

Abstract
Posterior reversible encephalopathy syndrome (PRES) is a well-documented pathology of the brain in systemic upsets. Majority of PRES cases present with edema in the cerebrum, most commonly in the territory of posterior circulation. It has been reported to show spinal cord involvement in a rare subgroup known as PRES with spinal cord involvement (PRES-SCI), with very limited existing literature even in adult patients. Our institution recently encountered a pediatric case with neurofibromatosis type I (NF 1) showing PRES with extensive reversible spinal cord changes. This case illustrates the features of this rare entity in the pediatric group of patients, and is the first reported case in NF 1 patients.
Keywords: Hypertensive crisis, neurofibromatosis, pediatric posterior reversible encephalopathy syndrome, posterior reversible encephalopathy syndrome, spinal cord edema
Introduction
Blood pressure fluctuation is known to cause acute changes in the brain in posterior reversible encephalopathy syndrome (PRES). Cerebral changes are commonly observed in occipital and parietal lobes, while other regions of the brain can also be affected in different patterns.[1] Our institution recently encountered a case with acute and reversible changes in both the brain and the spinal cord in an episode of hypertensive crisis, which is a very rare subset of PRES. We present the first reported case in a pediatric patient with neurofibromatosis type I (NF 1).
Case Report
A 4-year-old boy with unremarkable past health presented to a regional hospital with dehydration and tachycardia with history of recent increased fluid intake. Patient was cachexic with multiple café-au-lait spots. Neurological examination was unremarkable. On admission he had high blood pressure. Initial blood test results were nonspecific apart from some electrolyte disturbances. Chest radiograph showed increased cardiothoracic ratio (0.57) with clear lungs. Echocardiogram showed left ventricular hypertrophy with small left ventricular volume, but normal left ventricular systolic function. Pericardial effusion was detected. Bedside ultrasound examination showed absent right kidney. A computed tomography (CT) of brain performed for workup of diabetes insipidus showed brainstem swelling with obstructive hydrocephalus. He was then transferred to the pediatric intensive care unit of our institution, a tertiary referral center.
Urgent magnetic resonance imaging (MRI) of brain and spine [Figure 1] showed abnormal T1-weighted (T1W) hypointense and T2W hyperintense signal with swelling in medulla oblongata, anterior pons, and bilateral medial cerebellar hemispheres. Neither abnormal contrast enhancement nor restricted diffusion was noted. Swelling in brainstem caused obstructive hydrocephalus. No abnormal signal was seen in bilateral cerebrum. In the spine, there was diffuse swelling of whole spinal cord, most severe in cervical cord. Whole-cord generalized swelling with increase in T2W signal was noted. No abnormally enhancing focal lesion was evident. No evidence of leptomeningeal lesion was seen along the whole thecal sac.
Figure 1(A-E).
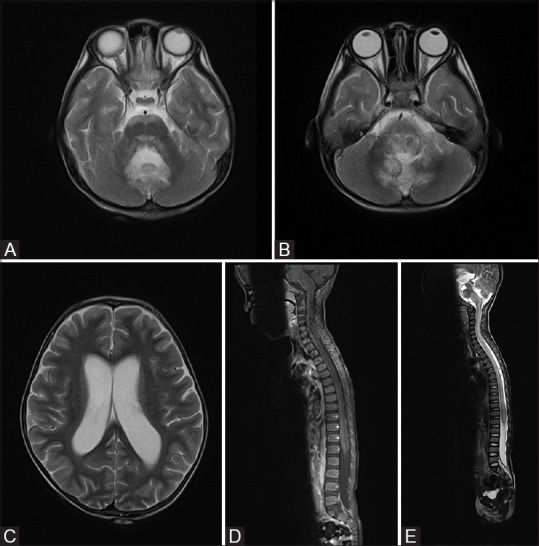
(A-C) First MRI brain showed T2W hyperintense signal in anterior pons, medulla and bilateral cerebellar hemispheres. Obstructive hydrocephalus is evident. No abnormal signal is seen in bilateral cerebrum. (D and E) First MRI spine showed generalized swelling and increase in T1W hypointense and T2W hyperintense signal affecting the whole spinal cord
Initial differential diagnoses included inflammatory causes as in longitudinally extensive myelitis such as neuromyelitis optica (NMO) associated with systemic lupus erythematosus (SLE) (presence of pericardial effusion); neoplastic causes including lymphoma or diffusely infiltrative ependymoma. Subsequent laboratory studies revealed negative immune markers, except elevated erythrocyte sedimentation rate, rendering systemic inflammatory conditions, including SLE and NMO unlikely. Cerebrospinal fluid (CSF) analysis also revealed no evidence of infection or oligoconal band. Neoplastic causes were not supported by serum tumor markers, as well as CT scan and metaiodobenzylguanidine scan, in particular for pheochromocytoma.
On CT scan, right renal agenesis was confirmed. Compensatory hypertrophy of left kidney was noted with poor enhancement. The patient had two left renal arteries in which the superior one showed focal narrowing in its proximal segment (0.3 cm in length) sparing ostium. Wedge-shaped renal parenchymal nonenhancement suggestive of infarcts was also noted. Ultrasonography Doppler of renal artery confirmed the diagnosis of renal artery stenosis [Figure 2]. Renal scintiscan study also showed slow uptake and retention of tracer in left kidney, compatible with renal artery stenosis. Focal lack of uptake confirming renal infarct was also evident.
Figure 2(A-D).
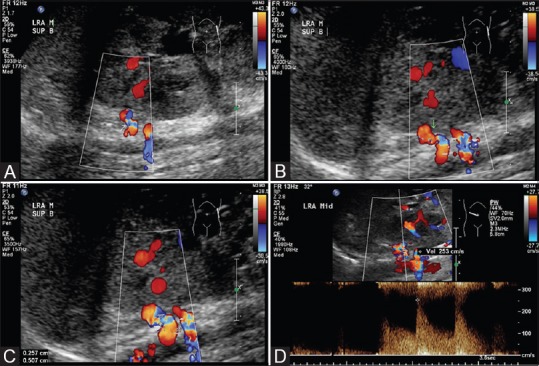
(Doppler USG study of renal arteries): (A-C) Doppler USG showing focal narrowing with aliasing (arrow) in superior branch of left renal artery. (D) Doppler USG showing elevated peak systolic velocity up to 253cm/s at the stenosis
Renal artery stenosis was identified as cause of persistent hypertension. To exclude underlying vasculitis causing renal artery stenosis, positive emission tomography–computed tomography was done, which was negative.
Considering the renal artery stenosis and multiple café-au-lait spots, NF 1 was suspected. Slit-lamp examination showed Lisch nodules in bilateral retina. Diagnosis of NF 1 was established by criteria by National Institutes of Health.[2] Radiograph of lower limbs of patient showed anterior bowing of left tibia with cortical thickening, suspected to be NF 1 associated bone lesion [Figure 3]. The patient had no first-degree relative suffering from NF 1.
Figure 3.
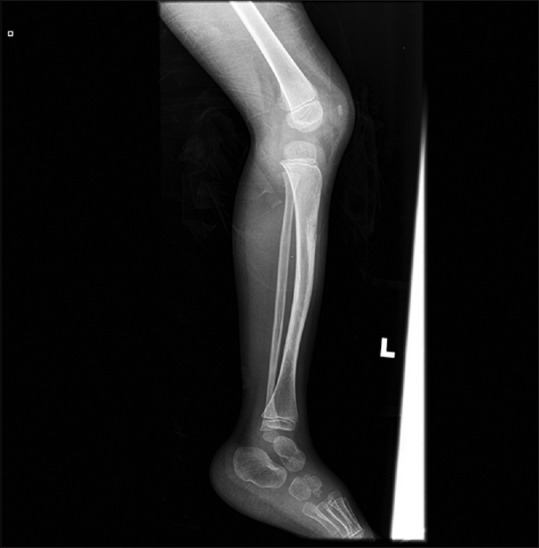
Lateral radiograph of left tibia and fibula of patient showing anterior bowing with increase in sclerosis and cortical thickening, suspected to be NF 1 related bone lesion
Patient's hypertension was difficult to control. It required titration of multiple antihypertensive medications. Initially, the patient was dependent on intravenous infusion of labetalol, infusion phentolamine, amlodipine, captopril, hydralazine, frusemide, minoxidil, and aldactone. Blood pressure slowly came down and he was able to take off phentolamine and changed to oral labetalol. Blood pressure was controlled on high side: systolic blood pressure of 140–180 mmHg and diastolic blood pressure 60–80 mmHg. Renal artery angioplasty was considered but not done in the acute setting due to high risk associated with small caliber artery and single kidney status. The plan was to delay the procedure to when the patient is older and bigger, giving a lower risk and higher rate of success in renal angioplasty.
After blood pressure was gradually controlled, serial CT brain scans showed gradual resolution of swelling in brainstem and cerebellum. Obstructive hydrocephalus also resolved. Follow-up MRI [Figure 4] was performed 6 weeks after the presentation. Abnormal spinal cord swelling and signal changes were resolved. Changes in brainstem and bilateral cerebellar hemispheres also resolved. Only minimal residual swelling of medulla was evident, with no abnormal signal. Hydrocephalus also resolved. The diagnosis of PRES precipitated by hypertensive crisis from NF 1-related renal artery stenosis was postulated. Patient remained clinically stable and was discharged.
Figure 4(A-C).
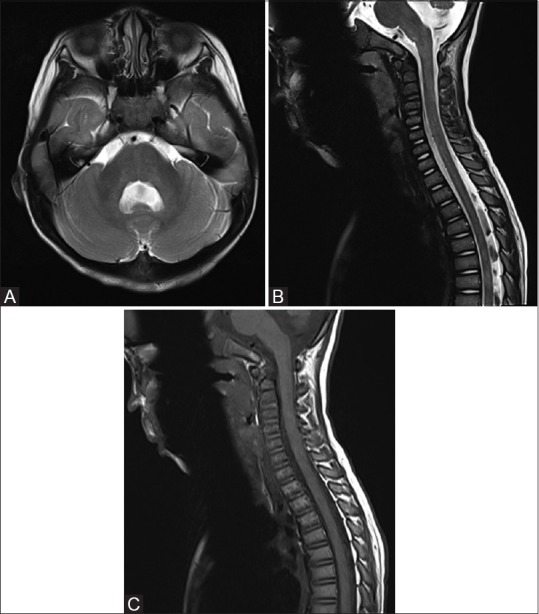
(A) Follow up MRI in 6 weeks showing resolution of abnormal signal and swelling in brainstem and cerebellum. (B and C) Follow up MRI spine in 6 weeks showing resolution of spinal cord abnormal signal and swelling
Discussion
Uncontrolled blood pressure is known to cause reversible changes in the brain in PRES. PRES can present in wide variety of patterns, most commonly as edema in territory supplied by posterior circulation. Three major patterns of involvement were identified by a study by Bartynski and Boardman,[1] namely the holohemispheric watershed, superior frontal sulcus, and dominant parietal-occipital patterns. Brainstem and basal ganglia are less commonly affected areas. The cause of cerebral edema caused by PRES is controversial, evidence suggesting local hypoperfusion and hyperperfusion both exist.[3] The cause of PRES is commonly perceived to be related to fluctuations in blood pressure. Despite the highly variable patterns of PRES involvement, reported changes had mainly been within the brain.
Several case reports had been published for cases with hypertensive crisis and reversible radiological changes in the cord, proposed to be a variant of PRES with spinal cord involvement. A total of around 10 such reported cases can be found in the existing literature. In 2014, Havenon et al.[4] reported two such cases and reviewed the literature of other six similar cases. They proposed the name PRES-SCI for this subset (PRES with spinal cord involvement). In their cohort of patients, abnormal T2W hyperintense confluent signal originated from cervicomedullary junction of spinal cord, some with entire cord involvement. All of them had elevated blood pressure with spinal cord changes showing improvement upon blood pressure control, like our case. Cases had also been reported in Japan[5] and Turkey,[6] in which elevated immunoglobulin G (IgG) levels in CSF were identified. These two cases lacked symptoms in association with the fulminant spinal cord changes, which is the “clinical radiologic dissociation” also observed in our case. Many of the reported cases had extreme hypertension related to renal diseases, which is also observed in our case.
It had been proposed that the posterior circulation has less sympathetic innervation[7] and hence less capacity to respond to fluctuations in blood pressure. Anterior spinal artery branches from the posterior circulation, which may explain why the upper spinal cord involvement is most commonly observed in PRES-SCI. NF patients can have various forms and extents of vasculopathies. Whether this would predispose NF patients to PRES-SCI is yet to be discovered. Also, for the pattern of intracranial reversible edematous changes in PRES, our case demonstrated reversible edematous changes in the medulla oblongata, anterior pons, and cerebellar hemispheres. This was quite unusual as compared with the pattern observed in most PRES cases, namely the holohemispheric watershed, superior frontal sulcus, and dominant parietal–occipital patterns.[1] Cause of this atypical pattern is uncertain, whether PRES-SCI shows a specific pattern of intracranial involvement, or this is a pattern common for NF 1, is yet to be discovered. Continual observation for more reported cases is required to make further postulations. Continual observations and more evidence are also required to raise awareness and aid diagnosis of this rare entity.
Our case also demonstrates the difficulty in diagnosing NF 1 in children, especially if solely dependent on radiological findings. NF 1 is mostly a clinical diagnosis using the criteria by a conference statement by the National Institutes of Health.[2] Molecular testing is rarely indicated for its diagnosis. However, as the constellations of NF 1 features differ between patients and development are age-dependent, diagnosis can be difficult, especially in very young patients. Café-au-lait spots are usually present since birth, which is the most obvious NF 1 feature in our patient. Skinfold freckling usually develops next. More specific feature of neurofibroma that can be picked up by MRI assessment usually develop after 10 years of age. This explains the lack of NF 1 features in the MRI brain of our patient, as they are yet to develop. The limitations of the usage of current diagnostic criteria in children below 8 years of age had been addressed by the existing literature[8] and modifications may be required. If the presentation is atypical or when the diagnosis is suspected in very young children when the clinical features are yet to develop, molecular testing may be required. NF 1 is caused by heterozygous loss-of-function pathogenic variants of the NF 1 gene. The pathogenic variants can be highly heterogeneous. Various molecular testing strategies are available, including the multistep variant detection protocol that identifies more than 95% of the NF 1 pathogenic variants.[9] Molecular testing is complex and advice from geneticists is usually required. The implication for radiologists is to keep high vigilance in this syndrome and molecular testing may be required to make the diagnosis early.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Bartynski W, Boardman J. Distinct imaging patterns and lesion distribution in posterior reversible encephalopathy syndrome. Am J Neuroradiol. 2007;28:1320–7. doi: 10.3174/ajnr.A0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neurofibromatosis. National Institutes of Health Consensus Statement Online. 1986;6:1–19. [PubMed] [Google Scholar]
- 3.Bartynski W. Posterior reversible encephalopathy syndrome, Part 1: Fundamental imaging and clinical features. Am J Neuroradiol. 2008;29:1036–42. doi: 10.3174/ajnr.A0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Havenon A, Joos Z, Longenecker L, Shah L, Ansari S, Digre K. Posterior reversible encephalopathy syndrome with spinal cord involvement. Neurology. 2014;83:2002–6. doi: 10.1212/WNL.0000000000001026. [DOI] [PubMed] [Google Scholar]
- 5.Nagato M, Takahashi Y, Yoshioka M, Nambu M. A case of hypertensive encephalopathy with extensive spinal lesions on MRI. Brain Dev. 2010;32:598–601. doi: 10.1016/j.braindev.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Yiş U, Karaoğlu P, Kurul S, Soylu A, Çakmakçi H, Kavukçu S. Posterior reversible leukoencephalopathy syndrome with spinal cord involvement in a 9-year-old girl. Brain Dev. 2016;38:154–7. doi: 10.1016/j.braindev.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Yasuda Y, Akiguchi I, Imai T, Sonobe M, Kage M. Hypertensive brainstem encephalopathy. Internal Med. 2003;42:1131–4. doi: 10.2169/internalmedicine.42.1131. [DOI] [PubMed] [Google Scholar]
- 8.DeBella K, Szudek J, Friedman J. Use of the National Institutes of Health Criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105:608–14. doi: 10.1542/peds.105.3.608. [DOI] [PubMed] [Google Scholar]
- 9.Friedman JM. Neurofibromatosis 1. 1998 [Updated Sep 4, 2014] In: Pagon RA, Adam MP, Ardinger HH, editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017. [PubMed] [Google Scholar]