

A high-resolution crystal structure of a pan-specific synthetic Fab selected using a phage-display system against the C-terminal domain of the nucleoprotein from the Ebola virus is reported.
Keywords: Fab structure, Ebola virus nucleoprotein, antibody–antigen interaction, synthetic antibody fragments, Ebola virus Bundibugyo strain
Abstract
The vast majority of platforms for the detection of viral or bacterial antigens rely on immunoassays, typically ELISA or sandwich ELISA, that are contingent on the availability of suitable monoclonal antibodies (mAbs). This is a major bottleneck, since the generation and production of mAbs is time-consuming and expensive. Synthetic antibody fragments (sFabs) generated by phage-display selection offer an alternative with many advantages over Fabs obtained from natural antibodies using hybridoma technology. Unlike mAbs, sFabs are generated using phage display, allowing selection for binding to specific strains or for pan-specificity, for identification of structural epitopes or unique protein conformations and even for complexes. Further, they can easily be produced in Escherichia coli in large quantities and engineered for purposes of detection technologies and other applications. Here, the use of phage-display selection to generate a pan-specific Fab (MJ20), based on a Herceptin Fab scaffold, with the ability to bind selectively and with high affinity to the C-terminal domains of the nucleoproteins (NPs) from all five known strains of the Ebola virus is reported. The high-resolution crystal structure of the complex of MJ20 with the antigen from the Bundibugyo strain of the Ebola virus reveals the basis for pan-specificity and illustrates how the phage-display technology can be used to manufacture suitable Fabs for use in diagnostic or therapeutic applications.
1. Introduction
Anthropogenic activities and climate change have led to the geographic spread of many viruses which until recently were considered to be highly endemic, such as Ebola virus (EBOV) and Zika virus (ZIKV), posing substantial threats to human health (Rochlin et al., 2016 ▸; Tabachnick, 2016 ▸; Bloom et al., 2017 ▸). One of the key tools to protect against this imminent danger are fast and accurate diagnostic methods (Chan et al., 2017 ▸). This was dramatically underscored during the recent epidemic of Ebola fever disease (EVD) in West Africa (Coltart et al., 2017 ▸), because the inability to effectively diagnose new cases prevented an adequate and timely response to the epidemic (Boisen et al., 2016 ▸).
There are currently five known major Ebola virus strains. The recent epidemic was caused by a variant of the Zaire strain (EBOV). The other four strains are Taï Forest (TAFV), Bundibugyo (BDBV), Reston (RESTV) and Sudan (SUDV) (Kuhn et al., 2013 ▸). EBOV, SUDV and BDBV have all caused large outbreaks of EVD in the past (Roddy et al., 2012 ▸; Feldmann & Geisbert, 2011 ▸), while TAFV has so far only been reported in one patient (Formenty et al., 1999 ▸). The five EBOV strains vary with respect to their corresponding fatality rates: RESTV is nonpathogenic to humans and SUDV and BDBV have fatality rates of about 40%, while EBOV has a fatality rate of up to 90%. The basis for this variation is not known. While the gold standard in diagnostics is virus isolation in cell culture, this is impractical for EVD as it requires biosafety level 4 and a long processing time. Reverse transcription polymerase chain reaction (RT-PCR) has been adopted as a reliable standard, but the deployment of the necessary equipment in remote areas of outbreak, along with the cost and time to readout, continues to present serious challenges (Broadhurst et al., 2016 ▸). A much better route would be a quick point-of-care assay, similar to a fertility or pregnancy test, which can reliably and quickly identify viral antigens in the bodily fluids of patients, typically blood (Broadhurst et al., 2016 ▸). The key components of a successful canonical diagnostic kit are antibodies used to detect the antigen. Typically, such kits use monoclonal antibodies (mAbs) obtained through hybridoma technology, whereby the initial antibody is generated through a natural process of immunization of animals, typically mice, rabbits and goats. The three currently available EVD rapid diagnostic tests (RDTs) which have received World Health Organization and/or Food and Drug Administration (FDA) Emergency Use Authorization status are all lateral flow immunoassays (LFIs) that utilize mAbs raised against three of the Ebola proteins: the glycoprotein (GP), the nucleoprotein (NP) and the matrix protein VP40 (Broadhurst et al., 2016 ▸). Detection of GP effectively allows the detection of circulating intact virus particles (viremia), while NP and VP40 are synthesized in infected cells and are detectable in the free form in the circulating blood of patients.
Unfortunately, the time and cost of producing natural mAbs for the purposes of production of diagnostics are substantial, and the quality and selectivity are unpredictable. These limitations can be overcome by using in vitro high-throughput selection methods using a single Fab template and synthetic combinatorial libraries that allow screening for high-affinity binders to a specific antigen. A powerful technology that has been developed in the past decade is that of fully automated high-throughput phage-display (HTPD) selection platforms. Besides the Fab format that we use, several alternative formats have been developed using other types of binding fragments (Miersch & Sidhu, 2012 ▸). In this regard, we note that other studies have employed a scheme involving a combination of immunization and phage display to generate variable heavy-chain (VhH) domains to identify specific binders that cross-react across several species of Ebola NP (Sherwood & Hayhurst, 2013 ▸; Changula et al., 2013 ▸).
We use a Fab phage-display process that is novel in its use of reduced genetic code libraries (Ye et al., 2007 ▸; Hornsby et al., 2015 ▸; Marcon et al., 2015 ▸; Fellouse et al., 2004 ▸). Additionally, the Fab template is the FDA-approved Herceptin scaffold, optimized for very high stability (T m ≃ 80°C) and overexpression in Escherichia coli cells, making it possible to produce large amounts of sFabs quickly and at low cost. The use of an invariant, recombinant scaffold makes it straightforward to move an sFab from one format to another and to engineer additional functionalities. It also makes it possible to design accessory proteins that function with essentially all sFabs in a generic manner. This creates an opportunity for the coupling of synthetic Fabs to new, sensitive detection systems, such as split-enzyme assays, in novel and easy-to-use ways.
We used HTPD to generate sFabs against one of the Ebola virus antigens circulating in the blood of infected patients, i.e. the nucleoprotein (NP). More specifically, the antigens used in the selection process were the recombinant C-terminal domains of NP (NPCt) from all five known strains of the virus (Baker et al., 2016 ▸; Dziubańska et al., 2014 ▸). Unlike the N-terminal domain of NP, NPCt is not known to bind any other proteins or nucleic acids, and therefore constitutes a suitable target for molecular recognition and diagnostics. The challenge, however, is the small size of the target domain of the antigen and the amino-acid sequence diversity between the strains, with pairwise identity ranging from 60 to 86% (Dziubańska et al., 2014 ▸). Our strategic objective was to test the possibility of obtaining pan-specific sFabs with potential applications in EVD diagnostics. We obtained one sFab, denoted MJ20, which binds with high affinity to the NPCTs from all five strains of the Ebola virus. Here, we describe the MJ20 Fab in complex with NPCt from BDBV, revealing the structural basis for specificity and illustrating the potential power of in vitro Fab selection for the design of diagnostics and therapeutics.
2. Materials and methods
2.1. sFab selection
NPCts from five different strains of the Ebola virus were expressed as biotinylated C-terminal SNAP-tag fusions to facilitate their immobilization onto streptavidin-coated paramagnetic beads. The NPCt construct contained residues 642–739 and the linker to the SNAP-tag was 12 residues in length. Covalent attachment of biotin to the SNAP-tag within the fusion proteins was achieved through an enzymatic reaction using commercially available BG-Biotin (SNAP-biotin; New England Biolabs). This avoids any negative effects on the structure of the fused target protein, while standard chemical biotinylation often impairs protein solubility. Results from MALDI-TOF mass spectrometry confirmed complete biotinylation (data not shown). A thrombin cleavage site was introduced between the SNAP-tag and the target antigens to provide a facile mechanism to release the tight-binding phages from the beads during the elution step of the biopanning process.
In this work, a synthetic phage-display library was used with a theoretical size of 1010 unique CDR sequences. Briefly, varying amounts of sequence diversity were introduced into four CDR loops, with CDR3HC > CDR3LC > CDR(1,2)HC. The CDR(1,2)LC loops were not diversified (Fellouse et al., 2007 ▸; Koide et al., 2007 ▸). The details of the conditions and reagents used in the biopanning steps have been reported elsewhere (Paduch et al., 2013 ▸). Using these protocols, four rounds of biopanning were performed. During the first round the antigen concentration was 100 nM; in each of the successive rounds the concentration was reduced (to 50, 20 and 10 nM) to increase stringency. After capture of the phage binders by the streptavidin-coated beads, the beads were rigorously washed to eliminate weak binders. The remaining high-affinity binders were then eluted from the beads by thrombin cleavage. The captured phages were then propagated in E. coli and the cycle was repeated.
The initial screening to identify high-affinity binders was performed using a single-point competitive phage ELISA procedure (Paduch et al., 2013 ▸; Mukherjee et al., 2015 ▸). This procedure is high-throughput, allowing many potential clones to be evaluated for each target. A total of more than 2500 single clones were screened and analysed using this method. Based on these results, 70 unique clones combined from the six independently targeted NPCt selections were carried forward to be analysed for binding and cross-reactivity. Because the Zaire strain was the subject of this study, a majority (total of 34) of the clones were chosen from the phage-display selection performed using this strain as the target. Owing to the highly conserved nature of the NPCt strains, the cross-reactivity between the sFabs tested was significant, with most displaying substantial binding to two or three of the strains. However, there were only two sFabs (MJ15 and MJ20) that showed binding across all five strains. Not surprisingly, based on epitope binning by surface plasmon resonance, both sFabs share a similar epitope on NPCt. However, because of its superior binding characteristics across all strains and its expression profile, MJ20 was selected to be carried forward for this study.
2.2. Protein expression and purification for crystallization studies
Recombinant MJ20 was expressed in E. coli strain BL21-CodonPlus (DE3)-RIPL cells (Stratagene) in a 3.5 l fermenter culture. Protein expression was induced at an OD600 of ∼1.5 by the addition of 1 mM IPTG and continued for 4 h at 37°C. The cells were harvested by centrifugation at 4000 rev min−1 for 30 min and frozen at −20°C. The pellet was suspended in 5 ml lysis buffer (20 mM Tris–HCl, 150 mM NaCl, 10% glycerol pH 7.4) per gram of biomass. The cells were disrupted with a Dounce homogenizer, a high-pressure homogenizer and by sonication (Branson Sonifier 450), followed by centrifugation at 35 000 rev min−1 for 55 min. The cleared lysate was loaded onto a gravitational column containing 1 ml SulfoLink Coupling Resin (Thermo Scientific) coupled with a SUMO-Protein-G-A1 variant (Bailey et al., 2014 ▸). After washing with buffer (50 mM Na2HPO4, 500 mM NaCl pH 7.4), the Fabs were eluted with 10 ml 100 mM glycine pH 2.6, neutralized to pH 7.5 and concentrated to ∼10 mg ml−1
The C-terminal domains (NPCts) of the nucleoproteins derived from five strains of the Ebola virus (EBOV, RESTV, SUDV, TAFV and BDBV) were expressed and purified using previously described protocols (Baker et al., 2016 ▸; Dziubańska et al., 2014 ▸). Briefly, cDNA constructs coding for the NPCt proteins were synthesized commercially using optimized codon frequencies for E. coli and subsequently cloned into the His6-MBP-Parallel1 vector (Sheffield et al., 1999 ▸). E. coli BL21-CodonPlus (DE3)-RIPL cells (Stratagene) were grown in Terrific Broth medium at 37°C until the OD600 reached ∼2.5. Expression was induced by the addition of 0.5 mM IPTG and continued for 18 h at 16°C. After cell lysis, MBP-NPCt fusion proteins were purified using nickel-affinity chromatography on Ni–NTA resin (Qiagen). The MBP tags were removed by rTEV protease cleavage and the respective NPCt was further purified to homogeneity using size-exclusion chromatography on a Superdex 75 column (GE Life Sciences).
2.3. Binding kinetics and thermodynamics
The kinetics and thermodynamics of the interactions of MJ20 with antigens were evaluated by surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC), respectively.
SPR measurements were performed at 20°C using a Biacore 3000 equipped with a Biacore NTA Sensor Chip. 30 response units (RU) of NPCt were immobilized on all four flow cells through Ni2+–His10-tag interaction. The first flow cell was used as a reference. For kinetics analysis, 150 µl of the sFab in EB buffer (10 mM HEPES, 200 mM NaCl, 50 µM EDTA, 0.01% Tween 20 pH 7.4) was injected over three flow cells at concentrations of 3.125, 6.25, 12.5, 25, 50 and 100 nM (30 µl min−1). For epitope mapping, 210 µl of the first sFab followed by the second sFab, or a mixture of both sFabs, was injected over three flow cells. Each of these analyses was made at 100 nM in EB buffer. For a negative control, the same sFab was injected twice followed by the sFab mixture. Data were collected at a rate of 2 Hz and were fitted and analysed using a 1:1 interaction model with the BiaEvaluation software (GE Healthcare).
Calorimetric measurements using a MicroCal iTC200 instrument (Malvern) were used to determine the thermodynamic parameters (ΔG, ΔH and ΔS) of interaction. All reactants were extensively dialyzed against the same buffer [100 mM HEPES, 150 mM NaCl, 5%(v/v) glycerol pH 7.5] and degassed before use. The molar concentration of a specific NPCt in the syringe was tenfold higher (100 µM) than that of MJ20 in the cell (10 µM). Titrations were carried out using 1.5 µl injections with 4 min intervals. To ensure that the titrant concentration was at its loading value, an initial injection of 0.5 µl was made. All titration data were collected at 25°C with a stirring speed of 700 rev min−1 and the reference power value of 6. The Origin software (MicroCal) was used to fit the binding isotherms by nonlinear regression using a single-site model and for calculation of the stoichiometry of the interaction (N), the equilibrium association constant (K a) and enthalpy (ΔH) and entropy (ΔS) changes.
2.4. Immunofluorescence staining
The components of the EBOV transcription/replication virus-like particle (trVLP) system were kindly provided by Dr Thomas Hoenen (Hoenen, 2018 ▸). pCAGGS-NP-FLAG was constructed by adding a FLAG-tag sequence at the C-terminus of NP. pCAGGS-NPΔCt-FLAG was then made by deleting the region corresponding to amino acids 642–739 from the parent plasmid. Standard molecular-cloning techniques were used for plasmid construction. HuH-7 cells were transfected with pCAGGS-NP-FLAG or trVLP components (pCAGGS-VP35, pCAGGS-VP30, pCAGGS-L, pCAGGS-T7 and p4cis-vRNA-RLuc) with pCAGGS-NP-FLAG or pCAGGS-NPΔCt-FLAG by TransIT-LT1 (Mirus). 48 h after transfection, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. The cells were stained with the primary antibodies MJ20, rabbit anti-FLAG antibody (Cell Signaling) and mouse anti-Ebola VP35 antibody (Kerafast) and the secondary antibodies goat anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody conjugated with Alexa Fluor 594, goat anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody conjugated with Alexa Fluor 594 (Invitrogen) and pre-adsorbed goat F(ab′)2 anti-human IgG-(Fab′)2 (DyLight 488; Abcam). After staining, cover slips were mounted using ProLong Gold Antifade Mountant with DAPI (Invitrogen). Pictures were taken using a Nikon Eclipse TE2000-E (Nikon) and processed with the ImageJ software.
2.5. Mutagenesis
The following EBOV NPCt variants were generated: Y652A, L656A, F678A, S679A, S681A, F731A, M732A and L735A. Primers were obtained from Genewiz, and the QuikChange method using Phusion High-Fidelity DNA Polymerase (New England Biolabs) was used to introduce the mutations. All mutations were verified at the DNA level through direct sequencing. The expression and purification of protein variants were performed as for the wild-type protein.
2.6. Thermal stability assay of NPCt variants
The midpoint of thermal denaturation (melting temperature, T m) was determined using scanning fluorescence with the aid of an RT-PCR instrument (StepOnePlus PCR System, Applied Biosystems) and the fluorophore SYPRO Orange (Life Technologies). Samples were prepared by mixing 16 µl protein (1 mg ml−1) in buffer (50 mM Tris, 300 mM NaCl pH 8.5) with 1× SYPRO Orange dye (as provided by the manufacturer) to give a total volume of 20 µl. Samples were transferred to 0.1 ml MicroAmp Fast Reaction tubes (Applied Biosystems) and scans were performed from 20 to 95°C.
2.7. Crystallization and structure determination
Purified NPCts mixed with MJ20 in a 1:1 molar ratio were incubated on ice for 4 h and the NPCt–MJ20 complexes were further purified to homogeneity by size-exclusion chromatography on a Superdex 75 column (GE Life Sciences) equilibrated with 20 mM Tris–HCl, 100 mM NaCl pH 7.5. The samples were screened at ∼10 mg ml−1 by the sitting-drop vapor-diffusion method at 16°C using commercially available screens (JCSG+, Midas and Morpheus II) and a Mosquito robot (TTP Labtech). Protein solution was mixed with precipitant solution in equal volume ratios and the total drop volume was 500 nl. All crystals were flash-cooled in liquid nitrogen directly from the crystallization drop, stored until data collection and then screened for diffraction quality.
Diffraction data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 19-ID and 19-BM beamlines at the Advanced Photon Source, Argonne National Laboratory, USA. Data were reduced, merged and scaled using HKL-3000 (Minor et al., 2006 ▸) and the structure was solved by molecular replacement. The search model for the BDBV NPCt–MJ20 complex was generated using the BALBES web server (Long et al., 2008 ▸), which automatically generates a search model using the PDB on the basis of sequence similarity. The molecular-replacement solution was refined using PHENIX (Adams et al., 2010 ▸). The model was completed manually (specifically, the CDR loops were built into the electron density) and additional adjustments were carried out with Coot (Emsley et al., 2010 ▸), which incorporates a MolProbity (Chen et al., 2010 ▸) stereochemistry check. The interface between the antigen and MJ20 was analysed using PISA (Baskaran et al., 2014 ▸).
3. Results and discussion
3.1. Binding kinetics, thermodynamics and selectivity
The kinetics and thermodynamics of the interaction of MJ20 with all five antigens were evaluated by SPR and ITC (Fig. 1 ▸, Table 1 ▸). The kinetics showed typical values, with k on in the region of ∼105 M −1 s−1 and k off in the region of ∼10−3 s−1. ITC revealed that binding by MJ20 showed an unfavorable large loss of entropy and was driven by a negative change in enthalpy (ΔH).
Figure 1.

Isothermal titration calorimetry analysis of the interaction of MJ20 with BDBV NPCt. Top panel, ITC raw titration; bottom panel, fitted affinity.
Table 1. Kinetic and thermodynamic parameters of binding between Ebola virus NPCts and MJ20 determined by surface plasmon resonance and isothermal titration calorimetry.
| NPCt | k on (M −1 s−1) | k off (s−1) | K d (nM) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) | ΔG (kcal mol−1) | K d (nM) |
|---|---|---|---|---|---|---|---|
| EBOV | 1.7 × 105 | 7.1 × 10−4 | 4.1 | −31.15 | 19.65 | −11.50 | 3.7 |
| BDBV | 2.6 × 105 | 4.3 × 10−3 | 16.6 | −26.23 | 15.86 | −10.37 | 23.4 |
| TAFV | 3.7 × 105 | 1.1 × 10−3 | 3.1 | −24.61 | 13.24 | −11.37 | 4.5 |
| SUDV | 1.8 × 105 | 3.6 × 10−3 | 20.3 | −18.26 | 8.34 | −9.91 | 51.0 |
| RESTV | 1.9 × 105 | 2.2 × 10−3 | 11.7 | −29.32 | 18.49 | −10.83 | 11.6 |
To test whether MJ20 could recognize EBOV NP expressed in mammalian cells, we performed immunofluorescence staining of HuH-7 cells transfected with a plasmid expressing NP alone or transfected with a set of plasmids encoding the entire EBOV transcription/replication virus-like particle (trVLP) system, which recapitulates EBOV infection and the production of infectious virus-like particles under biosafety level 2 containment (Biedenkopf & Hoenen, 2017 ▸; Hoenen et al., 2012 ▸). During EBOV infection, or transfection of the complete trVLP system, cells exhibit viral ‘inclusion bodies’ that are known to contain the viral proteins NP, VP35, VP30 and L as well as the viral RNA genome, which are the sites of EBOV replication and nucleocapsid formation (Hoenen et al., 2012 ▸; Nanbo et al., 2013 ▸). In addition, electron-microscopy studies have shown that structured nucleocapsids are present within the inclusion bodies (Watanabe et al., 2006 ▸). It has also been demonstrated that expression of NP alone results in the accumulation of NP in inclusion bodies (Groseth et al., 2009 ▸; Noda et al., 2011 ▸; Watanabe et al., 2006 ▸). As shown in Fig. 2 ▸(a), the expression of NP alone in HuH-7 cells was detected in typical inclusion bodies by MJ20, but transfection of the corresponding empty vector did not result in any staining by MJ20 (rows 1 and 2 in Fig. 2 ▸ a). MJ20 also recognized NP in inclusion bodies in the context of the complete trVLP system (row 3), in which productive transcription requiring assembled nucleocapsids takes place. Because NP was C-terminally tagged with the FLAG epitope, when full-length NP (NP-FLAG) was expressed MJ20 staining co-localized with anti-FLAG staining (rows 2 and 3 in Fig. 2 ▸ a), indicating that MJ20 indeed recognized the NP protein within cells. However, when NPΔCt (amino acids 1–641)-FLAG was expressed, strong anti-FLAG staining was detected but not MJ20 staining (row 4 in Fig. 2 ▸ a), demonstrating that MJ20 staining depends on the presence of the C-terminal domain of NP, as expected. In trVLP-transfected cells, staining was also detected with anti-VP35 antibody (Fig. 2 ▸ b). VP35 is known to co-localize with NP in inclusion bodies (Noda et al., 2011 ▸). As expected, VP35 staining co-localized with MJ20 staining when full-length NP was present with other trVLP components including VP35, demonstrating that MJ20 recognizes NP in the context of all of the components of the EBOV nucleocapsid. Therefore, MJ20 efficiently detected NP protein in immunofluorescence staining either with NP alone or in the context of all of the EBOV proteins under conditions in which intact nucleocapsids are present and in a manner that depends on the C-terminal domain of NP.
Figure 2.

Immunofluorescence staining of EBOV NP with MJ20. (a) HuH-7 cells were transfected with empty vector (pCAGGS) or plasmids encoding NP-FLAG or all trVLP components including the indicated NP constructs. They were stained as indicated with DAPI, MJ20 and anti-FLAG antibody. (b) Cells were transfected with the indicated plasmids and stained with DAPI, MJ20 and anti-VP35. MJ20 staining localizes in inclusion bodies, co-localizes with VP35 and depends on the expression of NP and NPCt.
3.2. Structure of the sFab MJ20–NPCt complex and the basis for the pan-specificity of MJ20
We were ultimately interested in how MJ20 is capable of binding with high affinity to the relatively small antigen and in a pan-specific manner. Although we were able to crystallize complexes of MJ20 with NPCt from RESTV, EBOV and BDBV, the latter yielded the best-quality crystals. Those used for data collection grew in the MIDAS screen (condition H11), i.e. 0.2 M ammonium formate, 10%(w/v) polyvinylpyrrolidone, 20%(w/v) PEG 4000. Crystals were prepared and data were collected as described in §2. Data-collection and refinement statistics are given in Table 2 ▸. The refined structure (Table 3 ▸) reveals details that rationalize the thermodynamic profile of the interaction and explain the pan-specificity of MJ20 (note that we also confirmed that the complexes of MJ20 with NPCt from EBOV and RESTV are isostructural to the BDBV NPCt–MJ20 complex; the significantly lower resolution precluded detailed refinement).
Table 2. Data-collection and refinement statistics for the NPCt–MJ20 complex.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P212121 |
| a, b, c (Å) | 45.41, 87.48, 130.40 |
| Resolution range (Å) | 50.00–1.75 (1.78–1.75) |
| 〈I/σ(I)〉 (at 1.75 Å) | 1.69 |
| Total No. of reflections | 422578 |
| No. of unique reflections | 53300 |
| Completeness (%) | 99.8 |
| Wilson B factor (Å2) | 27.5 |
| Multiplicity | 7.9 (8.0) |
| Refinement | |
| Resolution range (Å) | 42.88–1.75 (1.81–1.75) |
| No. of reflections (test set) | 53231 (1064) |
| R work/R free † | 0.175/0.202 |
| No. of atoms | |
| Protein | 8282 |
| Water | 567 |
| Average B ‡, all atoms (Å2) | 38.0 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.649 |
| Ramachandran plot | |
| Favored (%) | 97.4 |
| Allowed (%) | 2.6 |
| Disallowed (%) | 0.0 |
| PDB code | 5vkd |
R
cryst (the crystallographic R factor) = 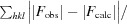
 and R
free =
where all reflections belong to a test set of randomly selected data.
and R
free =
where all reflections belong to a test set of randomly selected data.
Atomic displacement parameter (B factor).
Table 3. Thermodynamic parameters of interaction between EBOV NPCt mutants and MJ20 with the corresponding melting temperatures (T m) of each mutant.
Thermodynamics were evaluated using isothermal titration calorimetry, while scanning fluorescence was used to determine the melting temperatures (T m). Alanine substitution has little effect on the affinity for MJ20; however, the loss of the side chain influenced the stability of the antigen.
| sFab MJ20–EBOV NPCt complex | K d (nM) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) | ΔG (kcal mol−1) | T m (°C) |
|---|---|---|---|---|---|
| Wild type | 3.66 | −31.15 | 19.65 | −11.50 | 56.9 |
| Y652A | 18.0 | −30.49 | 19.95 | −10.54 | 50.9 |
| L656A | 10.0 | −29.72 | 18.81 | −10.91 | 52.7 |
| S679A | 7.41 | −30.36 | 19.26 | −11.10 | 56.0 |
| S681A | 12.0 | −28.28 | 17.47 | −10.81 | 57.3 |
| M732A | 32.0 | −29.90 | 16.70 | −10.20 | 54.0 |
| L735A | 103.5 | −28.10 | 18.57 | −9.52 | 47.8 |
In general, the crystal structure of the BDBV NPCt–MJ20 complex conforms to a canonical Fab–antigen interaction (Fig. 3 ▸ a). The elbow angle between the variable and constant domains (VD and CD) is 163°, which is within the typical range of the Herceptin scaffold, as shown by a survey of relevant structures in the PDB. The buried solvent-accessible area (BSA) between the antigen and the variable domain of MJ20 is ∼960 Å2, of which ∼830 Å2 is from the HC. This interface is dominated by CDR3HC (∼500 Å2 BSA), but it also includes small patches of CDR2HC (251 Å2) and CDR1HC (106 Å2). Importantly, of the 12 HC residues that bury more than 10 Å2 each, six are tyrosines (Tyr34, Tyr55, Tyr57, Tyr60, Tyr103 and Tyr105) and two are tryptophans (Trp104 and Trp110); collectively, these residues account for ∼550 Å2 of BSA. The interaction of CDR3LC with the antigen is marginal and involves only the side chains of Ser94 and Ser95, which bury ∼90 Å2.
Figure 3.

Crystal structure of the BDBV NPCt–MJ20 complex. (a) Ribbon representation of the complex, with NP colored green and the MJ20 light chain and heavy chain colored cyan and blue, respectively. CDR3HC, which forms the majority of contacts with the antigen, is shown as yellow sticks. The variable domain (V) and the constant domain (C) as well as the four complementarity-defining regions (CDRs) are marked. (b) The secondary structure of the CDR3HC loop is shown as sticks with the main chain colored green and the side chains in yellow. The conformation of each residue is indicated in superscript. The CDR3HC loop adopts a β-hairpin stabilized by backbone–backbone/side-chain hydrogen bonds (marked as dashed lines with their lengths in Å). Two NPCt residues (Tyr652 and Ser679) that form five intermolecular hydrogen bonds to CDR3HC are shown as magenta sticks.
That the key interaction is mediated by CDR3HC is not surprising given that this was allowed the highest variability in the selection process with respect to both amino-acid composition and length (up to 21 residues). Moreover, the similarities of the remaining CDRs to other known Fabs strongly suggest that CDR3HC is the most important contributor to the interface, with the other CDRs playing less significant roles. The CDR3HC of MJ20 is 14 residues in length; its secondary and tertiary features are shown in Fig. 3 ▸(b). It takes the shape of a β-hairpin stabilized by several backbone–backbone and backbone–side-chain hydrogen bonds, with the apical residue Gly108 in the ∊-conformation. Interaction with the antigen is primarily through the N-terminal arm of the hairpin, specifically the side chains of Tyr103, Trp104, Tyr105, His106 and Val107. Most of the contacts are distinctly apolar, which rationalizes the large negative enthalpy of the interaction with the antigen. There are only a few intermolecular hydrogen bonds: the hydroxyl of Tyr652NPCt engages the backbone amides of His106 and Val107 of CDR3HC, a main-chain (carbonyl) to main-chain (amide) hydrogen bond is formed between Ser679NPCt and His108CDR3HC, and the N∊1 atom of Trp104CDR3HC donates a hydrogen bond to the main-chain carbonyl of Ser679NPCt.
CDR3HC fits into a concave crevice in the antigen between the N-terminal α-helix hairpin and the C-terminal lobe of the protein. 16 residues on the surface of the antigen collectively account for ∼950 Å2 of BSA (Fig. 4 ▸ a). All but four of them (Gln653, Lys657, Thr658 and His738) are conserved among the five virus strains (Fig. 4 ▸ b). Several large side chains, including those of Tyr652, Gln653, Tyr668 and Phe678, interact exclusively with CDR3HC, while others (i.e. those of Thr658, Gln659, Gly660, Asp682, Met732, Gln736, His738 and Arg739) engage CDR1HC, CDR2HC and CDR3LC. Main-chain atoms mediate a significant number of interactions, which further rationalizes the pan-specificity of MJ20. A set of amino acids wedges between CDR3HC and the other CDR loops: these are Leu656, Lys657, Pro661, Ser681, Gly683 and Leu735.
Figure 4.

The topology and evolutionary conservation of the BDBV NPCt surface involved in the interaction with MJ20. (a) BDBV NPCt is represented schematically by a green ribbon, with the solvent-accessible surface (within the complex with MJ20) shown in a semitransparent manner. The N- and C-termini are marked for clarity. The MJ20-binding concave crevice is located centrally between the N-terminal α-helix hairpin and the C-terminal lobe of the protein. The residues forming the largest contacts with MJ20HC are represented by blue spheres, while eight conserved residues targeted by alanine scanning (see text) are shown as orange sticks. (b) The same view of the BDBV NPCt surface, showing amino-acid conservation among the five strains of the virus. The MJ20 interface (delineated by dashed lines) is shown again in the inset, with a stick representation of the critical seven amino acids from the CDR3HC loop of MJ20.
An important, unexpected feature of the complex is a large cavity buried at the interface containing a constellation of ordered water molecules (Fig. 5 ▸). Many of these waters are clearly defined in electron density owing to the high resolution of the data, with low temperature factors, filling the space between the antigen and the HC, mostly CDR3HC. The final atomic model contains approximately 15 such water molecules. Assuming that the average entropic cost of burying a water molecule at the interface is ∼1.5–2 kcal mol−1, the total −TΔS associated with trapping solvent within the sFab–antigen interface can be estimated at ∼25 kcal mol−1, which is in good agreement with the calorimetric measurements.
Figure 5.

Water molecules buried at the MJ20–antigen interface. The antigen is shown using a ribbon representation (top of the figure) and its molecular surface is shown. The variable domain of the MJ20 sFab is shown at the bottom, with the surface represented by a mesh. The 15 most buried water molecules are shown as gray spheres.
To determine the roles of selected antigen side chains in the interaction, we generated eight single-site mutants targeting the residues that bury significant surface upon binding MJ20. The variants (which were generated for the EBOV strain NPCt) were Y652A, L656A, F678A, S679A, S681A, F731A, M732A and L735A. Each protein was assessed by differential scanning fluorimetry to determine the midpoints of thermal denaturation. The F678A and F731A variants were found to be unfolded, as no denaturation transition was detected. Although partly exposed, both side chains are clearly vital to the integrity of the hydrophobic core of the antigen. The thermodynamic parameters of the interaction with MJ20 were then evaluated for the remaining variants (Table 3 ▸). Interestingly, none showed any dramatic impediment to the affinity for MJ20, even though in every case (with the exception of Ser681) the loss of the side chain significantly reduced the buried accessible surface. The two variants showing the most significant impairment of interaction with MJ20 were M732A (a tenfold decrease in affinity) and L735A (a 30-fold decrease). In both cases the side chains of the respective amino acids rather than the main-chain atoms make contact with MJ20.
4. Conclusion
Using an automated phage-display selection platform, we generated a synthetic Fab (MJ20) capable of binding to the C-terminal domains of the nucleoprotein antigens (NPCts) from all five known strains of the Ebola virus with dissociation constants (K d) of 3–50 nM. Analysis of the crystal structure of the complex of MJ20 with the NPCt derived from BDBV revealed that the pan-specificity of the sFab was primarily owing to the sequence and structure of the CDR3HC loop, which interacted with a specific structural feature on the surface of NPCt, i.e. a crevice between two subdomains of the antigen, and exploited numerous backbone interactions, thereby avoiding strain-specific amino acids on the surface of the antigen. This result attests to the potential of the in vitro selection of synthetic Fabs as a powerful tool in the custom design of reagents that could be used in diagnostic and therapeutic applications in lieu of naturally generated antibodies.
Supplementary Material
PDB reference: C-terminal domain of Ebola virus (Bundibugyo) nucleoprotein in complex with Fab fragment, 5vkd
Acknowledgments
Bacterial expression of the recombinant Fabs was carried out at the VCU Massey Cancer Center Biological Macromolecule Shared Resource, supported in part by funding from NIH–NCI Cancer Center Support Grant P30 CA016059. We are indebted to Dr Darrell Peterson for his assistance with this work. The X-ray data-collection experiments were performed on beamlines operated by the South-Eastern Region Collaborative Access Team (SER-CAT) of the Advanced Photon Source. Supporting institutions of SER-CAT may be found at http://www.ser-cat.org/members.html. We thank Ms Natalya Olekhnovich for technical assistance.
Funding Statement
This work was funded by Ben and Catherine Ivy Foundation grant . National Cancer Institute grant P30 CA016059. U.S. Department of Energy, Office of Biological and Environmental Research grant DE-AC02-06CH11357. U.S. Department of Energy, Office of Science grant DE-AC02-06CH11357.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Bailey, L. J., Sheehy, K. M., Hoey, R. J., Schaefer, Z. P., Ura, M. & Kossiakoff, A. A. (2014). J. Immunol. Methods, 415, 24–30. [DOI] [PMC free article] [PubMed]
- Baker, L. E., Ellena, J. F., Handing, K. B., Derewenda, U., Utepbergenov, D., Engel, D. A. & Derewenda, Z. S. (2016). Acta Cryst. D72, 49–58. [DOI] [PMC free article] [PubMed]
- Baskaran, K., Duarte, J. M., Biyani, N., Bliven, S. & Capitani, G. (2014). BMC Struct. Biol. 14, 22. [DOI] [PMC free article] [PubMed]
- Biedenkopf, N. & Hoenen, T. (2017). Methods Mol. Biol. 1628, 119–131. [DOI] [PubMed]
- Bloom, D. E., Black, S. & Rappuoli, R. (2017). Proc. Natl Acad. Sci. USA, 114, 4055–4059. [DOI] [PMC free article] [PubMed]
- Boisen, M. L., Hartnett, J. N., Goba, A., Vandi, M. A., Grant, D. S., Schieffelin, J. S., Garry, R. F. & Branco, L. M. (2016). Annu. Rev. Virol. 3, 147–171. [DOI] [PMC free article] [PubMed]
- Broadhurst, M. J., Brooks, T. J. & Pollock, N. R. (2016). Clin. Microbiol. Rev. 29, 773–793. [DOI] [PMC free article] [PubMed]
- Chan, J. F. W., Sridhar, S., Yip, C. C. Y., Lau, S. K. P. & Woo, P. C. Y. (2017). J. Microbiol. 55, 172–182. [DOI] [PMC free article] [PubMed]
- Changula, K., Yoshida, R., Noyori, O., Marzi, A., Miyamoto, H., Ishijima, M., Yokoyama, A., Kajihara, M., Feldmann, H., Mweene, A. S. & Takada, A. (2013). Virus Res. 176, 83–90. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Coltart, C. E. M., Lindsey, B., Ghinai, I., Johnson, A. M. & Heymann, D. L. (2017). Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160297. [DOI] [PMC free article] [PubMed]
- Dziubańska, P. J., Derewenda, U., Ellena, J. F., Engel, D. A. & Derewenda, Z. S. (2014). Acta Cryst. D70, 2420–2429. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Feldmann, H. & Geisbert, T. W. (2011). Lancet, 377, 849–862. [DOI] [PMC free article] [PubMed]
- Fellouse, F. A., Esaki, K., Birtalan, S., Raptis, D., Cancasci, V. J., Koide, A., Jhurani, P., Vasser, M., Wiesmann, C., Kossiakoff, A. A., Koide, S. & Sidhu, S. S. (2007). J. Mol. Biol. 373, 924–940. [DOI] [PubMed]
- Fellouse, F. A., Wiesmann, C. & Sidhu, S. S. (2004). Proc. Natl Acad. Sci. USA, 101, 12467–12472. [DOI] [PMC free article] [PubMed]
- Formenty, P., Boesch, C., Wyers, M., Steiner, C., Donati, F., Dind, F., Walker, F. & Le Guenno, B. (1999). J. Infect. Dis. 179, S120–S126. [DOI] [PubMed]
- Groseth, A., Charton, J. E., Sauerborn, M., Feldmann, F., Jones, S. M., Hoenen, T. & Feldmann, H. (2009). Virus Res. 140, 8–14. [DOI] [PMC free article] [PubMed]
- Hoenen, T. (2018). Methods Mol. Biol. 1604, 237–245. [DOI] [PubMed]
- Hoenen, T., Shabman, R. S., Groseth, A., Herwig, A., Weber, M., Schudt, G., Dolnik, O., Basler, C. F., Becker, S. & Feldmann, H. (2012). J. Virol. 86, 11779–11788. [DOI] [PMC free article] [PubMed]
- Hornsby, M. et al. (2015). Mol. Cell. Proteomics, 14, 2833–2847. [DOI] [PMC free article] [PubMed]
- Koide, A., Tereshko, V., Uysal, S., Margalef, K., Kossiakoff, A. A. & Koide, S. (2007). J. Mol. Biol. 373, 941–953. [DOI] [PMC free article] [PubMed]
- Kuhn, J. H. et al. (2013). Arch Virol. 159, 1229–1237.
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Marcon, E. et al. (2015). Nature Methods, 12, 725–731. [DOI] [PubMed]
- Miersch, S. & Sidhu, S. S. (2012). Methods, 57, 486–498. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Mukherjee, S., Ura, M., Hoey, R. J. & Kossiakoff, A. A. (2015). J. Mol. Biol. 427, 2707–2725. [DOI] [PMC free article] [PubMed]
- Nanbo, A., Watanabe, S., Halfmann, P. & Kawaoka, Y. (2013). Sci. Rep. 3, 1206. [DOI] [PMC free article] [PubMed]
- Noda, T., Kolesnikova, L., Becker, S. & Kawaoka, Y. (2011). J. Infect. Dis. 204, S878–S883. [DOI] [PMC free article] [PubMed]
- Paduch, M., Koide, A., Uysal, S., Rizk, S. S., Koide, S. & Kossiakoff, A. A. (2013). Methods, 60, 3–14. [DOI] [PMC free article] [PubMed]
- Rochlin, I., Faraji, A., Ninivaggi, D. V., Barker, C. M. & Kilpatrick, A. M. (2016). Nature Commun. 7, 13604. [DOI] [PMC free article] [PubMed]
- Roddy, P., Howard, N., Van Kerkhove, M. D., Lutwama, J., Wamala, J., Yoti, Z., Colebunders, R., Palma, P. P., Sterk, E., Jeffs, B., Van Herp, M. & Borchert, M. (2012). PLoS One, 7, e52986. [DOI] [PMC free article] [PubMed]
- Sheffield, P., Garrard, S. & Derewenda, Z. (1999). Protein Expr. Purif. 15, 34–39. [DOI] [PubMed]
- Sherwood, L. J. & Hayhurst, A. (2013). PLoS One, 8, e61232. [DOI] [PMC free article] [PubMed]
- Tabachnick, W. J. (2016). Annu. Rev. Virol. 3, 125–145. [DOI] [PubMed]
- Watanabe, S., Noda, T. & Kawaoka, Y. (2006). J. Virol. 80, 3743–3751. [DOI] [PMC free article] [PubMed]
- Ye, J. D., Tereshk, V., Frederiksen, J. K., Koide, A., Fellouse, F. A., Sidhu, S. S., Koide, S., Kossiakoff, A. A. & Piccirilli, J. A. (2007). J. Biomol. Struct. Dyn. 24, 688–689.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: C-terminal domain of Ebola virus (Bundibugyo) nucleoprotein in complex with Fab fragment, 5vkd