

Abstract
We recently demonstrated that offspring delivered to baboons deprived of estrogen during the second half of gestation exhibited insulin resistance prior to onset of puberty. Because gonadal hormones have a profound effect on insulin action and secretion in adults, we determined whether insulin resistance is retained after initiation of gonadal secretion of testosterone and estradiol. Glucose tolerance tests were performed in postpubertal baboon offspring of untreated and letrozole-treated animals (serum estradiol reduced >95 %). Basal fasting levels of insulin (P < 0.05) and peak 1 min and 1 + 3 + 5 min levels of glucose after glucose tolerance tests challenge (P < 0.03) were greater in offspring delivered to letrozole-treated, estrogen-deprived baboons than untreated animals. Moreover, the value for the HOMA-IR, an accepted index of insulin resistance, was 2-fold greater (P < 0.05) in offspring delivered to baboons treated with letrozole than in untreated animals. Collectively these results support the proposal that estrogen normally has an important role in programming mechanisms in utero within the developing fetus that lead to insulin sensitivity after birth.
Keywords: Estrogen, Insulin sensitivity, Offspring, Primate
Introduction
We recently demonstrated that baboon offspring delivered to mothers deprived of estrogen during the second half of gestation exhibited insulin resistance during their first three years of life, i.e. prior to onset of puberty [1]. Thus, the rise in serum glucose and plasma insulin levels induced by intravenous glucose challenge was greater in prepubertal offspring delivered to baboon mothers treated with the aromatase inhibitor letrozole, than in animals exposed in utero to the normal elevation in estrogen. Therefore, we proposed that estrogen normally has an important role in utero during primate pregnancy in programming mechanisms within the developing fetus that lead to insulin responsivity and the capacity to metabolize glucose in offspring after birth.
It is well established that in adult mammals, gonadal hormones significantly regulate pancreatic islet beta-cell integrity and function as well as the secretion and action of insulin important for the control of glucose metabolism [reviewed in 2, 3]. For example, androgens stimulate whereas estrogen inhibits oxidative stress-induced pancreatic beta cell apoptosis [4–6]. Estrogen has also been shown to improve glucose-stimulated insulin secretion [7], decrease the incidence of diabetes in postmenopausal women [8, 9] and restore insulin sensitivity and glucose metabolism in ovariectomized rhesus monkeys fed with a high-fat diet [10]. Estrogen receptor α is abundantly expressed in insulin-sensitive target tissues, including skeletal muscle [11–13] and estradiol stimulates insulin sensitivity and enhances glucose tolerance in skeletal muscle of adult mice [14–16].
Because gonadal hormones have a profound effect on insulin action and secretion, the present study determined whether the insulin resistance and glucose intolerance exhibited by offspring of estrogen suppressed mothers during the prepubertal period is retained after initiation of gonadal secretion of testosterone and estradiol. Therefore, in the present study, intravenous glucose tolerance tests (iv GTT) were performed in postpubertal baboon offspring born to mothers untreated or treated with the aromatase inhibitor letrozole.
Materials and methods
Animals
Female baboons (Papio anubis), originally obtained from the Southwest National Primate Research Center, San Antonio, TX, were housed individually in large primate cages in air-conditioned rooms with a 12 h/12 h light/dark lighting cycle and fed standard primate chow (Harlan Primate Diet, Madison, WI) twice daily, fresh fruits and vitamins daily and water ad libitum. Female baboons were paired with male baboons for 5 days at mid cycle and pregnancy confirmed by ultrasound. Pregnant baboons were then either untreated or treated daily between days 100 and 165–175 of gestation (term = 184 days) with the aromatase inhibitor letrozole (4,4-[1,2,3-triazol-1 yl-methylene]bis-benzonitrate, Novartis Pharma AG, Basel, Switzerland; 115 μg/kg body weight/day, via maternal sc injection in 1.0 ml sesame oil). Blood samples (2–3 ml) were obtained at 3–5 day intervals during the second half of gestation from a peripheral maternal saphenous vein after brief restraint and sedation with ketamine HCl (10 mg/kg body weight, im) and serum estradiol levels quantified by immulite radioimmunoassay [17]. On days 165–180 of gestation, offspring were either delivered spontaneously or were anesthetized with isoflurane and delivered by cesarean section to synchronize the timing of delivery. Baboon newborns that had been untreated (7 females, 4 males) or treated in utero with letrozole (7 females, 2 males) were then left with and nursed by their mothers for 8 months at which time they were weaned and placed in pairs in cages adjacent to their respective mothers and fed standard primate chow (Harlan Primate Diet) twice daily, fresh fruits and vitamins daily and water ad libitum.
Every 2–6 months thereafter, baboon offspring were briefly sedated with ketamine HCl (10 mg/kg body weight, im), body weight measured, and blood samples (2–3 ml) obtained following sedation with ketamine from a peripheral saphenous vein for the purpose of quantifying serum estradiol, testosterone, dehydroepiandrosterone and a panel of inflammatory cytokines. The use of baboons for this study was approved by the Institutional Animal Care and Use Committees of Eastern Virginia Medical School and the University of Maryland, School of Medicine.
Glucose tolerance test in postpubertal baboon offspring at 4¼–8 years of age
An iv glucose tolerance test was performed essentially as described previously [1], according to the established method of Overkamp et al. [18], sequentially (1–4 times) on each baboon offspring at 6–12 month intervals after onset of puberty between the ages of 4¼ (females)/5 (males) years and 8 years of age. Intravenous glucose tolerance test results in individual animals did not change between 4¼–8 years of age and thus the data obtained from the individual iv GTT performed was averaged to yield a single value for each animal. Briefly, baboons were fasted overnight and at 08 : 00 h the following morning sedated with ketamine HCl (initially 5–10 mg/kg body weight, im; then 2 mg/kg body weight, iv) and positioned on their left side on a 37 °C heating pad. Dextrose (0.25 g/kg body weight) was administered as a bolus injection via a 21 gauge needle into an antecubital vein at time 0 h. Blood samples (2.5 ml each) were obtained via a sterile catheter (21 gauge) inserted into a peripheral saphenous vein at −2 (i.e. basal fasting), 1, 3, 5, 10, 20, 40, 60 and 90 min before/after dextrose administration. Blood glucose levels were determined via an iStat Portable Clinical Analyzer (Model #210003, Abbott Labs, East Windsor, NJ) on 0.1 ml of blood and insulin determined in stored (−20 °C) plasma samples using a solid-phase chemiluminescent immunometric assay via an Immulite System (Siemens Healthcare Diagnostics, Tarrytown, NY) as described previously [1].
Expression of insulin signalling molecules in skeletal muscle before and after iv bolus injection of dextrose in baboon offspring at 12–14 years of age
Baboons (all female) were fasted overnight and at 08 : 00 h the following morning were sedated with ketamine HCl (0.1 mg/kg): propofol (0.2 mg/kg) infused (0.25 ml saline/min) via an antecubital vein. A biopsy of vastus lateralis skeletal muscle was then obtained approximately 10 min before and 30 min after iv administration at time 0 h of a bolus of dextrose (0.25 g/kg body weight) and frozen/stored (−80 °C). Blood samples (2.5 ml each) were obtained via a catheter (21 gauge) inserted into a peripheral saphenous vein at 0 (i.e. basal fasting), 1, 3, 5, 10, 20, and 30 min before/after dextrose administration and blood glucose and plasma insulin levels determined as outlined above.
Western immunoblot
Samples of skeletal muscle were homogenized on ice in phosphate-buffered saline containing 1 % cholic acid, 0.1 % SDS, 1 mM EDTA (Sigma-Aldrich, St Louis, MO) and a protease inhibitor cocktail, as described previously [1]. Briefly, samples (25–50 μg protein) were heated to 95° C in Laemmli buffer, cooled, centrifuged (800 × g), loaded onto 10 % SDS-polyacrylamide gels (PAGE) and electrophoresed using a Bio-Rad Mini-Protean electrophoresis chamber (Bio-Rad Laboratories, Richmond, CA) in 25 mM Tris (pH 8.3), 192 mM glycine and 0.1 % SDS. Proteins were wet-transferred onto an Immobilon-P membrane (Millipore Corp., Bedford, MA), blocked 1 h at room temperature with 5 % BSA in 10 mM Tris-HCl, pH 7.5, 150 M NaCl and 0.2 % Tween 20 (TBST), and then incubated overnight (4° C) with primary rabbit polyclonal antibodies to: anti-AKT (1 : 500 dilution, Cell Signaling, Danvers, MA), anti-pAKT-S473 (1 : 500, Cell Signaling), anti-pAKT-T308 (1 : 500, Abcam, Cambridge, MA), anti-Glut1 (1 : 500, Abcam), and anti-pGlut4-S488 (1 : 1000, Abcam). After washing in TBST, membranes were incubated for 1 h at room temperature with horseradish peroxidase (HRP)-labeled secondary antibody (Serotec, UK) in TBST containing 5 % BSA, washed in TBST, developed with enhanced chemiluminescence (GE Healthcare, Pittsburgh, PA) and exposed to Fuji Super RX medical x-ray film (Fujifilm Medical Systems, USA, Inc., Roselle, IL). Band intensities were quantified using Image J software (National Institutes of Health). Blots were then stripped and re-probed using HRP-conjugated GAPDH (1 : 5000, Abcam) as an internal loading control and results, arbitrary densitometric units/μg protein, expressed as a ratio to GAPDH. Specificity of the primary antibodies was determined previously [1].
Assay of serum steroids and cytokines
Serum levels of estradiol and testosterone were determined by radioimmunoassay using an automated chemiluminescent immunoassay system (Immulite; Siemens Healthcare Diagnostics, Deerfield, IL) as described previously [17, 19]. A nonhuman primate Cytokine Array Q1 Multiple ELISA (Ray Biotech, Norcross, GA) was used for simultaneous quantitative measurement of interleukin (IL)-1β, IL-5, IL-6, IL-16, IL-12p70, granulocyte macrophage-colony stimulating factor (GM-CSF), interferon gamma and tumor necrosis factor alpha (TNFα). Briefly, using reagents and materials provided by the manufacturer, serum samples from 5 untreated and 5 letrozole-treated 6–8 year-old offspring and cytokine standards were placed onto 75 × 25 mm glass slides pre-coated with identical antibody arrays. After blocking, samples were incubated overnight, washed to remove non-specific proteins, and then incubated with biotinylated detection antibodies and a streptavidin-conjugated fluor per manufacturer’s instructions. Slides were then shipped to Ray Biotech for detection and quantification of multiple signals using a fluorescence laser scanner. Computer generated signal intensities for standards and serum samples were created and used to construct standard curves for each cytokine and calculation of serum cytokine levels.
Statistical analysis
Data are expressed as mean ± SE and were analyzed by the Student t test for independent observations or the Mann-Whitney “t” test when standard deviations between the groups were different or by Two-Way ANOVA with replication using SAS statistical software (SAS Institutes).
Results
Serum steroid hormone levels
In untreated baboons, maternal peripheral serum estradiol levels rose from approximately 1.5 ng/ml on days 85–120 of gestation to approximately 4.0 ng/ml by day 175. Within 48–72 h of the onset of letrozole treatment on day 100, maternal serum estradiol levels decreased to and remained at approximately 0.1–0.2 ng/ml. Consequently, serum estradiol levels in maternal saphenous vein of mothers 2–3 days before delivery of offspring of the current study were >95 % lower (P < 0.001) in animals treated with letrozole (0.2 ± 0.1 ng/ml) than in animals untreated (3.7 ± 0.4 ng/ml; Table 1). In a contemporaneous group of baboons studied at the time of delivery on days 165–180 of gestation, serum estradiol concentrations in blood delivered to the fetus (i.e. umbilical vein) of letrozole-treated baboons (0.04 ± 0.01 ng/ml) were also only 5 % of that (P < 0.001) in untreated animals (0.68 ± 0.26 ng/ml, Table 1). In contrast, umbilical vein serum testosterone levels on days 165–175 in letrozole-treated baboons (3.2 ± 0.7 ng/ml) were 3-fold greater (P < 0.01) than in untreated controls (0.8 ± 0.2 ng/ml; Table 1).
Table 1.
Serum levels (ng/ml) of estradiol in maternal saphenous vein of baboons of the current study and of estradiol and testosterone in umbilical vein in late gestation in a contemporaneous group of baboons untreated or treated with letrozolea
| Maternal | Umbilical vein
|
||
|---|---|---|---|
| Estradiol | Estradiol | Testosterone | |
| Untreated | 3.7 ± 0.5 | 0.68 ± 0.26 | 0.8 ± 0.2 |
| Letrozole | 0.2 ± 0.1* | 0.04 ± 0.01* | 3.2 ± 0.7* |
P < 0.05 vs untreated
Baboons untreated (n = 11) or treated on days 100–165/175 of gestation (term = 184 days) with letrozole (115 μg/kg body weight/day via maternal sc injection, n = 9)
Postnatal development
Body weight of baboon offspring progressively increased (P < 0.01) throughout postnatal life in a comparable manner in animals untreated or treated in utero with letrozole (Tables 2 and 4). The greater than 1 : 1 ratio of female to male animals in the current study is not the result of a disproportionate loss of male vs female fetuses but rather reflects our concomitant interest in and study of ovarian development. Thus, as previously shown [19] at 3½–4 years of age, female offspring of untreated and letrozole-treated baboons exhibited onset of menstrual cycles characterized by comparable increases in serum levels of estradiol during the late follicular phase (range 75–125 pg/ml) and 24–48 h prior to the ovulatory surge (range 200–350 pg/ml) of LH. At 4½–5 years of age, male offspring of baboons untreated or treated with letrozole exhibited an increase in serum testosterone levels from <0.5 to >2 ng/ml, reflecting the onset of puberty and values remained relatively stable thereafter.
Table 2.
Body weight of and fasting blood glucose and plasma insulin levels in postpubertal baboon offspring
| Treatment | Age (yrs) | Body wt (kg) | Glucose (mg/dl) | Insulin (μIU/ml) | Ratio Glucose/Insulin |
|---|---|---|---|---|---|
| Untreated | 5.4 ± 0.3 | 13.4 ± 0.8 | 68 ± 2 | 6.6 ± 1.0 | 13.8 ± 3.4 |
| Letrozole | 5.7 ± 0.3 | 14.3 ± 1.2 | 67 ± 2 | 12.0 ± 2.2* | 6.8 ± 1.3** |
Values are expressed as means ± SE on the day of iv glucose tolerance test in offspring from baboons untreated (n = 11) or treated on days 100–165/175 of gestation (term = 184 days) with letrozole (115 μg/kg body weight/day via maternal sc injection, n = 9)
P < 0.03 vs. untreated (Student “t” test)
P < 0.03 vs. untreated (Mann-Whitney “t” test)
Table 4.
Body weight of and fasting blood glucose and plasma insulin levels, HOMA-IR and glucose and insulin AUC after iv bolus injection of dextrose in 12–14 year-old baboon offspring
| Treatment | Age (yrs) | Body wt (gm) | Glucose (mg/dl) | Insulin (μIU/ml) | Glucose AUC (mg/dl) | Insulin AUC (μIU/ml) | HOMA-IR |
|---|---|---|---|---|---|---|---|
| Untreated | 12.5 ± 0.8 | 15.5 ± 0.6 | 72 ± 2 | 10.5 ± 4.3 | 515 ± 21 | 253 ± 80 | 1.9 ± 0.8 |
| Letrozole | 13.0 ± 1.1 | 16.5 ± 0.3 | 74 ± 4 | 9.4 ± 3.2 | 498 ± 22 | 198 ± 57 | 1.8 ± 0.6 |
Values are expressed as means ± SE after an iv bolus of dextrose to female offspring from baboons untreated (n = 4) or treated on days 100–165/175 of gestation (term = 184 days) with letrozole (115 μg/kg body weight/day via maternal sc injection, n = 4)
Supplementary Table 1 shows that the mean (± SE) levels of proinflammatory and inflammatory cytokines were similar at 6–8 years of age in offspring of baboons untreated or treated in utero with letrozole.
Glucose tolerance test in 41/4–8 year-old postpubertal offspring
The results of iv GTT appeared to be comparable in male and female offspring and thus the data were combined and data presented as an overall mean for offspring untreated or treated in utero with letrozole.
As seen in Table 2, basal fasting levels of glucose immediately prior to the glucose (dextrose) tolerance test were not significantly different in offspring from baboons untreated (68 ± 2 mg/dl) or treated throughout the second half of gestation with letrozole (67 ± 2 mg/dl). However, basal fasting insulin levels were approximately 2-fold greater (P = 0.03) in offspring treated prenatally with letrozole (12.0 ± 2.2 μIU/ml) than in untreated animals (6.6 ± 1.0 μIU/ml). Thus, the fasting glucose:insulin ratio in untreated offspring (13.8 ± 3.4) was approximately 2-fold lower (P < 0.03) in offspring of letrozole-treated baboons (6.8 ± 1.3, Table 2).
The overall mean (± SE) patterns of glucose and insulin levels during the glucose challenge test in offspring of untreated and letrozole-treated baboons are shown in Fig. 1a, b. Within 1 min of a bolus iv injection of dextrose, blood glucose and plasma insulin increased to peak levels, then rapidly declined and were restored within 60–90 min to pre-challenge levels. However, in baboon offspring treated in utero with letrozole, the peak 1 min post challenge levels of glucose (213 ± 6 mg/dl, Fig. 1c) and the net elevation (i.e. 1 min peak minus fasting baseline) in glucose (144 ± 6 mg/dl; Fig. 1c) were greater (P < 0.03) than respective values in untreated animals (195 ± 5 mg/dl and 127 ± 6 mg/dl, respectively). Similarly, the peak 1 min level (Fig. 1d) and the net elevation in insulin (Fig. 1d) in offspring of letrozole-treated animals (104.0 ± 13.3 μIU/ml; 91.9 ± 12.3 μIU/ml, respectively) were approximately 35 % greater than respective values in untreated offspring (71.8 ± 10.7 μIU/ml and 65.2 ± 10.6 μIU) although the latter differences did not achieve statistical significance (P = 0.08 and P = 0.12, respectively).
Fig. 1.

a, b: Serum levels and patterns of blood glucose a and plasma insulin b before and after iv administration at time 0 min of a bolus of dextrose in 4¼–8 year-old postpubertal baboons delivered to mothers untreated (n = 11) or treated in utero with letrozole (n = 9) as described in legend to Table 1. Values are the means ± SE of the average of 1–4 glucose challenge tests performed longitudinally (i.e. every 6–12 months) in each animal at 4¼ to 8 years of postnatal life. c, d: Blood glucose c and plasma insulin d levels, expressed as 1 min peak and 1 min peak minus fasting baseline levels after administration of an iv bolus of dextrose for the baboon offspring in which the patterns of glucose and insulin are shown in Panels A and B. *P = 0.03 vs. untreated. #P = 0.08 vs. untreated
As seen in Table 3, when the 1, 3 and 5 min levels of glucose were averaged and corrected for baseline level and also calculated as area under the curve (AUC, 1–5 min), respective mean levels were greater (P = 0.06, P < 0.05, P = 0.07, respectively) in baboon offspring treated prenatally with letrozole (185 ± 5 mg/dl, 116 ± 4 mg/dl, 733 ± 20 mg/dl) than in offspring untreated. However, the mean 1, 3 and 5 min level of insulin, mean level of insulin at 1, 3 and 5 min minus fasting and insulin AUC (1–5 min) in offspring of letrozole-treated baboons (72.3 ± 9.6 μIU/ml; 60.2 ± 8.8 μIU/ml; and 281 ± 38 μIU/ml, respectively) were not significantly different from respective values in offspring of untreated animals (Table 3).
Table 3.
Blood glucose and plasma insulin levels in 4¼–8 year-old postpubertal baboon offspring after an iv glucose tolerance test
| Glucose (mg/dl)
|
Insulin (μIU/ml)
|
|||||
|---|---|---|---|---|---|---|
| Treatment | Mean 1 +3 +5 min | Mean 1 + 3 +5 min-fasting | Glucose (AUC)1–5 min | Mean 1 + 3+ 5 min | Mean 1 +3 + 5 min-fasting | Insulin (AUC) 1–5 min |
| Untreated | 174 ± 3 | 105 ± 3 | 692 ± 10 | 54.6 ± 11.6 | 48.3 ± 11.6 | 210 ± 44 |
| Letrozole | 185 ± 5a | 116 ± 4* | 733 ± 20b | 72.3 ± 9.6 | 60.2 ± 8.8 | 281 ± 38 |
Values are the means ± SE of the average levels of blood glucose and plasma insulin at 1, 3 and 5 min and area under curve (AUC) for glucose and insulin, after an iv bolus of dextrose to postpubertal baboon offspring untreated (n = 11) or treated in utero with letrozole (n = 9)
P < 0.05 vs. untreated
P = 0.06 vs. untreated
P = 0.07 vs. untreated
The overall mean value for the HOMA-IR, an accepted index of insulin resistance, was 2-fold greater (P < 0.05) in offspring delivered to baboons treated with the aromatase inhibitor letrozole (2.09 ± 0.47) than in untreated animals (1.11 ± 0.17, Fig. 2).
Fig. 2.
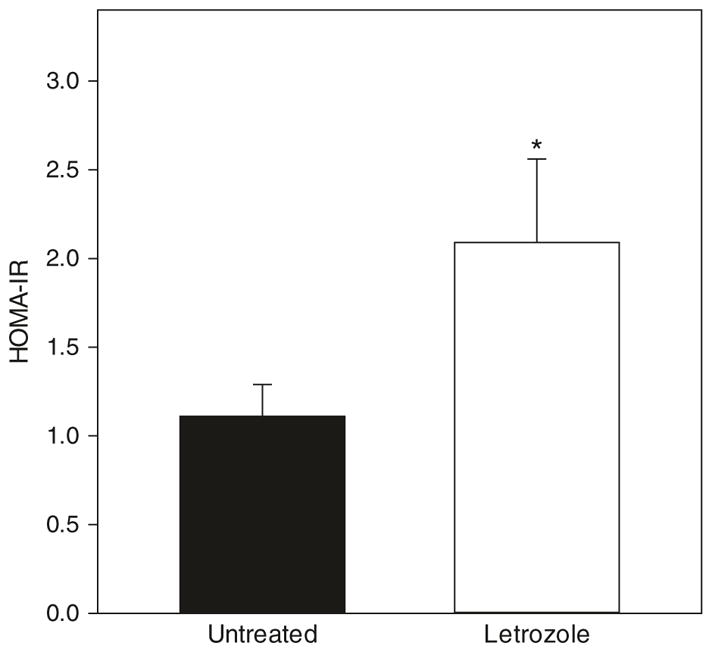
HOMA-IR (basal fasting glucose x basal fasting insulin levels ÷ 405) for the same 4¼ to 8 year-old baboon offspring in which serum glucose and insulin levels are shown in Fig. 1. *P < 0.05 vs. untreated
As seen in Table 4, basal fasting levels of glucose and insulin immediately prior to an iv bolus injection of dextrose were similar in 12–14 year-old offspring of baboons untreated (72 ± 2 mg/dl and 10.5 ± 4.3 μIU/ml) or treated throughout the second half of gestation with letrozole. Consequently, the HOMA-IR was also similar in the older offspring from baboons untreated or treated with letrozole. Table 4 also shows that the respective values for areas under the curve for glucose (515 ± 21 mg/dl) and insulin (253 ± 80 μIU/ml) were similar in 12–14 year-old baboon offspring untreated or treated prenatally with letrozole.
Skeletal muscle of 12–14 year-old baboon offspring untreated or treated with letrozole in utero expressed AKT phosphorylated at serine 473 (pAKTser) or threonine 308 (pAKTthr), total AKT, and Glut4 phosphorylated at serine 488 (pGlut4) and Glut1 and representative examples of these components of the insulin signalling pathway are shown in Supplementary Fig 1. As seen in Fig 3, skeletal muscle levels of total AKT, pAKTser, pAKTthr, pGlut4 and Glut1 expressed as a ratio to GAPDH were similar in the basal state and after dextrose/insulin challenge and not altered by letrozole treatment. However, two-way ANOVA confirmed that regardless of treatment in utero, compared to basal state, the ratio of pAKTser/total AKT was increased (P < 0.05) following the dextrose challenge.
Fig. 3.

Skeletal muscle insulin signaling molecule protein levels before (0) and 30 min (30) after bolus iv injection of dextrose to 12–14 year-old offspring delivered to baboons untreated (n = 4) or treated with letrozole (n = 4). Values are the means (± SE). * Regardless of treatment, mean value at 30 min exceeds that at 0 min (P < 0.05)
Discussion
We previously showed that offspring delivered to baboon mothers in which estrogen levels were suppressed during the second half of gestation exhibited glucose intolerance and insulin resistance during their first 3 years of life and thus prior to the onset of puberty and gonadal hormone secretion [1]. The results of the present study demonstrate that the offspring of estrogen-deprived baboons continue to exhibit insulin resistance/glucose intolerance after puberty. Thus, the peak rise in blood glucose levels induced by a glucose challenge in 41/4–8 year-old postpubertal offspring delivered to baboons in which estrogen had been suppressed throughout the second half of pregnancy was greater than in animals exposed in utero to the normal elevation in estrogen. In addition, the exaggerated rise in glucose levels of estrogen-suppressed baboon offspring was associated with higher basal fasting levels of plasma insulin and the HOMA-IR. Collectively, these results suggest that peripheral glucose uptake and/or metabolism were impaired, a condition consistent with insulin resistance. Therefore, because insulin resistance was maintained through age 8 in the postpubertal period and was not reversed by exposure of offspring to endogenous estradiol (females) or testosterone (males) production, we propose as originally put forth by our group [1] that estrogen normally has an important role in programming mechanisms in utero within the developing fetus that lead to insulin sensitivity and the capacity to metabolize glucose after birth.
Others have shown that ERα-null mice develop insulin resistance, glucose intolerance and decreased glucose uptake in skeletal muscle [20–24] and that administration of phytoestrogen to mice throughout gestation enhanced glucose tolerance in offspring at adulthood [25]. Moreover, impaired insulin sensitivity and glucose metabolism has previously been demonstrated in aromatase-null mice [26, 27] and male and female human offspring and thus also developed in an estrogen-suppressed intrauterine environment [28–30]. Estrogen treatment reversed insulin resistance and normalized serum insulin levels in aromatase-deficient mouse offspring [27].
The extent of insulin resistance based on peak glucose and insulin levels after iv glucose challenge and HOMA-IR elicited in offspring of baboon mothers deprived of estrogen in utero is comparable to findings in humans with pre-diabetes/diabetes. For example, in women with gestational diabetes, postpartum serum glucose levels after an oral GTT were 14 % greater than respective values in normal women and associated with impaired insulin signaling in skeletal muscle [31]. Similarly, in young adult women born preterm, a condition which increases risk for development of diabetes, the insulin sensitivity index was 11.9 % lower and insulin secretion 19.9 % higher than respective values in women born full term leading the authors to conclude that the latter indices of insulin resistance represent an early stage in the pathway to type 2 diabetes [32]. Ibanez et al. [33] also showed that the fasting insulin resistance index which is virtually identical to HOMA-IR was 50 % higher in women with type 2 diabetes or impaired glucose tolerance than in healthy non-diabetic women. Finally, estrogen deprived baboon offspring exhibited lower sensitivity to insulin soon after birth (1), whereas offspring born to untreated mothers remained insulin sensitive throughout the first 8 years of life then develop insulin resistance by 12–14 years of age. Although it remains to be determined whether offspring of estrogen-suppressed mothers will develop type 2 diabetes, humans with aromatase gene deficiency exhibit insulin resistance which frequently progresses to type 2 diabetes early in life [28].
Although we have proposed that estrogen programs mechanisms within the fetus important for glucose homeostasis after birth [1], testosterone levels were elevated in letrozole-treated baboon fetuses and androgens in high levels have been shown to impair insulin sensitivity [34]. Thus, maternal administration of testosterone during early gestation to rhesus monkeys and sheep elicited insulin resistance and glucose intolerance in the offspring [35–37]. However, key aspects of the testosterone-treated sheep model, notably adipocyte size, were not prevented by blocking androgen action, suggesting that the effects of prenatal exposure to excess androgen may in part be mediated by estrogen [38]. Therefore, although we propose that the elevated levels of estrogen in primate pregnancy have a major role in developing insulin sensitivity after birth, it is possible that the insulin resistance induced in offspring of letrozole-treated baboons results from a combination of estrogen-suppression and androgen excess and thus an alteration in the ratio of estrogen and androgen.
As reviewed by Del Prato and colleagues [39, 40], type 2 diabetes is a multifactorial disorder that results from prevalent insulin resistance associated with deficient insulin secretion or a prevalent defect in insulin secretion associated with impaired insulin action. Although there is an extremely high frequency of insulin resistance in type 2 diabetics, insulin resistance alone is insufficient to cause diabetes which ensues following impairment of beta cell function/insulin release. It is well established that in patients with impaired glucose tolerance (i.e. insulin resistant) or in the early stages of type 2 diabetes, first-phase insulin release is almost always lost, whereas second phase insulin secretion is often increased [41, 42]. In the current study, the basal levels of insulin, the HOMA-IR and levels of glucose 1–5 min after dextrose challenge in postpubertal offspring deprived of estrogen in utero significantly exceeded respective values in untreated offspring and collectively indicate that offspring deprived of estrogen in utero are glucose intolerant and insulin resistant. However, the levels of insulin 1–5 min post-dextrose challenge in letrozole-treated offspring were 35 % but not significantly higher than that achieved in untreated offspring that exhibited normal glucose tolerance. Thus, it is possible but remains to be determined that because of the continued resistance of peripheral tissues, e.g. skeletal muscle, to insulin over time in offspring deprived of estrogen in utero, the response of the pancreatic beta cell and thus first stage insulin release which occurs 1–10 min after intravenous glucose challenge has become compromised.
The results of the current study also indicate that the persistence of glucose intolerance and insulin resistance after onset of puberty in offspring of letrozole-treated baboons is likely not the result of an alteration in production of inflammatory cytokines which have been shown to promote insulin resistance and islet cell dysfunction in adult mammals [43, 44]. Thus, the serum levels of IL-1β, IL-5, IL-6, IL-16 and IL-12-P70 as well as GM-CSF, IFNy and TNFα in offspring of letrozole-treated animals were not different from respective values in untreated offspring. Although it is well established that increased food intake/adiposity promotes insulin resistance [45], it is also unlikely that the latter are the cause of insulin resistance in letrozole-treated offspring as growth and body weights were comparable in the untreated and letrozole-treated offspring. Stress is also not likely to be a causative factor as we have recently shown that serum cortisol levels are comparable in offspring born to mothers untreated or treated in utero with letrozole [46]. Because maternal insulin sensitivity and glucose tolerance were normal in letrozole-treated baboons [1], it appears that the defect in insulin sensitivity in offspring of letrozole-treated baboons is also not the result of a change in maternal glucose metabolism.
It is also apparent that by 12–14 years of age, although the number of baboons studied was relatively small, the offspring of untreated baboons have developed insulin resistance. Thus, the mean basal plasma insulin level and HOMA-IR in untreated 12–14 year-old offspring were comparable to respective values in offspring of letrozole-treated animals studied at 4¼–8 years of age. The factors contributing to this increase, remain to be determined but may simply reflect the fact that nonhuman primates and humans exhibit increased incidence of insulin resistance/diabetes with age [47, 48]. Although exercise promotes insulin sensitivity/glucose tolerance, it seems unlikely that the latter accounts for the age-related increase in HOMA-IR in the untreated baboon offspring, since the baboons are comparably housed and difference in exercise would presumably also impact offspring of letrozole-treated animals.
The insulin-dependent signaling pathways in skeletal muscle are upregulated via phosphotidylinositol-3-kinase which phosphorylates Akt at threonine 308, but only partially activates Akt and at serine 473 which fully activates Akt, thereby initiating events important for glucose uptake and metabolism [49, 50]. The results of the current study show that following glucose challenge (increased insulin), the ratio of the levels of skeletal muscle pAKTser to total AKT was significantly increased in 12–14 year-old offspring untreated or treated with letrozole, indicating that this key component of insulin signaling in skeletal muscle was responsive to insulin in both groups of baboons. Phosphorylation of pAKTser was also increased in insulin-sensitive and to a lesser degree in insulin-resistant adult baboons, 30–120 min during euglycemic insulin clamp [51]. However, levels of pGlut 4 which becomes functional following translocation to the membrane and Glut–1 which modulates basal glucose uptake were not altered. Chavez et al. [51] previously showed that expression of AKT, Glut-1 and Glut-4 was also similar in skeletal muscle before and 30–120 min during euglycemic insulin clamp of insulin sensitive and insulin resistant adult baboons. Presumably, the similar level of expression of these molecules and perhaps others throughout the network of pathways governing insulin action, in 12–14 year-old offspring of untreated and letrozole-treated baboons of the current study reflects the absence of a difference in insulin sensitivity in the latter two groups.
In summary, the present study shows that offspring delivered to baboons in which estrogen had been suppressed throughout the second half of pregnancy exhibit insulin resistance/glucose intolerance after puberty. Therefore, because insulin resistance of offspring deprived of estrogen in utero was maintained and not rectified by exposure to the increased levels of gonadal hormones at puberty, we propose as originally put forth by our group [1] that estrogen normally has an important role in programming mechanisms in utero within the developing fetus that lead to insulin sensitivity and the capacity to metabolize glucose after birth.
Supplementary Material
Acknowledgments
The secretarial assistance of Ms. Sandra Huband with preparation of the figures and computer word processing of the manuscript and the assistance of Ms. Marcia Burch and Ms. Lindsey Glenn with the cytokine assay are sincerely appreciated. We thank Novartis Pharma (Basel, Switzerland) for generously providing the aromatase inhibitor letrozole to conduct this study.
Funding
This research was supported by National Institutes of Health Research Grant R01 DK 93950.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12020-016-1145-9) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All baboons were cared for and used strictly in accordance with U.S. Department of Agriculture regulations and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition, National Academy Press, 2011). The Institutional Animal Care and Use Committees of the Eastern Virginia Medical School and the University of Maryland School of Medicine approved the experimental protocol employed in this study.
References
- 1.Maniu A, Aberdeen GW, Lynch TJ, Nadler J, Kim SO, Quon M, Pepe GJ, Albrecht ED. Estrogen deprivation in primate pregnancy leads to insulin resistance in offspring. J Endocrinol. 2016;230(2):171–183. doi: 10.1530/JOE-15-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu S, Mauvais-Jarvis F. Minireview: Estrogenic protection of beta-cell failure in metabolic diseases. Endocrinology. 2010;151:859–864. doi: 10.1210/en.2009-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kautzky-Willer A, Harreiter J, Pacini G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev. 2016;37:278–316. doi: 10.1210/er.2015-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A. 2006;103:9232–9237. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paik SG, Michelis MA, Kim YT, Shin S. Induction of insulin-dependent diabetes by streptozotocin. Inhibition by estrogens and potentiation by androgens. Diabetes. 1982;31:724–729. doi: 10.2337/diab.31.8.724. [DOI] [PubMed] [Google Scholar]
- 6.Nadal A, Alonso-Magdalena P, Soriano S, Quesada I, Ropero AB. The pancreatic beta-cell as a target of estrogens and xenoestrogens: Implications for blood glucose homeostasis and diabetes. Mol Cell Endocrinol. 2009;304:63–68. doi: 10.1016/j.mce.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Godsland IF. Oestrogens and insulin secretion. Diabetologia. 2005;48:2213–2220. doi: 10.1007/s00125-005-1930-0. [DOI] [PubMed] [Google Scholar]
- 8.Margolis KL, Bonds DE, Rodabough RJ, Tinker L, Phillips LS, Allen C, Bassford T, Burke G, Torrens J, Howard BV. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia. 2004;47:1175–1187. doi: 10.1007/s00125-004-1448-x. [DOI] [PubMed] [Google Scholar]
- 9.Karjalainen A, Paassilta M, Heikkinen J, Backstrom AC, Savolainen M, Kesaniemi YA. Effects of peroral and transdermal oestrogen replacement therapy on glucose and insulin metabolism. Clin Endocrinol (Oxf) 2001;54:165–173. doi: 10.1046/j.1365-2265.2001.01208.x. [DOI] [PubMed] [Google Scholar]
- 10.Wagner JD, Thomas MJ, Williams JK, Zhang L, Greaves KA, Cefalu WT. Insulin sensitivity and cardiovascular risk factors in ovariectomized monkeys with estradiol alone or combined with nomegestrol acetate. J Clin Endocrinol Metab. 1998;83:896–901. doi: 10.1210/jcem.83.3.4628. [DOI] [PubMed] [Google Scholar]
- 11.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 13.Wiik A, Ekman M, Johansson O, Jansson E, Esbjornsson M. Expression of both oestrogen receptor alpha and beta in human skeletal muscle tissue. Histochem Cell Biol. 2009;131:181–189. doi: 10.1007/s00418-008-0512-x. [DOI] [PubMed] [Google Scholar]
- 14.Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, Gustafsson JA, Dahlman-Wright K. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol. 2006;20:1287–1299. doi: 10.1210/me.2006-0012. [DOI] [PubMed] [Google Scholar]
- 15.Lundholm L, Bryzgalova G, Gao H, Portwood N, Falt S, Berndt KD, Dicker A, Galuska D, Zierath JR, Gustafsson JA, Efendic S, Dahlman-Wright K, Khan A. The estrogen receptor {alpha}-selective agonist propyl pyrazole triol improves glucose tolerance in ob/ob mice; potential molecular mechanisms. J Endocrinol. 2008;199:275–286. doi: 10.1530/JOE-08-0192e. [DOI] [PubMed] [Google Scholar]
- 16.Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150:2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- 17.Albrecht ED, Aberdeen GW, Pepe GJ. The role of estrogen in the maintenance of primate pregnancy. Am J Obstet Gynecol. 2000;182:432–438. doi: 10.1016/s0002-9378(00)70235-3. [DOI] [PubMed] [Google Scholar]
- 18.Overkamp D, Gautier JF, Renn W, Pickert A, Scheen AJ, Schmulling RM, Eggstein M, Lefebvre PJ. Glucose turnover in humans in the basal state and after intravenous glucose: a comparison of two models. Am J Physiol. 1997;273:E284–E296. doi: 10.1152/ajpendo.1997.273.2.E284. [DOI] [PubMed] [Google Scholar]
- 19.Pepe GJ, Lynch TJ, Albrecht ED. Regulation of baboon fetal ovarian development by placental estrogen: onset of puberty is delayed in offspring deprived of estrogen in utero. Biol Reprod. 2013;89(132):1–8. doi: 10.1095/biolreprod.112.107318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, Dahlman-Wright K, Nilsson S, Gustafsson JA, Efendic S, Khan A. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. 2006;49:588–597. doi: 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- 21.Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, Mohammad L, Henstridge DC, Febbraio MA, Hewitt SC, Korach KS, Bensinger SJ, Hevener AL. Myeloid-specific estrogen receptor alpha deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Natl Acad Sci U S A. 2011;108:16457–16462. doi: 10.1073/pnas.1104533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manrique C, Lastra G, Habibi J, Mugerfeld I, Garro M, Sowers JR. Loss of estrogen receptor alpha signaling leads to insulin resistance and obesity in young and adult female mice. Cardiorenal Med. 2012;2:200–210. doi: 10.1159/000339563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couse JF, Korach KS. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 24.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cederroth CR, Nef S. Fetal programming of adult glucose homeostasis in mice. PLoS One. 2009;4:e7281. doi: 10.1371/journal.pone.0007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S. 2000;97:12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, Onishi T, Sugiura T, Shizuta Y. Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol. 2003;176:237–246. doi: 10.1677/joe.0.1760237. [DOI] [PubMed] [Google Scholar]
- 28.Belgorosky A, Guercio G, Pepe C, Saraco N, Rivarola MA. Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm Res. 2009;72:321–330. doi: 10.1159/000249159. [DOI] [PubMed] [Google Scholar]
- 29.Rochira V, Madeo B, Zirilli L, Caffagni G, Maffei L, Carani C. Oestradiol replacement treatment and glucose homeostasis in two men with congenital aromatase deficiency: evidence for a role of oestradiol and sex steroids imbalance on insulin sensitivity in men. Diabet Med. 2007;24:1491–1495. doi: 10.1111/j.1464-5491.2007.02304.x. [DOI] [PubMed] [Google Scholar]
- 30.Zirilli L, Rochira V, Diazzi C, Caffagni G, Carani C. Human models of aromatase deficiency. J Steroid Biochem Mol Biol. 2008;109:212–218. doi: 10.1016/j.jsbmb.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 31.Barbour LA, McCurdy CE, Hernandez TL, Friedman JE. Chronically increased S6K1 is associated with impaired IRS1 signaling in skeletal muscle of GDM women with impaired glucose tolerance postpartum. J Clin Endocrinol Metab. 2011;96:1431–1441. doi: 10.1210/jc.2010-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kajantie E, Strang-Karlsson S, Hovi P, Wehkalampi K, Lahti J, Kaseva N, Jarvenpaa AL, Raikkonen K, Eriksson JG, Andersson S. Insulin sensitivity and secretory response in adults born preterm: the Helsinki study of very low birth weight adults. J Clin Endocrinol Metab. 2015;100:244–250. doi: 10.1210/jc.2014-3184. [DOI] [PubMed] [Google Scholar]
- 33.Ibanez L, Castell C, Tresserras R, Potau N. Increased prevalence of type 2 diabetes mellitus and impaired glucose tolerance in first-degree relatives of girls with a history of precocious pubarche. Clin Endocrinol (Oxf) 1999;51:395–401. doi: 10.1046/j.1365-2265.1999.00778.x. [DOI] [PubMed] [Google Scholar]
- 34.Golden SH, Dobs AS, Vaidya D, Szklo M, Gapstur S, Kopp P, Liu K, Ouyang P. Endogenous sex hormones and glucose tolerance status in postmenopausal women. J Clin Endocrinol Metab. 2007;92:1289–1295. doi: 10.1210/jc.2006-1895. [DOI] [PubMed] [Google Scholar]
- 35.Eisner JR, Dumesic DA, Kemnitz JW, Abbott DH. Timing of prenatal androgen excess determines differential impairment in insulin secretion and action in adult female rhesus monkeys. J Clin Endocrinol Metab. 2000;85:1206–1210. doi: 10.1210/jcem.85.3.6453. [DOI] [PubMed] [Google Scholar]
- 36.Bruns CM, Baum ST, Colman RJ, Eisner JR, Kemnitz JW, Weindruch R, Abbott DH. Insulin resistance and impaired insulin secretion in prenatally androgenized male rhesus monkeys. J Clin Endocrinol Metab. 2004;89:6218–6223. doi: 10.1210/jc.2004-0918. [DOI] [PubMed] [Google Scholar]
- 37.Padmanabhan V, Veiga-Lopez A, Abbott DH, Recabarren SE, Herkimer C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology. 2010;151:595–605. doi: 10.1210/en.2009-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cardoso RC, Veiga-Lopez A, Moeller J, Beckett E, Pease A, Keller E, Madrigal V, Chazenbalk G, Dumesic D, Padmanabhan V. Developmental programming: Impact of gestational steroid and metabolic milieus on adiposity and insulin sensitivity in prenatal testosterone-treated female sheep. Endocrinology. 2016;157:522–535. doi: 10.1210/en.2015-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Del Prato PS, Tiengo A. The importance of first-phase insulin secretion: implications for the therapy of type 2 diabetes mellitus. Diabetes Metab Res Rev. 2001;17:164–174. doi: 10.1002/dmrr.198. [DOI] [PubMed] [Google Scholar]
- 40.Del Prato PS, Marchetti P, Bonadonna RC. Phasic insulin release and metabolic regulation in type 2 diabetes. Diabetes. 2002;51(Suppl 1):S109–S116. doi: 10.2337/diabetes.51.2007.s109. [DOI] [PubMed] [Google Scholar]
- 41.Cerasi E, Luft R. The plasma insulin response to glucose infusion in healthy subjects and in diabetes mellitus. Acta Endocrinol (Copenh) 1967;55:278–304. doi: 10.1530/acta.0.0550278. [DOI] [PubMed] [Google Scholar]
- 42.Davies MJ, Rayman G, Grenfell A, Gray IP, Day JL, Hales CN. Loss of the first phase insulin response to intravenous glucose in subjects with persistent impaired glucose tolerance. Diabet Med. 1994;11:432–436. doi: 10.1111/j.1464-5491.1994.tb00302.x. [DOI] [PubMed] [Google Scholar]
- 43.Imai Y, Dobrian AD, Weaver JR, Butcher MJ, Cole BK, Galkina EV, Morris MA, Taylor-Fishwick DA, Nadler JL. Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes Metab. 2013;15(Suppl 3):117–129. doi: 10.1111/dom.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tersey SA, Bolanis E, Holman TR, Maloney DJ, Nadler JL, Mirmira RG. Minireview: 12-Lipoxygenase and Islet beta-Cell Dysfunction in Diabetes. Mol Endocrinol. 2015;29:791–800. doi: 10.1210/me.2015-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das A, Mukhopadhyay S. The evil axis of obesity, inflammation and type-2 diabetes. Endocr Metab Immune Disord Drug Targets. 2011;11:23–31. doi: 10.2174/187153011794982086. [DOI] [PubMed] [Google Scholar]
- 46.Pepe GJ, Maniu A, Aberdeen G, Lynch TJ, Albrecht ED. Estrogen regulation of fetal adrenal cortical zone-specific development in the nonhuman primate impacts adrenal production of androgen and cortisol and response to ACTH in females in adulthood. Endocrinology. 2016;157:1905–1913. doi: 10.1210/en.2015-2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tigno XT, Gerzanich G, Hansen BC. Age-related changes in metabolic parameters of nonhuman primates. J Gerontol A Biol Sci Med Sci. 2004;59:1081–1088. doi: 10.1093/gerona/59.11.1081. [DOI] [PubMed] [Google Scholar]
- 48.Muller DC, Elahi D, Tobin JD, Andres R. The effect of age on insulin resistance and secretion: a review. Semin Nephrol. 1996;16:289–298. [PubMed] [Google Scholar]
- 49.Carnagarin R, Dharmarajan AM, Dass CR. Molecular aspects of glucose homeostasis in skeletal muscle--A focus on the molecular mechanisms of insulin resistance. Mol Cell Endocrinol. 2015;417:52–62. doi: 10.1016/j.mce.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 50.Carnagarin R, Dharmarajan AM, Dass CR. PEDF attenuates insulin-dependent molecular pathways of glucose homeostasis in skeletal myocytes. Mol Cell Endocrinol. 2016;422:115–124. doi: 10.1016/j.mce.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 51.Chavez AO, Lopez-Alvarenga JC, Tejero ME, Triplitt C, Bastarrachea RA, Sriwijitkamol A, Tantiwong P, Voruganti VS, Musi N, Comuzzie AG, DeFronzo RA, Folli F. Physiological and molecular determinants of insulin action in the baboon. Diabetes. 2008;57:899–908. doi: 10.2337/db07-0790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.