of nuclear Nrf2 and decreased the levels of cytoplasmic Nrf2 in the TBI group compared with the vehicle-treated group (both P < 0.05; Figure 5A and 5B). This finding suggested that MitoQ promoted Nrf2 nuclear translocation.
Figure 5.
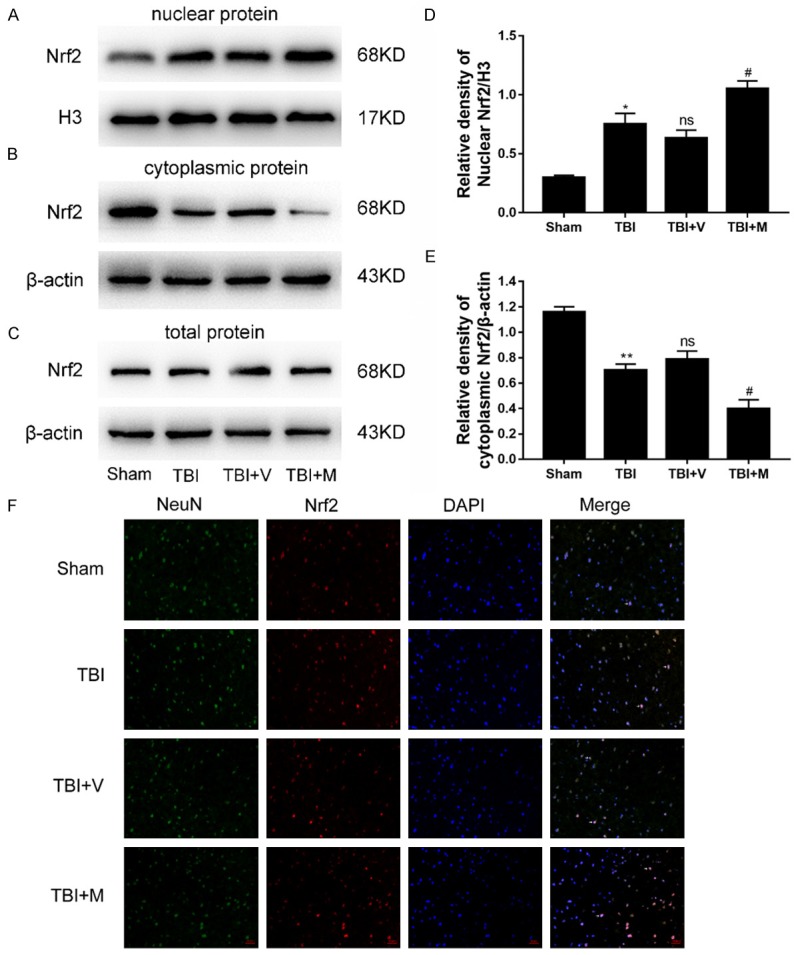
MitoQ accelerated translocation of Nrf2 from cytoplasm to nucleus. A-C. The levels of nuclear Nrf2, cytoplasmic Nrf2, and total Nrf2 were measured by western blot. D, E. The ratios of nuclear Nrf2 and cytoplasmic Nrf2. F. Representative immunofluorescence staining of Nrf2 after MitoQ treatment in mice with TBI. Double immunofluorescence analysis was performed with Nrf2 antibodies (red), NeuN (green), and DAPI (blue). The merged immunofluorescent images with NeuN, Nrf2 and DAPI represented for Nrf2 translocation from the cytoplasm to the nucleus. The white arrows indicate expressions and overlaps of NeuN, Nrf2 and DAPI. Scale bar =50 μm; Data are presented as mean ± SEM (n=6). * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. sham group; ns P > 0.05 vs. TBI group; # P < 0.05 and ## P < 0.01 vs. vehicle-treated group.
This effect was also confirmed by the immunofluorescence assay of Nrf2. As shown in (Figure 5F), only scattered Nrf2-immunopositive neurons were observed in the sham group, whereas numerous immunopositive-Nrf2 neurons were observed in the ipsilateral cortex following TBI. These observations suggested that TBI promotes Nrf2 nuclear translocation. However, the number of Nrf2-immunopositive neurons in the MitoQ-treated group were greatly increased compared with the vehicle-treated group. In short, these results demonstrated that MitoQ promotes Nrf2 translocation from the cytoplasm to the nucleus, thereby enhancing the binding ability to downstream proteins.
MitoQ upregulates the expression of Nrf2 downstream proteins
Previous studies have demonstrated that Nrf2 induces the expression of antioxidant enzymes such as HO-1 and Nqo1 via binding to the ARE. Accordingly, we hypothesized that effects of MitoQ might be mediated through the regulation of the Nrf2-ARE pathway. Therefore, we further investigated the expression of Nrf2 downstream proteins (HO-1 and Nqo1). The results revealed that the protein levels of HO-1 and Nqo1 were increased after TBI, compared with the sham group (P < 0.01 and P < 0.001, respectively; Figure 6). In addition, MitoQ administration further enhanced the expression of HO-1 and Nqo1, compared with vehicle-treated group (Figure 6A-D). These results reflected that the enhancement of the expression of Nrf2 downstream proteins by MitoQ through the activation of the Nrf2-ARE signaling pathway.
Figure 6.
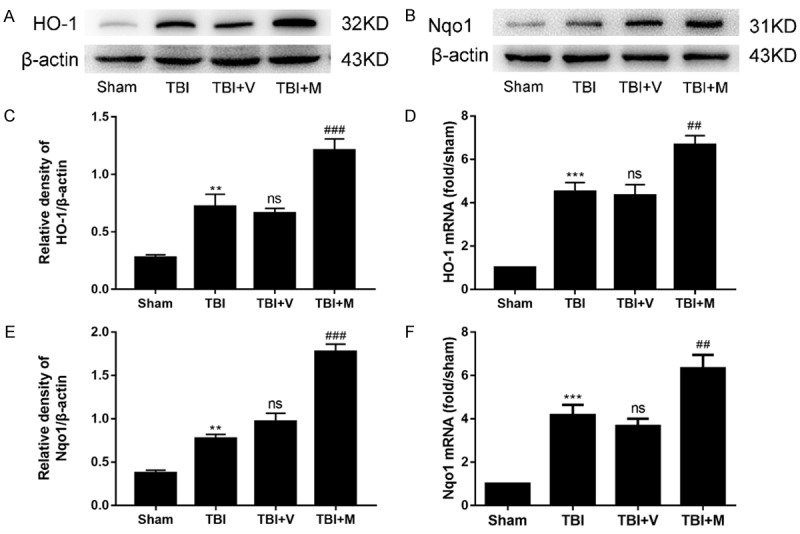
MitoQ upregulated the expression of Nrf2 downstream proteins in mice after TBI. A-D. The expression of HO-1 and Nqo1 was elevated following TBI. Moreover, MitoQ further upregulated their expression in the ipsilateral cortex after treatment with MitoQ (n=6). E, F. The mRNA expression of HO-1 and Nqo1 was enhanced after TBI and was further increased with MitoQ administration (n=3). Relative intensity means Nqo1 or HO-1/β-actin. Data are presented as mean ± SEM. ** P < 0.01, and *** P < 0.001 vs. sham group; ns P > 0.05 vs. TBI group; ## P < 0.01 and ### P < 0.001 vs. vehicle-treated group.
At the mRNA level, MitoQ also upregulated the expression of HO-1 and Nqo1 compared with vehicle-treated group (P < 0.001 and P < 0.01, respectively; Figure 6E and 6F), which is consistent with the protein changes. Thus, these results revealed that MitoQ further enhanced the expression of Nrf2 downstream factors at both protein and mRNA levels via the activation of the Nrf2-ARE signaling pathway.
Discussion
MitoQ is a highly effective mitochondrial-targeted antioxidant frequently used as a protectant in neurodegenerative diseases [22-24], ischemia-reperfusion injury [25-27], and cerebral ischemia [28]. The mitochondrial-targeted antioxidant MitoQ, which is unique and completely different from conventional antioxidants, can penetrate the mitochondria and exert an antioxidant effect by decreasing ROS production [29]. This helpful role encouraged us to consider the potential neuroprotective mechanism of MitoQ after TBI.
In the current study, we investigated the neuroprotection afforded by MitoQ in an experimental model of TBI and its related molecular mechanisms. This study showed unequivocally that MitoQ administration improved the neurological deficits, attenuated cerebral edema, and reduced apoptosis by attenuating the TBI-associated oxidative damage and suppressing the mitochondrial apoptotic pathway in mice after TBI. In addition, treatment with MitoQ upregulated the expression of the antioxidant enzymes GPx, SOD, HO-1 and Nqo1, via the activation of the Nrf2-ARE pathway. These results suggest that MitoQ exerts neuroprotective effects against TBI by means of the Nrf2-ARE pathway. To the best of our knowledge, this is the first study exploring the protective effects of MitoQ on TBI through the regulation of the activity of the Nrf2-ARE signaling pathway in a TBI model.
As is widely established, oxidative stress is an important factor in the pathophysiological changes after TBI [2,30,31]. Oxidative stress-induced mitochondrial damage causes generation of superfluous ROS [32,33]. Excessive ROS obstructs mitochondrial function and upsets the normal balance of endogenous oxidant and antioxidant mechanisms. The level of MDA, which indicates lipid peroxidation, begins to elevate immediately following TBI, and remains at an elevated level 24 and 48 h after injury [2]. Antioxidant enzymes (SOD and GPx) are used to eliminate metabolites produced by free radicals, transforming peroxides into innocuous substances [2]. In the present study, we found that the level of MDA in the injured cortex was clearly increased, while the level of SOD and GPx were decreased following TBI. On the other hand, MitoQ administration mitigated MDA levels and improved the activity of antioxidant enzymes including SOD and GPx compared with the vehicle-treated group after TBI. These results indicate that oxidative stress was induced by TBI. Furthermore, MitoQ notably alleviated the oxidative insult induced by TBI. These findings are consistent with previous studies [16,34-36]. According to these findings, we can infer that MitoQ exerts antioxidant effects to reduce oxidative damage after TBI.
Mitochondria play a pivotal role in the TBI through mitochondria-related apoptosis or by ROS production [5,37]. It has been well established that the pro-apoptotic protein Bax is normally located in the cytoplasm, while cytochrome c, which is located in the mitochondria inter membrane, could not be released from mitochondria [38,39]. In response to the stimulation of apoptosis, Bax is translocated from the cytoplasm to the mitochondria, thus increasing the permeability of the mitochondrial membrane, resulting in mitochondrial cytochrome c release. Cytochrome c liberated from the mitochondria into the cytoplasm induces the sequential activation of the caspase cascades, which culminates in apoptosis [40,41]. A similar phenomenon was found in our work. Our results suggest that Bax translocation into the mitochondrial membrane and cytochrome c release into the cytoplasm were enhanced by TBI, indicating that the mitochondrial-dependent apoptotic pathway was activated. These changes further triggered the activation of the intrinsic apoptosis pathway, leading to neuronal apoptosis [5]. These findings are also consistent with prior researches [34,38]. The effects were reversed by MitoQ following TBI. The results described above reveal that MitoQ provides neuroprotection in a mouse model of TBI by suppressing the mitochondrial-dependent apoptosis pathway.
The neuroprotective effects of MitoQ have been demonstrated in previous studies of neurodegenerative diseases and ischemia-reperfusion diseases [7,13,22,23]. Prior studies showed that Nrf2 is the key regulator in reducing oxidative stress, inflammatory response, and the accumulation of toxic metabolites, which are all involved in secondary brain injury after TBI [34,36]. Fewer studies have mentioned the relationship between MitoQ and Nrf2 [13,35,42,43]. Gonzalez et al. demonstrated that MitoQ-associated autophagy in breast cancer cells activated the Nrf2-Keap1 pathway and attenuated oxidative insult [42]. In a recent study, Li et al. reported that MitoQ administration increased the Nrf2 expression in diabetic kidney and mediated tubular mito-phagy, partly through the Nrf2-Keap1 pathways [35]. However, little is known about the specific molecular mechanisms associated with MitoQ in TBI. To ascertain whether the Nrf2 signaling pathway participates in the neuroprotection afforded by MitoQ in TBI, we examined the mechanisms whereby MitoQ modulates the Nrf2-ARE signaling pathway after TBI. The results suggested that Nrf2 expression in the nucleus were upregulated after TBI. Moreover, MitoQ treatment further increased the expression of nuclear Nrf2. This finding is in concordance with the classic pattern of Nrf2 activation in protein expression. The levels of cytoplasmic Nrf2 were dramatically decreased after TBI and was much lower following treatment with MitoQ. However, the level of Nrf2 relative to the total protein showed no considerable change in our study. Nrf2 activation promoted the expression of its downstream antioxidant genes. Our findings demonstrate that the effect of MitoQ on Nrf2 promotes its translocation from the cytoplasm to the nucleus, and regulates the levels of Nrf2 at the posttranslational level in this model. These data shed light on the effect of MitoQ on the activation of the Nrf2-ARE signal pathway, but the precise mechanism remains to be further elucidated.
Our study suffers from some limitations. First, an in vitro model of TBI should be used to determine whether MitoQ is involved in the protection against neuronal insult and oxidative damage. Second, a Nrf2 gene knockout mice model should be used to confirm the neuroprotective effects of MitoQ by activation of the Nrf2-ARE pathway. Additional comprehensive research remains to be further performed.
In conclusion, the results from the present study clearly demonstrated that MitoQ attenuated the oxidative stress by enhancing the expression and activity of antioxidant enzymes, and also suppressed the mitochondrial apoptotic pathway by inhibiting Bax translocation and cytochrome c release from the mitochondria in a TBI model. Furthermore, MitoQ administration resulted in activation of the Nrf2-ARE pathway. These findings indicate that MitoQ exhibits neuroprotective effects and has therapeutic potential for preventing secondary brain injury following TBI.
Acknowledgements
This study was grant-supported by the National Natural Science Fundation of China (No. 81371357, No. 81571162 and No. 81401026).
Disclosure of conflict of interest
None.
References
- 1.Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012;1822:675–684. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cornelius C, Crupi R, Calabrese V, Graziano A, Milone P, Pennisi G, Radak Z, Calabrese EJ, Cuzzocrea S. Traumatic brain injury: oxidative stress and neuroprotection. Antioxid Redox Signal. 2013;19:836–853. doi: 10.1089/ars.2012.4981. [DOI] [PubMed] [Google Scholar]
- 3.Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- 4.Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:14205–14218. doi: 10.3748/wjg.v20.i39.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robertson CL, Scafidi S, McKenna MC, Fiskum G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol. 2009;218:371–380. doi: 10.1016/j.expneurol.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 8.James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA, Murphy MP. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem. 2007;282:14708–14718. doi: 10.1074/jbc.M611463200. [DOI] [PubMed] [Google Scholar]
- 9.Smith RA, Adlam VJ, Blaikie FH, Manas AR, Porteous CM, James AM, Ross MF, Logan A, Cocheme HM, Trnka J, Prime TA, Abakumova I, Jones BA, Filipovska A, Murphy MP. Mitochondria-targeted antioxidants in the treatment of disease. Ann N Y Acad Sci. 2008;1147:105–111. doi: 10.1196/annals.1427.003. [DOI] [PubMed] [Google Scholar]
- 10.Wani WY, Gudup S, Sunkaria A, Bal A, Singh PP, Kandimalla RJ, Sharma DR, Gill KD. Protective efficacy of mitochondrial targeted antioxidant MitoQ against dichlorvos induced oxidative stress and cell death in rat brain. Neuropharmacology. 2011;61:1193–1201. doi: 10.1016/j.neuropharm.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Oyewole AO, Birch-Machin MA. Mitochondriatargeted antioxidants. FASEB J. 2015;29:4766–4771. doi: 10.1096/fj.15-275404. [DOI] [PubMed] [Google Scholar]
- 12.Rehman H, Liu Q, Krishnasamy Y, Shi Z, Ramshesh VK, Haque K, Schnellmann RG, Murphy MP, Lemasters JJ, Rockey DC, Zhong Z. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int J Physiol Pathophysiol Pharmacol. 2016;8:14–27. [PMC free article] [PubMed] [Google Scholar]
- 13.Yin X, Manczak M, Reddy PH. Mitochondriatargeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum Mol Genet. 2016;25:1739–1753. doi: 10.1093/hmg/ddw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 15.de Vries HE, Witte M, Hondius D, Rozemuller AJ, Drukarch B, Hoozemans J, van Horssen J. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med. 2008;45:1375–1383. doi: 10.1016/j.freeradbiomed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Yan W, Wang HD, Feng XM, Ding YS, Jin W, Tang K. The expression of NF-E2-related factor 2 in the rat brain after traumatic brain injury. J Trauma. 2009;66:1431–1435. doi: 10.1097/TA.0b013e318180f5c7. [DOI] [PubMed] [Google Scholar]
- 17.Cho HY, Reddy SP, Debiase A, Yamamoto M, Kleeberger SR. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic Biol Med. 2005;38:325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4:1328–1337. doi: 10.1038/nprot.2009.148. [DOI] [PubMed] [Google Scholar]
- 20.Xu J, Wang H, Ding K, Lu X, Li T, Wang J, Wang C, Wang J. Inhibition of cathepsin S produces neuroprotective effects after traumatic brain injury in mice. Mediators Inflamm. 2013;2013:187873. doi: 10.1155/2013/187873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beni-Adani L, Gozes I, Cohen Y, Assaf Y, Steingart RA, Brenneman DE, Eizenberg O, Trembolver V, Shohami E. A peptide derived from activity-dependent neuroprotective protein (ADNP) ameliorates injury response in closed head injury in mice. J Pharmacol Exp Ther. 2001;296:57–63. [PubMed] [Google Scholar]
- 22.Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis. 2010;20(Suppl 2):S609–631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solesio ME, Prime TA, Logan A, Murphy MP, Del Mar Arroyo-Jimenez M, Jordan J, Galindo MF. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim Biophys Acta. 2013;1832:174–182. doi: 10.1016/j.bbadis.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Ma T, Hoeffer CA, Wong H, Massaad CA, Zhou P, Iadecola C, Murphy MP, Pautler RG, Klann E. Amyloid beta-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci. 2011;31:5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross MF, Prime TA, Abakumova I, James AM, Porteous CM, Smith RA, Murphy MP. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem J. 2008;411:633–645. doi: 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- 26.Neuzil J, Widen C, Gellert N, Swettenham E, Zobalova R, Dong LF, Wang XF, Lidebjer C, Dalen H, Headrick JP, Witting PK. Mitochondria transmit apoptosis signalling in cardiomyocytelike cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox Rep. 2007;12:148–162. doi: 10.1179/135100007X200227. [DOI] [PubMed] [Google Scholar]
- 27.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 28.Ahmed E, Donovan T, Yujiao L, Zhang Q. Mitochondrial targeted antioxidant in cerebral ischemia. J Neurol Neurosci. 2015:6. doi: 10.21767/2171-6625.100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escribano-Lopez I, Diaz-Morales N, Rovira-Llopis S, de Maranon AM, Orden S, Alvarez A, Banuls C, Rocha M, Murphy MP, Hernandez-Mijares A, Victor VM. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016;10:200–205. doi: 10.1016/j.redox.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Rodriguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A. Oxidative stress in traumatic brain injury. Curr Med Chem. 2014;21:1201–1211. doi: 10.2174/0929867321666131217153310. [DOI] [PubMed] [Google Scholar]
- 32.Smith JA, Park S, Krause JS, Banik NL. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem Int. 2013;62:764–775. doi: 10.1016/j.neuint.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma. 2008;25:513–526. doi: 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- 34.Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 35.Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, Wang J, Qin Y, Liu Y, Tang C, He L, Greka A, Zhou Z, Liu F, Dong Z, Sun L. The mitochondriatargeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297–311. doi: 10.1016/j.redox.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, Wang H, Ding K, Zhang L, Wang C, Li T, Wei W, Lu X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol Med. 2014;71:186–195. doi: 10.1016/j.freeradbiomed.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Wang R, Liu YY, Liu XY, Jia SW, Zhao J, Cui D, Wang L. Resveratrol protects neurons and the myocardium by reducing oxidative stress and ameliorating mitochondria damage in a cerebral ischemia rat model. Cell Physiol Biochem. 2014;34:854–864. doi: 10.1159/000366304. [DOI] [PubMed] [Google Scholar]
- 38.Wei W, Wang H, Wu Y, Ding K, Li T, Cong Z, Xu J, Zhou M, Huang L, Ding H, Wu H. Alpha lipoic acid inhibits neural apoptosis via a mitochondrial pathway in rats following traumatic brain injury. Neurochem Int. 2015;87:85–91. doi: 10.1016/j.neuint.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Cahill J, Calvert JW, Marcantonio S, Zhang JH. p53 may play an orchestrating role in apoptotic cell death after experimental subarachnoid hemorrhage. Neurosurgery. 2007;60:531–545. doi: 10.1227/01.NEU.0000249287.99878.9B. discussion 545. [DOI] [PubMed] [Google Scholar]
- 40.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 41.Liu L, Sun T, Liu Z, Chen X, Zhao L, Qu G, Li Q. Traumatic brain injury dysregulates microRNAs to modulate cell signaling in rat hippocampus. PLoS One. 2014;9:e103948. doi: 10.1371/journal.pone.0103948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez Y, Aryal B, Chehab L, Rao VA. Atg7- and Keap1-dependent autophagy protects breast cancer cell lines against mitoquinoneinduced oxidative stress. Oncotarget. 2014;5:1526–1537. doi: 10.18632/oncotarget.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao VA, Klein SR, Bonar SJ, Zielonka J, Mizuno N, Dickey JS, Keller PW, Joseph J, Kalyanaraman B, Shacter E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J Biol Chem. 2010;285:34447–34459. doi: 10.1074/jbc.M110.133579. [DOI] [PMC free article] [PubMed] [Google Scholar]