

Abstract
As a component of collagen II, glycosaminoglycan (GAG) has a relatively close relationship with bone metabolism. GAG and collagen II have been proven to promote connection of the bone trabecular structure. However, the exact mechanism remains unknown. In this study, we aimed to determine the concrete effect and the mechanism of GAG and collagen II on glucocorticoid-induced osteoporosis. We implanted prednisolone pellets subcutaneously in mice to mimic glucocorticoid-induced osteoporosis. GAG was administered intragastrically every day for 60 days. The results demonstrated a protective effect of GAG and collagen II on glucocorticoid-induced osteoporosis. Trabecular number and connection density increased after treatment with GAG and collagen II. We generated bone marrow-derived macrophages to explore the effect of GAG and collagen II on osteoclast differentiation. We collected cell protein and RNA in the presence of macrophage colony-stimulating factor (M-CSF) and receptor activator for nuclear factor-κB ligand (RANKL) and found that GAG and collagen II inhibited the NF-κB and MAPK pathways, thereby down-regulating osteoclast differentiation molecules such as matrix metallopeptidase 9 (MMP 9) and nuclear factor of activated T-cells, cytoplasmic 1 (NFATc-1). Our findings suggest that GAG and collagen II may have therapeutic potential of patients with glucocorticoid-induced osteoporosis in clinical settings.
Keywords: Glucocorticoids, osteoporosis, GAG and collagen II, NF-κB and MAPK pathways
Introduction
Chronic kidney disease (CKD) is characterized by the progressive loss of kidney structure and function and is induced by various causes over a period of months or years, usually more than 3 months [1]. A prevalence study in China shows that 10.8% of Chinese suffer from CKD; thus, the number of CKD patients in China is estimated to be approximately 119.5 million [2]. Glucocorticoids (GCs) are one of the most common drugs used in CKD patients with their highly anti-inflammatory and immunosuppressive effects. However, long-term and high dose usage can renered patients to develop numerous adverse complications including GC-induced osteoporosis (GIOP) [3,4].
GIOP is the most common type of secondary osteoporosis and is characterized by decreased bone mass and strength as well as an increased fracture risk. The endogenous and physiological concentration of GCs plays a positive role in the differentiation and activation of osteoblasts and promotes the physiological bone formation process [5]. In contrast, patients receiving pharmacological doses of GCs suffer from a highly cumbersome experience. Some GC-sensitive patients cannot even walk or stand normally after long-term and high-dose GC therapies and thus have a low quality of life. Research showed that GCs damaged the balance among osteoblasts, osteocytes and osteoclasts activity, leading to osteoporosis. A decrease in the number and activities of osteoblasts and osteocytes or an increase in the number and activities of osteoclasts altered the balance between bone formation and absorption, finally resulting in osteoporosis [6-8].
The NF-κB pathway and its downstream molecules such as MMP 9, CTSK and NFATc-1 have been recognized to be the key regulators closely linked to GIOP [9-11]. Increased expression of these downstream molecules promoted bone marrow macrophage differentiation into osteoclasts and as a result led to osteoporosis.
Collagen II is a type of protein that mainly expresses in skin and bone. The components, collagen fibers, elastin and glycosaminoglycan (GAG), interweave with each other to form a network structure associated with bone density, thereby increasing bone mass and strength. GAG is especially used as an important marker for measuring collagen II and bone metabolism. However, the related mechanism is unknown.
In this study, we aimed to determine the effect of GAG and collagen II on the bone process during osteoclastogenesis. GAG and collagen II inhibited GC-induced osteoporosis by promoting osteoclast related molecules MMP 9, CTSK and NFATc-1 through the NF-κB and MAPK pathways, leading to osteoclast differentiation. GAG and collagen II inhibited GCs-mediated promotion of osteoclastogenesis, by which they repress GC-induced osteoporosis.
Materials and methods
Preparation of pure GAG and collagen II solution
The GAG and collagen II solution was prepared through GAG and collagen II tablets (Wuhan Huge Biotechnology Co., Ltd, China). The tablets were dissolved in distilled water at a concentration of 10% (10 g/100 ml); the PH valued was adjusted to 7.0. Finally, the solution was filtrated through a bacteria-proof filter to maintain sterility.
Animals and treatments
Male C57bl/6 mice (8 weeks old, weighing 25 g) were purchased from the Beijing Hua Fukang Laboratory Animal Technology Co., Ltd. (Beijing, China). Animals were acclimated to the housing environment in specific pathogen-free (SPF) conditions at the Tongji Medical College Animal Care Unit. Animals were kept in a 22°C temperature environment with a 12 h/12 h light/dark cycle for 10 weeks until they were 4.5 months old. Placebo and prednisolone slow-released pellets were purchased from Innovative Research of America (IRA, Florida, USA). Animals were randomly divided into four experimental groups: normal (no special intervention), placebo (mice were implanted with 7.5 mg placebo pellets subcutaneously, 60 days slow-released), prednisolone (mice were implanted with 7.5 mg prednisolone pellets subcutaneously, 60 days slow-released), and GAG (mice were implanted with same dose of slow-release prednisolone pellets subcutaneously and also received intragastric administration of GAG and collagen II solution); each group had 6-8 mice. We chose a total pellet dose of 7.5 mg and a 60-day release period for research. Mice were anesthetized by an intraperitoneal injection of 1% sodium pentobarbital (0.008 ml per gram body weight). Then, suitable pockets were created above the scapula to implant slow-released placebo and prednisolone pellets. After surgery, a GAG and collagen II solution was administered at a dose of 0.45 mg per gram body weight per day. Sixty days later, mice were sacrificed by cervical dislocation. We detected no pellets after 60 days. Femora, tibias and blood were harvested for further analysis. All experimental procedures were conducted in accordance with NIH guidelines and were approved by the Animal Care and Use Committee (ACUC) of Tongji Hospital.
Bone morphometric parameters
Bone morphometric parameters and micro-architectural properties of the left tibia were analyzed by micro computed tomography (μ-CT50; Scanco Medical, Basserdorf, Switzerland). Proximal tibias were adopted for 3D reconstruction by built-in software, trabecular parameters including bone volume/total volume ratio (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), trabecular space (Tb.Sp), bone surface/bone volume ratio (BS/BV), and connectivity density (Conn.D).
Bone pathological staining
Femurs were fixed in a 4% paraformaldehyde solution for 2 days. Then, they were decalcified in a 0.5 M EDTA (pH=8.0) solution for 3 weeks and embedded in paraffin wax. The paraffin-embedded femurs were sectioned in 5 μm sections, stained with hematoxylin and eosin (H&E), and visualized under a microscope (Olympus, Japan).
Western blotting analysis
Bone tissues were crushed in liquid nitrogen and homogenized in RIPA lysis buffer (Promoter, Wuhan, China) containing a protease inhibitor cocktail (Promoter, Wuhan, China) and phenylmethanesulfonyl fluoride (Promoter, Wuhan, China). Cell proteins were collected in the same way. Total protein concentrations were determined using a BCA assay kit following the manufacturer’s instructions (Promoter, Wuhan, China). Proteins were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skimmed milk in TBS with 0.1% Tween-20 for 1 h at 37°C and then probed with antibodies against OPG (dilution 1:1000, Bioworld, China), RANKL (dilution 1:1000, ProSci Inc., USA), MMP 9 (dilution 1:1000, Abcam, UK), NFATc-1 (dilution 1:1000, Abcam, UK), CTSK (dilution 1:1000; Abcam, UK), total and phosphorylated-65, -38, -ERK, and -JNK (dilution 1:1000, cell signaling technology, USA) and GAPDH (dilution 1:4000, Abbkine, China) at 4°C overnight. The blots were washed the next day, incubated with HRP-conjugated secondary antibodies for 1 h at 37°C, and visualized by enhanced chemiluminescence (ECL, BioRad, USA). The gray values of targeted bands were analyzed using ImageJ (NIH, USA).
Cell cultures
Primary bone marrow mononuclear cells were isolated from 4-week-old male C57bl/6 mice. Femurs and tibias were flushed with culture medium, and then bone marrow cells were collected into 10 cm-diameter dishes. The cells were cultured in α-MEM medium containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 U/mL streptomycin, and 30 ng/mL M-CSF (PeproTech, Rocky Hill, NJ). After 24 hours, floating cells were collected in the presence of M-CSF to continue culturing for another 3 days. Adherent cells were used as bone marrow derived macrophages (BMMs) for further use. BMMs were maintained in a humidified incubator (Thermo Scientific, USA), 5% CO2, and 95% air atmosphere at 37°C. The medium was replenished every two days.
Measurement of cell viability
Cell counting kit-8 (Promoter, Wuhan, China) was used to assess cell viability. BMMs were seeded at a density of 5 × 103 cells per well in 96-well plates. After adherence, cells were treated with different concentrations of GAG and collagen II (GAG: 0, 0.0015, 0.0075, 0.015, 0.15, 1.5 g/L) and dexamethasone (DXM: 0, 0.001, 0.01, 0.1, 1 μM) for 96 h. Then, the culture medium was replaced with complete medium with 10% CCK-8. After 1-4 h of incubation at 37°C, the absorbance at 450 nm was measured using a multimode reader.
TRAP staining
For detection of differentiation toward osteoclasts, BMMs were exposed to 100 ng/mL RANKL (PeproTech, Rocky Hill, NJ) and 30 ng/mL M-CSF. Cells were treated with different concentrations of GAG and collagen II (0, 0.0015, 0.0075, 0.015, 0.15, 1.5 g/L) and DXM (0.1 μM) for 96 h, then fixed with 4% paraformaldehyde and stained for tartrate-resistant acid phosphatase (TRAP) per the manufacturers’ instructions (Acid Phosphatase, Leukocyte (TRAP) Kit, Sigma, USA). TRAP-positive cells were counted as osteoclasts.
RNA preparation and RT-PCR
Total RNA was extracted from osteoclasts using Trizol reagent according to the manufacturer’s instructions (Invitrogen, USA). One microgram RNA was reverse transcribed into the first strand cDNA using the GoScript reverse transcription system (Promega, USA) in a 20 μL reaction system. The cycling parameters used were as follows: 40 cycle pf denaturation at 95°C for 15 s and annealing at 60°C for 60 s. Triplicated experiments for each sample were processed. Quantitative PCR was conducted using the SYBR master mix (Qiagen, Germany) on the Roche light 480II. The mRNA expression levels of several osteoclast-related markers, including MMP 9, CTSK, NFATc-1 and RANKL, were conducted via the comparative cycle threshold (Ct) method and normalized to the expression levels of GAPDH. The primer sequences for PCR are listed in Table 1.
Table 1.
The primer sequences
| Genes | Sequences | |
|---|---|---|
| NFATc-1 | Forward primer | 5’-CATCTTCGGCGTTTACTACAGG-3’ |
| Reverse primer | 5’-TCCACTTAGACTACTGCAAGCA-3’ | |
| c-fos | Forward primer | 5’-AGGGGCAAAGTAGAGCAGCTA-3’ |
| Reverse primer | 5’-CAATCTCAGTCTGCAACGCA-3’ | |
| MMP 9 | Forward primer | 5’-GCAGAGGCATACTTGTACCG-3’ |
| Reverse primer | 5’-TGATGTTATGATGGTCCCACTTG-3’ | |
| CTSK | Forward primer | 5’-CTCGGCGTTTAATTTGGGAGA-3’ |
| Reverse primer | 5’-CATATGGGAAAGCATCTTCAGAGTC-3’ | |
| GAPDH | Forward primer | 5’-CTGGAGAAACCTGCCAAGTA-3’ |
| Reverse primer | 5’-AAGAGTGGGAGTTGCTGTTG-3’ |
Statistical analysis
All data in the present study were reported as the means ± S.D. Comparisons between the two groups were accomplished using the unpaired t-test or nonparametric Mann-Whitney test. All data were analyzed using SPSS 18.0 and GraphPad Prism 5.0 software. P<0.05 was considered significant.
Results
The effect of GAG and collagen II on bone microarchitecture in GIOP mice
The bone microarchitectures of the left tibias were observed by micro-CT. A three-dimensional (3D) reconstruction technique was adopted to directly evaluate bone structure and bone mineral density. Trabecular parameters including BV/TV, Tb.N, Tb.Sp, and Conn.D were analyzed to estimate bone microstructure. Compared with the normal and placebo groups, the prednisolone group showed obvious bone loss in 3D reconstruction (Figure 1A). A significant decrease occurred in the measurements of BV/TV, Tb.N and Conn.D. Upon GAG and collagen II treatment, the parameters BV/TV, Tb.N and Conn.D increased notably, and the space between trabecula decreased (Figure 1B). The same result was shown by H&E staining of the femurs. GAG and collagen II improved the loss of bone trabecula caused by prednisolone (Figure 1C). Together, these results above suggest that prednisolone decreases bone mineral density (BMD) and sparse trabecular microarchitecture, which rendered the mice more susceptible to fracture. GAG and collagen II can protect mice from GCs-induced bone loss by increased bone trabecular and stronger microarchitecture.
Figure 1.
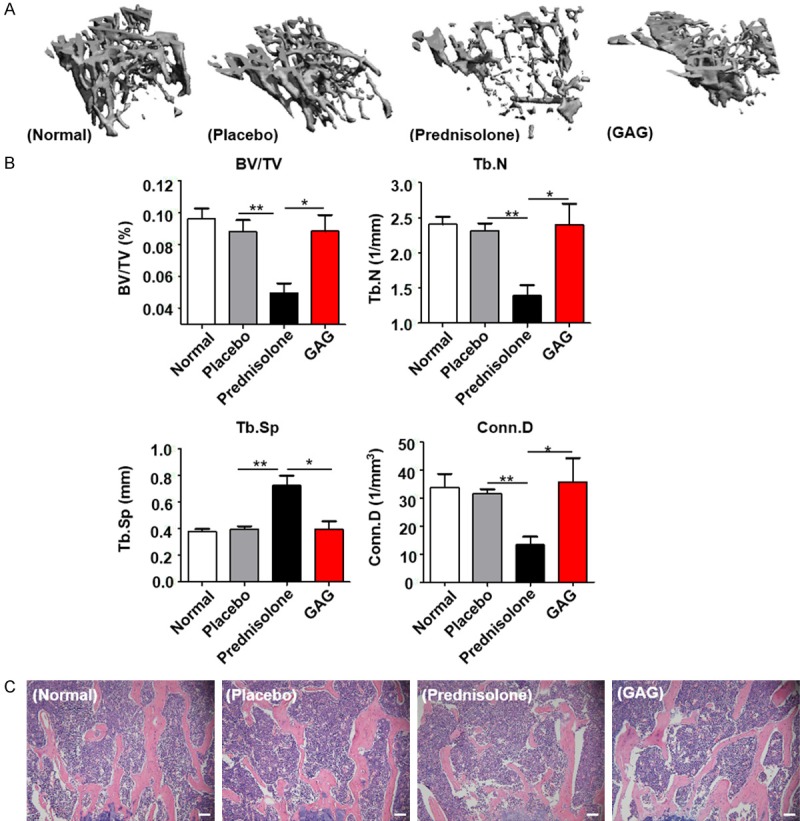
The effect of GAG and collagen II on bone microarchitecture in GIOP mice. Mice were implanted with 60 days slow-released 7.5 mg placebo pellets or prednisolone pellets subcutaneously, GAG and collagen II solution were administrated intragastricly. A. 3D reconstruction of proximal tibias by Micro-CT analysis from normal, placebo, prednisolone and GAG groups. B. Histograms showing the parameters of proximal tibias analyzed by Micro-CT: relative bone volume over total volume (BV/TV); trabecular number (Tb.N); trabecular space (Tb.Sp); connectivity density (Conn.D). C. Representative images of hematoxylin-eosin staining of femurs in different groups, scale bar: 20 μm. Data are presented as mean ± SD, n=5-7/group. Statistically significant differences between the treatment and control groups are indicated as *P<0.05, **P<0.01.
GAG and collagen II affected the expression of RANKL/OPG in GIOP mice
To investigate the possible mechanisms underlying GAG and collagen II attenuation of GC-induced osteoporosis, we first examined serum calcium (Ca2+) and phosphates, the biochemical markers. Mice treated with prednisolone pellets presented a low concentration of serum Ca2+ compared to that in normal and placebo groups. GAG and collagen II enhanced serum Ca2+ concentration. However, no significant difference was detected in serum phosphates. We next assessed protein levels of RANKL and OPG in the tibias and femora (Figure 2A). RANKL and its decoy receptor OPG were two key molecules to regulate the balance between bone resorption and formation. Increased expression of RANKL or decreased expression of OPG lead to further osteoclast differentiation and activity [12,13]. In GIOP mice, it was interestingly noted that prednisolone induced a significant oeverepression of RANKL rather than OPG (Figure 2B). The enhanced expression of RANKL and repressed OPG expression were alleviated in the GAG and collagen II group.
Figure 2.

GAG and Collagen II inhibited the expression of RANKL/OPG ratio in GIOP mice. A. Western blotting analysis of RANKL and OPG in GIOP mice. B. Histograms showing that prednisolone led to the activation of RANKL/OPG signaling pathway, thus promoting glucocorticoid-induced osteoporosis. GAG and Collagen II alleviated RANKL/OPG ratio in bone tissues and as a result protected mice from glucocorticoid-induced osteoporosis. All values are shown as mean ± SD, n=3-5/group, *P<0.05.
GAG and collagen II inhibited the differentiation of osteoclasts in BMMs
BMMs were then employed to dissect the link among GCs, GAG and collagen II and osteoporosis. DXM, GAG and collagen II affected BMMs independent of cytotoxicity (Figure 3A). It was noted that DXM dose-dependently induced osteoclast differentiation from 0 μM to 1 μM (Figure 3B). The numbers and areas of TRAP-positive (TRAP+) cells increased in parallel with the increase of DXM (Figure 3C). When GAG and collagen II were added, the numbers and areas of TRAP positive cells decreased without cytotoxicity (Figure 3D and 3E). Therefore, GCs promoted the differentiation and development of osteoclasts, which were inhibited by the administration of GAG and collagen II.
Figure 3.
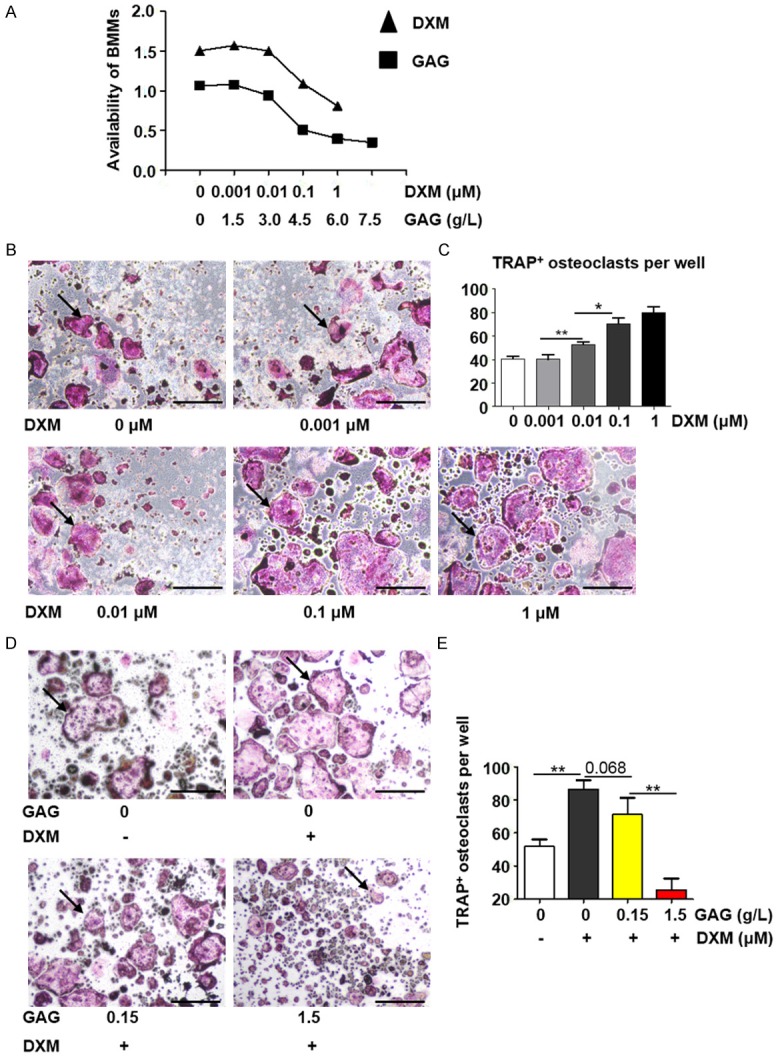
GAG and collagen II inhibited the differentiation of osteoclasts in BMMs. A. Different concentrations of DXM and GAG and Collagen II on the availability of BMMs. B. TRAP staining of BMMs with DXM (0, 0.001, 0.01, 0.1, 1 μM) and RANKL (100 ng/ml) for 4 days. Arrows indicated Trap-positive (TRAP+) multinuclear osteoclasts. C. Numbers of TRAP-positive cells per well. Data were presented as mean ± SD, n=3. D. Trap staining of BMMs with DXM (0.1 μM), GAG and Collagen II (0, 0.0015, 0.0075, 0.015, 0.15, 1.5 g/L) and RANKL (100 ng/ml) for 4 days. Arrows indicated TRAP+ multinuclear osteoclasts. E. Numbers of TRAP+ cells per well. Data are counted as mean ± SD, n=3. *P<0.05; **P<0.01. Scale bar: 500 μm.
GAG and collagen II inhibited RANKL-induced NF-κB and MAPK pathways
Given that the NF-κB pathway plays a critical role in RANKL-stimulated differentiation of osteoclasts [14], we, therefore, next examined the effect of GAG and collagen II on RANKL-induced activation of the NF-κB and MAPK pathways. As shown in Figure 4, GAG and collagen II inhibited the increase of phosphorylated p65 induced by DXM at 15 min. The same result was observed for the MAPK pathway, as manifested by the increase of phosphorylated p38, ERK and JNK upon DXM treatment in the presence of RANKL, which was attenuated by the administration of GAG and collagen II.
Figure 4.

GAG and collagen II inhibited RANKL-induced NF-κB and MAPK pathways. BMMs were cultured in medium with M-CSF (30 ng/ml), the cells were co-cultured with DXM, with or without GAG and Collagen II for 24 hours, then cells were stimulated with RANKL (100 ng/ml) for 0 min, 15 min, 30 min and 60 min. The protein levels of several genes were quantified by western blotting. A. Phosphorylated expression of p65 (p-p65) increased in 15 minutes and then decreased in 1 hour after DXM stimulation. The level of p-p65 decreased overall with the treatment of GAG and Collagen II. B. The subfamilies of MAPK pathway (p-ERK, p-JNK, p-p38) presented a similar trend to phosphorylated expression of p65.
GAG and collagen II repressed the expression of key molecules relevant to osteoclast differentiation
Finally, we detected an increase in the NF-κB and MAPK pathways in DXM administration, which was inhibited by GAG and collagen II. This result prompted us to examine its downstream signaling molecules NFATc-1 and c-fos. GAG and collagen II significantly inhibited the expression of NFATc-1 and c-fos both by Western blot and RT-PCR analysis. We then checked detected NFATc-1 targets MMP 9 and CTSK, and found that GAG and collagen II played a suppressive role in their expression. DXM activated the downstream genes NFATc-1, c-fos, MMP 9 and CTSK and, as a result, promoted osteoclast differentiation and the osteoporosis process. However, GAG and collagen II attenuated its effect, by which inhibiting GCs-induced osteoclast differentiation (Figure 5).
Figure 5.
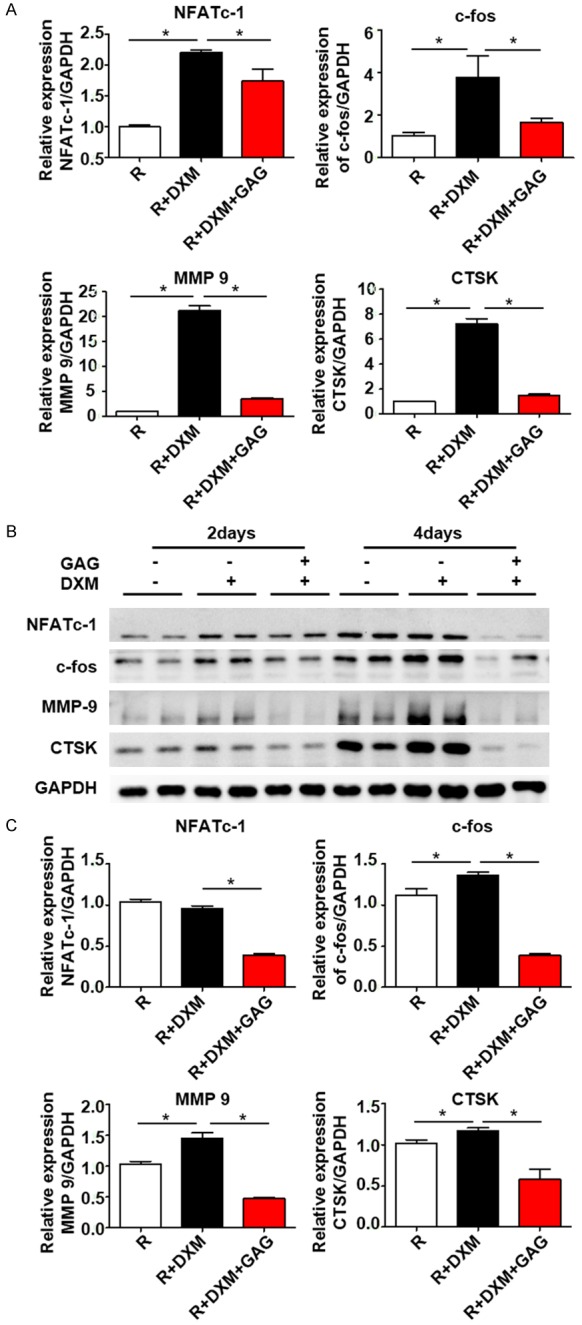
GAG and collagen II repressed the expression of key molecules relevant to osteoclast differentiation. BMMs were cultured in medium in the presence of RANKL (100 ng/ml) and M-CSF (30 ng/ml). DXM (0.1 μM) or GAG and Collagen II (1.5 g/L) were added into different groups, cells were cultured for 2 or 4 days. The mRNA expressions of several genes were quantified by RT-PCR. The protein levels were detected by western blotting assay. A. RT-PCR analysis of the transgenic levels of several markers connected with NF-κB and MAPK signaling pathways. B. Western blotting analysis of the protein expressions of NFATc-1, c-fos, MMP 9, CTSK. Data are presented as mean ± SD, n=3. C. Quantitation of NFATc-1, c-fos, MMP 9 and CTSK was normalized to GAPDH. *P<0.05.
Discussion
GAG and collagen II were considered as healthy food in China and have become well known for their effect on improving bone mass and strength. In ovariectomized rats, GAG and collagen II enhance femur biomechanical indexes such as maximal load, stress, elasticity modulus. However, their effect on GIOP remains unknown. This study mainly focused on the effect of GAG and collagen II in GIOP mice. It has been well recognized that endogenous and physiological doses of GCs regulate osteoblast differentiation and terminally inhibited osteoclast activity to maintain the balance between bone formation and resorption. Excessive GCs have an opposite effect by interrupting bone homeostasis and causing osteoporosis [15]. Since high doses of GC therapy cause secondary osteoporosis, which limited their clinical applications. RANKL is an essential factor that regulates the differentiation and activation of osteoclasts in GIOP. Upon ligation to its receptor RANK, RANKL initiates the recruitment of adaptor molecules, e.g., TRAFs, especially TRAF-6, and then activates the downstream NF-κB signaling pathway, subsequently regulating the gene expression of NFATc-1 and its target genes MMP 9 and CTSK and, as a result, promoting osteoclast differentiation and osteoporosis [16]. The increased expression of the RANKL or RANKL/OPG ratio alters bone homeostasis and finally causes osteoporosis [17,18]. Reduction of RANKL attenuates the differentiation of osteoclasts and bone resorption process. Previous studies suggested an effect of RANKL inhibition (OPG-Fc protein, RANKL inhibitor denosumab) on osteoclast differentiation, mainly in postmenopausal osteoporosis [12,19]. In our study, we demonstrated a reduction of the RANKL and RANKL/OPG ratio in prednisolone-treated mice, potentially explaining the protective effect of GAG and collagen II on GIOP.
NFATc-1 is a calcineurin and Ca2+ regulated transcription factor that is strongly induced by RANKL. During osteoclastogenesis, the ligation of RANKL to RANK leads to the dephosphorylation of NFATc-1 via the NF-κB signaling pathway and, consequently, translocation to the nucleus, enhancing the transcription and translation of osteoclast-associated genes such as MMP 9 and CTSK, thus affecting osteoclast differentiation and osteoporosis [20,21]. MMP 9 is a class of enzymes that belongs to the zinc-metalloproteinases family involved in the degradation of ECM. In bone tissues, MMP 9 is produced by osteoclasts, with the ability to degrade and remodel ECM in the skeleton, thus participating in bone resorption and remodeling in both physiological and pathological processes [22,23]. As a lysosomal cysteine protease expressed predominantly in osteoclasts, CTSK could induce the degradation of ECM also. Deficiency of CTSK causes disorder pycnodysostosis, as characterized by osteosclerosis [24].
We demonstrated that GAG and collagen II protect mice from GIOP by inhibition of the RANKL/OPG pathway. Elevated trabecular number and connectivity were detected with the treatment of GAG and collagen II. We further observed decreased expression of phosphorylated-65 and -38 when BMMs were treated with GAG and collagen II in the presence of RANKL. Consistently, down-regulations of downstream molecules NFATc-1, c-fos, MMP 9 and CTSK was noted in BMMs once treated with GAG and collagen II. The increased expression of OPG and decreased expression of RANKL in vivo and inhibition of the NF-κB and MAPK signaling pathways in vitro clearly demonstrate that GAG and collagen II protect mice from GIOP via the NF-κB and MAPK signaling pathways. Reduced expression of RANKL and enhanced OPG inhibited the NF-κB and MAPK signaling pathways, thus affecting transcriptions of NFATc-1 and c-fos, and finally leading to the down-regulation of MMP 9 and CTSK. As a result, osteoclast differentiation and ECM degradation were alleviated, thereby improving GC-induced osteoporosis.
In this study, we demonstrated a promoting effect of GCs on osteoclast differentiation. GAG and collagen II inhibited GC-induced osteoporosis via the NF-κB and MAPK pathways. GCs at a pharmacological dose induced osteoporosis as a result not only of osteoclasts but also of osteoblasts and osteocytes [25]. Excessive GCs suppressed the differentiation and proliferation of osteoblasts and promoted the apoptosis of osteoblasts [26]. Previous studies showed that the suppression of osteoblast differentiation and proliferation is correlated mainly with the reductions of beta-catenin and RUNX2 transcription factors relevant to the down-regulation of Wnt signaling [27]. Another key regulator was BMP2, which was proven to be inhibited in mice once treated with pharmacological dose of GCs. Decreased beta-catenin, RUNX2 or BMP2 caused the reduction in osteoblastogenesis and the differentiation of MSCs toward adipocytes [28]. Otherwise, GCs at therapeutic doses induced osteocyte autophagy and apoptosis, as well as the decrease in osteocyte viability [8,29]. With the activation of proapoptotic kinases or the production of autophagosomes in osteocytes, a cytotoxic environment was created, leading to cellular apoptosis and death. However, whether the suppression of autophagy in osteocyte weakened GIOP remains to be solved [30].
In summary, our study clearly showed that GAG and collagen II had a protective effect on glucocorticoid-induced osteoporosis. The associated mechanisms are associated with the reduction of RANKL expression and inhibition of the NF-κB and MAPK pathways. This study provided some theoretical basis and a potential therapy to attenuate GIOP in the clinical setting. However, additional studies are needed to assess whether GAG and collagen II affect osteoblasts or osteocytes, and to further explore the molecular mechanisms underlying the protective effect.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants 81570669, 81200521, 81570615) and the Wuhan health and family planning commission (grants 2016whzqnyxgg02).
Disclosure of conflict of interest
None.
References
- 1.Saunders MR, Cifu A, Vela M. Screening for chronic kidney disease. JAMA. 2015;314:615–616. doi: 10.1001/jama.2015.9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang L, Wang F, Wang L, Wang W, Liu B, Liu J, Chen M, He Q, Liao Y, Yu X, Chen N, Zhang JE, Hu Z, Liu F, Hong D, Ma L, Liu H, Zhou X, Chen J, Pan L, Chen W, Wang W, Li X, Wang H. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet. 2012;379:815–822. doi: 10.1016/S0140-6736(12)60033-6. [DOI] [PubMed] [Google Scholar]
- 3.Hofbauer LC, Rauner M. Minireview: live and let die: molecular effects of glucocorticoids on bone cells. Mol Endocrinol. 2009;23:1525–1531. doi: 10.1210/me.2009-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper MS, Zhou H, Seibel MJ. Selective glucocorticoid receptor agonists: glucocorticoid therapy with no regrets? J Bone Miner Res. 2012;27:2238–2241. doi: 10.1002/jbmr.1753. [DOI] [PubMed] [Google Scholar]
- 5.Henneicke H, Gasparini SJ, Brennan-Speranza TC, Zhou H, Seibel MJ. Glucocorticoids and bone: local effects and systemic implications. Trends Endocrinol Metab. 2014;25:197–211. doi: 10.1016/j.tem.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, Kirilov M, Mandic V, Takacz A, Schmidt-Ullrich R, Ostermay S, Schinke T, Spanbroek R, Zaiss MM, Angel PE, Lerner UH, David JP, Reichardt HM, Amling M, Schutz G, Tuckermann JP. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11:517–531. doi: 10.1016/j.cmet.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, Ross FP, Teitelbaum SL. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. 2006;116:2152–2160. doi: 10.1172/JCI28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 9.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 10.de Vrieze E, Sharif F, Metz JR, Flik G, Richardson MK. Matrix metalloproteinases in osteoclasts of ontogenetic and regenerating zebrafish scales. Bone. 2011;48:704–712. doi: 10.1016/j.bone.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Hou WS, Li Z, Gordon RE, Chan K, Klein MJ, Levy R, Keysser M, Keyszer G, Bromme D. Cathepsin k is a critical protease in synovial fibroblast-mediated collagen degradation. Am J Pathol. 2001;159:2167–2177. doi: 10.1016/S0002-9440(10)63068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocana A, Penninger JM, Vasavada RC. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-kappaB ligand pathway. Cell Metab. 2015;22:77–85. doi: 10.1016/j.cmet.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12:17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, Tuckermann JP. Molecular actions of glucocorticoids in cartilage and bone during health, disease, and steroid therapy. Physiol Rev. 2016;96:409–447. doi: 10.1152/physrev.00011.2015. [DOI] [PubMed] [Google Scholar]
- 16.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 17.Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA. 2004;292:490–495. doi: 10.1001/jama.292.4.490. [DOI] [PubMed] [Google Scholar]
- 18.Yin Y, Tang L, Chen J, Lu X. MiR-30a attenuates osteoclastogenesis via targeting DC-STAMP-c-Fos-NFATc1 signaling. Am J Transl Res. 2017;9:5743–5753. [PMC free article] [PubMed] [Google Scholar]
- 19.Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, Huang J, Dai W, Li C, Zheng C, Xu L, Chen H, Wang J, Li D, Siwko S, Penninger JM, Ning G, Xiao J, Liu M. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med. 2016;22:539–546. doi: 10.1038/nm.4076. [DOI] [PubMed] [Google Scholar]
- 20.Sundaram K, Nishimura R, Senn J, Youssef RF, London SD, Reddy SV. RANK ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp Cell Res. 2007;313:168–178. doi: 10.1016/j.yexcr.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231:241–256. doi: 10.1111/j.1600-065X.2009.00821.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Huang H, Zhao G, Yokoyama T, Vega H, Huang Y, Sood R, Bishop K, Maduro V, Accardi J, Toro C, Boerkoel CF, Lyons K, Gahl WA, Duan X, Malicdan MC, Lin S. ATP6V1H deficiency impairs bone development through activation of MMP9 and MMP13. PLoS Genet. 2017;13:e1006481. doi: 10.1371/journal.pgen.1006481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li G, Bu J, Zhu Y, Xiao X, Liang Z, Zhang R. Curcumin improves bone microarchitecture in glucocorticoid-induced secondary osteoporosis mice through the activation of microRNA-365 via regulating MMP-9. Int J Clin Exp Pathol. 2015;8:15684–15695. [PMC free article] [PubMed] [Google Scholar]
- 24.Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–1238. doi: 10.1126/science.273.5279.1236. [DOI] [PubMed] [Google Scholar]
- 25.Buehring B, Viswanathan R, Binkley N, Busse W. Glucocorticoid-induced osteoporosis: an update on effects and management. J Allergy Clin Immunol. 2013;132:1019–1030. doi: 10.1016/j.jaci.2013.08.040. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Li T, Fan J, Li T, Fan L, Wang S, Weng X, Han Q, Zhao RC. miR-216a rescues dexamethasone suppression of osteogenesis, promotes osteoblast differentiation and enhances bone formation, by regulating c-Cblmediated PI3K/AKT pathway. Cell Death Differ. 2015;22:1935–1945. doi: 10.1038/cdd.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou H, Mak W, Zheng Y, Dunstan CR, Seibel MJ. Osteoblasts directly control lineage commitment of mesenchymal progenitor cells through Wnt signaling. J Biol Chem. 2008;283:1936–1945. doi: 10.1074/jbc.M702687200. [DOI] [PubMed] [Google Scholar]
- 28.Salazar VS, Ohte S, Capelo LP, Gamer L, Rosen V. Specification of osteoblast cell fate by canonical Wnt signaling requires Bmp2. Development. 2016;143:4352–4367. doi: 10.1242/dev.136879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Cheng TS, Qin A, Pavlos NJ, Wang T, Song K, Wang Y, Chen L, Zhou L, Jiang Q, Takayanagi H, Yan S, Zheng M. Glucocorticoid impairs cell-cell communication by autophagy-mediated degradation of connexin 43 in osteocytes. Oncotarget. 2016;7:26966–26978. doi: 10.18632/oncotarget.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piemontese M, Onal M, Xiong J, Wang Y, Almeida M, Thostenson JD, Weinstein RS, Manolagas SC, O’Brien CA. Suppression of autophagy in osteocytes does not modify the adverse effects of glucocorticoids on cortical bone. Bone. 2015;75:18–26. doi: 10.1016/j.bone.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]