

Abstract
Ovarian cancer is one of the leading causes of cancer related deaths among women worldwide, with an overall 5-year survival of only 30-40%. Carbonic anhydrases are up-regulated in many types of cancer and play an important role in tumor progression and metastasis. Carbonic anhydrase 9 has been implicated as a potential anti-tumorigenic target. Topiramate (TPM) is a potent inhibitor of carbonic anhydrase isozymes, including carbonic anhydrase 9, and has been shown to have anti-tumorigenic activity in several cancer types. Our goal was to evaluate the effect of TPM on cell proliferation and to identify possible mechanisms by which TPM inhibits cell growth in ovarian cancer. TPM significantly inhibited ovarian cancer cell proliferation and induced cell cycle G1 arrest, cellular stress and apoptosis through the AKT/mTOR and MAPK pathways. TPM also exerted anti-metastatic effects by decreasing the adhesion and invasion of ovarian cancer cells and affecting the expression of critical regulators of the epithelial-mesenchymal transition (EMT). Our findings demonstrate that TPM has anti-tumorigenic effects in ovarian cancer and is worthy of further exploration in clinical trials.
Keywords: Ovarian cancer, carbonic anhydrases, topiramate, invasion
Introduction
Ovarian cancer is the most lethal gynecological cancer, with an estimated 22,440 new cases and 14,080 deaths in the U.S. in 2017 [1]. Due to the lack of effective screening methods, ovarian cancer is often diagnosed at a late stage, as reflected by the poor five-year overall survival (35%) [2]. Although most patients initially respond to standard treatment with surgical debulking and systemic platinum/taxane chemotherapy, the majority will suffer from a recurrence of disease and ultimately chemotherapy resistance [3,4]. Thus, developing new treatment options for ovarian cancer is urgently required [4].
The carbonic anhydrases (CAs) are a superfamily of metalloenzymes that efficiently catalyze the interconversion between CO2 and bicarbonate using a metal hydroxide nucleophilic mechanism. Sixteen different CAs have been identified, all of which have distinct tissue-specific expression, kinetic properties, and sensitivity to inhibitors [5]. CAs have varied activitiy based on the efficiency of proton transfer, differences in active site residues, quaternary structure and potential site localization [5-7]. Inhibition of CAs may have therapeutic implications for the treatment of edema, glaucoma, seizures, obesity, infectious diseases, arthritis, intracranial hypertension and cancer [5].
CA9 and CA12 belong to the cancer-associated transmembrane CA enzyme family, and are considered important enzymes related to the tumor microenvironment and oncogenic processes [8,9]. It has been confirmed that many types of cancer show strong expression of CA9 or co-expression of CA9 and CA12 in the peri-necrotic areas of tumors, supporting a concept that both isozymes are regulated by hypoxia and via hypoxia-induced factor 1α (HIF-1α) [10,11]. Expression of CA9 is absent in most normal tissues. Overexpression, in response to tumor hypoxia, is seen in a wide variety of solid tumors and contributes to hypoxia-related tumor acidosis and metastatic processes [12]. Overexpression of CA9 correlates with poor prognosis in ovarian cancer and has been associated with shortened disease specific survival [13]. Combined high expression of CA9 and VEGF has been linked to increased chemotherapy resistance and poor overall survival in high-grade serous ovarian carcinoma [14]. Pharmacologic interference of CA9 catalytic activity by monoclonal antibodies and small molecule inhibitors has been shown to disrupt pH regulation by cancer cells, reduce primary tumor growth, and inhibit cell migration and metastasis in preclinical models [13,15]. Overall, CA9 is being pursued as a marker for development of targeted systemic therapies and a prognostic indicator in cancer.
Topiramate (TPM) is a sulfamate-substituted monosaccharide with a complex pharmacologic mechanism of action that is effectively used to treat epilepsy and migraine headaches. TPM has been investigated for other indications including alcohol dependence, cocaine addiction, bipolar disorder, neuroprotection, binge eating disorder, post-traumatic stress disorder and obesity [16-22]. TPM has nearly ideal pharmacokinetics. This attribute combined with its complex pharmacodynamics translates into a drug with wide therapeutic utility [22]. While the molecular mechanisms of action for TPM are not fully understood, it has been confirmed that TPM has moderate potency in inhibiting carbonic anhydrase isozymes including CA9 [22,23]. Studies in mice with lung cancer have shown that TPM, like other CA9 inhibitors, has an inhibitory role in tumor progression, angiogenesis and metastases [24,25]. However, little is understood about the anti-tumorigenic effects of TPM in ovarian cancer. Thus, the aim of this study was to better understand the anti-tumorigenic potential of TPM in ovarian cancer by assessing the in vitro effect of TPM on proliferation, cell cycle progression, apoptosis, cellular stress and invasion in ovarian cancer cells.
Material and methods
Cell culture and reagents
The ovarian cancer cell lines, Hey, IGROV-1, OVCAR433, OVCAR5 and SKOV3, were used. The OVCAR433, OVCAR5 and SKOV3 were maintained in DMEM/F12 supplemented with 10% FBS, 300 mM L-glutamine, 5 μg/ml bovine insulin, 10,000 U/ml penicillin and 10,000 μg/ml streptomycin under 5% CO2. The Hey and IGROV-1 cells were cultured in RPMI 1640 with 5% FBS, 10,000 U/ml penicillin and 10,000 μg/ml streptomycin under 5% CO2. Topiramate, RNase and RIPA buffer were purchased from Sigma (St. Louis, MO). Antibodies to phosphorylated-S6 (Ser 235/236), phosphorylated-p42/44, phosphorylated-AKT, carbonic anhydrase IX (CA9), β-actin, pan-AKT, pan-p42/44 and pan-S6 were obtained from Cell Signaling Technology (Beverly, MA). The Annexin V FITC kit was purchased from BioVision (Mountain View, CA). Enhanced chemiluminescence western immunoblotting detection reagents were purchased from Amersham (Arlington Heights, IL). All other chemicals were purchased from Sigma.
Cell proliferation assay
The OVCAR5 and SKOV3 cells were plated and grown in 96-well plates at a concentration of 5000 cells/well for 24 hours. These cells were then treated with various concentrations of TPM for a period of 72 hours. After the addition of MTT dye (5 mg/mL), the 96-well plates were incubated for 1-2 hours at 37°C. 100 uL of DMSO was added to the plates in order to terminate the MTT reaction, and the plates were read by measuring absorption at 595 nm. The effect of TPM was calculated as a percentage of control cell growth obtained from PBS (1%) treated cells grown in the same 96-well plates. Each experiment was repeated three times to assess for consistency of results.
Colony formation assay
The OVCAR5 and SKOV3 cells growing in log phase were seeded (400 cells/well in a 6-well plate) in their standard growth medium. Cells were allowed to adhere for 24 hours, and then were treated with TPM for 24 hours. The cells were cultured at 37°C for 14 days with medium changes every third or fourth day. Cells were stained with 0.5% crystal violet, and colonies were counted under the microscope. Each experiment was repeated three times for consistency of results.
Cell cycle analysis
The effect of TPM on the cell cycle was measured using Cellometer (Nexcelom, Lawrence, MA, USA). Briefly, both cell lines were plated at 2.5 × 105 cells/well in 6-well plates and incubated overnight. Cells were then treated with TPM (from 1 to 1500 uM) for 24 hours. The cells were harvested and washed with PBS and centrifuged. The pellet was re-suspended and fixed in 90% pre-chilled methanol and stored at -20°C overnight. The cells were then washed with PBS and centrifuged, resuspended in 50 μl RNase A solution (250 μg/ml, 10 mM EDTA) followed by incubation for 30 minutes at 37°C. The cells were then stained with 50 μl of staining solution [containing 2 mg/ml Propidium iodide (Hayward, MA, USA), 0.1 mg/ml Azide (Sigma) and 0.05% Triton X-100 (Sigma)]. The final mixture was incubated for 15 minutes in the dark before being analyzed by Cellometer. The results were analyzed using FCS4 express software (Molecular Devices, Sunnyvale, CA). All experiments were performed in triplicate to assess for consistency of results.
Annexin V assay
The effect of TPM on apoptosis was assessed with the Annexin V FITC kit (Biolegend, San Diego, CA) and via Cellometer. Briefly, 2 × 105 cells/well were seeded into 6-well plates, incubated overnight and then treated with TPM with varying concentrations for 24 hours. The cells were then collected, washed with PBS and re-suspended in 100 ul of Annexin V and PI dual-stain solution (0.1 ug of Annexin V FITC and 1 μg of PI) for 15 min in the dark. Cells were then analyzed by Cellometer. The results were analyzed by FCS4 express software. All experiments were performed in triplicate to assess for consistency of response.
Reactive oxygen species (ROS) assay
The Fluorimetric Intracellular Total ROS Activity Assay Kit (AAT Bioquest) was used to detect alterations in the production of reactive oxygen species caused by TPM. Briefly, the OVCAR5 and SKOV3 cells (1.0 × 104 cells/well) were seeded into black 96-well plates. After 24 hours, the cells were treated with TPM and allowed to incubate overnight to induce ROS generation. DCFH-DA (20 μM) was then applied and allowed to incubate for 30 minutes. The fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a plate reader (Tecan). All experiments were performed in duplicate to assess for consistency of response.
Adhesion assay
Each well in a 96-well plate was coated with 100 ul laminin-1 (10 ug/ml) and incubated at 37°C for 1 hour. This fluid was then aspirated and 200 ul of blocking buffer was added to each well for 45-60 min at 37°C. The wells were then washed with PBS and the plate was allowed to chill on ice. To each well, 2.5 × 104 cells were added. PBS and varying concentrations of TPM were then added directly. The plate was then allowed to incubate at 37°C for 2 hours. After this period, the medium was aspirated and cells were fixed by directly adding 100 ul of 5% glutaraldehyde and incubating for 30 min at room temperature. Adherent cells were then washed with PBS and stained with 100 ul of 0.1% crystal violet for 30 minutes. The cells were then washed repeatedly with water, and 100 ul of 10% acetic acid was added to each well to solubilize the dye. After 5 minutes of shaking, the absorbance was measured at 570 nm using a micro-plate reader from Tecan. Each experiment was repeated three times for consistency of results.
Invasion assay
Cell invasion assays were performed using 96-well HTS transwells (Corning Life Sciences, Tewksbury, MA) coated with 0.5-1X BME (Trevigen Inc., Gaithersburg, MD). The OVCAR5 and SKOV3 cells (50,000/well) were starved for 12 hours and then seeded in the upper chambers of the wells in 50 μl FBS-free medium. The lower chambers were filled with 150 μl standard medium with TPM. The plate was incubated for 24 hours at 37°C to allow invasion into the lower chamber. After washing the upper and lower chambers with PBS once, 100 ul Calcein AM solution was added into lower chamber and incubated at 37°C for 30-60 minutes. The lower chamber plate was measured using a Tecan plate reader for reading fluorescence at EX/EM 485/520 nm. Each experiment was repeated three times for consistency of results.
Western blot analysis
Following treatment of cells with varying concentrations of TPM for 24 hours, total protein was extracted from the IGROV-1 and SKOV3 cells using RIPA buffer (Boston Bioproducts, Ashland, MA). Equal amounts (30 μg) of total protein were loaded and separated by 10-12% SDS-PAGE, then transferred to a PVDF membrane, blocked in 5% non-fat milk and incubated with a 1:1000 dilution of primary antibody at 4°C overnight. The membranes were then washed and incubated with the appropriate secondary antibodies for 1 hour at room temperature before development. The bands were developed and quantified using an Alpha Innotech Imaging System (San Leandro, CA, USA). After developing, the membranes were stripped or washed and re-probed using antibodies against total p42/44, AKT or pan-S6 and α-tubulin (for all proteins), respectively. Each experiment was repeated at least twice for consistency of results. α-Tubulin was used as the endogenous control.
Statistical analysis
Results were compared by Student’s t test, and data were expressed as mean ± S.E. Statistical significance was defined to be P < 0.05.
Results
TPM inhibits ovarian cancer cell proliferation
In order to determine the sensitivity of ovarian cancer cells to TPM, five ovarian cancer cell lines were treated with 0.01 to 3000 uM of TPM for 72 hours. Cell proliferation was determined by MTT assay and IC50 values were calculated from the dose response curves. TPM significantly inhibited cell proliferation in a dose-dependent fashion in all ovarian cancer cell lines. The IC50 values were in the range of 1.4 to 2.2 uM for the five cell lines (Figure 1A).
Figure 1.
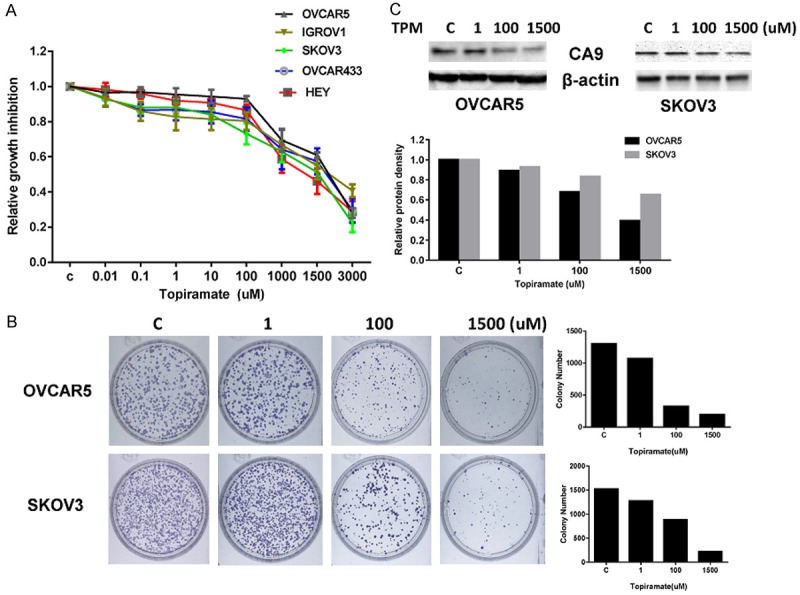
Effect of TPM on cell proliferation in ovarian cancer cells. The five ovarian cell lines were cultured in the presence of varying concentrations of TPM for 72 hours. Cell proliferation was determined by MTT assay (A). TPM inhibited cell proliferation in the five ovarian cancer cell lines. TPM significantly reduced colony formation in the OVCAR5 and SKOV3 cells after 14 days of treatment (B). Western blotting results showed that TPM decreased CA9 protein expression in a dose-dependent manner after 24 hours of treatment in the OVCAR5 and SKOV3 cells (C). Protein lysates (30 ug) were resolved by 12 % SDS-PAGE. β-actin was used as the loading control. Each experiment was performed three times. The results are shown as the mean ± SE of triplicate samples and are representative of three independent experiments.
Given that the colony formation assay is an excellent indicator of long term tumor cell survival, a colony formation assay was performed to investigate the long-term effect of TPM on cell growth in the OVCAR5 and SKOV3 cells. As shown in Figure 1B, the colony-forming ability of OVCAR5 and SKOV3 cells was reduced by 83.2% and 85.8%, respectively, after exposure to 1500 uM of TPM for 14 days. To test the effect of TPM on carbonic anhydrase isozymes, we assessed the effect of TPM on carbonic anhydrase 9 in the OVCAR5 and SKOV3 cells. Figure 1C demonstrates that TPM reduced the expression of CA9 in a dose- dependent manner in both cells as determined by Western blotting after 24 hours of treatment. These results suggest that TPM has inhibitory effects on the proliferation of ovarian cancer cells.
TPM induces cell cycle arrest in ovarian cancer cells
In order to evaluate the changes in cell cycle progression induced by TPM, an analysis of the different phases of the cell cycle after treatment with TPM was performed in the ovarian cancer cell lines. After 24 hours of exposure to TPM (1.5 mM), significant cell cycle G1 phase arrest was demonstrated in the ovarian cancer cells compared to controls (Figure 2A). G1 arrest increased from 48% in control cells to 57% in the TPM-treated OVCAR5 cells and 41 to 53% in the SKOV3 cells. In addition to an increased proportion of cells in G1 phase, we noted a concomitant decreased proportion of cells in G2 phase. Western blotting found that TPM treatment caused reduced protein expression of CDK4, CDK6 and cyclin D1 (Figure 2B). Although little is known regarding the effect of TPM on cell cycle progression, our data suggests that TPM induces changes in cell cycle progression through its effects on critical checkpoints in ovarian cancer cells.
Figure 2.
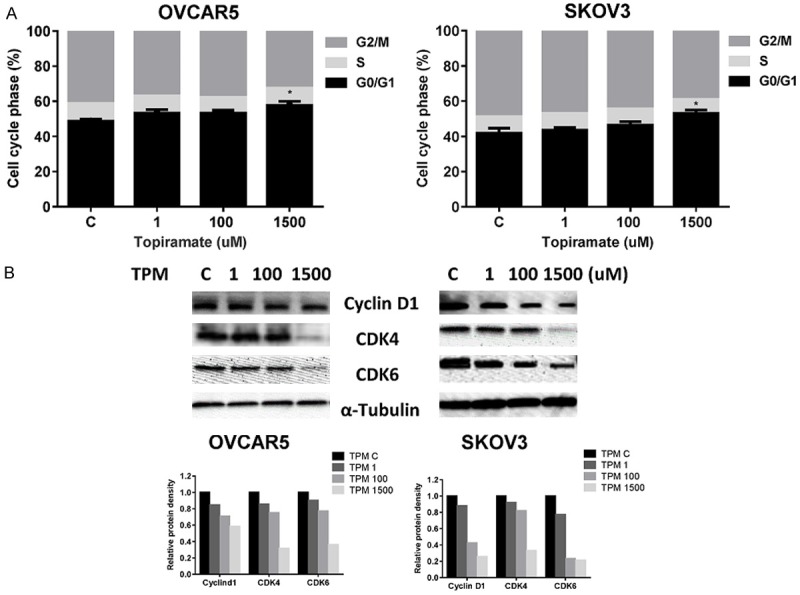
Effect TPM on cell cycle progression in ovarian cancer cells. The OVCAR5 and SKOV3 cells were treated with TPM at different doses for 24 hours. The effect of TPM on the distribution of cells in different phases of the cell cycle was analyzed by Cellometer. TPM induced cell cycle arrest in G1 phase in both cells (A). The OVCAR5 and SKOV3 cells were grown for 24 hours and then treated with TPM at the indicated concentrations for an additional 24 hours. Western blot analysis of the expression of the cell cycle related protein CDK4, CDK6 and cyclin D in both cells after being incubated with TPM. The results showed that TPM reduced the expression of CDK4, CDK6 and cyclin D in both cell lines (B). The results are shown as the mean ± SD and are representative of three independent experiments. (*P < 0.05).
TPM induces apoptosis in ovarian cancer cells
In order to determine whether the reduction of cell viability was due to increased apoptosis, we detected apoptotic cells by performing an Annexin-V and PI double staining assay. As shown in Figure 3A, the percentage of OVCAR5 and SKOV3 cells undergoing apoptosis significantly increased in a dose-dependent manner after 24 hours of TPM treatment when compared to controls. We next treated both cell lines with TPM for 18 hours, and found that TPM decreased the expression of BCL-1 and MCL-1 in a dose-dependent manner (Figure 3B). These results suggest that induction of apoptosis by TPM is one mechanism through which TPM inhibits cell proliferation.
Figure 3.
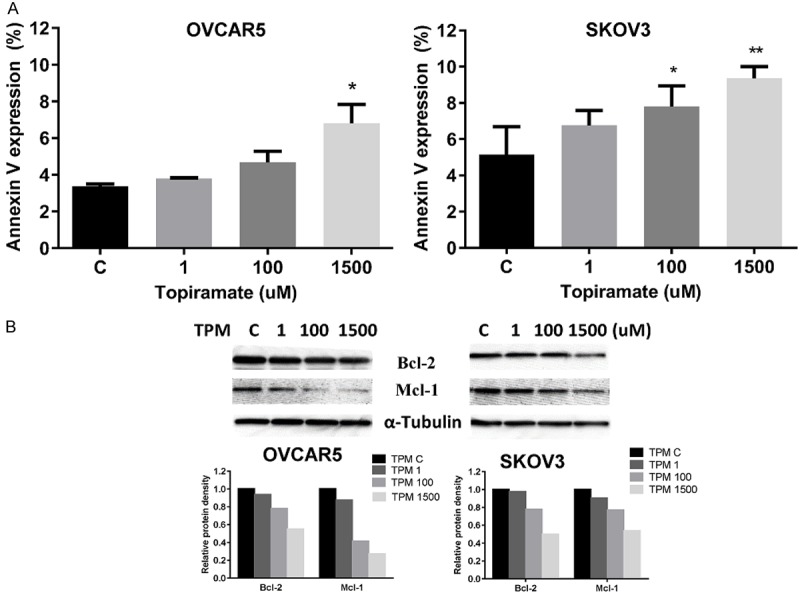
Effect TPM on apoptosis in ovarian cancer cells. The OVCAR5 and SKOV3 cells were treated with TPM for 24 hours. Quantification of apoptotic cells was detected using the Annexin-V FITC assay. TPM significantly increased Annexin V expression in both cell lines (A). Western blotting indicated treatment with TPM for 24 hours decreased the expression of the anti-apoptotic proteins, BCL-2 and MCL-1 (B). Protein lysates (30 ug) were resolved in 12% SDS-PAGE gels. β-actin was used as the loading control. Data are shown as mean + SEM of two experiments (*P < 0.05, **P < 0.01).
TPM induces cell stress in ovarian cancer cells
To investigate the involvement of oxidative stress in the anti-proliferative effect of TPM, intracellular ROS levels were examined by using the ROS fluorescence indicator DCF-DA. Treatment with TPM for 18 hours significantly increased ROS production in a dose-dependent manner in both the OVCAR5 and SKOV3 cells as seen in Figure 4A. The difference in ROS level between the TPM dose of 100 and 1500 μM and control was statistically different in both cell lines. Specifically, ROS production was increased by 12% in the OVCAR5 cells at a dose of 100 uM and by 19 % in the SKOV3 cells at a dose of 1500 uM.
Figure 4.
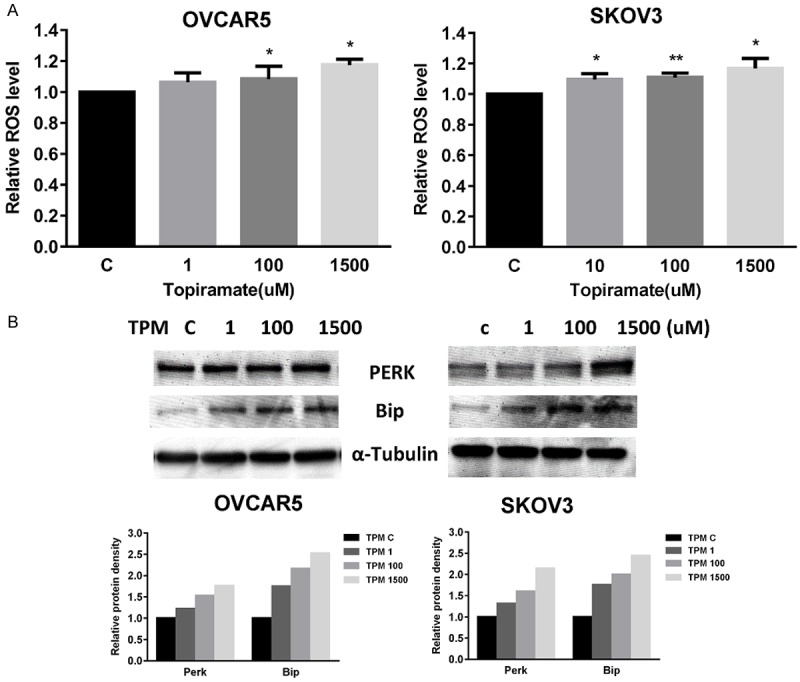
TPM induced cellular stress in ovarian cancer cells. The production of reactive oxygen species (ROS) in ovarian cancer cells was determined after incubating TPM-treated PC cells with 20,70-Dichlorofluorescein diacetate (DCFH-DA). TPM induced ROS production in a dose-dependent manner after 24 hours of treatment in OVCAR5 and SKOV3 cells (A). The expression of PERK and Bip after treatment of TPM for 24 hours was determined by western blotting. TPM induced PERK and Bip expression in a dose dependent manner (B). Data are shown as mean + SEM of two experiments. (*P < 0.05, **P < 0.01).
We next examined expression of cellular stress proteins following treatment with TPM using Western immunoblotting. As expected from our ROS findings, TPM increased the expression of the PERK and BIP proteins in a dose-dependent fashion after 24 hours of treatment (Figure 4B). These results suggest that the inhibition of cell proliferation by TPM in ovarian cancer cell lines also involves induction of cellular stress.
TPM inhibits adhesion and invasion in ovarian cancer cells
The effect of TPM on the migration of ovarian cancer cells was analyzed by adhesion and invasion assays. The OVCAR5 and SKOV3 cells treated with TPM had a reduced ability to adhere to laminin-1, as compared to control cells (Figure 5A). Similarly, the migratory capacity of the ovarian cancer cell lines was also reduced after treatment with TPM for 2 hours, as evaluated by a transwell assay (Figure 5B). Cell adhesion and invasion are mediated by a variety of membrane proteins as well as modulation of cytoskeletal assembly. To further analyze the effect of TPM on motility and migration of ovarian cancer cells, the expression levels of E-cadherin, a cell-cell adhesion glycoprotein that acts as an invasion suppressor, and Slug and Snail, proteins involved in epithelial-mesenchymal transitions and the regulation of E-cadherin, as well as Vimentin and B-catenin, were analyzed by Western blot analysis (Figure 5C). After 24 hours of treatment, TPM reduced Slug and Snail expression and increased E-cadherin expression in both cell liens. However, TPM decreased B-catenin and vimentin expression in the OVCAR5 cells and increased the expression of B-catenin and vimentin in the SKOV3 cells, suggesting that TPM inhibited cell invasion through different mechanisms in each cell line.
Figure 5.
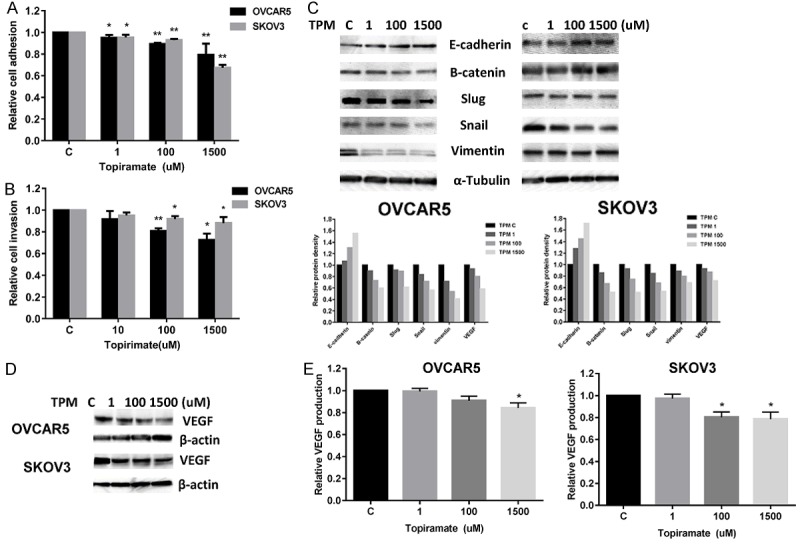
TPM inhibited adhesion and invasion in ovarian cancer cells. The OVCAR5 and SKOV3 cells were treated with varying concentrations of TPM and plated in laminin-coated 96-well plates for 2 hours. The effect of TPM on adhesion was assessed by a laminin-1 adhesion assay (A). Invasion was assessed using transwell invasion kits after 24 hours of treatment with TPM (B). TPM inhibited adhesion and invasion in the both cell lines. Western blotting results showed that TPM modulated expression of proteins related to migration/motility after 24 hours of treatment (C). In addition, VEGF expression in both cell lines was also assessed by western blot analysis (D). TPM reduced the production of VEGF in the media of OVCAR5 and SKOV3 cells after 24 hours of treatment (E). The results are shown as the mean ± SD and are representative of three independent experiments. (*P < 0.05, **P < 0.01).
We next investigated the effect of TPM on VEGF, a pro-angiogenic factor responsible for the migration and invasion of ovarian cancer cells. Western blotting results showed that TPM reduced the expression of VEGF in a dose-dependent manner after 24 hours of treatment (Figure 5D). The production of VEGF secretion in serum-free culture media was determined by ELISA assay. TPM notably decreased VEGF secretion in the media compared with controls in the OVCAR5 and SKOV3 cells (Figure 5E). Together, these results further support the role of TPM in the inhibition of angiogenesis and invasion in ovarian cancer cells.
Effect of TPM on the AKT/mTOR and MAPK pathways
The MAPK and mTOR pathways are involved in cellular growth regulation and survival and contribute to therapy induced tumor-growth suppression in ovarian cancer. To investigate the mechanisms underlying the inhibition of cell growth by TPM, we treated the cells with TPM at varying concentrations for 24 hours and evaluated the effect of different concentrations of TPM on the mTOR and MAPK pathways. TPM decreased phosphorylation of ribosomal protein S6 and AKT in a dose-dependent manner (Figure 6). Meanwhile, we also observed that TPM was able to reduce the level of phosphorylation of p42/44 in both ovarian cancer cell lines after 24 hours of treatment. These data suggest that TPM exerts its anti-tumorigenic activity via inhibition of the AKT/mTOR and MAPK signaling pathways in ovarian cancer cells.
Figure 6.
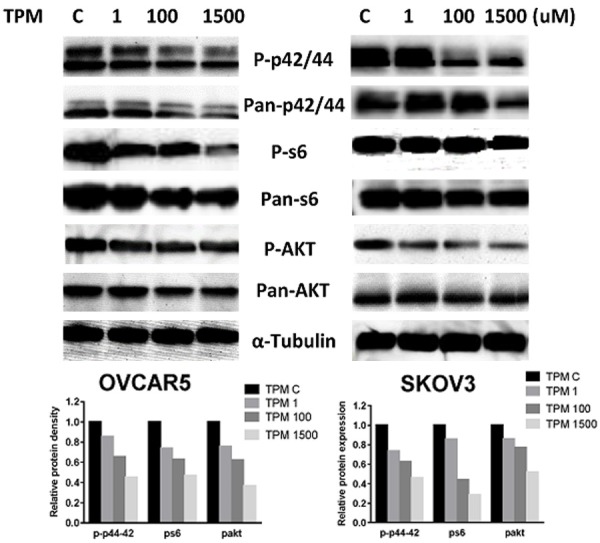
Effect of TPM on the AKT/mTOR/S6 and MAPK pathways in ovarian cancer cells. The OVCAR5 and SKOV3 cells were treated with TPM for 24 hours. Phosphorylated-AKT, pan-AKT, phosphorylated-S6, pan-S6, phosphorylated-p42/44 and pan-p42/44 were detected by Western immunoblotting. TPM decreased phosphorylation of AKT, S6 and p42/44 in both cell lines. The results are one of three independent experiments.
Discussion
TPM is an anti-epileptic drug with a broad spectrum of other clinical applications including the treatment of obesity, analgesia management, mood stabilization and cocaine addiction. In addition, TPM may have anti-tumorigenic effects via inhibition of CA9. However, to date few studies have explored the potential anti-tumorigenic activity of this drug [21]. In this study, we used ovarian cancer cell lines to explore the role of inhibition of CA9 by TPM in cancer cell growth. We found that TPM inhibited ovarian cancer cell proliferation, caused cell cycle G1 arrest and induced apoptosis in a dose-dependent manner. These findings were seen in parallel with decreased expression of CA9 in the ovarian cancer cells. Inhibition of CA9 by TPM resulted in increased cellular stress and inhibition of the MAPK and AKT/mTOR pathways. Moreover, TPM significantly decreased the ability of adhesion and invasion and reduced the production of VEGF in the ovarian cancer cells. Hence, these results suggest that TPM is an effective anti-tumorigenic and anti-metastatic agent, and that targeting and inhibiting CA activity may serve as a promising therapeutic strategy for the treatment of ovarian cancer.
CAs are important for the interconversion between carbon dioxide and bicarbonate. They are involved in crucial physiological processes related to the maintenance of pH and bicarbonate homeostasis. CAs also play a role in electrolyte secretion, biosynthetic reactions, bone metabolism, respiration, calcification and tumorigenesis [26]. Among the many isoforms of CA, CA9 and CA12 appear to be unique with respect to their association with different types of cancer. A recent meta-analysis showed that high expression of CA9 is an adverse prognostic marker and is associated with a higher risk of developing metastases in most solid tumors [27]. In ovarian cancer, mucinous and endometrioid carcinomas exhibited higher CA9 expression levels more frequently than serous and clear cell carcinomas [28]. Overexpression of CA9 was significantly associated with nuclear grade, tumor necrosis and poor prognosis [8,28,29]. Knockdown of CA9 expression by siRNA has shown to delay cell growth and to reduce primary tumor growth and proliferation in breast cancer models [30-32]. Reduction of CA9 activity by small molecule inhibitors significantly induced apoptosis and led to a significant increase in cell cycle G1 phase with a corresponding reduction in S phase in colon cancer and osteosarcoma cells [32-34]. TPM, as a potent inhibitor of CA2 and CA9, significantly inhibited primary tumor growth in C57BL/6 mice with Lewis lung carcinoma and reduced CA9 and CA2 mRNA expression in primary tumor tissues [25]. A recent study also found that TPM arrested the cell cycle in G1 phase and induced apoptosis in glioblastoma cells accompanied by significant downregulation in the expression of HSP70 and NFκB [35]. Our data found that the downregulation of CA9 induced by TPM in ovarian cancer cells resulted in inhibition of cell proliferation. Both cell cycle G1 phase arrest and an increased number of apoptotic cells were found in TPM-treated cells as compared to controls. This data suggests an inhibitory role for TPM in ovarian cancer cell growth and provides support for CA9 as an attractive target for anti-cancer therapies [8,27,31].
It has been established that TPM is capable of modulating the oxidant-antioxidant system [36]. There are some reports of the interactions between oxidative stress and TPM in tumor cells. Most of the available research of the antioxidant role of TPM is in brain and neurological cells [36]. Cardile et al reported that therapeutic range of TPM increases oxidative stress in astrocytes [37]. These studies suggest that there are both positive and negative regulatory effects of TPM on the antioxidant system [37,38]. In the current study, we found that TPM increased cellular ROS levels and induced the expression of the cellular stress proteins BIP and PERK after 24 hours of treatment, signifying that cellular stress contributed to a decrease in proliferation of ovarian cancer cells in addition to apoptosis and cell cycle G1 arrest.
The progression and invasion of ovarian cancer is thought to be the consequence of a series of unique biological events within the tumor microenvironment, in part due to aberrant metabolism of glucose and significantly higher amounts of lactic acid which induce extracellular acidification and leads to the breakdown of the extracellular matrix [39]. CA9 contributes to extracellular acidification by elevating the extracellular levels of carbon dioxide and protons as well as via a physical function to disrupt intercellular connections through competition with E-cadherin in binding with β-catenin [40,41]. The overexpression of exogenous human CA9 induces weakening of cell adhesions and augments cell motility by aberrant Rho-GTPase signal transduction in cervical cancer cells [40]. CA9 activity has also been linked to angiogenic pathways, and CA9 knockdown has been shown to enhance anti-VEGF therapy in vivo [28,33,42]. Furthermore, multivariate analysis revealed that mRNA expression of CA9 was strongly associated with the development of distant metastases in patients with cervical cancer [43]. Interestingly, extracellular acidification induced by CA9 is mandatory to elicit activation of stromal fibroblast delivered metalloprotease 2 and 9. Targeting CA9 is sufficient to impede epithelial-mesenchymal transition (EMT) and invasion in prostate cancer cells [44], which suggests the role of CA9 as a novel mechanism by which cancer-associated fibroblasts promote metastasis [45,46]. The combination of high expression of CA9 and VEGF was significantly associated with increased resistance to chemotherapy and poor overall survival in ovarian high-grade serous carcinoma, indicating that targeting of both CA9 and VEGF may be more effective than inhibiting either CA9 or VEGF alone [14]. TPM has been shown to inhibit angiogenesis by downregulation of VEGF, osteopontin, CA2 and CA9 in mice with Lewis lung carcinoma [24,25]. Treating ovarian cancer cells with TPM for 24 hours in our study resulted in decreased adhesion and invasion capabilities and was accompanied by decreased expression of Slug and Snail and increased expression of E-cadherin. In addition, we observed a significant decrease in VEGF levels in the cell culture media and cultured cells after treatment of TPM, as shown by ELISA assay and western blotting. The decrease in tumor-associated production of VEGF by TPM could be one of the major mechanisms by which it reduces angiogenesis in ovarian cancer cells.
Activation of the PI3K/AKT/mTOR and MAPK signaling pathways occurs frequently in ovarian cancer tumors and plays an integral role in mediating cell-cycle progression, cell survival, cell motility and angiogenesis [47,48]. The MAPK and AKT pathways are able to regulate CA9 activity induced by high cell density and hypoxia [28]. Furthermore, the MAPK pathway contributes to regulation of CA9 expression by modulating the activity of the transcription factor SP1 and p300, the transcriptional co-activator of HIF-1. Inhibition of MAPK by U0126 resulted in downregulation of HRE and PR1, two critical regulatory elements in the CA9 promoter [49]. The AKT pathway likely contributes to the activity of CA9 at the transcriptional level through reduced HIF-1 activity, which makes the effects cell-type specific [50]. Targeting mTOR activity by rapamycin significantly reduced the miRNA expression of CA9 in tumor cells in vitro [51]. Our results show that the MAPK and AKT/mTOR pathways were inhibited by TPM in a dose-dependent manner in both cell lines but to various extents. The effect of TPM on these signaling pathways suggest that the activity of CA9, with its role as a cross-talk-mediator with the AKT/mTOR and MAPK pathways, has a crucial role in ovarian cancer cell survival. Thus, TPM inhibits cell proliferation via multiple signaling pathways in ovarian cancer cells.
Currently, targeting CA9 with small molecule inhibitors or monoclonal antibodies is being studied clinically for use in the treatment of cancer and as adjuvant therapy in combination with cytotoxic agents [13,34,52,53]. The combination of CA9 inhibitors with conventional chemotherapy may yield even better efficacy than inhibition of CA9 alone [54]. Inhibition of CA9 with TPM is efficacious in decreasing tumor growth and inhibiting metastasis in preclinical tumor models without significant toxicity [25]. Given that there is wild-type expression of CA9 in ovarian cancer tissues and minimal expression of CA9 in normal tissues, and that CA9 is located on the external interface of tumor cells; CA9 may be an ideal clinical target for this disease [29]. Although fatigue, ataxia, dizziness, impairment of memory and attention, paraesthesias and somnolence have been associated with TPM use in epilepsy, the side effects of TPM are generally well tolerated, especially when compared with side effects of standard of cytotoxic treatments for ovarian cancer. Furthermore, the combination of TPM and phentermine has recently being approved by the FDA for the treatment of obesity, suggesting TPM is a safe medication for a long-term use [16,21]. Given that CA9 has been linked to chemotherapy and radiation resistance for several cancers, inhibition of CA9 may increase sensitivity to chemotherapy and radiation, particularly when used in combination with other agents [12,31,55,56]. Overall, the promising pre-clinical and clinical data make TPM a novel agent with promising potential in ovarian cancer treatment and certainly worthy of further investigation in clinical trials for ovarian cancer.
Acknowledgements
This work was generously supported by the She Rocks, the Steelman fund and National Natural Science Foundation of China (11672192).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Kritsch D, Hoffmann F, Steinbach D, Jansen L, Mary Photini S, Gajda M, Mosig AS, Sonnemann J, Peters S, Melnikova M, Thomale J, Durst M, Runnebaum IB, Hafner N. Tribbles 2 mediates cisplatin sensitivity and DNA damage response in epithelial ovarian cancer. Int J Cancer. 2017;141:1600–1614. doi: 10.1002/ijc.30860. [DOI] [PubMed] [Google Scholar]
- 3.Siddiqui MK, Tyczynski J, Pahwa A, Fernandes AW. Objective response rate is a possible surrogate endpoint for survival in patients with advanced, recurrent ovarian cancer. Gynecol Oncol. 2017;146:44–51. doi: 10.1016/j.ygyno.2017.03.515. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Martin A, du Bois A. Factors to consider and questions to ask in the management of recurrent ovarian cancer: a focus on the role of trabectedin + pegylated liposomal doxorubicin. Expert Rev Anticancer Ther. 2016;16:3–10. doi: 10.1080/14737140.2016.1243477. [DOI] [PubMed] [Google Scholar]
- 5.Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov. 2017;12:61–88. doi: 10.1080/17460441.2017.1253677. [DOI] [PubMed] [Google Scholar]
- 6.Supuran CT. Carbonic anhydrases--an overview. Curr Pharm Des. 2008;14:603–614. doi: 10.2174/138161208783877884. [DOI] [PubMed] [Google Scholar]
- 7.Akocak S, Alam MR, Shabana AM, Sanku RK, Vullo D, Thompson H, Swenson ER, Supuran CT, Ilies MA. PEGylated bis-sulfonamide carbonic anhydrase inhibitors can efficiently control the growth of several carbonic anhydrase IX-expressing carcinomas. J Med Chem. 2016;59:5077–5088. doi: 10.1021/acs.jmedchem.6b00492. [DOI] [PubMed] [Google Scholar]
- 8.Hynninen P, Vaskivuo L, Saarnio J, Haapasalo H, Kivela J, Pastorekova S, Pastorek J, Waheed A, Sly WS, Puistola U, Parkkila S. Expression of transmembrane carbonic anhydrases IX and XII in ovarian tumours. Histopathology. 2006;49:594–602. doi: 10.1111/j.1365-2559.2006.02523.x. [DOI] [PubMed] [Google Scholar]
- 9.Eom KY, Jang MH, Park SY, Kang EY, Kim SW, Kim JH, Kim JS, Kim IA. The expression of carbonic Anhydrase (CA) IX/XII and lymph node metastasis in early breast cancer. Cancer Res Treat. 2016;48:125–132. doi: 10.4143/crt.2014.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vullo D, Innocenti A, Nishimori I, Pastorek J, Scozzafava A, Pastorekova S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the transmembrane isozyme XII with sulfonamides-a new target for the design of antitumor and antiglaucoma drugs? Bioorg Med Chem Lett. 2005;15:963–969. doi: 10.1016/j.bmcl.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 11.Logsdon DP, Grimard M, Luo M, Shahda S, Jiang Y, Tong Y, Yu Z, Zyromski N, Schipani E, Carta F, Supuran CT, Korc M, Ivan M, Kelley MR, Fishel ML. Regulation of HIF1alpha under Hypoxia by APE1/Ref-1 impacts CA9 expression: dual targeting in patient-derived 3D pancreatic cancer models. Mol Cancer Ther. 2016;15:2722–2732. doi: 10.1158/1535-7163.MCT-16-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tafreshi NK, Lloyd MC, Bui MM, Gillies RJ, Morse DL. Carbonic anhydrase IX as an imaging and therapeutic target for tumors and metastases. Subcell Biochem. 2014;75:221–254. doi: 10.1007/978-94-007-7359-2_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget. 2012;3:84–97. doi: 10.18632/oncotarget.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams E, Martin S, Moss R, Durrant L, Deen S. Co-expression of VEGF and CA9 in ovarian high-grade serous carcinoma and relationship to survival. Virchows Arch. 2012;461:33–39. doi: 10.1007/s00428-012-1252-9. [DOI] [PubMed] [Google Scholar]
- 15.Gieling RG, Williams KJ. Carbonic anhydrase IX as a target for metastatic disease. Bioorg Med Chem. 2013;21:1470–1476. doi: 10.1016/j.bmc.2012.09.062. [DOI] [PubMed] [Google Scholar]
- 16.Donegan S, Dixon P, Hemming K, Tudur-Smith C, Marson A. A systematic review of placebo-controlled trials of topiramate: how useful is a multiple-indications review for evaluating the adverse events of an antiepileptic drug? Epilepsia. 2015;56:1910–1920. doi: 10.1111/epi.13209. [DOI] [PubMed] [Google Scholar]
- 17.Goh ET, Morgan MY. Review article: pharmacotherapy for alcohol dependence - the why, the what and the wherefore. Aliment Pharmacol Ther. 2017;45:865–882. doi: 10.1111/apt.13965. [DOI] [PubMed] [Google Scholar]
- 18.Pigott K, Galizia I, Vasudev K, Watson S, Geddes J, Young AH. Topiramate for acute affective episodes in bipolar disorder in adults. Cochrane Database Syst Rev. 2016;9:CD003384. doi: 10.1002/14651858.CD003384.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh M, Keer D, Klimas J, Wood E, Werb D. Topiramate for cocaine dependence: a systematic review and meta-analysis of randomized controlled trials. Addiction. 2016;111:1337–1346. doi: 10.1111/add.13328. [DOI] [PubMed] [Google Scholar]
- 20.McElroy SL, Guerdjikova AI, Mori N, Munoz MR, Keck PE. Overview of the treatment of binge eating disorder. CNS Spectr. 2015;20:546–556. doi: 10.1017/S1092852915000759. [DOI] [PubMed] [Google Scholar]
- 21.Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat. 2013;23:725–735. doi: 10.1517/13543776.2013.790957. [DOI] [PubMed] [Google Scholar]
- 22.Shank RP, Maryanoff BE. Molecular pharmacodynamics, clinical therapeutics, and pharmacokinetics of topiramate. CNS Neurosci Ther. 2008;14:120–142. doi: 10.1111/j.1527-3458.2008.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winum JY, Scozzafava A, Montero JL, Supuran CT. Sulfamates and their therapeutic potential. Med Res Rev. 2005;25:186–228. doi: 10.1002/med.20021. [DOI] [PubMed] [Google Scholar]
- 24.Ma B, Xiang Y, Li T, Yu HM, Li XJ. Inhibitory effect of topiramate on Lewis lung carcinoma metastasis and its relation with AQP1 water channel. Acta Pharmacol Sin. 2004;25:54–60. [PubMed] [Google Scholar]
- 25.Ma B, Pan Y, Song Q, Tie L, Zhang Y, Xiao Y, Zhang J, Han J, Xu Y, Xiang Y, Yu HM, Li XJ. The effect of topiramate on tumor-related angiogenesis and on the serum proteome of mice bearing Lewis lung carcinoma. Eur J Pharmacol. 2011;663:9–16. doi: 10.1016/j.ejphar.2011.04.056. [DOI] [PubMed] [Google Scholar]
- 26.Winum JY, Vullo D, Casini A, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozymes I and II and transmembrane, tumor-associated isozyme IX with sulfamates including EMATE also acting as steroid sulfatase inhibitors. J Med Chem. 2003;46:2197–2204. doi: 10.1021/jm021124k. [DOI] [PubMed] [Google Scholar]
- 27.van Kuijk SJ, Yaromina A, Houben R, Niemans R, Lambin P, Dubois LJ. Prognostic significance of carbonic anhydrase IX expression in cancer patients: a meta-analysis. Front Oncol. 2016;6:69. doi: 10.3389/fonc.2016.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choschzick M, Oosterwijk E, Muller V, Woelber L, Simon R, Moch H, Tennstedt P. Overexpression of carbonic anhydrase IX (CAIX) is an independent unfavorable prognostic marker in endometrioid ovarian cancer. Virchows Arch. 2011;459:193–200. doi: 10.1007/s00428-011-1105-y. [DOI] [PubMed] [Google Scholar]
- 29.Kim K, Park WY, Kim JY, Sol MY, Shin DH, Park DY, Lee CH, Lee JH, Choi KU. Prognostic relevance of the expression of CA IX, GLUT-1, and VEGF in ovarian epithelial cancers. Korean J Pathol. 2012;46:532–540. doi: 10.4132/KoreanJPathol.2012.46.6.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robertson N, Potter C, Harris AL. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res. 2004;64:6160–6165. doi: 10.1158/0008-5472.CAN-03-2224. [DOI] [PubMed] [Google Scholar]
- 31.Mahon BP, Pinard MA, McKenna R. Targeting carbonic anhydrase IX activity and expression. Molecules. 2015;20:2323–2348. doi: 10.3390/molecules20022323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Tsoi H, Li X, Wang H, Gao J, Wang K, Go MY, Ng SC, Chan FK, Sung JJ, Yu J. Carbonic anhydrase IV inhibits colon cancer development by inhibiting the Wnt signalling pathway through targeting the WTAP-WT1-TBL1 axis. Gut. 2016;65:1482–1493. doi: 10.1136/gutjnl-2014-308614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIntyre A, Patiar S, Wigfield S, Li JL, Ledaki I, Turley H, Leek R, Snell C, Gatter K, Sly WS, Vaughan-Jones RD, Swietach P, Harris AL. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin Cancer Res. 2012;18:3100–3111. doi: 10.1158/1078-0432.CCR-11-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perut F, Carta F, Bonuccelli G, Grisendi G, Di Pompo G, Avnet S, Sbrana FV, Hosogi S, Dominici M, Kusuzaki K, Supuran CT, Baldini N. Carbonic anhydrase IX inhibition is an effective strategy for osteosarcoma treatment. Expert Opin Ther Targets. 2015;19:1593–1605. doi: 10.1517/14728222.2016.1086339. [DOI] [PubMed] [Google Scholar]
- 35.Kaur T, Manchanda S, Saini V, Lakhman SS, Kaur G. Efficacy of Anti-epileptic drugs in the treatment of tumor and its associated epilepsy: an in vitro perspective. Ann Neurosci. 2016;23:33–43. doi: 10.1159/000443554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naziroglu M, Yurekli VA. Effects of antiepileptic drugs on antioxidant and oxidant molecular pathways: focus on trace elements. Cell Mol Neurobiol. 2013;33:589–599. doi: 10.1007/s10571-013-9936-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cardile V, Pavone A, Renis M, Maci T, Perciavalle V. Effects of Gabapentin and Topiramate in primary rat astrocyte cultures. Neuroreport. 2001;12:1705–1708. doi: 10.1097/00001756-200106130-00037. [DOI] [PubMed] [Google Scholar]
- 38.Cardenas-Rodriguez N, Coballase-Urrutia E, Rivera-Espinosa L, Romero-Toledo A, Sampieri A 3rd, Ortega-Cuellar D, Montesinos-Correa H, Floriano-Sanchez E, Carmona-Aparicio L. Modulation of antioxidant enzymatic activities by certain antiepileptic drugs (valproic acid, oxcarbazepine, and topiramate): evidence in humans and experimental models. Oxid Med Cell Longev. 2013;2013:598493. doi: 10.1155/2013/598493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen M, Petignat P. Purified autoantibodies against glucose-regulated protein 78 (GRP78) promote apoptosis and decrease invasiveness of ovarian cancer cells. Cancer Lett. 2011;309:104–109. doi: 10.1016/j.canlet.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Shin HJ, Rho SB, Jung DC, Han IO, Oh ES, Kim JY. Carbonic anhydrase IX (CA9) modulates tumor-associated cell migration and invasion. J Cell Sci. 2011;124:1077–1087. doi: 10.1242/jcs.072207. [DOI] [PubMed] [Google Scholar]
- 41.Svastova E, Zilka N, Zat’ovicova M, Gibadulinova A, Ciampor F, Pastorek J, Pastorekova S. Carbonic anhydrase IX reduces E-cadherinmediated adhesion of MDCK cells via interaction with beta-catenin. Exp Cell Res. 2003;290:332–345. doi: 10.1016/s0014-4827(03)00351-3. [DOI] [PubMed] [Google Scholar]
- 42.Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, Harris AL. Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res. 2001;61:7992–7998. [PubMed] [Google Scholar]
- 43.Kim JY, Shin HJ, Kim TH, Cho KH, Shin KH, Kim BK, Roh JW, Lee S, Park SY, Hwang YJ, Han IO. Tumor-associated carbonic anhydrases are linked to metastases in primary cervical cancer. J Cancer Res Clin Oncol. 2006;132:302–308. doi: 10.1007/s00432-005-0068-2. [DOI] [PubMed] [Google Scholar]
- 44.Fiaschi T, Giannoni E, Taddei ML, Cirri P, Marini A, Pintus G, Nativi C, Richichi B, Scozzafava A, Carta F, Torre E, Supuran CT, Chiarugi P. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle. 2013;12:1791–1801. doi: 10.4161/cc.24902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chadwick AL, Howell A, Sotgia F, Lisanti MP. Carbonic anhydrase 9 (CA9) and redox signaling in cancer-associated fibroblasts: therapeutic implications. Cell Cycle. 2013;12:2534–2535. doi: 10.4161/cc.25842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bose P, Dort JC, Brockton NT. Identifying the stromal cell type that contributes to tumor aggressiveness associated with carbonic anhydrase IX. Cell Cycle. 2013;12:2535–2536. doi: 10.4161/cc.25843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol. 2015;137:173–179. doi: 10.1016/j.ygyno.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Miller CR, Oliver KE, Farley JH. MEK1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol Oncol. 2014;133:128–137. doi: 10.1016/j.ygyno.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 49.Kaluz S, Kaluzova M, Stanbridge EJ. The role of extracellular signal-regulated protein kinase in transcriptional regulation of the hypoxia marker carbonic anhydrase IX. J Cell Biochem. 2006;97:207–216. doi: 10.1002/jcb.20633. [DOI] [PubMed] [Google Scholar]
- 50.Shafee N, Kaluz S, Ru N, Stanbridge EJ. PI3K/Akt activity has variable cell-specific effects on expression of HIF target genes, CA9 and VEGF, in human cancer cell lines. Cancer Lett. 2009;282:109–115. doi: 10.1016/j.canlet.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dayan F, Bilton RL, Laferriere J, Trottier E, Roux D, Pouyssegur J, Mazure NM. Activation of HIF-1alpha in exponentially growing cells via hypoxic stimulation is independent of the Akt/mTOR pathway. J Cell Physiol. 2009;218:167–174. doi: 10.1002/jcp.21584. [DOI] [PubMed] [Google Scholar]
- 52.Dittrich C, Zandvliet AS, Gneist M, Huitema AD, King AA, Wanders J. A phase I and pharmacokinetic study of indisulam in combination with carboplatin. Br J Cancer. 2007;96:559–566. doi: 10.1038/sj.bjc.6603606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siebels M, Rohrmann K, Oberneder R, Stahler M, Haseke N, Beck J, Hofmann R, Kindler M, Kloepfer P, Stief C. A clinical phase I/II trial with the monoclonal antibody cG250 (RENCAREX(R)) and interferon-alpha-2a in metastatic renal cell carcinoma patients. World J Urol. 2011;29:121–126. doi: 10.1007/s00345-010-0570-2. [DOI] [PubMed] [Google Scholar]
- 54.Pastorekova S, Ratcliffe PJ, Pastorek J. Molecular mechanisms of carbonic anhydrase IXmediated pH regulation under hypoxia. BJU Int. 2008;101(Suppl 4):8–15. doi: 10.1111/j.1464-410X.2008.07642.x. [DOI] [PubMed] [Google Scholar]
- 55.Betof AS, Rabbani ZN, Hardee ME, Kim SJ, Broadwater G, Bentley RC, Snyder SA, Vujaskovic Z, Oosterwijk E, Harris LN, Horton JK, Dewhirst MW, Blackwell KL. Carbonic anhydrase IX is a predictive marker of doxorubicin resistance in early-stage breast cancer independent of HER2 and TOP2A amplification. Br J Cancer. 2012;106:916–922. doi: 10.1038/bjc.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakamura A, Osonoi T, Terauchi Y. Relationship between urinary sodium excretion and pioglitazone-induced edema. J Diabetes Investig. 2010;1:208–211. doi: 10.1111/j.2040-1124.2010.00046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]