

Abstract
RNA polymerase (Pol) III has a specialized role in transcribing the most abundant RNAs in eukaryotic cells, tRNAs, along with other ubiquitous small non-coding RNAs, many of which have functions related to the ribosome and protein synthesis. The high energetic cost of producing these RNAs and their central role in protein synthesis underlies the robust regulation of Pol III transcription in response to nutrients and stress by growth regulatory pathways. Downstream of Pol III, signaling impacts posttranscriptional processes affecting tRNA function in translation and tRNA cleavage into smaller fragments that are increasingly attributed with novel cellular activities. In this review, we consider how nutrients and stress controls Pol III transcription via its factors and its negative regulator, Maf1. We highlight recent work showing that the composition of the tRNA population and the function of individual tRNAs is dynamically controlled, and that unrestrained Pol III transcription can reprogram central metabolic pathways.
Keywords: Pol III transcription, signaling, metabolism, tRNA modification, tRNA fragments
INTRODUCTION
In rapidly growing cells the synthesis of rRNAs, tRNAs and other small non-coding RNAs by RNA polymerases (Pols) I and III accounts for ~80% of the nucleotides consumed in nuclear gene transcription. Pol III transcription contributes about one quarter of this amount. The high energetic cost of synthesizing these molecules is thought to underlie their robust, coordinate regulation (especially repression) which conserves metabolic energy during periods of nutrient deprivation and stress and allows resources to be optimally balanced between cell survival and homeostatic functions versus growth. New research shows that regulatory changes in Pol III transcription are not evenly distributed across tRNA genes or the mature tRNA population since nutrients and stress can also affect tRNA processing, maturation and modification. The net result is a different tRNA profile and different tRNA functionality. In this review, we summarize current understanding about how changes in cell signaling impact Pol III transcription and affect the production and function of tRNA molecules along with their truncated derivatives. We focus on studies in yeast and mammalian systems to emphasize the broadly conserved nature of these responses which, in higher eukaryotes, help to shape normal and disease cell states.
NUTRIENT AND STRESS SIGNALING TO THE RNA POLYMERASE III SYSTEM
The ability of yeast cells to alter ribosome and tRNA synthesis in response to growth rate and nutrient levels has been known since the mid-1970s (1, 2). However, it was not until the mid to late 1990s that compelling evidence for the coordinate transcriptional control of these processes began to emerge. Independent studies on the secretory pathway and Target of Rapamycin (TOR) signaling found that genetic or chemical inhibition of these processes led to transcriptional repression of Pols I and III along with repression of ribosomal protein (RP) gene transcription by Pol II (3–7). Thus, common signaling molecules (TOR kinase and components of the cell integrity pathway, respectively) were found to mediate the coordinate transcriptional responses to nutrients and perturbations of cellular function. Identifying the targets of these pathways in the corresponding transcription systems continues to the present day. For Pol III, an early success was the identification of Maf1, a master regulator in this system whose function is essential for modulating transcription with changing nutritional, environmental and cellular stress conditions (8, 9). While not essential for viability, yeast cells lacking Maf1 are largely blocked in their ability to repress Pol III transcription (see the sidebar titled The Pol III Transcription Machinery). These properties appear to be conserved in higher eukaryotes. MAF1 is encoded by a single gene in mice and humans and, as in yeast, a whole body knockout of Maf1 in mice is viable and fertile (10). Mammalian cells in culture require MAF1 for repression of Pol III transcription following serum starvation and treatments with TOR Complex 1 (TORC1) inhibitors or DNA damaging agents (11, 12) and in mouse tissues for the down-regulation of Pol III transcription following an overnight fast (10; Bonhoure et al. submitted).
Mechanism of Repression by Maf1
Initial genetic and biochemical studies in yeast identified Pol III and Brf1, a subunit of the initiation factor TFIIIB, as direct targets of Maf1 and showed that the binding of Maf1 to these molecules inhibits two discrete steps in transcription: Binding to Brf1 inhibits its recruitment by the promoter-bound assembly factor TFIIIC while binding to Pol III blocks its recruitment to TFIIIB-TFIIIC-DNA complexes (11, 12). Both of these inhibitory interactions are conserved in mammals. Additional biochemical studies in yeast and human systems established that preinitiation complexes containing Pol III and stalled elongation complexes are protected from inhibition by Maf1 (13, 14). These complexes are able to initiate or resume transcription, respectively, to generate full-length products despite the presence of excess Maf1. Factor-independent transcription on an elongation scaffold or a 3′ tailed template is also unaffected by Maf1 (15). Consistent with this, Pol III was shown to bind various model templates and Maf1 simultaneously. The central conclusion from these studies is that Maf1 can bind to Pol III engaged with DNA in a manner that is compatible with transcription. Further mechanistic insight emerged with the finding that Pol III molecules undergoing facilitated recycling, a process in which the polymerase terminates transcription and reinitiates without dissociating from the template (16), are resistant to repression by Maf1 (14). Together, these observations support the view that Maf1 binding to Pol III after it dissociates from DNA inhibits subsequent promoter binding. However, a different type of Maf1 interaction is apparently possible during elongation and facilitated recycling: The function of actively transcribing Pol III-Maf1 complexes is apparently unaffected under these conditions but the repressor is poised to inhibit transcription the moment the polymerase dissociates from the DNA (Figure 1). One can infer from this that events governing Pol III dissociation from DNA, facilitated recycling and the functions of initiation factors are likely important in Maf1-dependent control of transcription (see below).
Figure 1.
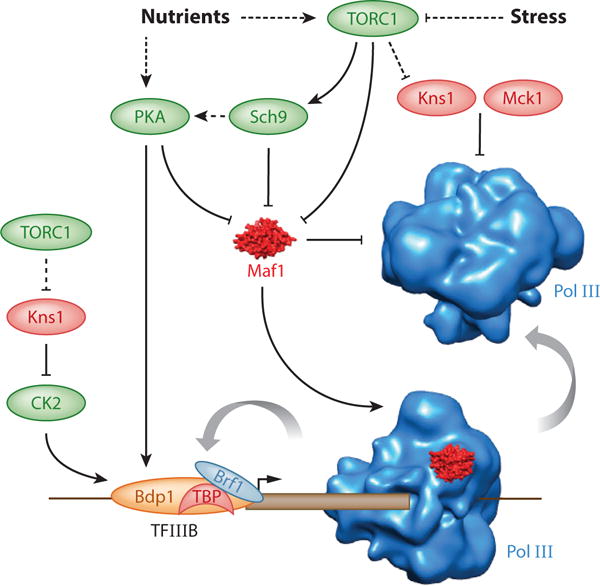
Signaling to the RNA polymerase III system in yeast. The scheme shows the known signaling kinases that mediate the response to nutrients and stress and their targets. Kinases that function to promote or repress transcription are shown in green and red ovals, respectively. Direct and indirect interactions/effects are represented by solid and dashed lines, respectively. Surface models of human Maf1 (red) and the yeast PolIII-Maf1 complex (15) are shown. Pol III is depicted on a tRNA gene with Maf1 positioned according to Vannini et al. (15). Large arrows indicate alternate fates of Pol III at the terminator, i.e. Maf1-resistant recycling or dissociation from the DNA. Dephosphorylation of Maf1 is known to involve PP4 and PP2A (not shown). Rpc53 phosphorylation by Kns1 and Mck1 is represented targeting free Pol III, but could also occur on the transcribing polymerase.
Initial structural insight into the Maf1-Pol III interaction has come from low resolution (~19 Å) cryoEM studies of Pol III and a Pol III-Maf1 complex (15). The difference density map could accommodate the X-ray structure of Maf1 and together with nanogold labeling studies, the repressor was located on top of the clamp domain of the largest Pol III subunit (Figure 2a). This location partially overlaps density assigned to the Rpc82-Rpc34-Rpc31 subcomplex in a Pol III-DNA-RNA complex. Thus, Maf1 binding to Pol III is thought to reposition the Rpc82-Rpc34-Rpc31 subcomplex leading to unfavorable interactions between its Rpc34 subunit and the Brf1 subunit of TFIIIB and hence impaired Pol III recruitment. Clearly, an important goal for the future will be to solve atomic resolution structures of the Pol III-Maf1 complex with and without an elongation scaffold. Comparison of these structures with recently solved high resolution structures of the corresponding complexes lacking Maf1 (17) will reveal molecular details of transcriptional repression by Maf1.
Figure 2.
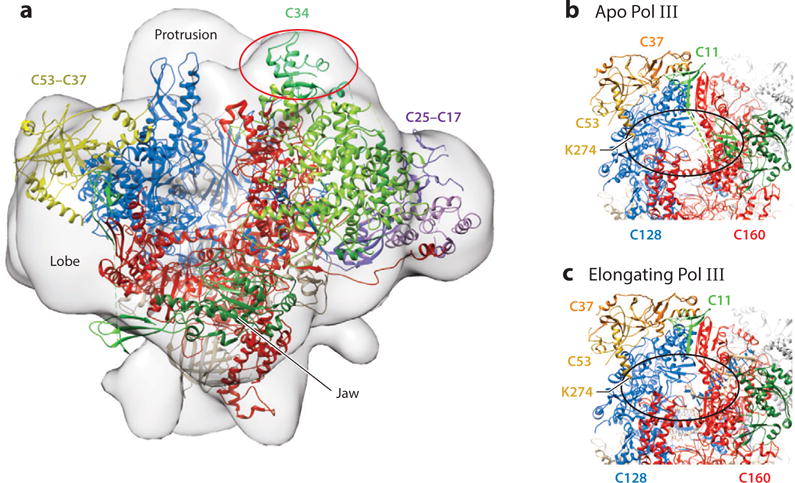
Structures of yeast Pol III. A Pol III-Maf1 complex. The 18.5Å resolution map (EMD-1755) is presented in the front view (15). The 4.7Å cryoEM structure of apo Pol III in the open form (17) was fitted into the envelope of the Pol III-Maf1 complex using Chimera. After fitting, 1806 out of 38434 atoms remained outside the contour. The DNA binding cleft comprising Rpc160 (red) and Rpc128 (blue) is viewed from the perspective of downstream DNA with the Wall in the rear. CryoEM difference density attributed to Maf1 (15) is shown in the oval (red) and lies behind the ribbon structure of Rpc34 (green) which is entirely outside the envelope in this fit. Rpc53/37 (yellow) and Rpc25/17 (purple) are shown for orientation. B and C. CryoEM structures of apo and elongating Pol III forms indicate mobility of the Rpc11 C-terminal zinc-binding domain. The Rpc53-Rpc37 subcomplex is positioned on the lobe of Rpc128. The two zinc ribbon domains of Rpc11 in the apo structure are connected by a linker (dashed line) which was not resolved. Density corresponding to the C-terminal domain of Rpc11 and the linker are missing in the elongating complex. Density for Rpc53 begins at K274 as shown. Photocrosslinking data place the Rpc53 phosphosites at amino acids 224, 228 and 232 within reach of Rpc160 and Rpc128, predicted to be within the oval. It is proposed that phosphorylation of these sites may affect the dynamics of the Rpc11 linker and its C-terminal zinc ribbon domain.
Phosphoregulation of Maf1
The phosphorylation state of Maf1 regulates its interaction with Pol III in different species. All of the mapped phosphorylation sites that are known to be biologically important (i.e. are differentially modified by nutrients or stress and participate in regulated transcription) are located in a region of variable sequence between two phylogenetically conserved segments (termed A and B)(8). Yeast Maf1 contains seven phosphosites in this region. Under optimal growth conditions where Maf1 is inactive, six of these sites are redundantly phosphorylated by protein kinase A (PKA) and Sch9, the TOR-regulated ortholog of mammalian S6 kinase, while the seventh site is targeted selectively by Sch9. Based on a wide variety of studies, work by multiple groups has established that Maf1 phosphorylation and Pol III transcription in yeast is modulated by the Ras/PKA pathway and by the rapamycin-sensitive TORC1 complex (11, 12). In addition, crosstalk between these pathways allows TORC1 to control the interaction of regulatory and catalytic PKA subunits and thereby regulate PKA activity towards some of its substrates, among them, Maf1 (18) (Figure 1). Other regulatory inputs into the Pol III system are provided by crosstalk with other signaling molecules (e.g. protein kinase CK2, see below) and other pathways such as the cell integrity pathway. However, the connectivity in this signaling network is not fully defined. Indeed for many stresses (e.g. DNA damage, oxidative stress, stress on the endoplasmic reticulum, etc.) it is not known how the signals are transduced to Maf1.
Like many transcriptional activators and repressors that mediate nutrient and stress responses in yeast (19), Maf1 shuttles between the nucleus and the cytoplasm. Its steady state distribution in these compartments is determined by several factors including (i) a strong nuclear localization sequence (NLS) which is inhibited by regulatory region phosphorylation, attributed to cytoplasmic PKA isoforms and Sch9, (ii) a second relatively weaker but constitutive NLS and (iii) the exportin Msn5 which returns Maf1 to the cytoplasm after its phosphorylation in the nucleus. The localization of Maf1 is also affected by ssd1-d, a defective allele of SSD1 that causes pleiotropic effects through the disruption of polarized mRNA trafficking and translational repression (20, 21). As reviewed elsewhere (11, 12), nucleocytoplasmic shuttling is thought to fine-tune the function of Maf1 since its phosphoregulation occurs normally when the protein is confined to nucleus by deletion of MSN5. Kinases that phosphorylate Maf1 in the nucleus likely include the Tpk2 isoform of PKA which is predominantly located in this compartment in unstressed cells (22) and TORC1 which is found in the yeast nucleolus associated with the 35S promoter and 5S rDNA (20). The localization of Tor1 to these sites is dependent on an NLS and a putative DNA binding motif (HLH). Interestingly, while Tor1 is not detected on tRNA genes in yeast, a rapamycin-resistant TOR1 allele bearing mutations in the NLS or the HLH showed diminished Maf1 phosphorylation and reduced transcription of both 5S rRNA and tRNA genes. Together with immunofluorescence studies, it was proposed that nucleolar TORC1 regulates nucleoplasm-to-nucleolus transport of Maf1 and helps maintain high levels of Pol III transcription by directing inactive phosphorylated Maf1 protein into the nucleoplasm. In the same study, immunopurified TORC1 was shown to phosphorylate Maf1 in vitro. However, to date the phosphorylation sites have not been mapped and the relative contributions of TORC1 and Tpk2 in phosphorylating nuclear Maf1 are unknown. Nonetheless, the yeast findings are instructive in light of work on TORC1, Maf1 and Pol III-transcribed genes in human cells.
The regulatory region of human Maf1 contains at least three sites that are differentially phosphorylated in response to nutrient signaling and alkylating DNA damage (23–25). None of these sites occur within PKA consensus motifs. Indeed, rather than employing PKA or intermediary kinases, Maf1 in human cells is directly phosphorylated by mTORC1 in the nucleus. Supporting this conclusion, in vitro phosphorylation of human Maf1 by immunopurified mTORC1 was shown to be dependent on the catalytic activity of mTOR and was rapamycin-sensitive (23) while the interaction of Maf1 with mTOR in the nucleus was shown by proximity ligation assays (PLA)(25). In addition, mTOR is associated with all types of Pol III-transcribed genes in mammals (24, 25) and its interaction with TFIIIC, demonstrated by immunoprecipitation and PLA data, provides a mechanism for mTORC1 recruitment, at least for 5S and tRNA genes (25). A large number of Pol III loci have now been associated with TORC1 following an mTOR ChIP-seq study in mouse liver. A comparison of the results with one of several published Pol III ChIP-seq datasets revealed a high degree of overlap with Pol III-occupied genes (26). With the exception of Pol III-occupied SINEs which generally lack mTOR, all other Pol III-transcribed genes appear to be mTOR targets. Notably, the function of mTOR as a direct regulator of gene transcription also extends to many protein coding genes including those involved in insulin receptor signaling, cancer and immune signaling, the proteasome and metabolic genes. mTOR occupancy of its target genes appears to be rapamycin-sensitive (25, 26) although not all studies support this conclusion (24). In any event, the presence of mTORC1 at Pol III-transcribed genes under growth promoting conditions is likely to result in the local inactivation of Maf1 and raises the possibility that mTORC1 may also target the polymerase and/or initiation factors.
Phosphoregulation of Pol III and TFIIIB is mechanistically linked to Maf1-dependent repression in yeast
The first indication that repression of Pol III transcription involved more than simply dephosphorylating Maf1 in the nucleus was suggested by studies with alanine substitution mutants (Maf1 6SA and 7SA) that prevent most or all of the known regulatory phosphorylation. The mutant proteins accumulated in the nucleus and interacted with the polymerase but transcription in otherwise optimal growth conditions was either not affected or only minimally reduced (27, 28). However, transcription was still robustly repressed under growth inhibitory conditions. This suggested that pro-growth signaling towards other components of the transcription machinery was preventing repression and/or that the modification of additional targets under repressing conditions was needed to achieve strong repression. Subsequent research has found evidence for both of these scenarios.
Differential phosphorylation of the Pol III subunit Rpc53, which along with Rpc37 comprises its heterodimeric TFIIF-like subcomplex, correlates with Maf1 dephosphorylation under all tested repressing conditions (29). Rpc53 phosphoregulation is attributed to the action of two protein kinases, the Cdc-like kinase Kns1 and the glycogen synthase kinase 3 family member Mck1. Both of these kinases function downstream of TORC1 with Kns1 increasing in abundance, becoming activated by autophosphorylation and accumulating in the nucleus after rapamycin treatment. Kns1 phosphorylates a single site in Rpc53 to prime successive phosphorylation events by Mck1 at neighboring residues. This process was reconstituted in vitro and mutation of the corresponding phosphosites abolished the phosphorylation in vitro and in vivo. Importantly, deletion of KNS1 and MCK1 attenuated transcriptional repression by Pol III as well as Pol I and RP gene transcription demonstrating that these kinases are part of the coordinate response of ribosome and tRNA synthesis to nutrient deprivation. However, analysis of the triple phosphosite to alanine mutant in Rpc53 (termed M1A) showed no effect on repression of Pol III transcription. Confirmation of the regulatory significance of these modifications required the M1A mutant to be assayed in the context of a hypomorphic RPC11 allele, known to be defective in facilitated recycling (29, 30). Since facilitated recycling is resistant to repression by Maf1 (14), the M1A mutant is thought to antagonize this process in a mutant rpc11 strain that is sensitized to these changes. Recent cryoEM structures of Pol III support the finding that the Rpc53-Rpc37 subcomplex stabilizes Rpc11 on the enzyme but the N-terminal two-thirds of Rpc53, which includes the phosphoregulated sites, is missing in the structures (17). Photocrosslinking data show that the Rpc53 phosphosites are in close proximity to Rpc160 and Rpc128 (31) and in light of the position of these subunits relative to Rpc53 and Rpc11 it is conceivable that phosphorylation of Rpc53 may affect facilitated recycling by changing the dynamics of Rpc11 interactions with the core polymerase (17). In this regard it is interesting that in the open and closed apo Pol III structures the N-terminal and C-terminal zinc-binding domains of Rpc11 are well resolved although the linker between them is not (Figure 2b and 2c). In contrast, only the N-terminal zinc-binding domain can be seen in the elongating Pol III complex consistent with the mobility of the C-terminal domain (17). Further examination of this model is needed bearing in mind that Rpc53 phosphorylation could also affect other documented activities of the Rpc53-Rpc37 subcomplex such as promoter opening, polymerase elongation and termination (30, 32, 33).
A systematic examination of differential phosphorylation among the 17 subunits of Pol III and the three subunits of TFIIIB identified two additional proteins whose phosphorylation state changes in response to TORC1 activity. The Rpc82 subunit of the polymerase and the Bdp1 subunit of TFIIIB are both dephosphorylated under repressing conditions (34). Analysis of phos-tag acrylamide gels suggested that the change in phosphorylation involves ~20% of the total amount of each protein. In the case of Rpc82, this could potentially impact the function of 400-500 polymerase molecules, roughly equal to the number of Pol III-transcribed genes in yeast (35). Rpc82 phosphosite mapping and functional studies are needed to evaluate these changes. For Bdp1, multiple phosphopeptides have been reported but only four sites account for the bulk of the phosphorylation in the protein under optimal growth conditions. In vitro and in vivo studies suggest that these sites are targeted by pro-growth kinases, specifically PKA, Sch9 and CK2. In functional studies, the quadruple phosphosite to alanine mutant (Bdp1-4SA) did not affect normal or repressed levels of transcription in a wild-type strain. However, in a strain containing a hypomorphic Maf1-myc allele that limits the extent of repression by lowering the affinity of Maf1 for the polymerase, the Bdp1-4SA mutation enabled robust repression. Thus, weakening the Maf1-Pol III interaction exposed the functional importance of dephosphorylating Bdp1 under repressing conditions. It follows that Bdp1 phosphorylation opposes Maf1-dependent repression. How this is achieved mechanistically is not known at the present time. However, in keeping with the role of facilitated recycling in achieving high levels of Pol III transcription and the resistance of this process to repression by Maf1 (14, 16), it has been proposed that Bdp1 phosphorylation may promote polymerase rebinding to the TFIIIB-DNA complex during facilitated recycling (34)(Figure 1).
Integration of CK2 into TOR pathway regulation of Pol III transcription
As a pro-growth serine/threonine protein kinase, CK2 has generally been associated with positive effects on Pol III transcription in yeast and mammals although its effect on transcription in mitotic HeLa cell extracts serves as a notable exception (36–41). To date, the principal target of CK2 in these systems is the initiation factor TFIIIB. In vitro activity of CK2 against all three subunits of TFIIIB (TBP, Brf1 and Bdp1) has been reported but attributing the modifications of these subunits to altered Pol III function in a physiological context has been challenging. CK2 has historically been considered a constitutive enzyme and its catalytic function, which is essential for viability, impacts myriad cellular processes (42). Thus, acute perturbations of CK2 activity by either genetic or chemical means can result in secondary effects in cells (43). Nonetheless, CK2 has been implicated in Pol III transcriptional regulation during the cell cycle in humans (40) and following DNA damage (37) or carbon source switching in yeast (41). Consistent with its regulatory function, CK2 is associated with Pol III loci in multiple species (40, 41, 44) but the basis for its dynamic control of Pol III transcription has been unclear. Recently, studies in yeast have found that CK2 is a downstream target of TORC1 (34). Subunit gene deletions of CK2, which is an αα′ββ′ heterotetramer comprising two catalytic α subunits and two regulatory β subunits, are rapamycin-sensitive. This phenotype is explained by the finding that the Cdc-like kinase Kns1, which is activated upon inhibition of TORC1 (discussed above), phosphorylates a single site in vitro and in vivo on one of the CK2 regulatory subunits (Ckb1)(Figure 1). The change in Ckb1 phosphorylation correlates with reduced occupancy of CK2 on tRNA genes, dephosphorylation of functionally important CK2 sites in Bdp1 and repression of Pol III transcription (34, 45). Considering the functional buffering of regulatory changes seen with Rpc53 and Bdp1, it is not surprising that the preceding effects of CK2 phosphorylation were not induced by a phosphomimetic mutation or attenuated by an alanine substitution at the Ckb1 phosphosite. It appears that unmasking the effect of Ckb1 phosphosite mutants on Pol III transcription will require an appropriately sensitized strain. The question of how CK2 phosphorylation might affect its function is still open although the possibility that the catalytic and regulatory subunits dissociate under repressing conditions appears to have been excluded (34). The high stoichiometry of phosphorylation of Ckb1 under many different repressing conditions suggests that the function of close to half of the CK2 in the cell may be altered. Taking CK2 structural and electrostatic considerations into account, it has been proposed that the modification may affect CK2 interactions with a subset of its many substrates (34).
Nutrient-dependent SUMOylation
The Pol III system has been associated with the small ubiquitin-like modifier (SUMO) since the first studies on the SUMOylome in yeast (46–49). These reports provided evidence for SUMOylation of Pol III subunits and transcription factors (Brf1) under normal growth conditions as well as an indication that SUMOylation of the polymerase might be dynamically regulated in response to stress (48). More recently, important progress has been made validating and expanding these observations and there is a growing appreciation of the functional significance of SUMOylation in Pol III transcription. SUMO Chip-seq studies in human fibroblasts and in yeast have revealed that the genes most prominently associated with SUMO are involved in cell growth and include Pol I- and Pol III-transcribed genes as well as RP genes (50, 51). Curiously, subsequent experiments arrived at opposite conclusions about the role of SUMO at these loci in the different organisms. The reasons for the divergent conclusions are not clear but may reflect different functions of SUMO in different contexts, e.g. the maintenance of homeostasis in cultured human cells versus an acute stress response in yeast. In fibroblasts, SUMOylation is associated with active Pol III loci and is at least partly due to direct modifications of chromatin since UBC9, the E2 conjugating enzyme for SUMO, colocalizes with SUMOylated proteins at these loci. Knockdown of UBC9 and SUMO led to up-regulation of Pol I- and Pol III-transcribed genes along with RP genes and other growth control genes. Thus, it was concluded that SUMO normally works to limit the expression of these genes (50). Recently, deep profiling of the human SUMO proteome has found that SUMOylation in the Pol III system, like many other nuclear processes, is pervasive and responsive to proteotoxic stress (52). Multiple SUMOylation sites have been mapped in the majority of subunits representing each of the functional entities, i.e. TFIIIC, SNAPc, TFIIIB and Pol III. The large number of modification sites (157 mapped sites in 23 proteins) poses significant challenges for future functional studies.
Studies in yeast revealed UBC9-dependent SUMOylation at the start sites of Pol III-transcribed genes and at the promoters of RP and snoRNA genes under normal growth conditions (51) and led to experiments on the role of SUMO in the coregulation of these genes by TORC1. Transcriptome analysis and short RNA sequencing showed the expected rapid decrease in tRNA and RP gene transcription after rapamycin treatment and similar dramatic reductions in the expression of these genes at the permissive temperature in a ubc9-1 mutant strain. For Pol III-transcribed genes, the decrease in transcription in response to rapamycin also correlated with reduced SUMOylation of these loci. For RP genes, the relationship between SUMO ChIP signals and gene expression was more complex. Nonetheless, the overall results support the global conclusion that SUMO positively regulates the transcription of tRNA and RP genes in yeast (51, 53).
Analysis of the yeast SUMOylome under nitrogen-rich and nitrogen-limiting conditions revealed differential SUMOylation of multiple Pol III components (53). Notably, the SUMOylation of three polymerase subunits, Rpc128, Rpc82 and Rpc53, was significantly reduced with nitrogen limitation or rapamycin treatment whereas for Brf1 and several TFIIIC subunits the opposite response was observed. While these changes were measured in total cell extracts, the patterns reflect changes in the physical associations of the factors and polymerase with DNA, i.e. dissociation of the polymerase, retention of Brf1 and increased occupancy of TFIIIC upon nutrient starvation as determined from genome-wide occupancy data (54). Investigation of the role of SUMOylation in these processes is likely to be informative. The mechanistic implications of SUMOylation in the Pol III system are currently best understood for Rpc82 where lysine to arginine substitutions of four predicted modification sites (rpc82-4KR) abolished its SUMOylation. This mutant allele showed reduced assembly and/or stability of Pol III and a reduced interaction with Brf1 implying a defect in polymerase recruitment. These findings are consistent with reduced tRNA gene transcription in unstressed rpc82-4KR cells, an effect that was not dependent on Maf1. The role of Rpc82 SUMOylation in Maf1-dependent repression, if any, is not yet clear. Future studies to address this important question will benefit from the sensitivity and coverage of small RNA sequencing.
Human Maf1 is SUMOylated on lysine 35 and the corresponding K35R mutant has diminished interactions with Pol III and diminished function in repression (55). However, this modification is apparently insensitive to rapamycin, at least in COS7 and 293T cells. Thus, while the function of Maf1 appears to be impacted by its SUMOylation, evidence for a direct regulatory role is currently lacking. Examination of a wider range of physiological perturbations in different cell lines might help to address this issue.
SUMOylation can affect the function of transcription factors both positively and negatively by multiple mechanisms (56). Thus, coupled with multiple sites and targets, the regulation of Pol III transcription by SUMO is likely to be complex. Current data affirm this view with apparently opposing effects of SUMOylation on different polymerase subunits (55). Aside from its potential to control various steps in Pol III transcription, SUMOylation could also modulate the concentrations of Pol III components. Of particular interest is the possible involvement of phosphorylation-dependent SUMOylation (57) since this may allow the integration of specific SUMOylation events into current models of Maf1-dependent repression (Figure 1).
DIURNAL REGULATION OF POL III TRANSCRIPTION
Recent reports show that ribosome synthesis, protein synthesis and Pol III transcription are rhythmically regulated by the circadian clock (58–61), the internal time-keeping system that adapts cell physiology and behavior to day and night cycles (62–64). Circadian regulation of Pol III transcription overlays the feeding-fasting cycle of Pol III transcriptional control in clear but subtle ways.
Diurnal regulation of Pol III transcription has been examined in mouse liver using Pol III ChIP-seq as a proxy for Pol III transcription (60). The experiments compared control mice to (i) arrhythmic mice that lacked a core clock component, ARNTL, (ii) mice that were fed at constant intervals during the day and night and (iii) mice that were missing the MAF1 repressor. Initial experiments showed that rhythmic expression of core clock-regulated genes was lost in Arntl KO mice, but not in constantly fed mice, as expected. Conversely, constantly fed mice lost the normal diurnal fluctuations in blood insulin levels and liver AKT-TORC1 signaling which were retained in Arntl KO mice. These two conditions together enabled effects on Pol III occupancy to be interpreted in terms of a response to either the core circadian clock or nutrients since food was only provided to control and Arntl KO mice at night. The results showed that high nighttime Pol III occupancy in control and Arntl KO mice occurs in response to feeding. In addition, control and constantly fed mice showed higher Pol III occupancy at the end of the day, before lights out and access to food, than in the morning. This increase was absent in the clock-deficient mice. Together, these findings suggest an anticipatory circadian response that ramps up Pol III transcription at the end of the day in preparation for active nighttime feeding, as is typical for nocturnal rodents. Thus, Pol III transcription appears to follow a diurnal cycle with clock-dependent anticipatory changes as well as changes that occur upon feeding. The timing of these changes correlates with the accumulation of ribosomes and with protein synthetic activity which is maximal during the night (61).
Loss of MAF1 does not significantly perturb the expression of clock-controlled genes. However, Pol III occupancy in Maf1 KO mice fed only at night increases significantly during both the day and night compared to control mice, consistent with the loss of the fasting-feeding response in these animals. Closer examination of Pol III occupancy in Maf1 KO mice at the beginning and the end of the day revealed an anticipatory rise before nighttime and food availability suggestive of core clock-dependent regulation. Thus, the findings support a clock-dependent but Maf1-independent mechanism for increasing Pol III occupancy in the anticipatory period before dark followed by a Maf1-dependent response to feeding. The latter is regulated by insulin via PI3K/AKT/TORC1 signaling but the mechanisms employed by the circadian clock to affect Pol III occupancy are unknown. Extensive analysis of diurnal rhythms in mouse liver by transcriptome and ribosome profiling and nuclear proteomics has found little evidence for rhythmic expression of the Pol III transcription machinery (58, 59, 65). Thus, posttranslational mechanisms employing, for example, any one of several protein kinases inferred by phosphoproteomics to be active from the middle to the end of the day (65), are perhaps more likely.
DEREGULATION OF POL III TRANSCRIPTION IN CANCER
Given the central role of 5S rRNA and tRNAs in protein synthesis and cell growth, the activation of Pol III transcription by transforming DNA tumor viruses, cellular oncogenes, tumor suppressors and oncogenic signaling pathways has been extensively studied in an effort to understand the complex relationship between Pol III gene transcription and cancer. Research in this area has focused mostly on the PTEN-PI3K-AKT and Ras-ERK pathways and the RB, p53 and Myc proteins which impinge directly on Pol III (and Pol I) transcription. Many excellent reviews of these studies are available and should be consulted for a full appreciation of the accumulated knowledge (66–69).
Early seminal work on Pol III transcription in cancer cell lines and biochemical mechanism-of-action studies motivated efforts to examine the importance of deregulating Pol III transcription for cell transformation and tumorigenesis. Subsequently it was shown that reducing Brf1 expression to offset Pol III gene activation in Myc-transformed Rat1a fibroblasts inhibited anchorage-independent growth and tumor formation in mice (70). Thus, enhanced Pol III transcription in these cells is required for tumorigenesis. Similar results have been obtained by stable overexpression of Maf1 in PTEN-deficient glioblastoma cells and in Huh-7, HepB3 and other liver cancer cell lines (71–73). Conversely, shRNA knockdown of Maf1 in several of the same cell lines showed the opposite behavior with increased cell growth, proliferation and invasiveness (73). Attributing these Maf1-dependent changes to effects on Pol III transcription is complicated, however, by the potential for Maf1 to also influence transcription of specific Pol II genes (71–73). Indeed, in the preceding studies, the effect of Maf1 overexpression on proliferation and invasiveness could be explained by changes in PTEN expression (73). Further studies are needed to elaborate the activation mechanisms which appear to be part of a Maf1-PTEN feedforward loop wherein Maf1 activation of PTEN expression leads to repression of PI3K-AKT signaling and to a FoxO1-mediated increase in Maf1 abundance (74).
Altered tRNA levels promote disease progression in cancer
Logically, the ability to sustain an increase in growth rate as tumor cells proliferate requires the upregulation of Pol III transcripts. Indeed, this appears to be the case as reflected by increased total tRNA levels (elevated 1.2-1.4 fold) and relative increases in individual tRNA isoacceptors measured by microarray in breast cancer and myeloma cell lines and breast tumors versus controls (75, 76). Interestingly, quantitative differences across the tRNA population in these experiments revealed selective effects with some isoacceptors being more dramatically affected than others. Similar observations have been made across different human tissues (77). In other studies, tRNA microarrays have been used to assess gene expression changes in hundreds of samples representing various states of proliferation, differentiation, transformation and senescence (78). Collectively, these studies indicate that the composition of the tRNA population, i.e. the tRNA profile, is not static but changes depending on the cell type and most notably, on the proliferative state of the cells (77, 78).
What is the biological importance of changing tRNA profiles? The answer is complex since it relates to the extent to which the codon usage of the transcriptome and the closely correlated abundance of the tRNA decoders function to fine-tune translation of different classes of genes (78). This functional relationship between codon usage, tRNA abundance and protein level is generally well accepted in bacteria and yeast but remains controversial in mammals (79–83). However, if the question is more narrowly defined to ask whether the levels of particular tRNA isoacceptors are important for translation of specific mRNAs, then interesting examples come to light.
Using a splinted-ligation methodology, tRNA profiling of poorly metastatic breast cancer cell lines along with their highly metastatic derivatives identified correlated changes in tRNA abundance in the metastatic cells (84). Two tRNAs (tRNAGluUUC and tRNAArgCCG) were prominently upregulated in these cells. shRNA knockdown of these tRNAs in metastatic cells or their overexpression in the parental cell lines had reciprocal effects on the metastatic phenotype in mice and on cell invasion in vitro. Subsequent ribosome profiling and proteomic analysis of the tRNA overexpressing cells revealed enrichment of the respective codons among genes that had relatively higher ribosome occupancy and higher protein levels. The function of two proteins, EXOSC2 and GRIPAP1, selected as potential mediators of the metastatic phenotype in tRNAGluUUC overexpressing cells was confirmed and the expression of EXOSC2 was clinically associated with invasive breast cancer. Thus, the work shows that tRNA levels can modulate the expression of proteins whose mRNAs are enriched in the corresponding codons and that these changes promote disease progression in metastatic breast cancer cells (84).
The unique role of initiator methionine tRNA (tRNAiMet) in translation raises the possibility that its overexpression, which is documented in numerous cancers, might increase the rate of protein synthesis globally or have preferential effects on the expression of genes that promote tumor development (78, 85). Indeed, in Drosophila, an extra copy of tRNAiMet is sufficient to increase protein synthesis, larval growth rate and body size (86). However, analogous experiments in transgenic mice where tRNAiMet expression was increased 1.3-1.5 fold from two extra copies of the gene (2+tRNAiMet mice), found no effect on size, bodyweight or the rate of protein synthesis in MEFs derived from these animals (87). Although these gross metrics were unchanged, when 2+tRNAiMet mice were used to establish subcutaneous allografts of transformed melanoblasts, the resulting tumors were larger and more vascularized than in control animals. Thus, elevated tRNAiMet expression in the host animal promotes tumor growth and angiogenesis. These phenotypes were explained by a highly selective effect of tRNAiMet on the expression of secretory proteins, especially type II collagen, in 2+tRNAiMet fibroblasts, leading to the production of an extracellular matrix that favors the migration of endothelial cells and angiogenesis.
In a separate but related study, tRNAiMet overexpression was shown to drive cell migration of fibroblasts in vitro and to increase melanoblast migration during embryogenesis in 2+tRNAiMet mice (88). Further, when crossed with INK4a-/- mice expressing a melanocyte-specific N-Ras mutant, isolated melanocytes from tRNAiMet overexpressing animals migrated faster than controls. Cell migration and invasiveness in vitro was also increased in melanoma cell lines engineered to increase tRNAiMet expression, as was metastasis in xenograft models. In many of these assays the effects of tRNAiMet overexpression were found to be achieved without changes in cell growth or proliferation. Thus, the results point to selective effects on the translational program, potentially related to increased secretory pathway flux, as having an important role in tumor cell migration and metastasis (87, 88).
In cancer cells, elevated levels of both initiator and elongator tRNAs have now been shown to have a significant impact on gene expression and disease progression, especially metastasis. Considering these few examples and the fact that changes in the tRNA profile between normal and transformed cells involve differences in absolute and relative levels of many tRNAs (75, 78), it seems likely that additional contributions of tRNAs to gene expression in cancer will be uncovered.
Cellular perturbations have differential effects on Pol III gene transcription
How does cell transformation lead to disproportionate effects on the expression of different tRNAs? Given the many steps in tRNA biogenesis (89), there are multiple opportunities for preferential expression at the level of the gene and beyond. Current work suggests that quantitative differences in Pol III gene transcription can be achieved by multiple mechanisms: (i) The frequent amplification of genomic regions in cancer can increase tRNA gene dosage leading to differences in expression (84); (ii) Changes to the chromatin landscape in transformed cells result in the restriction and/or expansion of access to the underlying DNA (90) and may affect the number and type of transcriptionally active Pol III loci. Notably, since only about one-half to two-thirds of the ~500 human tRNA genes are transcriptionally active (91–95), there is significant potential to change the tRNA profile through the activation of otherwise silent genes. Evidence that the expressed tRNA gene repertoire changes with cell state includes a comparative study of adipose-derived stem cells and their induced pluripotent derivatives where an additional 85 tRNA genes were found to be occupied by Pol III (96); (iii) other less-well defined mechanisms also contribute to differential expression. For example, quantitative differences, primarily within a set of ~300 tRNA genes, were observed in mouse tissue development (97). Nutrient limitation in both yeast and human cells shows selective effects on Pol III occupancy/transcription (95, 98). Serum starvation of human fibroblasts results in dramatic reductions in Pol III occupancy for a large majority of Pol III-transcribed genes. These changes are dependent on MAF1 and are strongly correlated with substantially lower levels of nascent transcripts, measured by ethynyl uridine pulse-labeling and deep sequencing. However, a group of 49 genes that have generally high Pol III occupancies were found to be relatively resistant to serum starvation and only moderately sensitive to Maf1 knockdown. Most tRNA isotypes are represented in this group along with genes containing type III promoter elements. Similar findings have been made in yeast using in vivo crosslinking and analysis of cDNA (CRAC) to examine nascent Pol III transcripts following a switch from glucose medium at 30°C to glycerol medium at 37°C (98). A continuum of sensitivity to repression by Maf1 was found and a group of low responsive genes was identified. As in humans, many but not all of the genes in this group were strongly expressed under optimal growth conditions. Together these studies suggest that a small number of Pol III-transcribed genes are relatively protected from repression under acute stress conditions and thus may serve a maintenance function, ensuring a low basal level of Pol III output during the stress response. The mechanisms that determine the differential sensitivity to repression are presently unknown.
POST-TRANSCRIPTIONAL REGULATION OF TRNA FUNCTION
As the largest class of Pol III transcripts, tRNAs have been well-studied with regard to the steps required to generate functional molecules. Each nascent tRNA transcript is subject to multiple processing steps: 5′ leader and 3′ trailer extensions are cleaved by RNase P and RNase Z respectively, introns (where present) are removed, CCA is added to the 3′ end and multiple nucleotide modifications are introduced prior to aminoacylation and function in translation (99). Many of these steps are regulated by stress and nutrients (89) and aberrantly processed and unmodified tRNAs are degraded by nuclear and cytoplasmic RNA surveillance pathways (100)(Figure 3). Despite the complexity of signaling to regulate tRNA transcription and the competing dynamics of processing and degradation to generate mature tRNAs, the total amount of mature tRNA remains relatively stable under acute stress (82). However, in response to a variety of cell stresses tRNAs accumulate in the nucleus (99), become differentially modified (101), are rapidly clipped at the 3′-end to remove CCA (102) and are cleaved in the anticodon loop and elsewhere to generate tRNA half-molecules and tRNA-related fragments (tRFs). These novel tRNA-derived products represent new classes of non-coding RNAs with emerging regulatory roles (102, 103).
Figure 3.
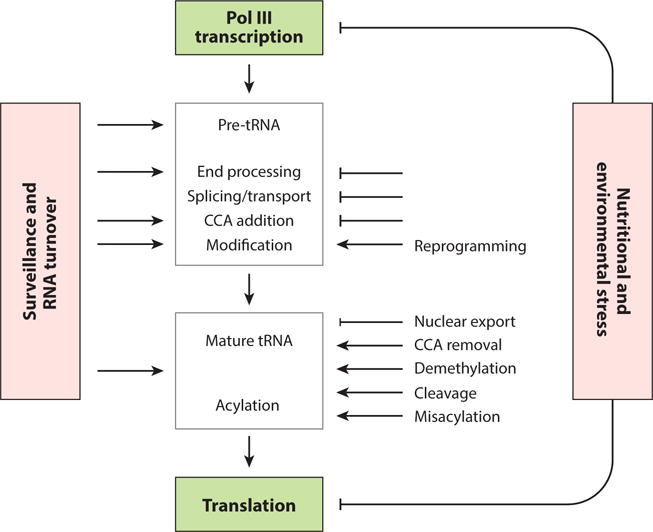
Regulation of tRNA biosynthesis and function. The steps in tRNA biogenesis from transcription to function in translation are listed. Steps known to be inhibited or altered by nutrients and/or stress are indicated along with processes known to be subject to tRNA quality control/ surveillance and RNA turnover.
Regulation of tRNA maturation and transport
High-resolution transcriptome studies indicate that a significant fraction of nascent tRNA transcripts are degraded prior to processing and maturation. Pulse-chase experiments show that approximately 50% of the precursor tRNAs synthesized in unstressed wild-type yeast cells growing in nutrient-replete conditions are degraded by the nuclear exosome and therefore do not contribute to the mature tRNA pool (104, 105). Thus, a kinetic competition appears to exist between tRNA processing and the nuclear RNA degradation pathway (106).
Recent reviews detail the many steps in tRNA processing that are subject to stress and nutritional regulation (89, 99). The essential RNA component of RNase P is transcribed by Pol III in both yeast (RPR1) and mammals (RppH1). Rpr1 and RppH1 synthesis is subject to Maf1-mediated repression (95; Moir and Willis, unpublished data) and thus may limit tRNA production under some conditions. 3′-end maturation of tRNA is more complex requiring endonuclease cleavage by RNaseZ, the stabilizing functions of La protein bound to the 3′ terminal oligo U stretch in the nascent transcript and addition of CCA nucleotides to the mature end. Other exonucleases (Rex1 and Rrp6, the latter functioning independently of the exosome) and the nuclear Lsm2-8 complex also contribute to 3′-end maturation in competition with degradation by the RNA nuclear surveillance pathway (107, 108). In higher eukaryotes, the La protein is phosphoregulated in response to growth conditions and is subject to proteolysis in response to DNA damage (89). In yeast, the activity of both Rex1 and Cca1 (the CCA addition enzyme) become limiting in unfavorable growth conditions (109, 110).
Cytoplasmic tRNAs are constitutively transported from the cytoplasm back into the nucleus under normal growth conditions by the retrograde nuclear import pathway (89, 99, 111). In yeast, this pathway is required for pre-tRNA splicing, wybutosine modification of tRNAPhe and nuclear degradation or repair of a subset of tRNAs. In its absence, the translation of numerous proteins involved in amino acid biosynthesis is compromised. tRNAs also rapidly accumulate in the nucleus of both yeast and mammalian cells in response to nutrient deprivation (89). In yeast, two karyopherins, Los1 and Msn5 (homologs of exportins T and 5, respectively) directly bind and transport tRNAs out of the nucleus. The function of these proteins is differentially regulated by a range of cell stressors and nutrients and results in their relocation to the cytoplasm where they are unable to access their nuclear cargo (89). How different signaling pathways induce the relocalization of these proteins in unknown. It is also unclear whether tRNA import is constitutive and only tRNA export is regulated by growth conditions, or whether both import and export are regulated.
tRNA modifications: dynamic consequences for structure and function
Nucleoside modifications on tRNA are abundant and varied with chemically distinct modifications identified at multiple positions. In general, modifications in the D and T arms contribute to tRNA folding and stability of the tertiary structure while modifications in and around the anticodon loop contribute to coding accuracy, wobble-base recognition and reading frame fidelity through stabilization or increased flexibility in codon-anticodon base-pairing (101, 112, 113). Hypomodified tRNAs can be subjected to a second cycle of CCA addition, or polyadenylated and then degraded by the tRNA quality control surveillance machinery (100). tRNA modifications in otherwise sequence-identical isoacceptors can be both heterogeneous and substoichiometric (114). The modification enzymes themselves can be limiting for function (115) and combinations of non-essential modifications can stabilize specific tRNAs and protect them from degradation (116).
tRNA modifications are dynamically controlled in response to environmental conditions and stress (82, 89, 101, 113, 117). Chemicals that induce mechanistically different stress responses generate global tRNA modification patterns that are unique and specific for each chemical. Modifications vary throughout the yeast cell cycle (113), between tumor and adjacent normal human tissue and are more abundant in proliferating human cell lines than in human tissue (118). Alterations in tRNA modification profiles have been shown to shift translation towards the expression of codon-biased genes required for specific cellular responses and to limit codon-specific translational pausing by altering wobble base modifications (113). Hydrogen peroxide treatment of yeast produces a global elevation of m5C, 2′-O-methylcytidine (Cm), and N2,N2-dimethylguanosine (m22G) modifications and deletion of the enzymes that catalyze these modifications results in hypersensitivity to peroxide stress (113). Moreover, the peroxide-induced increase in m5C modification at the wobble position in tRNALeuCAA results in the selective expression of UUG codon-enriched mRNAs (119). Proteins involved in translation were overrepresented in this group and included the UUG codon-biased RP gene RPL22A, but not its unbiased paralog RPL22B. Since the enzyme responsible for m5C modification (Trm4) and RPL22A were required for the normal response to oxidative stress, the findings link the protein composition of the ribosome with differential translation during the stress response. How or whether the activity of Trm4 is increased by hydrogen peroxide or unmethylated C34-containing tRNALeuCAA is specifically targeted for degradation or cleavage is not known. In another example, DNA damage-induced TRM9-dependent increases in wobble mcm5U modifications of tRNAArgUCU and tRNAGlnUUG are associated with enhanced translation of stress response proteins enriched with AGA and GAA codons (113). Similarly, in mammalian cells, a TRM9-like methyltransferase (Alkbh8) induced by reactive oxygen species (ROS) is required for wobble uridine modifications of selenocysteine tRNA that increase its translational efficiency and enhance the expression of selenocysteine-containing proteins involved in ROS detoxification (113).
Recently, ALKBH1 was identified as a demethylase for N1-methyladenosine (m1A), a modification present in most tRNAs at position 58 (120). Induction of ALKBH1 expression by low glucose in HeLa cells reduces translation initiation and elongation by decreasing the levels of tRNAiMet and reducing the affinity of numerous tRNAs for the elongation factor eEF1A, respectively. In sum, tRNA modifications can be dynamically placed and, in some cases removed, allowing fine-tuning of translation in response to environmental and stress conditions.
Regulating the functional tRNA pool and generating tRNA second messengers
The CCA termini present on all matured tRNAs can be rapidly removed in response to oxidative stress by the action of Angiogenin (ANG), a vertebrate-specific protein with weak RNase activity (102). Removal of CCA offers the potential for reversible inhibition of translation since the 3′-end can be repaired to restore tRNA aminoacylation and translation (82). ANG and its functional homolog in yeast, Rny1, also cleave a small fraction of mature cytoplasmic tRNA in the anticodon loop. This conserved response to a variety of cell stresses generates 5′- and 3′-tRNA half molecules (121–125). ANG abundance is regulated by stress-responsive transcription, the protein relocalizes from the nucleus to the cytoplasm under stress and its activity is controlled by binding of an inhibitor, RNH1 (126). The mechanism by which Rny1 accesses its substrates, whether by Rny1 relocalization from the vacuole or by targeting tRNA to the vacuole has not been resolved (127–129).
tRNA halves can also be detected in the absence of overt stress (127, 130). For example, 5′-tRNA halves derived from tRNAGlyGCC are especially abundant in human plasma and urine (131), in mouse blood cells and in hematopoietic tissues where their profile is age-dependent (130). Mammalian sperm are enriched for 5′-tRNA halves, particularly tRNAGlu and tRNAGly (132, 133). Shorter tRNA fragments have also been identified that map to the 5′- and 3′-ends of mature and pre-tRNAs and internal sequences (134).
tRNA halves and tRFs do not appear to be debris from tRNA degradation pathways: The patterns of tRNA cleavage vary with the type and intensity of stress and they are increasingly connected to specific cellular processes (135). Respiratory syncytial virus (RSV) infection, but not all virus infections, leads to elevated levels of 5′-tRNA halves, a subset of which (tRNAGluCTC, tRNAGlyCCC and tRNALysCTT) promote RSV replication (136)(137). tRNA half molecules (SHOT RNAs) are also elevated in estrogen receptor-positive breast cancer and androgen receptor-positive prostate cancer cell lines where they are thought to be produced in response to sex hormone-signaling pathways. These 5′- and 3′-halves possess a 2’,3′ cyclic phosphate and an amino acid at the 3′-end, respectively, indicating ANG cleavage of mature aminoacylated tRNA (138). The 5′-tRNA halves of tRNALysCUU and tRNAHisGUG in these cancer cell lines are implicated in promoting cell proliferation.
ANG-mediated tRNA cleavage induced by stress or treatment of cells with recombinant ANG reduces global protein synthesis and enhances stress granule (SG) formation to promote cell survival (126). These effects are attributed to a subset of tRFs (5′-tiRNAAla and 5′-tiRNACys) with a critical 5′-oligoguanine motif that (i) displace the eIF4F cap-binding complex from the 5′-end of mRNAs, thereby inhibiting initiation of translation, and (ii) interact with the translational repressor, YBX1, for packaging of tiRNA-inhibited mRNAs into SGs. YBX1 also features in the function of tRFs induced by hypoxia in breast cancer and normal cells (139). In this study, stress-induced cleavage of several tRNAs produced internally truncated tRFs that retain the anticodon stem and loop. These tRFs were found to compete with endogenous oncogenic transcripts for YBX1 binding, resulting in mRNA destabilization, loss of gene expression and suppression of growth, invasion and metastasis. 5′-tRFs in mouse sperm have been associated with intergenerational inheritance of gene expression and metabolic phenotypes. Male mice fed low protein or high fat diets exhibit changes in the profile of tRFs in their sperm. When tRFs from the sperm of high fat fed mice are injected into normal zygotes, the F1 offspring are glucose intolerant after weaning and dietary challenge (140) and gene expression changes are seen in early embryos and in islets of the F1 mice. Similar experiments with sperm tRFs from mice fed a low protein diet were found to cause repression of embryonic gene expression regulated by the LTR of the endogenous retroelement MERVL (132). Together these studies identify tRFs as a paternal epigenetic factor.
METABOLIC IMPACT OF TRANSCRIPTION BY POL III
In yeast, there is general agreement that the regulation of ribosome and tRNA synthesis is critical for metabolic economy and cellular fitness (141–143). In line with this, recent studies in mice have found that a failure to appropriately repress Pol I or Pol III transcription is energetically wasteful and results in the expenditure of metabolic energy that would otherwise be stored as fat. Thus, defective repression of Pol I transcription seen in a liver-specific knockout of nucleomethylin, a subunit of the energy-dependent nucleolar silencing complex (eNoSC), is associated with resistance to diet-induced obesity and non-alcoholic fatty liver disease (144). These phenotypes are also seen in mice with a whole body knockout of Maf1 (10).
Current work on the mechanisms by which loss of Maf1 leads to a lower body weight and a lean phenotype has identified three contributing factors: Reduced food intake, reduced metabolic efficiency and a slightly smaller body size (10). The food intake phenotype, a 10-15% reduction (normalized for body weight) in the Maf1 KO has not been explored. The difference in the overall growth rate, which is apparent in the body length of animals at >6 months of age, may be due in part to a small decrease in global translation. Data supporting this conclusion include in vivo puromycin-labeling of nascent liver polypeptides and ribosome profiling (Bonhoure et al. submitted). Notably, mRNAs containing 5′-terminal oligopyrimidine (TOP) sequences, which are characteristic of ribosomal and other proteins involved in protein synthesis, were among a group of mRNAs whose translation efficiency was significantly reduced in Maf1 KO liver. The mechanism(s) responsible for these translational effects have yet to be determined.
A major factor contributing to the lean phenotype of Maf1 KO mice is thought to be their inefficient metabolism (10). At a whole body level, this is demonstrated by the reduced ability of Maf1 KO mice to convert calories into biomass under paired-feeding conditions (i.e. normalized food intake) and by the increased energy expenditure of the mice which are not more active than wild-type mice. Thus, Maf1 KO mice have an intrinsically greater demand for metabolic energy. Experiments show that this demand is satisfied without affecting glucose homeostasis or insulin sensitivity by increased lipolysis in white adipose tissue (WAT) and increased autophagy and lipophagy in liver (10). These processes provide fatty acids and amino acids for oxidative metabolism and other key reactions (Figure 4). Targeted metabolite profiling shows that the levels of many amino acids are elevated in Maf1 KO liver and muscle while the levels of many long acylcarnitine species are dramatically reduced in liver, consistent with increased β-oxidation (10; I. Willis, unpublished data).
Figure 4.
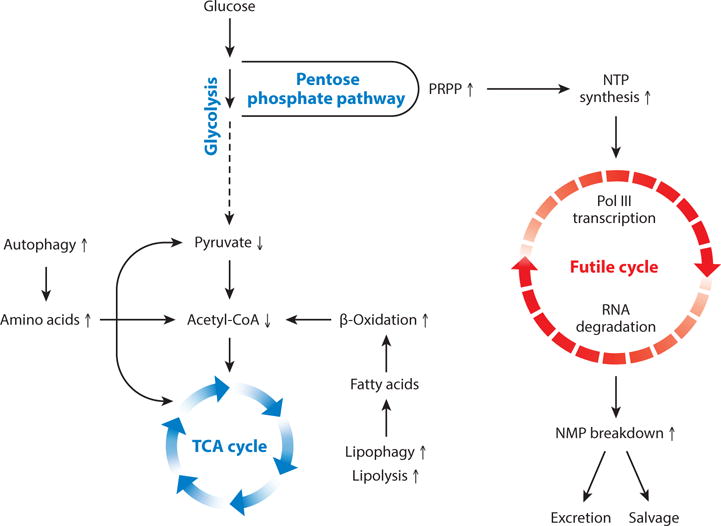
Schematic model of metabolic inefficiency in Maf1 KO mice. A futile cycle of Pol III transcription and RNA turnover is thought to reprogram central metabolism as outlined in the text. Arrows indicate how metabolic processes and select metabolite levels are changed in Maf1 KO liver. Increased ATP synthesis to power de novo nucleotide synthesis is thought to result from increased flux through the TCA cycle.
What is driving enhanced catabolism in these mice? Pol II transcriptome profiling in WAT and liver of wild-type and Maf1 KO mice reveals few differences and provides no real clues to the mechanism (10; Bonhoure et al. submitted). Moreover, changes in the expression of specific Pol II genes that have been noted in cultured cells following Maf1 knockdown and overexpression (e.g. Tbp, Egr1, Pten, Fasn, Acc1) are not seen in Maf1 KO transcriptome or ribosome profiling data (see Johnson and Stiles (74) for further discussion of these changes). In contrast, short-lived precursor tRNAs, which reflect Pol III transcription, are elevated ~3-10 fold across a range of Maf1 KO tissues in the fasted state (10). Similar dramatic increases in precursor tRNAs are seen in the same tissues in the fed state with the exception of liver where the increases are lower (Bonhoure et al. submitted). However, in both fed and fasted liver, the vast majority of active Pol III loci exhibit significantly higher occupancy by Pol III in the Maf1 KO (145; Bonhoure et al. submitted). Similar findings have been obtained in independent Pol III ChIP-seq experiments conducted in mouse liver throughout the diurnal cycle (60). Together these data support and extend a conclusion drawn recently from studies of Maf1-dependent regulation and serum starvation in IMR90hTert cells (95): MAF1 functions chronically during cell growth in serum-replete conditions and throughout feeding-fasting and diurnal cycles in mouse tissue to significantly limit Pol III transcription.
Considering that Pol III transcription is chronically elevated in virtually all cells in Maf1 KO mice, it is especially striking that the level of total mature tRNA in numerous tissues as well as individual mature tRNAs are not significantly altered (10). Thus, the increased synthesis of tRNA (and likely other Pol III transcripts) in the knockout must be largely matched by increased RNA turnover. These observations form the basis of the hypothesis that a futile cycle of RNA synthesis and turnover drives energy expenditure in Maf1 KO mice (10)(Figure 4). A key prediction of this hypothesis is that wasteful metabolism due to unrestrained transcription by Pol III must be sustained by increased nucleotide synthesis. To achieve this, the flux of metabolites through central metabolic pathways is expected to change since nucleotide concentrations in cells are robustly maintained in a relatively narrow range, largely through allosteric mechanisms (146). Thus, changes are expected in the concentrations of some of the metabolites in the affected pathways (147). Remarkably, targeted profiling identified 43 significantly altered metabolites at the interface of glycolysis and the TCA cycle, in the urea cycle and the pentose phosphate pathway as well as de novo nucleotide synthesis and nucleotide turnover pathways, including known rate-limiting intermediates and allosteric regulators (I. Willis, unpublished data). These findings demonstrate the capacity of Pol III transcription to reprogram metabolism and support the concept of futile cycling of Pol III transcripts as a mechanism for achieving metabolic inefficiency.
CONCLUDING REMARKS
The control of Pol III transcription and the downstream processing and function of its transcripts by nutrient and stress signaling pathways plays a vital role in determining cellular phenotype. Indeed, current work suggests that manipulating Pol III transcription and the functions of its non-coding RNA products has the potential to alter outcomes in cancer and metabolic disease. However, as we have noted throughout this review, there are still many areas where our knowledge, especially of the regulatory mechanisms, is limited. The ongoing challenge will be to develop our understanding in these important areas while considering how to exploit opportunities for possible therapeutic intervention.
TERMS AND DEFINITIONS.
TORC1: Multi-subunit TOR kinase complex that regulates cell growth by nutrients, growth factors, energy, redox and diverse cellular stresses.
PTEN: Lipid phosphatase and tumor suppressor that limits phosphatidylinositol-3,4,5-trisphosphate (PIP3) levels to negatively regulate AKT signaling.
AKT: Serine/Threonine protein kinase activated by phosphatidylinositol-3-kinase and phosphoinositide-dependent kinase 1 for control of cell survival and pro-growth signaling.
Lsm2-8 complex: the protein chaperone complex for the Pol III-transcribed U6 snRNA.
SINE: Short Interspersed Nuclear Element, an abundant retrotransposon evolved from Pol III-transcribed genes that encoded tRNA, 5S RNA or 7SL RNA.
PKA: Protein kinase A, activated by cAMP, regulates cell growth in yeast in response to glucose.
THE POL III TRANSCRIPTION MACHINERY.
Pol III transcription of tRNA genes requires a six-subunit assembly factor (TFIIIC) for binding to intragenic A and B block promoter elements and a three-subunit initiation factor (TFIIIB) comprising the TATA box-binding protein TBP, the TFIIB-paralog Brf1 and Bdp1. TFIIIB is stably assembled onto DNA upstream of the transcription start site and recruits the 17 subunit polymerase (148). Preinitiation complex assembly on 5S RNA genes employs a different intragenic promoter architecture, requiring an additional factor (TFIIIA) to direct the binding of TFIIIC. A third class of Pol III promoters found in metazoans utilizes upstream rather than intragenic sequence elements and replaces TFIIIA and TFIIIC with SNAPc for initiation complex assembly. SNAPc-dependent genes utilize a different TFIIB-paralog, Brf2, in the initiation factor (149, 150). Pol III contains a ten-subunit catalytic core found in all nuclear RNA polymerases plus additional sub-complexes and protein domains that have structural and functional homology to general transcription factors for Pol II (151). The core subunit Rpc11, similar to TFIIS, has intrinsic RNA cleavage activity for error-correction and is required for facilitated reinitiation (30). The Rpc37/53 subunits are related to TFIIF and contribute to promoter opening, Pol III termination and reinitiation via interactions with Rpc11 (17, 32). The Rpc82/34/31 subcomplex, related to the Pol II initiation factor TFIIE, is positioned over the clamp domain of the largest core subunit, Rpc160, and functions in polymerase recruitment through interactions between Brf1 and Rpc34 (17, 152). Pol III transcription elongation is slowed by the Rpc37/53/11 subcomplex to facilitate termination through interactions of Rpc37 and the non-template strand within an oligo(dT) terminator sequence (33).
Acknowledgments
We thank the members of our lab for their contributions to our understanding in this work and apologize to our colleagues whose research could not be cited due to space limitations. Our work is funded by grant GM120358 from the National Institutes of Health.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Waldron C, Lacroute F. Effect of growth rate on the amounts of ribosomal and transfer ribonucleic acids in yeast. J Bacteriol. 1975;122(3):855–65. doi: 10.1128/jb.122.3.855-865.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludwig R, Oliver SG, McLaughlin CS. The regulation of RNA synthesis in yeast II: amino acids shift-up experiments. Mol Gen Genet. 1977;158(2):117–22. doi: 10.1007/BF00268303. [DOI] [PubMed] [Google Scholar]
- 3.Zaragoza D, Ghavidel A, Heitman J, Schultz MC. Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol Cell Biol. 1998;18(8):4463–70. doi: 10.1128/mcb.18.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powers T, Walter P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1999;10(4):987–1000. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizuta K, Warner JR. Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol Cell Biol. 1994;14(4):2493–502. doi: 10.1128/mcb.14.4.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nierras CR, Warner JR. Protein kinase C enables the regulatory circuit that connects membrane synthesis to ribosome synthesis in Saccharomyces cerevisiae. J Biol Chem. 1999;274(19):13235–41. doi: 10.1074/jbc.274.19.13235. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Moir RD, Sethy-Coraci IK, Warner JR, Willis IM. Repression of ribosome and tRNA synthesis in secretion-defective cells is signaled by a novel branch of the cell integrity pathway. Mol Cell Biol. 2000;20(11):3843–51. doi: 10.1128/mcb.20.11.3843-3851.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pluta K, Lefebvre O, Martin NC, Smagowicz WJ, Stanford DR, et al. Maf1p, a negative effector of RNA polymerase III in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21(15):5031–40. doi: 10.1128/MCB.21.15.5031-5040.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Upadhya R, Lee J, Willis IM. Maf1 is an essential mediator of diverse signals that repress RNA polymerase III transcription. Mol Cell. 2002;10(6):1489–94. doi: 10.1016/s1097-2765(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 10.Bonhoure N, Byrnes A, Moir RD, Hodroj W, Preitner F, et al. Loss of the RNA polymerase III repressor Maf1 confers obesity resistance. Genes Dev. 2015;29(9):934–47. doi: 10.1101/gad.258350.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moir RD, Willis IM. Regulation of Pol III transcription by nutrient and stress signaling pathways. Biochim Biophys Acta. 2013;1829(3–4):361–75. doi: 10.1016/j.bbagrm.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boguta M. Maf1, a general negative regulator of RNA polymerase III in yeast. Biochim Biophys Acta. 2013;1829(3–4):376–84. doi: 10.1016/j.bbagrm.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Desai N, Lee J, Upadhya R, Chu Y, Moir RD, Willis IM. Two steps in Maf1-dependent repression of transcription by RNA polymerase III. J Biol Chem. 2005;280(8):6455–62. doi: 10.1074/jbc.M412375200. [DOI] [PubMed] [Google Scholar]
- 14.Cabart P, Lee J, Willis IM. Facilitated recycling protects human RNA polymerase III from repression by Maf1 in vitro. J Biol Chem. 2008;283(52):36108–17. doi: 10.1074/jbc.M807538200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vannini A, Ringel R, Kusser AG, Berninghausen O, Kassavetis GA, Cramer P. Molecular basis of RNA polymerase III transcription repression by Maf1. Cell. 2010;143(1):59–70. doi: 10.1016/j.cell.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Dieci G, Sentenac A. Facilitated recycling pathway for RNA polymerase III. Cell. 1996;84(2):245–52. doi: 10.1016/s0092-8674(00)80979-4. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann NA, Jakobi AJ, Moreno-Morcillo M, Glatt S, Kosinski J, et al. Molecular structures of unbound and transcribing RNA polymerase III. Nature. 2015;528(7581):231–36. doi: 10.1038/nature16143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soulard A, Cremonesi A, Moes S, Schütz F, Jenö P, Hall MN. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol Biol Cell. 2010;21(19):3475–86. doi: 10.1091/mbc.E10-03-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong YT, Koh JLY, Friesen H, Duffy SK, Cox MJ, et al. Yeast proteome dynamics from single cell imaging and automated analysis. Cell. 2015;161(6):1413–24. doi: 10.1016/j.cell.2015.04.051. [DOI] [PubMed] [Google Scholar]
- 20.Wei Y, Tsang CK, Zheng XFS. Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 2009;28(15):2220–30. doi: 10.1038/emboj.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurischko C, Kuravi VK, Herbert CJ, Luca FC. Nucleocytoplasmic shuttling of Ssd1 defines the destiny of its bound mRNAs. Mol Microbiol. 2011;81(3):831–49. doi: 10.1111/j.1365-2958.2011.07731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tudisca V, Recouvreux V, Moreno S, Boy-Marcotte E, Jacquet M, Portela P. Differential localization to cytoplasm, nucleus or P-bodies of yeast PKA subunits under different growth conditions. Eur J Cell Biol. 2010;89(4):339–48. doi: 10.1016/j.ejcb.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, et al. mTORC1 directly phosphorylates and regulates human Maf1. Mol Cell Biol. 2010;30(15):3749–57. doi: 10.1128/MCB.00319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, et al. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J Biol Chem. 2010;285(20):15380–92. doi: 10.1074/jbc.M109.071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Natl Acad Sci U S A. 2010;107(26):11823–8. doi: 10.1073/pnas.1005188107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaveroux C, Eichner LJ, Dufour CR, Shatnawi A, Khoutorsky A, et al. Molecular and genetic crosstalks between mTOR and ERRα are key determinants of rapamycin-induced nonalcoholic fatty liver. Cell Metab. 2013;17(4):586–98. doi: 10.1016/j.cmet.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Moir RD, Lee J, Haeusler RA, Desai N, Engelke DR, Willis IM. Protein kinase A regulates RNA polymerase III transcription through the nuclear localization of Maf1. Proc Natl Acad Sci U S A. 2006;103(41):15044–9. doi: 10.1073/pnas.0607129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huber A, Bodenmiller B, Uotila A, Stahl M, Wanka S, et al. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009;23(16):1929–43. doi: 10.1101/gad.532109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Moir RD, McIntosh KB, Willis IM. Tor signaling regulates ribosome and tRNA synthesis via LAMMER/Clk and GSK-3 family kinases. Mol Cell. 2012;45(6):836–43. doi: 10.1016/j.molcel.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landrieux E, Alic N, Ducrot C, Acker J, Riva M, Carles C. A subcomplex of RNA polymerase III subunits involved in transcription termination and reinitiation. EMBO J. 2006;25(1):118–28. doi: 10.1038/sj.emboj.7600915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.C-C Wu, Y-C Lin, H-T Chen. The TFIIF-like Rpc37/53 dimer lies at the center of a protein network to connect TFIIIC, Bdp1, and the RNA polymerase III active center. Mol Cell Biol. 2011;31(13):2715–28. doi: 10.1128/MCB.05151-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kassavetis GA, Prakash P, Shim E. The C53/C37 subcomplex of RNA polymerase III lies near the active site and participates in promoter opening. J Biol Chem. 2010;285(4):2695–706. doi: 10.1074/jbc.M109.074013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arimbasseri AG, Maraia RJ. Mechanism of transcription termination by RNA polymerase III utilizes a non-template strand sequence-specific signal element. Mol Cell. 2015;58(6):1124–32. doi: 10.1016/j.molcel.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-Casalongue ME, Lee J, Diamond A, Shuldiner S, Moir RD, Willis IM. Differential phosphorylation of a regulatory subunit of protein kinase CK2 by target of rapamycin complex 1 signaling and the Cdc-like kinase Kns1. J Biol Chem. 2015;290(11):7221–33. doi: 10.1074/jbc.M114.626523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hellerstedt ST, Nash RS, Weng S, Paskov KM, Wong ED, et al. Curated protein information in the Saccharomyces genome database. Database (Oxford) 2017;2017(1) doi: 10.1093/database/bax011. http://www.yeastgenome.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghavidel A, Schultz MC. Casein kinase II regulation of yeast TFIIIB is mediated by the TATA-binding protein. Genes Dev. 1997;11(21):2780–9. doi: 10.1101/gad.11.21.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghavidel A, Schultz MC. TATA binding protein-associated CK2 transduces DNA damage signals to the RNA polymerase III transcriptional machinery. Cell. 2001;106(5):575–84. doi: 10.1016/s0092-8674(01)00473-1. [DOI] [PubMed] [Google Scholar]
- 38.Johnston IM, Allison SJ, Morton JP, Schramm L, Scott PH, White RJ. CK2 forms a stable complex with TFIIIB and activates RNA polymerase III transcription in human cells. Mol Cell Biol. 2002;22(11):3757–68. doi: 10.1128/MCB.22.11.3757-3768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu P, Wu S, Hernandez N. A minimal RNA polymerase III transcription system from human cells reveals positive and negative regulatory roles for CK2. Mol Cell. 2003;12(3):699–709. doi: 10.1016/j.molcel.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 40.Hu P, Samudre K, Wu S, Sun Y, Hernandez N. CK2 phosphorylation of Bdp1 executes cell cycle-specific RNA polymerase III transcription repression. Mol Cell. 2004;16(1):81–92. doi: 10.1016/j.molcel.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 41.Graczyk D, Debski J, Muszyńska G, Bretner M, Lefebvre O, Boguta M. Casein kinase II-mediated phosphorylation of general repressor Maf1 triggers RNA polymerase III activation. Proc Natl Acad Sci U S A. 2011;108(12):4926–31. doi: 10.1073/pnas.1010010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: from birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66(11–12):1817–29. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moir RD, Lee J, Willis IM. Recovery of RNA polymerase III transcription from the glycerol-repressed state: revisiting the role of protein kinase CK2 in Maf1 phosphoregulation. J Biol Chem. 2012;287(36):30833–41. doi: 10.1074/jbc.M112.378828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H-S, Mukhopadhyay R, Rothbart SB, Silva AC, Vanoosthuyse V, et al. Identification of a BET family bromodomain/casein kinase II/TAF-containing complex as a regulator of mitotic condensin function. Cell Rep. 2014;6(5):892–905. doi: 10.1016/j.celrep.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J, Moir RD, Willis IM. Differential phosphorylation of RNA polymerase III and the initiation factor TFIIIB in Saccharomyces cerevisiae. PLoS ONE. 2015;10(5):e0127225. doi: 10.1371/journal.pone.0127225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panse VG, Hardeland U, Werner T, Kuster B, Hurt E. A proteome-wide approach identifies sumoylated substrate proteins in yeast. J Biol Chem. 2004;279(40):41346–51. doi: 10.1074/jbc.M407950200. [DOI] [PubMed] [Google Scholar]
- 47.Wohlschlegel JA, Johnson ES, Reed SI, Yates JR. Global analysis of protein sumoylation in Saccharomyces cerevisiae. J Biol Chem. 2004;279(44):45662–8. doi: 10.1074/jbc.M409203200. [DOI] [PubMed] [Google Scholar]
- 48.Zhou W, Ryan JJ, Zhou H. Global analyses of sumoylated proteins in Saccharomyces cerevisiae. induction of protein sumoylation by cellular stresses. J Biol Chem. 2004;279(31):32262–8. doi: 10.1074/jbc.M404173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hannich JT, Lewis A, Kroetz MB, Li S-J, Heide H, et al. Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J Biol Chem. 2005;280(6):4102–10. doi: 10.1074/jbc.M413209200. [DOI] [PubMed] [Google Scholar]
- 50.Neyret-Kahn H, Benhamed M, Ye T, Le Gras S, Cossec J-C, et al. Sumoylation at chromatin governs coordinated repression of a transcriptional program essential for cell growth and proliferation. Genome Res. 2013;23(10):1563–79. doi: 10.1101/gr.154872.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chymkowitch P, Nguéa AP, Aanes H, Koehler CJ, Thiede B, et al. Sumoylation of Rap1 mediates the recruitment of TFIID to promote transcription of ribosomal protein genes. Genome Res. 2015;25(6):897–906. doi: 10.1101/gr.185793.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hendriks IA, Lyon D, Young C, Jensen LJ, Vertegaal ACO, Nielsen ML. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat Struct Mol Biol. 2017;24(3):325–36. doi: 10.1038/nsmb.3366. [DOI] [PubMed] [Google Scholar]
- 53.Chymkowitch P, Nguéa PA, Aanes H, Robertson J, Klungland A, Enserink JM. TORC1-dependent sumoylation of Rpc82 promotes RNA polymerase III assembly and activity. Proc Natl Acad Sci U S A. 2017;114(5):1039–44. doi: 10.1073/pnas.1615093114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts DN, Stewart AJ, Huff JT, Cairns BR. The RNA polymerase III transcriptome revealed by genome-wide localization and activity-occupancy relationships. Proc Natl Acad Sci U S A. 2003;100(25):14695–700. doi: 10.1073/pnas.2435566100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohira AD, Chen C-Y, Allen JR, Johnson DL. Covalent small ubiquitin-like modifier (SUMO) modification of Maf1 protein controls RNA polymerase III-dependent transcription repression. J Biol Chem. 2013;288(26):19288–95. doi: 10.1074/jbc.M113.473744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosonina E, Akhter A, Dou Y, Babu J, Sri Theivakadadcham VS. Regulation of transcription factors by sumoylation. Transcription. 2017:e131. doi: 10.1080/21541264.2017.1311829. 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, et al. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci U S A. 2006;103(1):45–50. doi: 10.1073/pnas.0503698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jouffe C, Cretenet G, Symul L, Martin E, Atger F, et al. The circadian clock coordinates ribosome biogenesis. PLoS Biol. 2013;11(1):e1001455. doi: 10.1371/journal.pbio.1001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Atger F, Gobet C, Marquis J, Martin E, Wang J, et al. Circadian and feeding rhythms differentially affect rhythmic mRNA transcription and translation in mouse liver. Proc Natl Acad Sci U S A. 2015;112(47):E6579–88. doi: 10.1073/pnas.1515308112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mange F, Praz V, Migliavacca E, Willis IM, Schütz F, et al. Diurnal regulation of RNA polymerase III transcription is under the control of both the feeding-fasting response and the circadian clock. Genome Res. 2017;27(6):973–84. doi: 10.1101/gr.217521.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sinturel F, Gerber A, Mauvoisin D, Wang J, Gatfield D, et al. Diurnal oscillations in liver mass and cell size accompany ribosome assembly cycles. Cell. 2017;169(4):651–63.e14. doi: 10.1016/j.cell.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schibler U, Gotic I, Saini C, Gos P, Curie T, et al. Clock-talk: interactions between central and peripheral circadian oscillators in mammals. Cold Spring Harb Symp Quant Biol. 2015;80:223–32. doi: 10.1101/sqb.2015.80.027490. [DOI] [PubMed] [Google Scholar]
- 63.Asher G, Sassone-Corsi P. Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell. 2015;161(1):84–92. doi: 10.1016/j.cell.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 64.Atger F, Mauvoisin D, Weger B, Gobet C, Gachon F. Regulation of mammalian physiology by interconnected circadian and feeding rhythms. Front Endocrinol (Lausanne) 2017;8:42. doi: 10.3389/fendo.2017.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang J, Mauvoisin D, Martin E, Atger F, Galindo AN, et al. Nuclear proteomics uncovers diurnal regulatory landscapes in mouse liver. Cell Metab. 2017;25(1):102–17. doi: 10.1016/j.cmet.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005;6(1):69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- 67.White RJ. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008;24(12):622–9. doi: 10.1016/j.tig.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 68.Gjidoda A, Henry RW. RNA polymerase III repression by the retinoblastoma tumor suppressor protein. Biochim Biophys Acta. 2013;1829(3–4):385–92. doi: 10.1016/j.bbagrm.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grewal SS. Why should cancer biologists care about tRNAs? tRNA synthesis, mRNA translation and the control of growth. Biochim Biophys Acta. 2015;1849(7):898–907. doi: 10.1016/j.bbagrm.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Johnson SAS, Dubeau L, Johnson DL. Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation. J Biol Chem. 2008;283(28):19184–91. doi: 10.1074/jbc.M802872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palian BM, Rohira AD, Johnson SAS, He L, Zheng N, et al. Maf1 is a novel target of pPTEN and PI3K signaling that negatively regulates oncogenesis and lipid metabolism. PLoS Genet. 2014;10(12):e1004789. doi: 10.1371/journal.pgen.1004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson SS, Zhang C, Fromm J, Willis IM, Johnson DL. Mammalian Maf1 is a negative regulator of transcription by all three nuclear RNA polymerases. Mol Cell. 2007;26(3):367–79. doi: 10.1016/j.molcel.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 73.Li Y, Tsang CK, Wang S, Li X-X, Yang Y, et al. Maf1 suppresses AKT-mTOR signaling and liver cancer through activation of PTEN transcription. Hepatology. 2016;63(6):1928–42. doi: 10.1002/hep.28507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson DL, Stiles BL. Maf1, a new PTEN target linking RNA and lipid metabolism. Trends Endocrinol Metab. 2016;27(10):742–50. doi: 10.1016/j.tem.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37(21):7268–80. doi: 10.1093/nar/gkp787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou Y, Goodenbour JM, Godley LA, Wickrema A, Pan T. High levels of tRNA abundance and alteration of tRNA charging by bortezomib in multiple myeloma. Biochem Biophys Res Commun. 2009;385(2):160–4. doi: 10.1016/j.bbrc.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2(12):e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014;158(6):1281–92. doi: 10.1016/j.cell.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 79.Subramaniam AR, Pan T, Cluzel P. Environmental perturbations lift the degeneracy of the genetic code to regulate protein levels in bacteria. Proc Natl Acad Sci U S A. 2013;110(6):2419–24. doi: 10.1073/pnas.1211077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quax TEF, Claassens NJ, Söll D, van der Oost J. Codon bias as a means to fine-tune gene expression. Mol Cell. 2015;59(2):149–61. doi: 10.1016/j.molcel.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Novoa EM, Ribas de Pouplana L. Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. 2012;28(11):574–81. doi: 10.1016/j.tig.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 82.Kirchner S, Ignatova Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet. 2015;16(2):98–112. doi: 10.1038/nrg3861. [DOI] [PubMed] [Google Scholar]
- 83.Rudolph KLM, Schmitt BM, Villar D, White RJ, Marioni JC, et al. Codon-driven translational efficiency is stable across diverse mammalian cell states. PLoS Genet. 2016;12(5):e1006024. doi: 10.1371/journal.pgen.1006024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016;165(6):1416–27. doi: 10.1016/j.cell.2016.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pavon-Eternod M, Gomes S, Rosner MR, Pan T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA. 2013;19(4):461–6. doi: 10.1261/rna.037507.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rideout EJ, Marshall L, Grewal SS. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci U S A. 2012;109(4):1139–44. doi: 10.1073/pnas.1113311109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Clarke CJ, Berg TJ, Birch J, Ennis D, Mitchell L, et al. The initiator methionine tRNA drives secretion of type II collagen from stromal fibroblasts to promote tumor growth and angiogenesis. Curr Biol. 2016;26(6):755–65. doi: 10.1016/j.cub.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Birch J, Clarke CJ, Campbell AD, Campbell K, Mitchell L, et al. The initiator methionine tRNA drives cell migration and invasion leading to increased metastatic potential in melanoma. Biology open. 2016;5(10):1371–9. doi: 10.1242/bio.019075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang H-Y, Hopper AK. Multiple layers of stress-induced regulation in tRNA biology. Life(Basel, Switzerland) 2016;6(2) doi: 10.3390/life6020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357(6348):eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Canella D, Praz V, Reina JH, Cousin P, Hernandez N. Defining the RNA polymerase III transcriptome: genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 2010;20(6):710–21. doi: 10.1101/gr.101337.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moqtaderi Z, Wang J, Raha D, White RJ, Snyder M, et al. Genomic binding profiles of functionally distinct RNA polymerase III transcription complexes in human cells. Nat Struct Mol Biol. 2010;17(5):635–40. doi: 10.1038/nsmb.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barski A, Chepelev I, Liko D, Cuddapah S, Fleming AB, et al. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat Struct Mol Biol. 2010;17(5):629–34. doi: 10.1038/nsmb.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, et al. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol. 2010;17(5):620–28. doi: 10.1038/nsmb.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Orioli A, Praz V, Lhôte P, Hernandez N. Human Maf1 targets and represses active RNA polymerase III genes by preventing recruitment rather than inducing long-term transcriptional arrest. Genome Res. 2016;26(5):624–35. doi: 10.1101/gr.201400.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alla RK, Cairns BR. RNA polymerase III transcriptomes in human embryonic stem cells and induced pluripotent stem cells, and relationships with pluripotency transcription factors. PLoS ONE. 2014;9(1):e85648. doi: 10.1371/journal.pone.0085648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmitt BM, Rudolph KLM, Karagianni P, Fonseca NA, White RJ, et al. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA-tRNA interface. Genome Res. 2014;24(11):1797–807. doi: 10.1101/gr.176784.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Turowski TW, Leśniewska E, Delan-Forino C, Sayou C, Boguta M, Tollervey D. Global analysis of transcriptionally engaged yeast RNA polymerase III reveals extended tRNA transcripts. Genome Res. 2016;26(7):933–44. doi: 10.1101/gr.205492.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hopper AK. Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics. 2013;194(1):43–67. doi: 10.1534/genetics.112.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Megel C, Morelle G, Lalande S, Duchêne A-M, Small I, Maréchal-Drouard L. Surveillance and cleavage of eukaryotic tRNAs. Int J Mol Sci. 2015;16(1):1873–93. doi: 10.3390/ijms16011873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gu C, Begley TJ, Dedon PC. tRNA modifications regulate translation during cellular stress. FEBS Lett. 2014;588(23):4287–96. doi: 10.1016/j.febslet.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Czech A, Wende S, Mörl M, Pan T, Ignatova Z. Reversible and rapid transfer-RNA deactivation as a mechanism of translational repression in stress. PLoS Genet. 2013;9(8):e1003767. doi: 10.1371/journal.pgen.1003767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kumar P, Kuscu C, Dutta A. Biogenesis and function of transfer RNA-related fragments (tRFs) Trends Biochem Sci. 2016;41(8):679–89. doi: 10.1016/j.tibs.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gudipati RK, Xu Z, Lebreton A, Séraphin B, Steinmetz LM, et al. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell. 2012;48(3):409–21. doi: 10.1016/j.molcel.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D. Transcriptome-wide analysis of exosome targets. Mol Cell. 2012;48(3):422–33. doi: 10.1016/j.molcel.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Porrua O, Libri D. RNA quality control in the nucleus: the angels’ share of RNA. Biochim Biophys Acta. 2013;1829(6–7):604–11. doi: 10.1016/j.bbagrm.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 107.Maraia RJ, Lamichhane TN. 3′ processing of eukaryotic precursor tRNAs. Wiley Interdiscip Rev RNA. 2011;2(3):362–75. doi: 10.1002/wrna.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Skowronek E, Grzechnik P, Späth B, Marchfelder A, Kufel J. tRNA 3′ processing in yeast involves tRNAse Z, Rex1, and Rrp6. RNA. 2014;20(1):115–30. doi: 10.1261/rna.041467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Foretek D, Wu J, Hopper AK, Boguta M. Control of Saccharomyces cerevisiae pre-tRNA processing by environmental conditions. RNA. 2016;22(3):339–49. doi: 10.1261/rna.054973.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Foretek D, Nuc P, Żywicki M, Karlowski WM, Kudla G, Boguta M. Maf1-mediated regulation of yeast RNA polymerase III is correlated with CCA addition at the 3′ end of tRNA precursors. Gene. 2017;612:12–8. doi: 10.1016/j.gene.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kramer EB, Hopper AK. Retrograde transfer RNA nuclear import provides a new level of tRNA quality control in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2013;110(52):21042–7. doi: 10.1073/pnas.1316579110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Agris PF. Decoding the genome: a modified view. Nucleic Acids Res. 2004;32(1):223–38. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Endres L, Dedon PC, Begley TJ. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015;12(6):603–14. doi: 10.1080/15476286.2015.1031947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schaefer M, Kapoor U, Jantsch MF. Understanding RNA modifications: the promises and technological bottlenecks of the “epitranscriptome”. Open Biol. 2017;7(5) doi: 10.1098/rsob.170077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arimbasseri AG, Blewett NH, Iben JR, Lamichhane TN, Cherkasova V, et al. RNA polymerase III output is functionally linked to tRNA dimethyl-G26 modification. PLoS Genet. 2015;11(12):e1005671. doi: 10.1371/journal.pgen.1005671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, et al. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell. 2006;21(1):87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 117.Duechler M, Leszczyńska G, Sochacka E, Nawrot B. Nucleoside modifications in the regulation of gene expression: focus on tRNA. Cell Mol Life Sci. 2016;73(16):3075–95. doi: 10.1007/s00018-016-2217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dong C, Niu L, Song W, Xiong X, Zhang X, et al. tRNA modification profiles of the fast-proliferating cancer cells. Biochem Biophys Res Commun. 2016;476(4):340–5. doi: 10.1016/j.bbrc.2016.05.124. [DOI] [PubMed] [Google Scholar]
- 119.Chan CTY, Pang YLJ, Deng W, Babu IR, Dyavaiah M, et al. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu F, Clark W, Luo G, Wang X, Fu Y, et al. ALKB1-mediated tRNA demethylation regulates translation. Cell. 2016;167(3):816–28.e16. doi: 10.1016/j.cell.2016.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yamasaki S, Ivanov P, Hu G-F, Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185(1):35–42. doi: 10.1083/jcb.200811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fu H, Feng J, Liu Q, Sun F, Tie Y, et al. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009;583(2):437–42. doi: 10.1016/j.febslet.2008.12.043. [DOI] [PubMed] [Google Scholar]
- 123.Saikia M, Krokowski D, Guan B-J, Ivanov P, Parisien M, et al. Genome-wide identification and quantitative analysis of cleaved tRNA fragments induced by cellular stress. J Biol Chem. 2012;287(51):42708–25. doi: 10.1074/jbc.M112.371799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mesitov MV, Soldatov RA, Zaichenko DM, Malakho SG, Klementyeva TS, et al. Differential processing of small RNAs during endoplasmic reticulum stress. Sci Rep. 2017;7:46080. doi: 10.1038/srep46080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thompson DM, Parker R. The RNAse Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J Cell Biol. 2009;185(1):43–50. doi: 10.1083/jcb.200811119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lyons SM, Fay MM, Akiyama Y, Anderson PJ, Ivanov P. RNA biology of angiogenin: current state and perspectives. RNA Biol. 2017;14(2):171–8. doi: 10.1080/15476286.2016.1272746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Thompson DM, Parker R. Stressing out over tRNA cleavage. Cell. 2009;138(2):215–219. doi: 10.1016/j.cell.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 128.Luhtala N, Parker R. Structure-function analysis of Rny1 in tRNA cleavage and growth inhibition. PLoS ONE. 2012;7(7):e41111. doi: 10.1371/journal.pone.0041111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huang H, Kawamata T, Horie T, Tsugawa H, Nakayama Y, et al. Bulk RNA degradation by nitrogen starvation-induced autophagy in yeast. EMBO J. 2015;34(2):154–68. doi: 10.15252/embj.201489083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dhahbi JM, Spindler SR, Atamna H, Yamakawa A, Boffelli D, et al. 5′ tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC Genomics. 2013;14:298. doi: 10.1186/1471-2164-14-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yeri A, Courtright A, Reiman R, Carlson E, Beecroft T, et al. Total extracellular small RNA profiles from plasma, saliva, and urine of healthy subjects. Sci Rep. 2017;7:44061. doi: 10.1038/srep44061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351(6271):391–96. doi: 10.1126/science.aad6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peng H, Shi J, Zhang Y, Zhang H, Liao S, et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res. 2012;22(11):1609–12. doi: 10.1038/cr.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Keam SP, Hutvagner G. tRNA-derived fragments (tRFs): emerging new roles for an ancient RNA in the regulation of gene expression. Life (Basel, Switzerland) 2015;5(4):1638–51. doi: 10.3390/life5041638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gebetsberger J, Polacek N. Slicing tRNAs to boost functional ncRNA diversity. RNA Biol. 2013;10(12):1798–806. doi: 10.4161/rna.27177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhou J, Liu S, Chen Y, Fu Y, Silver AJ, et al. Identification of two novel functional tRNA-derived fragments induced in response to respiratory syncytial virus infection. J Gen Virol. 2017;98(7):1600–10. doi: 10.1099/jgv.0.000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang Q, Lee I, Ren J, Ajay SS, Lee YS, Bao X. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol Ther. 2013;21(2):368–79. doi: 10.1038/mt.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Honda S, Kirino Y. SHOT-RNAs: a novel class of tRNA-derived functional RNAs expressed in hormone-dependent cancers. Molecular & Cellular Oncology. 2016;3(2):e1079672. doi: 10.1080/23723556.2015.1079672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Goodarzi H, Liu X, Nguyen HCB, Zhang S, Fish L, Tavazoie SF. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161(4):790–802. doi: 10.1016/j.cell.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen Q, Yan M, Cao Z, Li X, Zhang Y, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016;351(6271):397–400. doi: 10.1126/science.aad7977. [DOI] [PubMed] [Google Scholar]
- 141.Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24(11):437–40. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 142.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11(12):4241–57. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353(6306):aaf1420. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Oie S, Matsuzaki K, Yokoyama W, Tokunaga S, Waku T, et al. Hepatic rRNA transcription regulates high-fat-diet-induced obesity. Cell Rep. 2014;7(3):807–20. doi: 10.1016/j.celrep.2014.03.038. [DOI] [PubMed] [Google Scholar]
- 145.Bonhoure N, Bounova G, Bernasconi D, Praz V, Lammers F, et al. Quantifying ChIP-seq data: a spiking method providing an internal reference for sample-to-sample normalization. Genome Res. 2014;24(7):1157–68. doi: 10.1101/gr.168260.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lane AN, Fan TW-M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43(4):2466–85. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Geiduschek EP, Kassavetis GA. The RNA polymerase III transcription apparatus. J Mol Biol. 2001;310(1):1–26. doi: 10.1006/jmbi.2001.4732. [DOI] [PubMed] [Google Scholar]
- 149.Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002;16(20):2593–2620. doi: 10.1101/gad.1018902. [DOI] [PubMed] [Google Scholar]
- 150.Dumay-Odelot H, Durrieu-Gaillard S, El Ayoubi L, Parrot C, Teichmann M. Contributions of in vitro transcription to the understanding of human RNA polymerase III transcription. Transcription. 2014;5(1):e27526. doi: 10.4161/trns.27526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Khatter H, Vorländer MK, Müller CW. RNA polymerase I and III: similar yet unique. Curr Opin Struct Biol. 2017;47:88–94. doi: 10.1016/j.sbi.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 152.Khoo S-K, Wu C-C, Lin Y-C, Lee J-C, Chen HT. Mapping the protein interaction network for TFIIIB-related factor Brf1 in the RNA polymerase III preinitiation complex. Mol Cell Biol. 2014;34(3):551–559. doi: 10.1128/MCB.00910-13. [DOI] [PMC free article] [PubMed] [Google Scholar]